

Abstract
Background
Exercise intolerance is a cardinal symptom in right (RV) and left ventricular (LV) failure. The underlying skeletal muscle contributes to increased morbidity in patients. Here, we compared skeletal muscle sarcopenia in a novel two‐stage model of RV failure to an established model of LV failure.
Methods
Pulmonary artery banding (PAB) or aortic banding (AOB) was performed in weanling rats, inducing a transition from compensated cardiac hypertrophy (after 7 weeks) to heart failure (after 22–26 weeks). Cardiac function was characterized by echocardiography. Skeletal muscle catabolic/anabolic balance and energy metabolism were analysed by histological and biochemical methods, real‐time PCR, and western blot.
Results
Two clearly distinguishable stages of left or right heart disease with a comparable severity were reached. However, skeletal muscle impairment was significantly more pronounced in LV failure. While the compensatory stage resulted only in minor changes, soleus and gastrocnemius muscle of AOB rats at the decompensated stage demonstrated reduced weight and fibre diameter, higher proteasome activity and expression of the muscle‐specific ubiquitin E3 ligases muscle‐specific RING finger 1 and atrogin‐1, increased expression of the atrophy marker myostatin, increased autophagy activation, and impaired mitochondrial function and respiratory chain gene expression. Soleus and gastrocnemius muscle of PAB rats did not show significant changes in muscle weight and proteasome or autophagy activation, but mitochondrial function was mildly impaired as well. The diaphragm did not demonstrate differences in any model or disease stage except for myostatin expression, which was altered at the decompensated stage in both models. Plasma interleukin (IL)‐6 and angiotensin II were strongly increased at the decompensated stage (AOB > > PAB). Soleus and gastrocnemius muscle itself demonstrated an increase in IL‐6 expression independent from blood‐derived cytokines only in AOB animals. In vitro experiments in rat skeletal muscle cells suggested a direct impact of IL‐6 and angiotensin II on distinctive atrophic changes.
Conclusions
Manifold skeletal muscle alterations are more pronounced in LV failure compared with RV failure despite a similar ventricular impairment. Most of the catabolic changes were observed in soleus or gastrocnemius muscle rather than in the constantly active diaphragm. Mitochondrial dysfunction and up‐regulation of myostatin were identified as the earliest signs of skeletal muscle impairment.
Keywords: Left heart failure, Right heart failure, Mitochondria, Proteasome, Muscle wasting
Introduction
Muscle wasting occurs in the elderly (primary sarcopenia) or during the course of various chronic diseases (secondary sarcopenia). These include cancer, renal failure, chronic obstructive pulmonary disease (COPD), sepsis, trauma, or heart failure (HF). Among the main mechanisms involved in muscle wasting are alterations in mitochondrial function and biogenesis, increased oxidative stress, and disturbances in protein balance by anabolic and catabolic processes. The loss of muscle mass results from excessive protein breakdown by the ubiquitin–proteasome system (UPS) and autophagy or via forkhead box O (FOXO)‐mediated proteolysis. 1 , 2 , 3 Two muscle‐specific ubiquitin ligases, muscle‐specific RING finger 1 (MuRF1) and atrogin‐1, are induced during the atrophy process and catalyze the ubiquitination and degradation of the myofibrillar apparatus or of proteins involved in protein synthesis, respectively. 4 , 5 In addition to the activation of catabolic factors, a down‐regulation in anabolic signalling pathways can also lead to a net loss of muscle mass. One of the most important anabolic factors in muscle is the insulin‐like growth factor 1 (IGF‐1). Accordingly, a reduced protein synthesis due to a reduced IGF‐1 signalling or reduced IGF‐1 expression occurs in HF patients. 6 , 7 Elevated pro‐inflammatory cytokines such as tumour necrosis factor‐α (TNF‐α), interleukin (IL)‐6 and IL‐1β, or members of the transforming growth factor (TGF)‐β family such as myostatin and activin A also contribute to the catabolic metabolism, finally resulting in muscle wasting and cardiac cachexia in HF. 8 , 9 Myostatin is predominantly synthesized and expressed in skeletal muscle and negatively regulates muscle mass by modulating muscle growth, differentiation, and renewal. 10 , 11 TGF‐β family members stimulate the activation of the transcription factors SMAD2 and SMAD3, which contribute together with FOXO1, FOXO3, or NFKB to the increased expression of genes that influence muscular atrophy (atrogenes) such as MuRF1, atrogin‐1, or autophagy‐related genes.
The prevalence of a loss of muscle mass and muscle function is higher in patients suffering from HF (~20% in elderly patients) compared with healthy elderly people. 12 Even in less advanced stages of HF, skeletal muscle abnormalities such as skeletal muscle atrophy, altered fibre composition, and metabolic disturbances occur. 13 , 14 However, these alterations are significantly exacerbated in HF with reduced ejection fraction compared with HF with preserved ejection fraction. 15 In addition to these data derived from HF patients with left ventricular (LV) dysfunction, diseases resulting in a right ventricular (RV) dysfunction such as COPD or pulmonary artery hypertension are also associated with exercise intolerance. Originally mainly attributed to respiratory dysfunction and low RV cardiac output (CO), numerous studies demonstrated abnormalities in skeletal muscle also in these patients. COPD affects both respiratory and limb muscles, although the latter seem to be more severely affected. 16 The fact that respiratory muscles in COPD are chronically exposed to higher workloads and must remain active was suggested to induce a training‐like positive adaptation and render them more fatigue resistant. 16 Quadriceps weakness occurs in one‐third of COPD patients even at early disease stages, and quadriceps strength is a useful predictor of mortality in patients with COPD. 17 , 18 An up‐regulation of the atrogin‐1 and MuRF1, UPS activation, diminished protein kinase B signalling in skeletal muscle, and elevated levels of pro‐inflammatory mediators such as TNF‐α, IL‐6, or IL‐1β have been described in patients as well as animals with pulmonary artery hypertension. 19 In summary, RV and LV dysfunction lead to progressive changes in skeletal muscle, putatively involving similar mechanisms. Therefore, the aim of the present study was to directly compare the effects of cardiac dysfunction on skeletal muscles in a novel two‐stage model of RV failure after pulmonary artery banding (PAB) to an established two‐stage model of LV failure after AOB. In addition, we intended to investigate potential differences in atrophic processes between limb muscles and respiratory muscles with the latter being potentially more resistant to sarcopenia due to training‐like effects. A priori, we hypothesized that there are common but also distinct systemic and local markers of catabolic/anabolic balance, mitochondrial function, and inflammation in these two different models of HF. In addition, we expected a differential response of the constantly active diaphragm and occasionally active soleus muscle in these models.
Methods
Ascending aortic banding and pulmonary artery banding model
Ascending aortic banding (AOB) and PAB was performed in 6‐week‐old male hybrid rats (Wistar Kyoto × Lewis rats) as described before. 20 In brief, an anterolateral thoracotomy was performed under isoflurane anaesthesia in fully ventilated rats, and a Titanium Clip® (Teleflex Medical GmbH, Fellbach, Germany) with an inner diameter of 0.6 mm was placed around the ascending aorta, or a clip with an inner diameter of 1.1 mm was placed around the pulmonary artery, respectively. Sham‐operated animals served as age‐matched controls. At the time of surgery, the clip was not constricting. However, as the animals grow, the clip became constricting, resulting in a stage of compensatory LV (AOB model) or RV hypertrophy (PAB model) 7 weeks after surgery. A stage of beginning LV failure was reached 24–26 weeks after AOB and a stage of beginning RV failure 20–22 weeks after PAB. Two‐dimensional and M‐mode echocardiographic examinations were performed in accordance with the criteria of the American Society of Echocardiography using a Vevo2100 system (FUJIFILM VisualSonics Inc., Toronto, Canada), evaluating both cardiac geometry and function. The animals were also inspected for clinical markers of HF, including tachypnoea, pleural effusion, enlarged liver, and ascites. All animal experiments conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and were performed according to the regional authorities and ethics committees for animal research. The experiments were registered under Number G14‐2017.
Cell culture model
Adult rat skeletal muscle cells (RSkMCs) were obtained from Sigma‐Aldrich and cultured in RSkMC growth medium (Sigma‐Aldrich Chemie GmbH, Schnelldorf, Germany). In order to induce differentiation of RSkMC myoblasts into RSkMC myotubes, cells were grown on collagen‐coated cell culture dishes until they reached confluency and afterwards cultured in RSkMC differentiation growth medium (Sigma) for another 7 days. Stimulation of all cells was performed in Opti‐MEM (Thermo Fisher Scientific GmbH, Dreieich, Germany) utilizing IL‐6 (tebu‐bio), angiotensin II (Ang II) (tebu‐bio GmbH, Offenbach, Germany), the angiotensin II receptor type 1 (AT1) receptor antagonist losartan (Tocris Bioscience, Bristol, UK), or the STAT3 inhibitor Stattic (tebu‐bio) at the concentrations indicated in the according figures.
Enzyme‐linked immunosorbent assay
Plasma brain natriuretic peptide (BNP), IL‐6, Ang II, TNF‐α, and IL‐1β concentrations were measured using a commercial enzyme‐linked immunosorbent assay (Rat BNP ELISA Kit, RayBiotech, Norcross, GA, USA; Rat IL‐6, BioVendor GmbH, Kassel, Germany; Angiotensin II Human/Rat, BioVendor; Rat TNF‐α, BioVendor; and Rat IL‐1β, BioVendor) according to the manufacturer's instructions.
Measurement of apoptosis
Skeletal muscle tissue was lyzed in a buffer containing 50 mM HEPES, pH 7.5, 0.1% CHAPS, 2 mM dithiothreitol, 0.1% Nonidet P‐40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 2 μg/mL leupeptin, and 2 μg/mL pepstatin A at 4°C. After sonication and centrifugation, protein concentration was measured with the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of tissue homogenate protein (10 μg) were transferred to a white 96‐well microplate, and caspase 3 activity was detected with the luminometric Caspase‐Glo® 3/7 Assay (both Promega GmbH, Mannheim Germany) on a GloMax™ 96 Plate luminometer.
Determination of muscle fibre type and cross section
Eight micrometre cryosections were prepared from frozen tissue. Sections were incubated in pre‐incubation solution (pH 10.4) containing 90 mM CaCl2, 100 mM glycine, and 3.7% formaldehyde for 5 min. After rinsing the sections, they were incubated in a solution (pH 9.4) containing 40 mM glycine, 20 mM CaCl2, and 2.5 mM ATP at 37°C for 90 min. Thereafter, sections were washed and rinsed in a 2% CoCl2 solution for 3 min. After staining in a 1% Azure A solution for 30 s, sections were rinsed continuously under tap water for 5 min, dehydrated, and mounted. This staining method allows a discrimination between three fibre types, one slow‐twitch fibre, type I (white fibres), and two fast‐twitch fibre types, types IIA (light blue) and IIB (dark blue). Two regions of soleus muscle, diaphragm, and gastrocnemius muscle (deep, red portion) were photographed at ×20 magnification without visual field overlap. All muscle fibres delineated by entire fibre boundaries were counted and measured for cross‐sectional area. A random sample of 150 fibres from each muscle was analysed, and cross‐sectional area was calculated with the software ImageJ (National Institutes of Health, Bethesda, MD, USA). All area measurements were performed by two operators who were blinded to group assignment.
Respirometric measurements
Immediately before oxygraphic measurements, skeletal muscle fibres were permeabilized for 30 min with saponin. After permeabilization, the fibres were washed to remove saponin and adenine nucleotides. We used the high‐resolution OROBOROS® oxygraph, a two‐chamber respirometer with a Peltier thermostat, and integrated electromagnetic stirrers. Bundles of fibres (5–10 mg) were transferred into the oxygraph chambers. The measurements were performed at 30°C in 1.42 mL incubation medium consisting of 75 mM mannitol, 25 mM sucrose, 100 mM KCl, 10 mM KH2PO4, 0.5 mM EDTA, 5 mM MgCl2, 20 mM Tris–HCl, and 1 mg/mL BSA (pH 7.4) using different substrates: 10 mM pyruvate + 2 mM malate and 10 mM succinate + 5 μM rotenone. The weight‐specific oxygen consumption was calculated as the time derivative of the oxygen concentration (DATGRAPH Analysis software, OROBOROS®). The rate of state 3 respiration was determined following the addition of 5 mM ADP. State 4 respiration was measured after the addition of 1.8 mM atractyloside. The respiratory control index was calculated as ratio between state 3 to state 4 respiratory rates for each addition of ADP. For the analysis of uncoupled respiration, 2,4‐dinitrophenol was added in a two‐step titration up to 60 μM. The difference between atractylate and antimycin A respiration (45 μM antimycin A) indicates the leak respiration. 21
Mitochondrial enzyme activity
For the determination of mitochondrial enzyme activity, small pieces of frozen tissue were homogenized (1/30 w/v) in a solution containing 50 mM of Tris buffer (pH 7.5), 100 mM potassium chloride, 5 mM MgCl2, and 1 mM EDTA using a glass/glass homogenizer (Kontes Glass Co., Vineland, NJ, USA, 2 mL, 0.025 mm clearance). Enzyme activities were assayed at 30°C spectrophotometrically using a Cary 50 spectrophotometer (Varian Inc., Palo Alto, CA, USA) and related to total protein content in the same sample. Assays were run in duplicates with two different quantities of sample. The analysis of the mitochondrial respiratory chain complexes nicotinamide adenine dinucleotide:coenzyme Q1 oxidoreductase (complex I), nicotinamide adenine dinucleotide:cytochrome c oxidoreductase (complex I + III), succinate:cytochrome c oxidoreductase (complex II + III), ubiquinone:cytochrome c oxidoreductase (complex III), cytochrome c oxidase (complex IV), and citrate synthase as a mitochondrial marker enzyme was performed as described previously. 22
RNA isolation, reverse transcription PCR, and real‐time PCR
Total RNA was isolated from soleus muscle, diaphragm, gastrocnemius muscle (deep, red portion), and RSkMCs with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Prior to cDNA synthesis, integrity and quality of the RNA was confirmed by gel electrophoresis, and the concentration was determined by measuring UV absorption. Reverse transcription of RNA samples (500 ng total RNA) was carried out for 30 min at 42°C using the SuperScript™ III First‐Strand cDNA Synthesis Kit (Thermo Fisher Scientific GmbH, Dreieich, Germany). Reverse transcription PCR was performed for rat MuRF1, atrogin‐1, myostatin, peroxisome proliferator‐activated receptor‐γ coactivator 1α (PGC‐1α), IGF‐1, IL‐1β, IL‐6, TNF‐α, and 18S rRNA. Amplification products were subjected to electrophoresis through 1.5% agarose gels, stained with GelRed (VWR International, Darmstadt, Germany), and visualized with the Fusion FX7 imaging system (Peqlab Biotechnologie GmbH, Erlangen, Germany). PCR products were excised from the gel, purified, and directly sequenced. Real‐time PCR (qPCR) and data analysis were performed using the Mx3000P Multiplex Quantitative PCR System (Stratagene, San Diego, CA, USA) as described previously. 23 Each assay was performed in triplicate, and validation of the PCR runs was assessed by evaluation of the melting curve of the PCR products (primer sequences in Table 1). Threshold cycles (CT) of target genes were normalized to the housekeeping gene 18S rRNA (18S rRNA Control kit, Yakima Yellow®–Eclipse® Dark Quencher, Eurogentec Deutschland GmbH, Köln, Germany). The resulting ΔCT values were compared with sham animals (7 weeks after surgery), and relative mRNA expression was calculated by R = 2−ΔΔCT.
Table 1.
Primer sequences
| GenBank accession # | Forward primer | Reverse primer | |
|---|---|---|---|
| MuRF1 | NM_080903.1 | CTCCTTGTGCAAGGTGTTCG | AGTCTGAACTCGGTCGTTCC |
| Atrogin‐1 | NM_133521.1 | AGCTTGTGCGATGTTACCCA | GGTGAAAGTGAGACGGAGCA |
| Myostatin | NM_019151.1 | TATCACGCTACCACGGAAACAA | ATCCACAGCTGGGCCTTTAC |
| PGC‐1α | NM_031347.1 | CCGAGAATTCATGGAGCAAT | GTGTGAGGAGGGTCATCGTT |
| IGF‐1 | NM_001082477.2 | GGACGCTCTTCAGTTCGTGT | GATCACAGCTCCGGAAGCAA |
| IL‐1β | NM_031512.2 | CTATGTCTTGCCCGTGGAGC | CGTCATCATCCCACGAGTCA |
| IL‐6 | NM_012589.2 | CACAAGTCCGGAGAGGAGAC | TCTGACAGTGCATCATCGCT |
| TNF‐α | NM_012675.3 | TGATCCGAGATGTGGAACTGG | CGATCACCCCGAAGTTCAGTA |
| AT1 | NM_030985.4 | CCCACTCAAGCCTGTCTACG | CTGGCTTCTGTCAGGGCATTA |
| AT2 | NM_012494.3 | TTTGCCATCCTCCTGGGATT | GCCTTGGAGCCAAGTAATGG |
| 18S rRNA | NR_046237 | GTTGGTGGAGCGATTTGTCTG | GCTCAATCTCGGTGGCTGA |
AT1, angiotensin II receptor type 1; AT2, angiotensin II receptor type 2; IGF‐1, insulin‐like growth factor 1; IL, interleukin; MuRF1, muscle‐specific RING finger 1; PGC‐1α, peroxisome proliferator‐activated receptor‐γ coactivator 1α; TNF‐α, tumour necrosis factor‐α.
Western blotting
Tissue or RSkMCs were homogenized in a buffer containing 50 mM Tris HCl, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% sodium deoxycholate, and protease and phosphatase inhibitor cocktails (Sigma); 25 μg of protein was loaded on a SDS‐PAGE gel and transferred to a nitrocellulose membrane. Following blocking, filters were incubated with antibodies directed against MuRF1 (ECM Biosciences, Köln, Germany), atrogin‐1 (ECM Biosciences), myostatin (Santa Cruz Biotechnology, Heidelberg, Germany), ubiquitin (Abcam, Cambridge, UK), LC3A/B (Cell Signaling), NDUFS1 (Thermo Fisher Scientific), ND1 (Abcam), cytochrome oxidase (COX) I (Thermo Fisher Scientific), COX IV (Cell Signaling Technology, Danvers, MA, USA), total OXPHOS (Abcam), pSTAT3 (Cell Signaling), and GAPDH (Abcam). After incubation with peroxidase‐conjugated secondary antibody, blots were subjected to the enhanced chemiluminescent detection method with the Fusion FX7 imaging system (Peqlab).
Adipokine array
The expression profile of 19 different cytokines in soleus muscle was analysed using a cytokine expression array (Rat Cytokine Antibody Array C1, RayBiotech). For this purpose, blood was cleared from skeletal muscle circulation via cannulation of the femoral artery and infusion of Krebs–Henseleit buffer. Frozen tissue was homogenized in lysis buffer according to the manufacturer's instructions, and 500 μg protein was utilized per array. Blocking, hybridization of the array filters, washing conditions, and chemiluminescent detection with the Fusion FX7 imaging system (Peqlab) were performed according to the manufacturer's instructions. Measurements of the intensity of signals on the array membrane were performed using Quantity One® software (Bio‐Rad Laboratories GmbH, Neuberg, Germany). Once the raw numerical densitometry data were extracted, the background was subtracted, the data were normalized to the positive control signals, and the relative cytokine expression was determined.
Proteasome activity
The peptidase activities of the proteasome in the cytosolic fraction of muscle tissue were determined as recently described. 24 Chymotrypsin‐like and trypsin‐like hydrolyzing activities were assayed using the peptides Suc‐LLVY‐aminoluciferin and Z‐LRR‐aminoluciferin and the Proteasome‐Glo Assay System (Promega). In brief, 10 μg of cytosolic proteins was incubated with reaction buffer and the respective labelled peptide in white 96‐well microplate for 60 min. Dual measurements with or without the addition of 30 μM of the irreversible proteasomal inhibitor adamantane‐acetyl‐(6‐aminohexanoyl)3‐(leucinyl)3‐vinyl‐(methyl)‐sulfone (AdaAhx3L3VS, Santa Cruz Biotechnology) were carried out. The resulting luminescence was measured on a GloMax™ 96 Plate Luminometer. Only the proportion of the reaction that could be inhibited by AdaAhx3L3VS was regarded as proteasomal activity.
Statistical analysis
All data are presented as mean ± standard error of the mean. Statistical analyses were performed with SigmaStat 3.5 software (Systat Software, GmbH, Erkrath, Germany), using analysis of variance with post hoc analysis. P values of <0.05 were considered statistically significant.
Results
Rat model of right ventricular or left ventricular hypertrophy or failure
Here, we established a novel model of slowly developing RV hypertrophy and transition to RV failure and compared this with our well‐established, two‐stage model of LV hypertrophy. 20 In the initial cohort, we also compared sham animals 22 weeks after surgery with sham animals 26 weeks after surgery (data now shown). Because no significant difference in any of the parameters was observed, they were thereafter considered as one group (named sham 22–26 weeks). Seven weeks after surgery, an RV (PAB model) or LV (AOB model) hypertrophy had developed, which resulted in a significant increase in RV weight but not LV weight in the PAB model and an exclusive increase in LV weight in the AOB model (Figure 1A). Accordingly, echocardiography showed an increased RV wall thickness in the PAB model and an increased LV wall thickness in the AOB model (Table 2). Interestingly, the interventricular septum showed only in the AOB model a mild hypertrophy (Table 2). At that time point, no functional impairment or increase in diameter of any of the ventricles was observed (Figure 1 and Table 2). The LV of AOB animals even demonstrated a mild increase in LV stroke volume (SV) and CO at this stage (Table 2). However, 22 weeks after PAB, parameters of RV function (tricuspid annular plane systolic excursion and RV fractional area change) showed a strong decline (Figure 1B), RV diameters were increased (Table 2), and plasma BNP (Table 3) was elevated. The higher liver weight in PAB rats indicated a beginning RV decompensation (Figure 1A). However, this did not result in an impairment of LV SV or CO in PAB animals (Table 2). In AOB animals, LV function (LV EF% and LV FS%) showed a significant impairment 26 weeks after surgery (Figure 1B), which was associated with an increased LV diameter, reduced SV and CO (Table 2), and strongly increased plasma BNP levels (Table 3). The increased lung weight indicated a beginning LV decompensation (Figure 1A). Interestingly, at this stage of beginning ventricular decompensation, hypertrophy of the interventricular septum was observed in both the AOB and PAB rats (Table 2).
Figure 1.
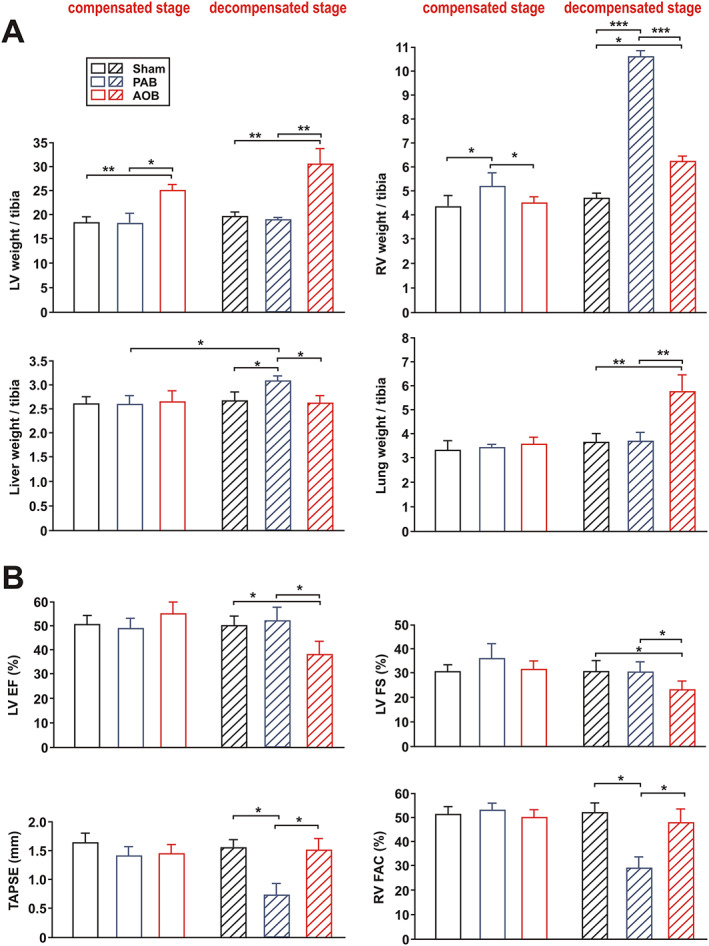
Animal characteristics in the two‐stage ascending aortic banding (AOB) and pulmonary artery banding (PAB) models. (A) Left ventricular (LV) weight, right ventricular (RV) weight (weight in mg per tibia in mm), liver weight (weight in g × 10 per tibia in mm), and lung weight (weight in g × 100 per tibia in mm) in sham animals, AOB animals, and PAB animals 7 weeks after surgery (compensated stage) or 22/26 weeks after surgery (decompensated stage). At the decompensated stage, results from sham animals 22–26 weeks after surgery are shown. (B) LV ejection fraction (LVEF), LV fractional shortening (LVFS), tricuspid annular plane systolic excursion (TAPSE), and RV fractional area change (RV FAC). Groups are the same as in (A). All data are mean ± standard error of the mean. n = 10–14 animals per group. * P < 0.05; ** P < 0.01; and *** P < 0.001.
Table 2.
Additional parameters obtained by echocardiography
| Sham | PAB | AOB | Sham | PAB | AOB | |
|---|---|---|---|---|---|---|
| 7 weeks | 7 weeks | 7 weeks | 22–26 weeks | 22 weeks | 26 weeks | |
| (n = 14) | (n = 14) | (n = 14) | (n = 20) | (n = 10) | (n = 10) | |
| LVEDD (mm) | 6.44 ± 0.06 | 6.73 ± 0.04 | 6.47 ± 0.05 | 6.62 ± 0.09 | 5.79 ± 0.12 * | 8.73 ± 0.08 * , §§ |
| RVEDD (mm) | 2.51 ± 0.15 | 2.22 ± 0.21 | 2.10 ± 0.09 | 2.43 ± 0.18 | 5.85 ± 0.23 ** | 2.81 ± 0.14 §§ |
| LVPWT (mm) | 1.35 ± 0.11 | 1.27 ± 0.04 | 1.72 ± 0.08 * , § | 1.42 ± 0.16 | 1.45 ± 0.12 | 2.01 ± 0.13 * , § |
| IVSD (mm) | 0.95 ± 0.03 | 1.02 ± 0.04 | 1.13 ± 0.02 * | 0.98 ± 0.02 | 1.25 ± 0.09 * , † | 1.64 ± 0.04 ** , § |
| RVWTD (mm) | 0.30 ± 0.02 | 0.42 ± 0.05 * | 0.33 ± 0.04 | 0.33 ± 0.04 | 0.52 ± 0.06 ** , † | 0.35 ± 0.03 §§ |
| LV SV (μL) | 143.4 ± 9.9 | 145.1 ± 6.3 | 181.7 ± 15.2 * , § | 151.2 ± 8.5 | 148.6 ± 8.2 | 121.7 ± 12.1 * , † , § |
| LV CO (mL/min) | 46.7 ± 1.2 | 46.2 ± 1.9 | 58.6 ± 2.5 * , § | 47.4 ± 0.9 | 47.8 ± 3.3 | 33.9 ± 1.2 ** , †† , § |
AOB, ascending aortic banding; PAB, pulmonary artery banding.
Echocardiographic evaluation of LVEDD (left ventricular end‐diastolic diameter), RVEDD (right ventricular end‐diastolic diameter), LVPWT (left ventricular posterior wall thickness in diastole), IVSD (interventricular septum in diastole), RVWTD (right ventricular wall thickness in diastole), LV SV (stroke volume = left ventricular end‐diastolic − left ventricular end‐systolic volume), and LV CO (cardiac output = stroke volume × heart rate).
P < 0.05.
P < 0.01 vs. respective age‐matched sham.
P < 0.05.
P < 0.01 vs. respective 7 week animals.
P < 0.05.
P < 0.01 vs. respective age‐matched PAB animals.
Table 3.
Plasma parameters at the end of the study
| Sham | PAB | AOB | Sham | PAB | AOB | |
|---|---|---|---|---|---|---|
| 7 weeks | 7 weeks | 7 weeks | 22–26 weeks | 22 weeks | 26 weeks | |
| (n = 14) | (n = 14) | (n = 14) | (n = 20) | (n = 10) | (n = 10) | |
| BNP (pg/mL) | 4.25 ± 2.06 | 36.62 ± 8.17 ** | 40.39 ± 8.05 ** | 10.54 ± 9.43 | 137.93 ± 21.76 *** , † | 126.45 ± 17.71 *** , † |
| TNF‐α (pg/mL) | 8.85 ± 3.26 | 17.29 ± 3.79 * | 17.88 ± 3.51 * | 8.04 ± 2.08 | 20.06 ± 2.44 * | 42.73 ± 4.13 * , † , § |
| IL‐6 (pg/mL) | 4.11 ± 1.25 | 9.43 ± 2.31 * | 10.27 ± 2.68 * | 5.38 ± 1.21 | 11.79 ± 2.74 * | 19.65 ± 2.72 ** , † , § |
| IL‐1β (pg/mL) | 26.31 ± 6.83 | 31.07 ± 6.36 | 34.14 ± 8.59 | 28.15 ± 6.29 | 52.63 ± 6.76 * , † | 62.08 ± 7.14 * , † |
| Angiotensin II (pg/mL) | 15.79 ± 3.81 | 24.83 ± 8.12 * | 43.49 ± 6.10 ** , § | 17.94 ± 5.74 | 35.18 ± 8.09 * | 102.33 ± 5.98 *** , †† , §§ |
AOB, ascending aortic banding; BNP, brain natriuretic peptide; IL, interleukin; PAB, pulmonary artery banding; TNF‐α, tumour necrosis factor‐α.
Plasma concentrations of the indicated mediators as determined by enzyme‐linked immunosorbent assay.
P < 0.05.
P < 0.01.
P < 0.001 vs. respective age‐matched sham.
P < 0.05.
P < 0.01 vs. respective 7 week animals.
P < 0.05.
P < 0.01 vs. respective age‐matched PAB animals.
Basic skeletal muscle characteristics in both models
Soleus and gastrocnemius muscle did not show significant difference in the sarcopenia index (muscle weight/body weight) between the groups 7 weeks after surgery. However, at the decompensated stage, a significant drop in muscle weight in animals with LV failure but not in animals with RV failure occurred in both muscles (Figure 2A). Furthermore, a positive correlation between LV CO and soleus weight (all animals: r = 0.34, P < 0.01; only animals at decompensated stage: r = 0.70, P < 0.001) or gastrocnemius weight (all animals: r = 0.38, P < 0.001; only animals at decompensated stage: r = 0.63, P < 0.001) was observed. The weight of the diaphragm, however, was not altered in any of the groups at any time point (Figure 2A) and did not show a correlation to CO (all animals: r = −0.14, P = 0.23).
Figure 2.
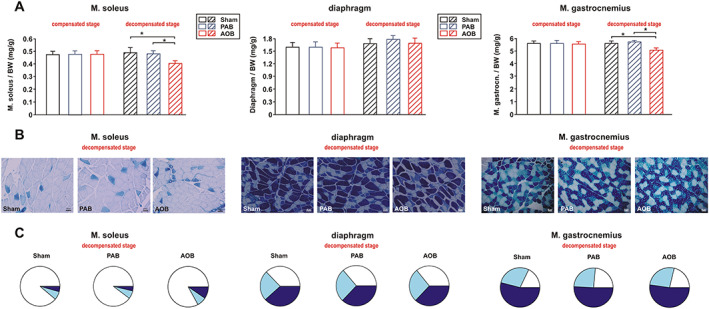
Muscle weights (sarcopenia index) and muscle fibre types. (A) Differences in the weight of soleus muscle, diaphragm, and gastrocnemius muscle normalized per body weight (BW) in sham animals, ascending aortic banding (AOB) animals, and pulmonary artery banding (PAB) animals 7 weeks after surgery (compensated stage) or 22/26 weeks after surgery (decompensated stage). All data are mean ± standard error of the mean. n = 10–14 animals per group. * P < 0.05. (B) Representative results from ATPase staining in soleus muscle, diaphragm, and gastrocnemius muscle to distinguish between type I fibres (white), type IIA fibres (light blue), and type IIB fibres (dark blue). The size bar indicates 50 μm. (C) Analysis of the percentage of the according fibre types at the decompensated stage depicted as a pie chart. n = 6 animals per group.
Further analyses of fibre number, type, and cross‐sectional area revealed a significant decrease in the number (Figure 2B and 2C) and diameter of type I fibres (Table 4) in the soleus muscle of AOB but not PAB rats at the decompensated stage. In addition, the percentage of type IIB fibres was significantly increased in soleus muscle at this stage compared with sham and PAB animals (sham 4.41 ± 0.50%, AOB 9.51 ± 0.23%, and PAB 3.75 ± 0.61%) as shown in Figure 2B. No difference in soleus fibre composition or cross‐sectional area (Table 4) was observed at the compensated stage. Unlike soleus muscle, which consists mainly of slow‐twitch type I fibres, 25 the diaphragm contains a higher amount of fast‐twitch fibres. 26 As shown in Figure 2B and 2C, all three fibre types showed an equal distribution in all animal groups at the compensated and decompensated stage. Interestingly, cross sections of type IIB fibres in diaphragm were significantly larger, while type I fibres were significantly smaller compared with soleus muscle (Table 4). Gastrocnemius muscle contains a superficial white portion with 1–10% type I fibres and a deep red portion with 20–40% type I fibres. 25 , 27 We utilized the deep, red portion of gastrocnemius muscle in all our analyses. Gastrocnemius muscle showed a reduced diameter of type I fibres in AOB but not PAB rats at the decompensated stage (Table 4). No significant alterations in fibre composition were observed (Figure 2B and 2C).
Table 4.
Fibre diameter
| Sham | PAB | AOB | Sham | PAB | AOB | |
|---|---|---|---|---|---|---|
| 7 weeks | 7 weeks | 7 weeks | 22–26 weeks | 22 weeks | 26 weeks | |
| (n = 14) | (n = 14) | (n = 14) | (n = 20) | (n = 10) | (n = 10) | |
| Soleus, type I | 1.75 ± 0.02 | 1.78 ± 0.02 | 1.77 ± 0.03 | 1.73 ± 0.03 | 1.70 ± 0.02 | 1.37 ± 0.02 * , † , § |
| Soleus, type IIA | 0.97 ± 0.12 | 0.99 ± 0.02 | 1.01 ± 0.13 | 0.96 ± 0.07 | 1.01 ± 0.05 | 0.95 ± 0.04 |
| Soleus, type IIB | 0.91 ± 0.07 | 0.95 ± 0.12 | 0.94 ± 0.11 | 0.92 ± 0.06 | 0.87 ± 0.04 | 0.89 ± 0.08 |
| Diaphragm, type I | 0.93 ± 0.01 | 0.95 ± 0.03 | 0.89 ± 0.02 | 0.87 ± 0.02 | 0.89 ± 0.02 | 0.86 ± 0.01 |
| Diaphragm, type IIA | 0.73 ± 0.02 | 0.85 ± 0.07 | 0.79 ± 0.07 | 0.82 ± 0.02 | 0.79 ± 0.03 | 0.80 ± 0.06 |
| Diaphragm, type IIB | 1.62 ± 0.05 | 1.57 ± 0.08 | 1.58 ± 0.07 | 1.68 ± 0.06 | 1.68 ± 0.05 | 1.66 ± 0.04 |
| Gastrocnemius, type I | 0.63 ± 0.05 | 0.69 ± 0.08 | 0.71 ± 0.07 | 0.69 ± 0.05 | 0.60 ± 0.07 | 0.54 ± 0.06 * , † |
| Gastrocnemius, type IIA | 0.53 ± 0.07 | 0.54 ± 0.09 | 0.55 ± 0.13 | 0.55 ± 0.07 | 0.59 ± 0.05 | 0.57 ± 0.11 |
| Gastrocnemius, type IIB | 0.68 ± 0.07 | 0.69 ± 0.12 | 0.70 ± 0.11 | 0.72 ± 0.06 | 0.70 ± 0.11 | 0.73 ± 0.08 |
AOB, ascending aortic banding; PAB, pulmonary artery banding.
Changes in fibre diameter are displayed as relative units as obtained by the software ImageJ.
P < 0.05 vs. respective age‐matched sham.
P < 0.05 vs. respective 7 week animals.
P < 0.05 vs. respective age‐matched PAB animals.
Catabolic and anabolic parameters in soleus muscle
No significant change in proteasome activity (Figure 3A) or the expression of the muscle‐specific ubiquitin E3 ligases MuRF1 and atrogin‐1 was observed 7 weeks after surgery in soleus muscle (Figure 3D). Similarly, myostatin, a well‐known negative regulator of muscle growth and protein synthesis, was also not altered (Figure 3D). However, at the stage of decompensation, proteasome activity (chymotrypsin like as well as trypsin like) was strongly increased in AOB rats but not altered in PAB rats in the soleus muscle (Figure 3A). Accordingly, we detected an increased MuRF1, atrogin‐1, and myostatin protein expression in the soleus muscle of AOB rats. The respective soleus muscle of the PAB rats did not show significant changes in MuRF1 or atrogin‐1 protein expression, while myostatin showed a mild increase (Figure 3D). qPCR analyses revealed an increased MuRF1 and myostatin mRNA expression in the soleus muscle of AOB animals only (Supporting Information, Figure S1A). Protein ubiquitination appeared very low in all skeletal muscle samples (Figure S2). At the compensated stage, no difference was observed (Figure S2A), while the decompensated stage resulted in an increased protein ubiquitination in the PAB and in the AOB model with strongest effects observed in the latter samples (Figure S2B). Muscle homogenates were also probed with an antibody detecting the autophagy marker light chain 3. No difference in the conversion to LC3‐II was observed 7 weeks after surgery in any of the groups. At the stage of decompensation, we observed a strong increase in LC3‐II, suggesting autophagy activation, mainly in the AOB rats (Figure 4A). In addition to activation of autophagy, apoptotic cell death may also have contributed to the observed loss of muscle mass in HF. However, we did not find evidence for increased apoptotic cell death as deduced from the measurement of caspase 3 activity in any of the groups (Figure 4B). Compared with sham animals, mRNA expression of the anabolic factor IGF‐1 was significantly reduced in the soleus muscle of AOB and of PAB animals at the stage of cardiac decompensation but not at the compensatory stage (Figure S1A).
Figure 3.
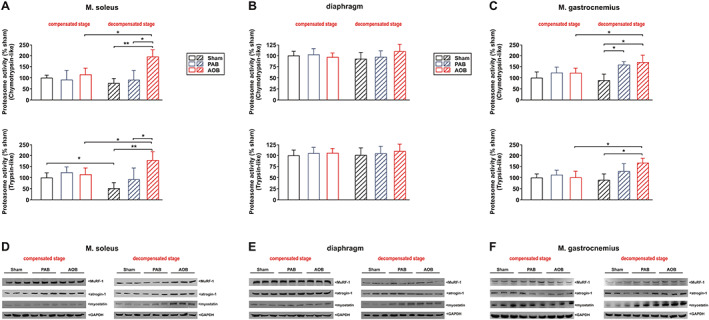
Proteasome activation and expression. (A) Differences in chymotrypsin‐like and trypsin‐like proteasome activity in soleus muscle. (B) Differences in chymotrypsin‐like and trypsin‐like proteasome activity in diaphragm. (C) Differences in chymotrypsin‐like and trypsin‐like proteasome activity in gastrocnemius muscle. Data are normalized to 7‐week‐old sham animals. All data are mean ± standard error of the mean. n = 8 animals per group. * P < 0.05; ** P < 0.01. (D) Representative western blots from soleus muscle at both disease stages. (E) Representative western blots from diaphragm at both disease stages. (F) Representative western blots from gastrocnemius muscle at both disease stages. Homogenates of skeletal muscle were probed with antibodies detecting muscle‐specific RING finger 1 (MuRF1), atrogin‐1, and myostatin. GAPDH served as a loading control. n = 6 samples per group. AOB, ascending aortic banding; PAB, pulmonary artery banding.
Figure 4.
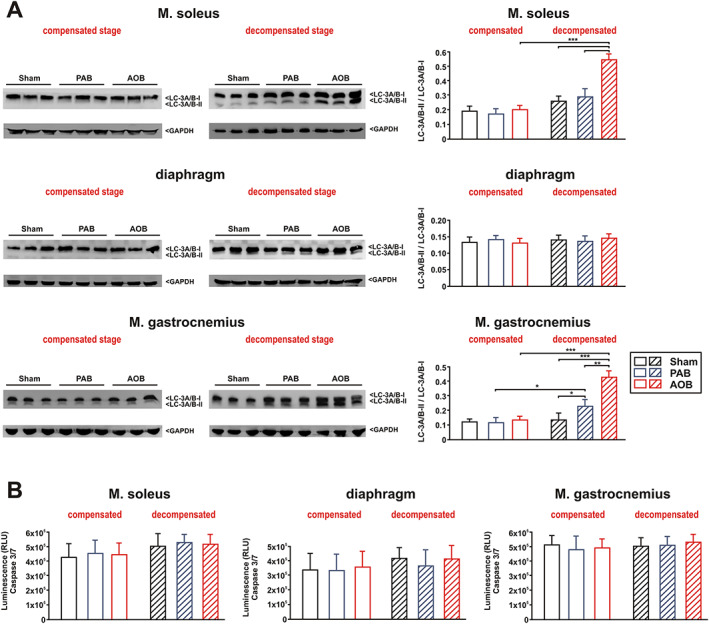
Autophagy and apoptosis. (A) Representative western blots and densitometry of protein data from soleus muscle, diaphragm, and gastrocnemius muscle at both disease stages. Homogenates of skeletal muscle were probed with an antibody detecting the autophagy marker light chain 3 (LC3, isoform A + B). The conversion of the LC3‐I form to the lower migrating form, LC3‐II, was used as an autophagy indicator. GAPDH served as a loading control. n = 6 samples per group. ** P < 0.01. (B) Ten micrograms of tissue homogenate was transferred to a 96‐well microplate, and caspase 3/7 activity was detected in sham animals, ascending aortic banding (AOB) animals, and pulmonary artery banding (PAB) animals 7 weeks after surgery (compensated stage) or 22/26 weeks after surgery (decompensated stage). All data are mean ± standard error of the mean. n = 8 samples per group.
Catabolic and anabolic parameters in diaphragm
Compared with sham animals, no significant change in proteasome activity was observed at the compensatory stage or the stage of decompensation in the diaphragm in AOB and PAB rats (Figure 3B). Similarly, the expression of MuRF1, atrogin‐1, or myostatin was not altered 7 weeks after surgery in any of the groups (Figure 3E). A significant increase in myostatin protein expression, however, was observed at the decompensated stage in the PAB as well as in the AOB model (Figure 3E). MuRF1 and atrogin‐1 were not altered (Figure 3E). qPCR analyses revealed a comparable increase in myostatin mRNA expression in the diaphragm of AOB and PAB animals at the stage of decompensation (Figure S1B). No significant alterations in protein ubiquitination (Figure S2), autophagy activation (Figure 4A), or caspase 3 activation (Figure 4B) were observed in the diaphragm in any model or disease stage. Compared with sham animals, mRNA expression of IGF‐1 was mildly reduced in the diaphragm of AOB animals at the stage of cardiac decompensation but not at the compensatory stage (Figure S1B). No difference in IGF‐I mRNA was observed in the PAB model (Figure S1B).
Catabolic and anabolic parameters in gastrocnemius muscle
Compared with sham animals, a significant change in proteasome activity (chymotrypsin like as well as trypsin like) was observed at the stage of decompensation in gastrocnemius muscle in AOB rats (Figure 3C). PAB animals demonstrated an increase in chymotrypsin‐like proteasome activity (Figure 3C) at the stage of decompensation. Accordingly, we detected an increased MuRF1, atrogin‐1, and myostatin protein expression in AOB rats at this stage (Figure 3F). qPCR analyses revealed an increased myostatin mRNA expression in the gastrocnemius muscle of AOB and PAB animals at both disease stages (Figure S1C). Protein ubiquitination was strongly increased at the decompensated stage in the PAB and AOB models with strongest effects observed in the latter samples (Figure S2B). No difference in the conversion to LC3‐II was observed 7 weeks after surgery in any of the groups (Figure 3A). At the stage of decompensation, we observed a mild increase in LC3‐II in PAB samples and a strong increase in LC3‐II in AOB samples (Figure 3A). No change in caspase 3 activity was detected in any of the gastrocnemius samples (Figure 3B). Compared with sham animals, mRNA expression of the anabolic factor IGF‐1 was significantly reduced in gastrocnemius muscle of AOB and PAB animals at the stage of cardiac decompensation but not at the compensatory stage (Figure S1C).
Mitochondrial parameters
At the compensated stage of LV or RV hypertrophy, we did not observe a significant change in the enzyme activity of citrate synthase, as an indicator of mitochondrial content, or the activity of the respiratory chain complexes I–III in soleus muscle (Figure 5A). Only the activity of COX (complex IV) was mildly reduced in AOB rats 7 weeks after surgery (Figure 5A). At the stage of cardiac decompensation, citrate synthase activity and the activity of complex I and complex IV of the respiratory chain, both containing mitochondrial‐encoded subunits, were significantly reduced in the soleus muscle of AOB and of PAB rats as shown in Figure 5A. Pyruvate‐dependent state 3 respiration, measured in saponin‐skinned muscle fibres of soleus muscle, did not differ 7 weeks after surgery but showed a significant decrease at the decompensated stage in the AOB and PAB models (Figure 5B). Accordingly, the respiratory control index was significantly lower in these samples (sham 4.72 ± 0.55; PAB 3.65 ± 0.87; AOB 3.09 ± 0.49, both P < 0.05 vs. sham). In general, mitochondrial functional impairments appeared more serious in AOB rats compared with PAB rats. Succinate‐dependent state 3 respiration (Figure 5B), resting respiration, and leak respiration (not shown) were not significantly different between the groups at any disease stage.
Figure 5.
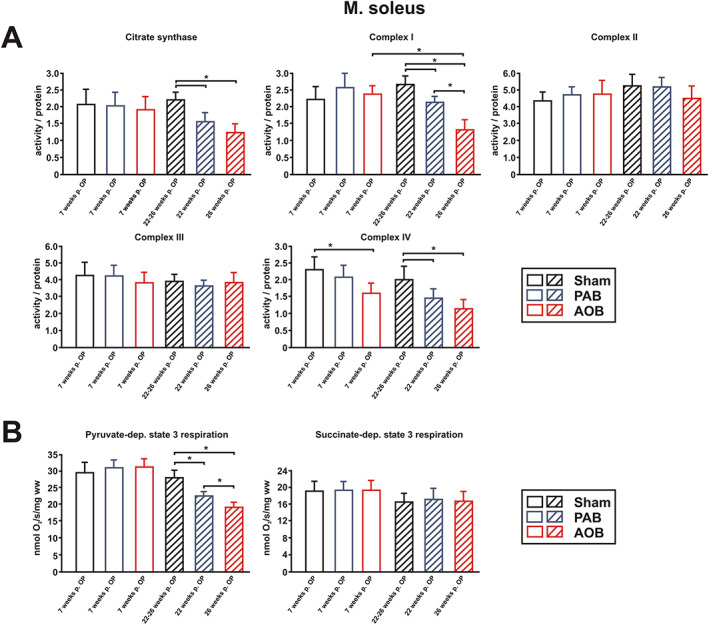
Mitochondrial function in soleus muscle. (A) Citrate synthase activity and the activity of complex I, complex II, complex III, and complex IV of the respiratory chain were measured in soleus muscle lysates. All values were normalized to total protein content of the samples. (B) Active rates of respiration (state 3) were measured in saponin‐skinned fibres of soleus muscle in the presence of 5 mM ADP and either 10 mM pyruvate + 2 mM malate or 10 mM succinate in the presence of 5 μM rotenone. All data are mean ± standard error of the mean. n = 6 samples per group. * P < 0.05. AOB, ascending aortic banding; PAB, pulmonary artery banding.
Disturbances in mitochondrial gene expression are known to have an impact on mitochondrial respiratory chain function. Accordingly, no significant changes in the expression of respiratory chain complexes I–V were observed in soleus muscle at the compensatory stage, when utilizing the MitoProfile total OXPHOS antibody cocktail (Figure 6A). At the decompensated stage, a mild decrease in complex IV protein expression was observed in the AOB rats (Figure 6A), which may in part explain the functional changes observed (Figure 5). However, no change was observed for complex I protein expression despite reduced complex I enzyme activity and reduced pyruvate‐dependent respiration. In addition to the total OXPHOS antibody cocktail, we therefore also utilized separate antibodies detecting nuclear‐encoded subunits and mtDNA‐encoded subunits of complex I and complex IV of the respiratory chain. As shown in Figure 6B, no difference in these proteins was observed in soleus muscle at the compensatory stage. However, at the decompensated stage, a lower expression of the mtDNA‐encoded subunits of complex I (ND1) and complex IV (COX I) was observed in soleus muscle of AOB rats, while the according nuclear‐encoded subunits of complex I (NDUFS1) and complex IV (COX IV) were not altered (Figure 6B). PAB rats did not demonstrate any change in the expression of these subunits of the respiratory chain in soleus muscle. Compared with sham animals, mRNA expression of the mitochondrial transcriptional coactivator PGC‐1α was not altered in soleus muscle (Figure S1A).
Figure 6.
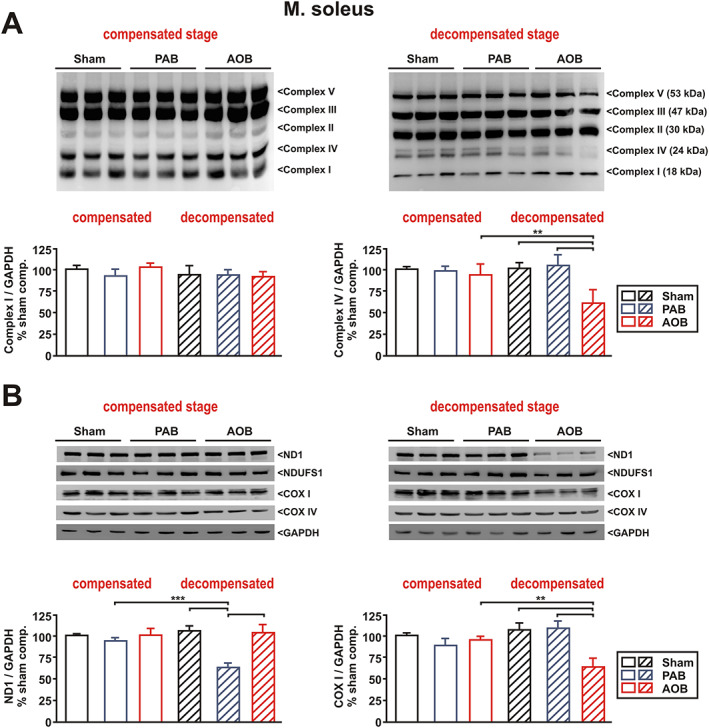
Mitochondrial respiratory chain protein expression. (A) Representative blots and densitometry of selected protein data (complex I and complex IV) from soleus muscle at both disease stages. (B) Representative blots and densitometry of selected protein data (ND1 and COX I) from soleus muscle at both disease stages. Homogenates of skeletal muscle were probed with antibodies detecting mitochondrial respiratory chain complexes I–V (total OXPHOS), the complex I proteins ND1 and NDUFS1, and the complex IV proteins COX I and COX IV. GAPDH served as a loading control. n = 6 samples per group. ** P < 0.01; *** P < 0.001. AOB, ascending aortic banding; PAB, pulmonary artery banding.
In the diaphragm, pyruvate‐dependent state 3 respiration and succinate‐dependent state 3 respiration were not significantly different between the groups at any disease stage (Figure S4). Measurements of respiratory chain enzyme activities suggested a lower activity of citrate synthase, complexes II–IV in PAB animals at the decompensated stage compared with the respective animals at the compensated stage (Figure S3A), No significant changes in protein expression of respiratory chain complexes I–V (Figure S5A and S5B) or PGC‐1α mRNA expression (Figure S1B) were observed in the diaphragm at the compensatory stage or at the decompensated stage.
Gastrocnemius muscle on the other hand mimicked many of the changes observed in soleus muscle. At the stage of cardiac decompensation, citrate synthase activity and the activity of complexes I–IV of the respiratory chain were significantly reduced in sham, PAB, and AOB animals with lowest enzyme activities observed in the respective AOB rats (Figure S4A). Pyruvate‐dependent state 3 respiration was reduced in PAB as well in AOB animals at the decompensated stage (Figure S4B). However, a lower expression of the mtDNA‐encoded subunits of complex I (ND1) and complex IV (COX I) was only observed in AOB rats at the decompensated stage (Figure S6B). Unlike soleus muscle or diaphragm, we observed an increased expression of PGC‐1α mRNA in all animals 22–26 weeks after surgery (Figure S1C).
Inflammatory mediators
Plasma concentrations of various inflammatory mediators were different among the groups. A 95–100% increase in TNF‐α in PAB and AOB rats was detectable at the compensatory stage with a further increase at the decompensated stage in AOB rats only (Table 3). Similarly, the cytokine IL‐6 increased in PAB (130%) and AOB (150%) rats at the compensatory stage. However, at the decompensated stage, only the AOB rats demonstrated a further, significant increase in plasma IL‐6 by ~230% (Table 3). In contrast, IL‐1β did not show a significant increase at the compensatory stage but increased significantly in PAB (90%) and AOB (120%) rats at the decompensated stage (Table 3). Protein expression of selected cytokines was also analysed in blood‐free perfused soleus muscle in order to estimate inflammatory activation of the tissue independent from circulating cytokines. These array analyses demonstrated increased IL‐1β, IL‐6, and TNF‐α protein expression in soleus muscle of AOB rats at the decompensated stage compared with sham animals (Table 5). In addition, other cytokines such as granulocyte–macrophage colony‐stimulating factor, interferon‐γ, monocyte chemoattractant protein‐1, or macrophage inflammatory protein‐3α (Table 5) were significantly elevated suggesting strong inflammatory tissue activation in the AOB rats but not in the PAB rats. PAB animals showed a minor elevation in TNF‐α protein expression compared with AOB animals (Table 5). Unlike the strong differences observed in cytokine plasma levels (Table 3) or tissue protein expression (Table 5), only minor changes were detectable in skeletal muscle cytokine mRNA expression. TNF‐α mRNA expression did not show any difference among the groups in soleus muscle (Figure S1A). At the compensatory stage, IL‐6 and IL‐1β mRNA did not differ in soleus muscle. However, IL‐6 mRNA showed a significant increase at the decompensated stage in AOB rats but not in PAB rats, while IL‐1β mRNA was increased in PAB and AOB rats (Figure S1A). In the diaphragm, we observed only a mild increase in IL‐1β mRNA expression at the decompensated stage in AOB rats (Figure S1B). None of the other cytokines showed an expressional mRNA change in the diaphragm (Figure S1B). Gastrocnemius muscle revealed an increased IL‐6 mRNA expression in AOB animals at the decompensated stage (Figure S1C). Interestingly, Ang II, which has been suggested to play a central role in the development of skeletal muscle abnormalities in HF, showed a moderate increase in the plasma in PAB rats by 60% but an increase by 170% in AOB rats at the compensatory stage (Table 3). Cardiac decompensation resulted in a moderate further increase in the PAB rats (90%) and in a tremendous augmentation (>400%) of plasma Ang II in AOB rats (Table 3). Furthermore, a negative correlation between plasma Ang II and CO (all animals: r = −0.39, P < 0.001; only animals at decompensated stage: r = −0.84, P < 0.0000001) was observed.
Table 5.
Cytokine array
| Sham | PAB | AOB | |
|---|---|---|---|
| 22–26 weeks | 22 weeks | 26 weeks | |
| (n = 4) | (n = 4) | (n = 4) | |
| CINC‐2 | 0.56 ± 0.17 | 0.39 ± 0.08 | 0.42 ± 0.19 |
| CINC‐3 | 0.69 ± 0.19 | 0.61 ± 0.04 | 0.61 ± 0.16 |
| CNTF | 0.34 ± 0.19 | 0.40 ± 0.12 | 0.28 ± 0.18 |
| Fractalkine | 0.12 ± 0.12 | 0.15 ± 0.10 | 0.16 ± 0.14 |
| GM‐CSF | 0.20 ± 0.19 | 0.39 ± 0.14 | 1.06 ± 0.16 * , § |
| IFN‐γ | 0.45 ± 0.06 | 0.31 ± 0.11 | 0.95 ± 0.17 * , § |
| IL‐1α | 0.94 ± 0.08 | 0.76 ± 0.21 | 1.22 ± 0.13 § |
| IL‐1β | 0.19 ± 0.05 | 0.23 ± 0.08 | 0.97 ± 0.19 ** , § |
| IL‐4 | 0.99 ± 0.03 | 0.87 ± 0.24 | 1.02 ± 0.25 |
| IL‐6 | 0.23 ± 0.02 | 0.33 ± 0.19 | 1.01 ± 0.26 * , § |
| IL‐10 | 1.13 ± 0.18 | 0.96 ± 0.28 | 1.20 ± 0.26 |
| Leptin | 0.61 ± 0.13 | 0.41 ± 0.36 | 0.45 ± 0.13 |
| LIX | 0.91 ± 0.15 | 0.76 ± 0.27 | 1.43 ± 0.30 |
| MCP‐1 | 0.67 ± 0.21 | 1.19 ± 0.48 | 1.40 ± 0.09 ** |
| MIP‐3α | 0.18 ± 0.08 | 0.35 ± 0.14 | 0.89 ± 0.22 * |
| NGF‐β | 0.23 ± 0.05 | 0.30 ± 0.27 | 0.43 ± 0.22 |
| TIMP‐1 | 0.86 ± 0.11 | 0.90 ± 0.40 | 1.29 ± 0.38 |
| TNF‐α | 0.31 ± 0.08 | 0.91 ± 0.13 * | 2.01 ± 0.36 *** , § |
| VEGF‐A | 0.77 ± 0.21 | 0.81 ± 0.23 | 1.06 ± 0.32 |
AOB, ascending aortic banding; CINC, cytokine‐induced neutrophil chemoattractant; CNTF, cytokine‐induced neutrophil chemoattractant; GM‐CSF, granulocyte–macrophage colony‐stimulating factor; IFN‐γ, interferon‐γ; IL, interleukin; LIX, lipopolysaccharide‐induced CXC chemokine; MCP‐1, monocyte chemoattractant protein‐1; MIP‐3α, macrophage inflammatory protein‐3α; NGF‐β, nerve growth factor‐β; PAB, pulmonary artery banding; TIMP, tissue inhibitor of metalloproteinase‐1; TNF‐α, tumour necrosis factor‐α; VEGF‐A, vascular endothelial growth factor A.
Relative cytokine expression in the soleus muscle as determined by an antibody array.
P < 0.05.
P < 0.01.
P < 0.001 vs. sham.
P < 0.05 vs. PAB animals.
Impact of interleukin‐6 and angiotensin II on atrophic changes observed in skeletal muscle
We performed in vitro experiments in adult RSkMCs to obtain a deeper insight into the role of IL‐6 and Ang II in triggering distinctive atrophic changes observed in soleus and gastrocnemius muscle. Treatment of RSkMC myoblasts with increasing concentrations of IL‐6 resulted in a mild increase in MuRF1 and atrogin protein expression but a strong, clearly dose‐dependent increase of myostatin protein expression and LC‐3A/B activation (Figure 7A). Similarly, a mild increase in protein ubiquitination at the highest IL‐6 concentrations was detectable (Figure 7B). Differentiated RSkMCs revealed an even stronger increase in myostatin, LC‐3A/B, or ubiquitination (Figure 7A and 7B). Treatment of RSkMC myoblasts with increasing concentrations of Ang II resulted in a mild increase in myostatin protein expression and LC‐3A/B activation only at the highest Ang II concentration (1 μM), while all other parameters remained largely unaffected by Ang II (Figure 7C and 7D). However, differentiated RSkMCs revealed an increased MuRF1 and myostatin protein expression, LC‐3A/B activation (Figure 7C), and ubiquitination (Figure 7D) already at lower Ang II concentrations (10–100 nM). As there are conflicting studies relating to Ang II receptor expression in skeletal muscles and muscle cells, 28 we investigated the expressional levels of both angiotensin receptors. As shown in Figure S7, differentiated RSkMCs express significantly higher levels of AT1 mRNA than undifferentiated RSkMCs, similar to the levels observed in skeletal muscle (Figure S7), suggesting that the differences observed between myoblasts and myotubes in response to Ang II may be caused by the low AT1 expression in myoblasts. No major differences in angiotensin II receptor type 2 mRNA expression were observed between the samples (Figure S7). Inhibition of the IL‐6 effects by interfering with its downstream signalling molecule STAT3 resulted in a reduced MuRF1 and myostatin protein expression, LC‐3A/B activation, and ubiquitination in myoblasts and myotubes (Figure 8A and 8B). The AT1 receptor antagonist losartan on the other hand reduced MuRF1 and myostatin protein expression, LC‐3A/B activation, and ubiquitination mainly in myotubes (Figure 8A and 8B).
Figure 7.
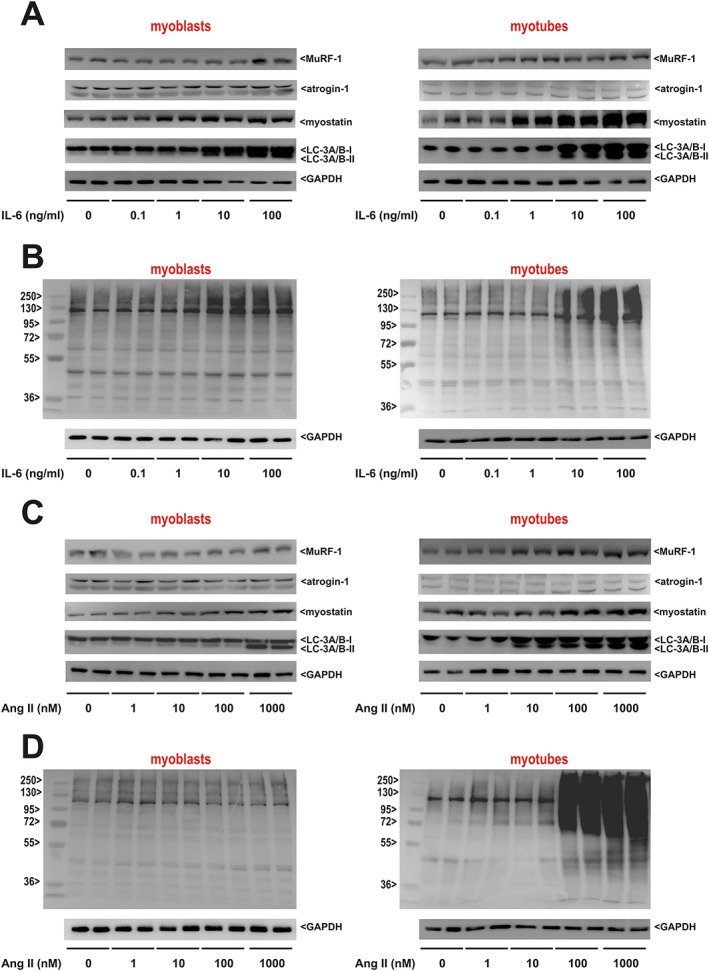
Impact of interleukin‐6 (IL‐6) and angiotensin II (Ang II) on mediators involved in muscular atrophy and autophagy. (A) Rat skeletal muscle cell (RSkMC) myoblasts (left panel) or RSkMC myotubes (right panel) were treated with increasing concentrations of IL‐6 as indicated. Homogenates of RSkMC were probed with antibodies detecting muscle‐specific RING finger 1 (MuRF1), atrogin‐1, myostatin, and light chain 3 (LC3) (isoform A + B). GAPDH served as a loading control. (B) RSkMC myoblasts (left panel) or RSkMC myotubes (right panel) were treated as described previously and probed with an antibody detecting ubiquitin. A molecular weight marker to estimate the size of the ubiquitinated proteins is included. GAPDH served as a loading control. (A, B) n = 8 samples per group, four independent experiments. (C) RSkMC myoblasts (left panel) or RSkMC myotubes (right panel) were treated with increasing concentrations of Ang II as indicated. Homogenates of RSkMC were probed with antibodies detecting MuRF1, atrogin‐1, myostatin, and LC3 (isoform A + B). GAPDH served as a loading control. (D) RSkMC myoblasts (left panel) or RSkMC myotubes (right panel) were treated as described previously and probed with an antibody detecting ubiquitin. A molecular weight marker to estimate the size of the ubiquitinated proteins is included. GAPDH served as a loading control. (C, D) n = 8 samples per group, four independent experiments.
Figure 8.
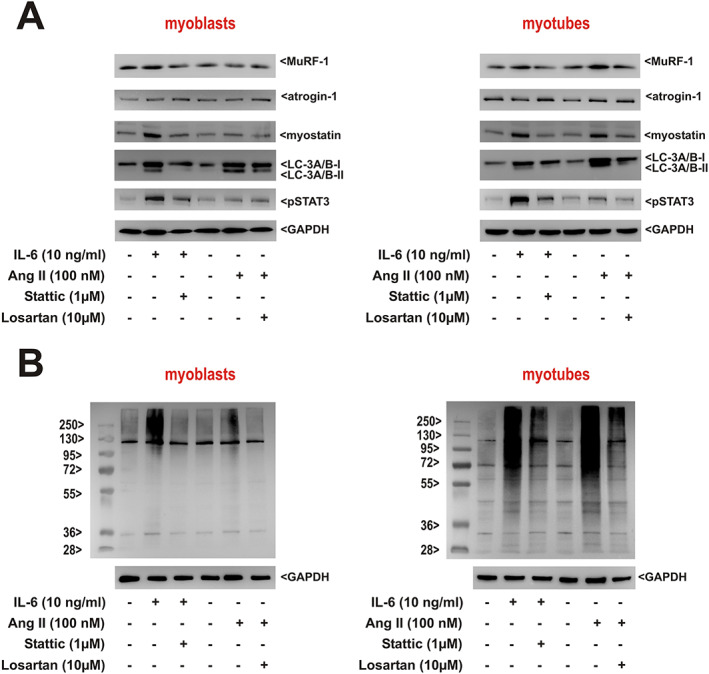
Role of interleukin‐6 (IL‐6) and angiotensin II (Ang II) pathway activation in rat skeletal myoblasts and myotubes in vitro. (A) Rat skeletal muscle cells (RSkMC) myoblasts (left panel) or RSkMC myotubes (right panel) were treated with Stattic (1 μM) or losartan (10 μM) for 1 h before adding IL‐6 (10 ng/mL) or Ang II (100 nM). Homogenates of RSkMC were probed with antibodies detecting muscle‐specific RING finger 1 (MuRF1), atrogin‐1, myostatin, light chain 3 (LC3) (isoform A + B), and pSTAT3. GAPDH served as a loading control. (B) RSkMC myoblasts (left panel) or RSkMC myotubes (right panel) were treated as described previously and probed with an antibody detecting ubiquitin. A molecular weight marker to estimate the size of the ubiquitinated proteins is included. GAPDH served as a loading control. (A, B) n = 8 samples per group, four independent experiments.
Discussion
Novel animal model
In many cardiac diseases, LV and RV overload develops gradually. The well‐established model of AOB in weanling rats allows such a gradual disease development of the LV, development of concentric hypertrophy, and later on transition to LV failure as shown previously by our group. 20 However, no comparable model of gradual increase in RV pressure exists in rats so far. Among the established models of RV overload, PAB in adult rats and monocrotaline administration impose an acute and strong increase in RV overload. 29 On the other hand, the model of chronic exposure to hypoxia induces pulmonary hypertension, which does not reach the same severity as in humans 29 and therefore needs to be combined with the tyrosine kinase inhibitor SU5416. 30 Here, we successfully established a novel model of slowly developing RV hypertrophy and transition to RV failure and compared this with the two‐stage model of LV hypertrophy. 20 In both models, the stage of compensatory hypertrophy was characterized by a preserved systolic function of the respective ventricle, a moderate increase in plasma BNP, and exclusive hypertrophy of the overloaded ventricle without affecting the respective other heart chamber. Disease progression resulted in dilation of the overloaded ventricle, a further increase of BNP and liver (PAB model) or lung congestion (AOB model), indicating the transition to RV or LV failure. The decline in the respective ventricular function amounted to ~20–30% in the AOB model and even 40–50% in the PAB model. Systemic plasma BNP was similarly increased in rats undergoing PAB or AOB surgery at the decompensated stage, suggesting that a comparable magnitude of HF was reached in both models.
Distinct effects of right ventricular and left ventricular failure on skeletal muscle protein balance
Left ventricular failure induced marked disturbances in anabolic and catabolic processes in soleus and gastrocnemius muscle, resulting in reduced muscle weight and fibre diameter. However, this shift towards increased proteasome or autophagy activation was largely missing in soleus and gastrocnemius muscle of PAB animals, although others have described the occurrence of muscle atrophy in patients and animals suffering from RV failure. 19 , 31 We cannot rule out that small variations between the models such as observational period or clip size may have contributed to the differences observed in skeletal muscle in RV and LV failure, although echocardiographic data and plasma BNP suggested that a similar degree of cardiac failure was reached in both models. However, the RV and the LV differ in many aspects: (i) signalling responses in the mechanically stressed RV appear to display several unique features. 32 (ii) The anatomy, myocardial ultrastructure, and coronary physiology of the RV reflect a high‐volume, low‐pressure pump unlike the LV. 33 (iii) The relative increase in afterload that the RV needs to withstand in severe pulmonary hypertension surpasses several fold the relative increase in LV afterload even in severe systemic hypertension. In addition, there are also a number of pathophysiological differences between both models. First, LV SV and CO were significantly reduced in the AOB model but not in the PAB model. In addition, among the circulating mediators known to be involved in sarcopenia, plasma Ang II and IL‐6 as well as soleus and gastrocnemius muscle IL‐6 mRNA expression were significantly higher in AOB compared with PAB animals at the decompensated stage. The renin–angiotensin–aldosterone system (RAAS) regulates blood pressure and fluid/electrolyte balance. Reduced renal blood flow and sodium delivery to the distal tubule leads to renin release, finally resulting in increased Ang II. In HF with reduced CO, activation of the RAAS serves as a compensatory mechanism to maintain CO. This can be envisioned for the AOB animals at the decompensated stage with their significantly reduced CO, resulting in high Ang II levels and the subsequent known effects of Ang II on skeletal muscle. PAB animals with their normal LV CO and thus unchanged systemic circulation may therefore not have experienced a comparable activation of the RAAS. Ang II infusion has been shown to reduce hindlimb muscle weights, muscle cross‐sectional area, and type I fibres in healthy, young mice. 34 Furthermore, signs of skeletal muscle apoptosis, disturbed protein balance, and impaired mitochondrial respiratory chain function were detectable. 34 In rodent models, an increase in systemic Ang II also leads to reduced protein synthesis via inhibition of the IGF‐1 signalling pathway and activation of the UPS. 28 Our in vitro data support the notion that Ang II induces profound and diverse atrophic changes in differentiated skeletal muscle cells. The Ang II‐induced wasting effects have been suggested to be indirect, involving inflammatory cytokines such as IL‐6 or TNF‐α and reactive oxygen species. 35 Thus, the high Ang II levels may also have contributed to the observed sarcopenic changes via increased expression of IL‐6 protein in skeletal muscle and high plasma IL‐6 in the AOB rats. Indeed, circulating Ang II correlated to IL‐6 plasma levels (r = 0.52, P = 0.00000102) but not to the other cytokines. Furthermore, Ang II infusion was shown to raise circulating IL‐6, an effect suggested being essential for Ang II‐induced muscle wasting via reduced IGF‐I signalling. 36 Our data obtained in RSkMC myoblasts and myotubes in vitro provide evidence for the major impact of IL‐6 on distinctive atrophic changes observed in skeletal muscle in vivo. Skeletal muscle cells themselves are a source of IL‐6, induced by contraction or by high circulating Ang II levels as shown before. 36 , 37 Interestingly, a correlation between circulating IL‐6 and IL‐6 mRNA expression in soleus muscle (r = 0.74, P = 1.305E − 014) and gastrocnemius muscle (r = 0.81, P = 1.41E − 014) was observed in our animals, supporting the hypothesis of muscle‐derived IL‐6. This is further corroborated by the cytokine array data from blood‐free soleus muscle, showing a strong IL‐6 tissue expression in AOB rats.
Distinct effects of left ventricular failure on limb muscles and diaphragm
However, this also leads to the question why the diaphragm was barely affected despite high systemic Ang II levels. We observed only an increase in myostatin mRNA and protein expression and a minor decline in IGF‐I mRNA without signs of increased proteasomal activation, autophagy, apoptosis, or mitochondrial dysfunction in the diaphragm at the decompensated stage. Ang II induces diaphragm muscle wasting within 1 day in mice. 38 Similarly, in a mouse model of myocardial infarction‐induced HF diaphragm fibre atrophy, contractile dysfunction and impaired mitochondrial function have been demonstrated. 39 However, in a rat model of myocardial infarction‐induced HF, the diaphragm was largely unaffected, while soleus muscle presented with muscle atrophy, proteasome activation, oxidative stress, and mitochondrial impairments. 15 Therefore, species differences between mouse and rat may play a role.
Furthermore, there are major differences between the constantly active diaphragm and the occasionally active soleus and gastrocnemius muscle, which may render both limb muscles more susceptible to muscle wasting than the diaphragm. These other influencing factors include differences in fibre composition, in perfusion and oxygen supply, in contraction activity, and in muscle regeneration. Skeletal muscle wasting and remodelling during HF can be attributed to secondary consequences of the disease, for example, inflammation, and also to disuse. 40 Accordingly, limb muscles are affected by HF‐induced inactivity, which exacerbates atrophy and exercise intolerance. The extent of muscle atrophy and activation of the UPS appears to be related to the reduction in muscle activation as shown in animals and humans. 40 , 41 While a reduced level of activity favours the signalling of muscle protein breakdown, exercise training in HF patients reduces muscular inflammation and increases antioxidant defence in limb muscle 42 , 43 , 44 and attenuates muscle wasting in animals. 45 On the other hand, HF has been reported to result in training‐like effects in the constantly active diaphragm due to increased breathing work during pulmonary congestion, resulting in a fibre shift and increased oxidative capacity. 46
The soleus muscle has a high proportion of slow‐twitch type I fibres (>80%), which are rich in mitochondria, myoglobin, and capillary supply, resulting in high oxidative capacity and fatigue resistance, while the diaphragm contains a higher amount of fast‐twitch, type II fibres (60–80%) with a lower concentration of mitochondria, myoglobin, and capillaries. 25 , 26 In type I muscles such as the soleus muscle, microvascular oxygen exchange dynamics appear to more unfavourable than in muscles consisting mainly of type II fibres. 47 The neuroendocrine overactivation with increased circulating Ang II, norepinephrine, endothelin‐1, and vasopressin in HF results in vasoconstriction in order to maintain CO but subsequently impairs skeletal muscle perfusion. Studies suggest that under these conditions, the diaphragm and other respiratory muscles can ‘steal’ perfusion from the locomotory muscles. 48 In addition, a pathologically limited CO and reduced perfusion may also result in an imbalance between oxygen supply and oxygen demand in skeletal muscle, possibly causing hypoxia‐driven reduction of muscle protein synthesis and activation of proteolysis. 49 Interestingly, myostatin, which emerged as a very sensitive and early indicator of skeletal muscle disturbances in our study, was shown to contribute to hypoxia‐driven muscle wasting, 50 possibly through inhibition of the protein kinase B/mammalian target of rapamycin pathway. 51 Finally, intramuscular and intermuscular heterogeneity in satellite cells, the muscle cell type required for muscle regeneration, was shown to exist. 52 , 53 At least in vitro experiments suggest that satellite cell‐derived myoblasts of the diaphragm behave differently than those of the hindlimb with a higher proliferative capacity and myogenic differentiation of diaphragm cultures. 52 , 54 All the previously mentioned characteristics may compensate some of the deleterious HF effects and help in part to explain the differences between soleus muscle and diaphragm in our study. Finally, variations in fibre composition 25 , 27 may have contributed to the subtle differences between soleus and gastrocnemius muscle observed in our study.
Skeletal muscle mitochondrial dysfunction as an early indicator
At the stage of cardiac decompensation, citrate synthase activity and the activity of several respiratory chain complexes as well as pyruvate‐dependent state 3 respiration were significantly reduced in the soleus and gastrocnemius muscle of AOB and of PAB rats. Thus, unlike the parameters of skeletal muscle protein balance, PAB animals were affected as well, although less seriously than AOB animals. In a mouse model of Ang II‐induced muscle wasting, it has been suggested that mitochondrial dysfunction is an early sign of HF‐associated muscle disease, while manifest muscle atrophy occurs later. 34 Furthermore, a time and dose dependency was clearly shown. While a low‐dose, short‐term infusion of Ang II (50 ng/kg/min for 1 week) induced mitochondrial dysfunction and oxidative stress, higher doses (1000 ng/kg/min for 4 weeks) induced additional atrophy. 34 , 55 This suggests that more severe stages of HF, associated with long‐term exposure of high Ang II levels, are more likely to result in muscle wasting. Thus, the increased Ang II plasma levels in the PAB rats may have contributed to the mitochondrial dysfunction, although they were not high enough to induce muscle wasting. Disturbances in mitochondrial gene expression can have an impact on mitochondrial respiratory chain function. Complex I and complex IV of the respiratory chain both contain mitochondrial‐encoded subunits unlike for example complex II. Mitochondrial DNA is more susceptible to ROS‐induced damage because it lacks repair mechanisms and protective histones of the nuclear genome. Therefore, the transcription of mitochondrial‐encoded genes may be altered under circumstances of increased oxidative stress more easily than nuclear‐encoded genes. This could explain the lower expression of the mtDNA‐encoded subunits of complex I and complex IV observed in soleus and gastrocnemius muscle of AOB rats, while the according nuclear‐encoded subunits were not altered. However, other factors in addition to altered gene expression must have contributed because PAB animals demonstrated mitochondrial functional impairment without altered mitochondrial protein expression. These factors include alterations in mitochondrial dynamics (fusion, fission, mitophagy, and biogenesis), mitochondrial trafficking/transport, and abnormal size and morphology, which were not investigated in the present study.
Potential implications of the study
Mitochondrial dysfunction and up‐regulation of myostatin were identified as the earliest signs of skeletal muscle impairment in HF, even in the absence of atrophy. Both may serve as markers to estimate the onset and progression of HF‐associated alterations in skeletal muscle. However, the potential value of myostatin as a biomarker of sarcopenia is controversial. 56 , 57 Only studies with serial muscle biopsies and their biochemical characterization can clarify the succession of alterations occurring on the way to apparent muscle wasting. This may also pave the way to an early therapeutic targeting of the involved pathways such as altered mitochondrial function or impaired protein balance and help to delay or even prevent muscle wasting. Currently, resistance exercise has emerged as an option to treat primary (i.e. age associated) and secondary sarcopenia (disease related), at least in part by improving antioxidant defence, mitochondrial function, and protein turnover. 42 , 43 , 44 In addition to resistance exercise, the management of sarcopenia is recommended to include a protein intake of 1 to 1.5 g/kg/day. 58 Future therapeutic strategies to improve muscle oxidative function and protein balance may include anti‐inflammatory and antioxidant strategies, but direct, targeted interference with deleterious pathways can also be envisioned. Accordingly, it was just recently shown that small‐molecule inhibitors of MuRF1 (compound ID#704946) partly reversed muscle atrophy and dysfunction in a mouse model of HF through protection of mitochondrial function. 39
In summary, our study shows more pronounced soleus and gastrocnemius muscle sarcopenia in LV failure compared with RV failure despite a similar functional impairment of the respective ventricle. This suggests a major impact of impaired systemic circulation. The constantly active diaphragm did not show major sarcopenic changes. Mitochondrial dysfunction and up‐regulation of myostatin were identified as the earliest signs of skeletal muscle impairment in HF. In addition to the manifold markers of skeletal muscle atrophy observed in our study, the potential impairment in skeletal muscle contractile function in these animals requires further investigations in future studies.
Conflict of interest
None declared.
Funding
This study was in part funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer 268555672—SFB 1213, Project B03.
Supporting information
Data S1. Supporting Information
Figure S1: Changes in mRNA expression in rat skeletal muscle
Figure S2: Changes in protein ubiquitination in rat skeletal muscle
Figure S3: Mitochondrial function in diaphragm
Figure S4: Mitochondrial function in gastrocnemius muscle
Figure S5: Respiratory chain protein expression in diaphragm
Figure S6: Respiratory chain protein expression in gastrocnemius muscle
Figure S7: Comparison of angiotensin receptor mRNA expression in RSkMC and in selected rat skeletal muscles
Acknowledgements
We appreciate the technical assistance of B. Reuter. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 59 Open access funding enabled and organized by Projekt DEAL.
Knapp F., Niemann B., Li L., Molenda N., Kracht M., Schulz R., and Rohrbach S. (2020) Differential effects of right and left heart failure on skeletal muscle in rats, Journal of Cachexia, Sarcopenia and Muscle, 11, 1830–1849, 10.1002/jcsm.12612
[Correction added on 07 November 2020, after first online publication: Projekt Deal funding statement has been added.]
References
- 1. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 2. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458–471. [DOI] [PubMed] [Google Scholar]
- 3. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1‐dependent ubiquitylation. J Cell Biol 2009;185:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lagirand‐Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet‐Pichon S, Tintignac LA, et al. The initiation factor eIF3‐f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J 2008;27:1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, et al. Insulin‐like growth factor‐1 (IGF‐1) inversely regulates atrophy‐induced genes via the phosphatidylinositol 3‐kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 2005;280:2737–2744. [DOI] [PubMed] [Google Scholar]
- 7. Hambrecht R, Schulze PC, Gielen S, Linke A, Mobius‐Winkler S, Yu J, et al. Reduction of insulin‐like growth factor‐I expression in the skeletal muscle of noncachectic patients with chronic heart failure. J Am Coll Cardiol 2002;39:1175–1181. [DOI] [PubMed] [Google Scholar]
- 8. Reid MB, Li YP. Tumor necrosis factor‐α and muscle wasting: a cellular perspective. Respir Res 2001;2:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci U S A 2005;102:18117–18122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down‐regulating MyoD expression. J Biol Chem 2002;277:49831–49840. [DOI] [PubMed] [Google Scholar]
- 11. McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self‐renewal. J Cell Biol 2003;162:1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fulster S, Tacke M, Sandek A, Ebner N, Tschope C, Doehner W, et al. Muscle wasting in patients with chronic heart failure: results from the studies investigating co‐morbidities aggravating heart failure (SICA‐HF). Eur Heart J 2013;34:512–519. [DOI] [PubMed] [Google Scholar]
- 13. Mancini DM, Walter G, Reichek N, Lenkinski R, McCully KK, Mullen JL, et al. Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation 1992;85:1364–1373. [DOI] [PubMed] [Google Scholar]
- 14. Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long‐term heart failure. Circulation 1990;81:518–527. [DOI] [PubMed] [Google Scholar]
- 15. Seiler M, Bowen TS, Rolim N, Dieterlen MT, Werner S, Hoshi T, et al. Skeletal muscle alterations are exacerbated in heart failure with reduced compared with preserved ejection fraction: mediated by circulating cytokines? Circ Heart Fail 2016;9. [DOI] [PubMed] [Google Scholar]
- 16. Barreiro E, Jaitovich A. Muscle atrophy in chronic obstructive pulmonary disease: molecular basis and potential therapeutic targets. J Thorac Dis 2018;10:S1415–S1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seymour JM, Spruit MA, Hopkinson NS, Natanek SA, Man WD, Jackson A, et al. The prevalence of quadriceps weakness in COPD and the relationship with disease severity. Eur Respir J 2010;36:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Swallow EB, Reyes D, Hopkinson NS, Man WD, Porcher R, Cetti EJ, et al. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 2007;62:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marra AM, Arcopinto M, Bossone E, Ehlken N, Cittadini A, Grunig E. Pulmonary arterial hypertension‐related myopathy: an overview of current data and future perspectives. Nutr Metab Cardiovasc Dis 2015;25:131–139. [DOI] [PubMed] [Google Scholar]
- 20. Rohrbach S, Yan X, Weinberg E, Hasan F, Bartunek J, Marchionni MA, et al. Neuregulin in cardiac hypertrophy in rats with aortic stenosis: differential expression of erbB2 and erbB4 receptors. Circulation 1999;100:407–412. [DOI] [PubMed] [Google Scholar]
- 21. Kunz WS, Kuznetsov AV, Schulze W, Eichhorn K, Schild L, Striggow F, et al. Functional characterization of mitochondrial oxidative phosphorylation in saponin‐skinned human muscle fibers. Biochim Biophys Acta 1993;1144:46–53. [DOI] [PubMed] [Google Scholar]
- 22. Scheubel RJ, Tostlebe M, Simm A, Rohrbach S, Gellerich FN, Silber RE, et al. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J Am Coll Cardiol 2002;40:2174–2181. [DOI] [PubMed] [Google Scholar]
- 23. Niemann B, Chen Y, Teschner M, Li L, Silber RE, Rohrbach S. Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria. J Am Coll Cardiol 2011;57:577–585. [DOI] [PubMed] [Google Scholar]
- 24. Strucksberg KH, Tangavelou K, Schroder R, Clemen CS. Proteasomal activity in skeletal muscle: a matter of assay design, muscle type, and age. Anal Biochem 2010;399:225–229. [DOI] [PubMed] [Google Scholar]
- 25. Staron RS, Kraemer WJ, Hikida RS, Fry AC, Murray JD, Campos GE. Fiber type composition of four hindlimb muscles of adult Fisher 344 rats. Histochem Cell Biol 1999;111:117–123. [DOI] [PubMed] [Google Scholar]
- 26. Orliaguet G, Langeron O, Coirault C, Fratea S, Coriat P, Riou B. Effects of dantrolene on rat diaphragm muscle during postnatal maturation. Anesthesiology 2001;94:468–474. [DOI] [PubMed] [Google Scholar]
- 27. Morales‐Lopez JL, Aguera E, Miro F, Diz A. Variations in fibre composition of the gastrocnemius muscle in rats subjected to speed training. Histol Histopathol 1990;5:359–364. [PubMed] [Google Scholar]
- 28. Yoshida T, Tabony AM, Galvez S, Mitch WE, Higashi Y, Sukhanov S, et al. Molecular mechanisms and signaling pathways of angiotensin II‐induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int J Biochem Cell Biol 2013;45:2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maarman G, Lecour S, Butrous G, Thienemann F, Sliwa K. A comprehensive review: the evolution of animal models in pulmonary hypertension research; are we there yet? Pulmonary circulation 2013;3:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taraseviciene‐Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death‐dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 2001;15:427–438. [DOI] [PubMed] [Google Scholar]
- 31. Vinke P, Bowen TS, Boekschoten MV, Witkamp RF, Adams V, van Norren K. Anti‐inflammatory nutrition with high protein attenuates cardiac and skeletal muscle alterations in a pulmonary arterial hypertension model. Sci Rep 2019;9:10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015;131:1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dell'Italia LJ. Anatomy and physiology of the right ventricle. Cardiol Clin 2012;30:167–187. [DOI] [PubMed] [Google Scholar]
- 34. Kadoguchi T, Kinugawa S, Takada S, Fukushima A, Furihata T, Homma T, et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol 2015;100:312–322. [DOI] [PubMed] [Google Scholar]
- 35. Tabony AM, Yoshida T, Galvez S, Higashi Y, Sukhanov S, Chandrasekar B, et al. Angiotensin II upregulates protein phosphatase 2Cα and inhibits AMP‐activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension 2011;58:643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL‐6 and serum amyloid A synergy mediates angiotensin II‐induced muscle wasting. J Am Soc Nephrol: JASN 2009;20:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hiscock N, Chan MH, Bisucci T, Darby IA, Febbraio MA. Skeletal myocytes are a source of interleukin‐6 mRNA expression and protein release during contraction: evidence of fiber type specificity. FASEB J 2004;18:992–994. [DOI] [PubMed] [Google Scholar]
- 38. Rezk BM, Yoshida T, Semprun‐Prieto L, Higashi Y, Sukhanov S, Delafontaine P. Angiotensin II infusion induces marked diaphragmatic skeletal muscle atrophy. PLoS ONE 2012;7:e30276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams V, Bowen TS, Werner S, Barthel P, Amberger C, Konzer A, et al. Small‐molecule‐mediated chemical knock‐down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J Cachexia Sarcopenia Muscle 2019;10:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Degens H, Alway SE. Control of muscle size during disuse, disease, and aging. Int J Sports Med 2006;27:94–99. [DOI] [PubMed] [Google Scholar]
- 41. Reid MB. Response of the ubiquitin‐proteasome pathway to changes in muscle activity. Am J Physiol Regul Integr Comp Physiol 2005;288:R1423–R1431. [DOI] [PubMed] [Google Scholar]
- 42. Gielen S, Adams V, Mobius‐Winkler S, Linke A, Erbs S, Yu J, et al. Anti‐inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol 2003;42:861–868. [DOI] [PubMed] [Google Scholar]
- 43. Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, et al. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation 2005;111:1763–1770. [DOI] [PubMed] [Google Scholar]
- 44. Bowen TS, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 2015;6:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Souza RW, Piedade WP, Soares LC, Souza PA, Aguiar AF, Vechetti‐Junior IJ, et al. Aerobic exercise training prevents heart failure‐induced skeletal muscle atrophy by anti‐catabolic, but not anabolic actions. PLoS ONE 2014;9:e110020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tikunov B, Levine S, Mancini D. Chronic congestive heart failure elicits adaptations of endurance exercise in diaphragmatic muscle. Circulation 1997;95:910–916. [DOI] [PubMed] [Google Scholar]
- 47. Behnke BJ, Delp MD, McDonough P, Spier SA, Poole DC, Musch TI. Effects of chronic heart failure on microvascular oxygen exchange dynamics in muscles of contrasting fiber type. Cardiovasc Res 2004;61:325–332. [DOI] [PubMed] [Google Scholar]
- 48. Smith JR, Hageman KS, Harms CA, Poole DC, Musch TI. Effect of chronic heart failure in older rats on respiratory muscle and hindlimb blood flow during submaximal exercise. Respir Physiol Neurobiol 2017;243:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Favier FB, Britto FA, Freyssenet DG, Bigard XA, Benoit H. HIF‐1‐driven skeletal muscle adaptations to chronic hypoxia: molecular insights into muscle physiology. Cell Mol Life Sci 2015;72:4681–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hayot M, Rodriguez J, Vernus B, Carnac G, Jean E, Allen D, et al. Myostatin up‐regulation is associated with the skeletal muscle response to hypoxic stimuli. Mol Cell Endocrinol 2011;332:38–47. [DOI] [PubMed] [Google Scholar]
- 51. Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, et al. Down‐regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 2009;150:286–294. [DOI] [PubMed] [Google Scholar]
- 52. Day K, Shefer G, Richardson JB, Enikolopov G, Yablonka‐Reuveni Z. Nestin‐GFP reporter expression defines the quiescent state of skeletal muscle satellite cells. Dev Biol 2007;304:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Randolph ME, Pavlath GK. A muscle stem cell for every muscle: variability of satellite cell biology among different muscle groups. Front Aging Neurosci 2015;7:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stuelsatz P, Shearer A, Li Y, Muir LA, Ieronimakis N, Shen QW, et al. Extraocular muscle satellite cells are high performance myo‐engines retaining efficient regenerative capacity in dystrophin deficiency. Dev Biol 2015;397:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Inoue N, Kinugawa S, Suga T, Yokota T, Hirabayashi K, Kuroda S, et al. Angiotensin II‐induced reduction in exercise capacity is associated with increased oxidative stress in skeletal muscle. Am J Physiol Heart Circ Physiol 2012;302:H1202–H1210. [DOI] [PubMed] [Google Scholar]
- 56. Scharf G, Heineke J. Finding good biomarkers for sarcopenia. J Cachexia Sarcopenia Muscle 2012;3:145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arrieta H, Hervas G, Rezola‐Pardo C, Ruiz‐Litago F, Iturburu M, Yanguas JJ, et al. Serum myostatin levels are higher in fitter, more active, and non‐frail long‐term nursing home residents and increase after a physical exercise intervention. Gerontology 2019;65:229–239. [DOI] [PubMed] [Google Scholar]
- 58. Bauer J, Morley JE, Schols A, Ferrucci L, Cruz‐Jentoft AJ, Dent E, et al. Sarcopenia: a time for action. An SCWD position paper. J Cachexia Sarcopenia Muscle 2019;10:956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Figure S1: Changes in mRNA expression in rat skeletal muscle
Figure S2: Changes in protein ubiquitination in rat skeletal muscle
Figure S3: Mitochondrial function in diaphragm
Figure S4: Mitochondrial function in gastrocnemius muscle
Figure S5: Respiratory chain protein expression in diaphragm
Figure S6: Respiratory chain protein expression in gastrocnemius muscle
Figure S7: Comparison of angiotensin receptor mRNA expression in RSkMC and in selected rat skeletal muscles