

Abstract
Background
Our evolutionary history is defined, in part, by our ability to survive times of nutrient scarcity. The outcomes of the metabolic and behavioural adaptations during starvation are highly efficient macronutrient allocation, minimization of energy expenditure, and maximized odds of finding food. However, in different contexts, caloric deprivation is met with vastly different physiologic and behavioural responses, which challenge the primacy of energy homeostasis.
Methods
We conducted a literature review of scientific studies in humans, laboratory animals, and non‐laboratory animals that evaluated the physiologic, metabolic, and behavioural responses to fasting, starvation, protein‐deficient or essential amino acid‐deficient diets, and cachexia. Studies that investigated the changes in ingestive behaviour, locomotor activity, resting metabolic rate, and tissue catabolism were selected as the focus of discussion.
Results
Whereas starvation responses prioritize energy balance, both protein malnutrition and cachexia present existential threats that induce unique adaptive programmes, which can exacerbate the caloric insufficiency of undernutrition. We compare and contrast the behavioural and metabolic responses and elucidate the mechanistic pathways that drive state‐dependent alterations in energy seeking and partitioning.
Conclusions
The evolution of energetically inefficient metabolic and behavioural responses to protein malnutrition and cachexia reveal a hierarchy of metabolic priorities governed by discrete regulatory networks.
Keywords: Starvation, Cachexia, Metabolism, Evolution, Protein malnutrition
[Correction added on 28 October 2020 after first online publication: Summary figure has been added in this current version.]
Introduction
Appropriate nutrient consumption is an essential process in sustaining normal biological processes. Both small and extreme fluctuations in caloric intake result in changes in metabolism and behaviour in effort to maintain organismal homeostasis. During calorically plentiful states, organisms activate energy‐consuming anabolic pathways and satiety behaviours, while states of caloric deprivation result in energy‐liberating catabolic pathways and foraging behaviours. 1 , 2 These metabolic programmes and behaviours are evolutionarily conserved responses that were shaped to endure periods of famine. 3 Specifically, starvation results in metabolic adaptations that decrease energy expenditure and conserve protein stores in order to preserve organ function, while increasing appetite and foraging behaviours in attempt to correct the underlying nutritional deficiency. However, in the context of protein‐specific malnutrition or disease‐associated cachexia, the metabolic and behavioural responses to nutrient insufficiency differ from simple starvation. For example, in both protein malnutrition and cachexia, catabolic processes are activated, and macronutrient intake is paradoxically suppressed, violating the rule of energy conservation. While decades of research demonstrate that the physiologic and behavioural responses activated during starvation serve to spare energy stores and restrict energy expenditure, recent discoveries regarding the physiology and behavioural neuroscience of protein malnutrition and cachexia reveal additional levels of metabolic regulation that lend insight into the pressures that guided metabolic pathway evolution.
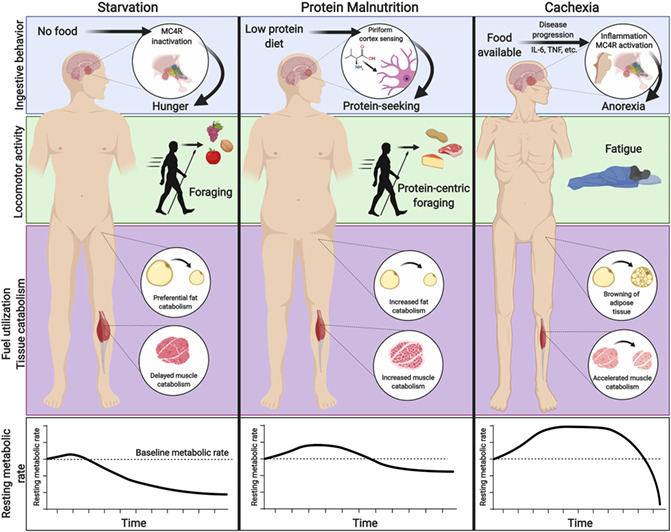
Simple starvation, defined herein as pure caloric deficit in an otherwise healthy organism, activates programmes that prioritize metabolic efficiency, thereby promoting resilience and survival. Less is known about protein malnutrition and cachexia, and recent evidence suggests that the metabolic programmes of these states in response to caloric deficit are broadly inefficient. Furthermore, these three states of nutrient deprivation result in unique behavioural responses that either complement or contradict the overall nutrient requirement of the organism (Figure 1). In this review, we will discuss the disparate physiologic responses of the metabolic states of simple starvation, protein malnutrition, and cachexia, with a particular focus on our current understanding of metabolism, neurophysiology, and behavioural outputs.
Figure 1.
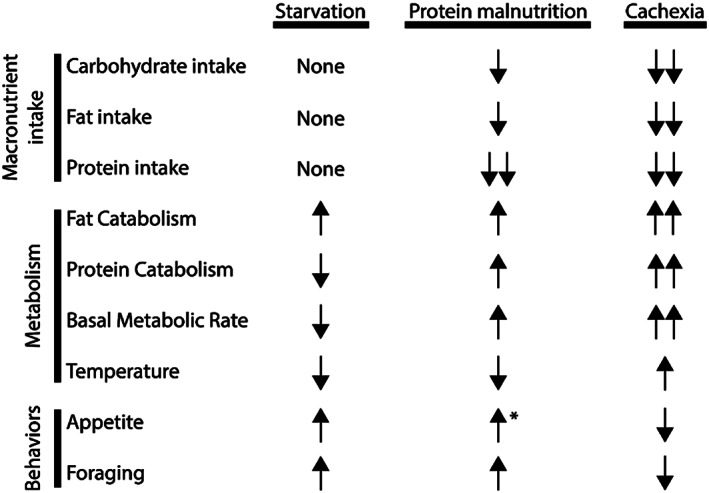
An overview of macronutrient intake, tissue metabolism, and behavioural changes observed during simple starvation, protein deficiency, and cachexia. *Increased appetite to protein‐rich foods, yet active rejection of protein‐poor foods.
Starvation
Metabolic homeostasis is maintained by a well‐described network involving regions in the hypothalamus and brainstem that respond to hormonal and metabolic signals of both short‐term and long‐term energy supply and engage appropriate behavioural and physiologic programmes. 4 Under physiologic conditions, mammals are able to match cumulative energy intake with energy expenditure with exceptional precision because of tight control of tissue metabolism and feeding behaviours by the central nervous system. 5 , 6 Because nutrient scarcity represented an existential threat to organisms throughout their evolution, these systems prioritize efficiency and energy storage to maximize both countermeasures against and resilience to undernutrition. This response is defined behaviourally by an increase in appetite and foraging behaviour and metabolically by a decrease in basal metabolism and preferential catabolism of adipose over lean tissue. 7 , 8 Although this imbalanced response to nutrient availability likely made humans more vulnerable to obesity in the context of high nutrient availability, it ensures that the long‐term effects of nutrient insufficiency are minimized. In this review, we use simple starvation to introduce the homeostatic response to nutritional insufficiency, which will then serve as our comparator when discussing the respective situations of protein malnutrition and cachexia.
Ingestive behaviour
Secreted peripheral factors alter neuronal activity and feeding behaviours during starvation, with decades of research demonstrating the influence of gut‐secreted and fat‐secreted neuropeptides on the mediobasal hypothalamus (MBH). 9 Here, we will briefly discuss the well‐studied endocrine molecules ghrelin and leptin that are known to play roles in driving behaviours of feeding at least in part through their direct, yet opposing, mechanisms on hypothalamic neurons. Other peripherally secreted hormones that influence food intake under physiologic conditions are summarized in Table 1. During fasting and starvation, the stomach releases the peptide hormone ghrelin, the only known circulating hormone that stimulates appetite. After secretion by the stomach, acylation of ghrelin is required for its binding to its receptor, the growth‐hormone‐secretagogue receptor, and for its ability to cross the blood–brain barrier. 10 Once in the brain, ghrelin stimulates appetite through interaction with appetite‐regulating neurons in the MBH. These include neurons that inhibit food intake [pro‐opiomelanocortin (POMC) neurons], and appetite‐stimulating neurons expressing neuropeptide Y and agouti‐related protein (AgRP), known collectively as the melanocortin system. The melanocortin system exerts many of its effects through regulation of activity at the Type 4 melanocortin receptor (MC4R), which is expressed in numerous brain regions. POMC neurons release the MC4R agonist neurotransmitter alpha‐melanocyte stimulating hormone, whereas AgRP neurons directly inhibit POMC neuronal activity and also release the MC4R inverse agonist AgRP at most MC4R expressing neurons. Ghrelin induces food intake primarily via activation of neurons expressing neuropeptide Y/AgRP neurons in the arcuate nucleus of the MBH, 11 , 12 , 13 which in turn send projections to numerous nuclei within the hypothalamus, including the paraventricular, ventromedial, dorsomedial, and lateral hypothalamus, as well as nuclei outside of the hypothalamus, including the nucleus of tractus solitarii and parabrachial nucleus (PBN). 14 Conversely, the anorexigenic adipokine leptin is markedly reduced during starvation. 15 Leptin functions as a long‐term signal of energy status, with circulating levels proportional to total adipose stores. 16 Whereas the presence of leptin is permissive of normal caloric intake and neuroendocrine function, a fall in leptin levels signals a loss of long‐term energy stores. Correspondingly, decreased leptin is associated with decreased anorexigenic POMC neuronal activity, thereby triggering hunger and accompanying physiologic responses during starvation. 17 This neuroendocrine interplay between rising levels of ghrelin and falling levels of leptin synergistically increases appetite during starvation. This intricate balance between upregulation of orexigenic and downregulation of anorexigenic molecules is a unifying theme of starvation neurophysiology.
Table 1.
Additional endocrine molecules that mediate food intake
| Hormone | Source | Signalling mechanism(s) |
|---|---|---|
| Cholecystokinin (CCK) | Enteroendocrine cells of the duodenum and jejunum | Peripheral vagal afferent receptors and transmission of signals to nucleus of the solitary tract; Melanocortin system 198 |
| Glucagon‐like peptide‐1 (GLP1) | L cells of distal small and large intestine | nucleus of the solitary tract in the brainstem and the paraventricular nucleus of the hypothalamus; glucose regulation 199 , 200 |
| Peptide YY (PYY) | endocrine L cells of the gut | Hypothalamic melanocortin system*; Aversive response; protein‐dependent satiety 201 , 202 |
| Glucocorticoids | Adrenal gland | Unclear, but potentially permissive in the orexigenic effect of AgRP in the hypothalamus 203 , 204 |
| Insulin | Endocrine pancreas | Hypothalamic melanocortin system 205 , 206 |
Locomotor activity
The regulation of activity in response to starvation is somewhat more complex than that of appetite. On one hand, voluntary activity increases energy usage and exacerbates energy debt in the absence of food intake. Yet, for nearly all of human existence, survival depended on the ability to forage or to efficiently locate, acquire, and consume food. 18 As such, foraging is deemed an obligate life history strategy, and a species' ability to recognize when foraging is beneficial or detrimental is a part of its evolutionary code. 19 In general, starvation increases foraging behaviours when the likelihood of a meal is increased, yet limits foraging and movement when prey or food is limited. 20 , 21 During calorie deprivation, hyperactivity and increased foraging behaviour are readily observed in rodents, wherein they exhibit stereotypic food anticipatory activity in the hours preceding mealtime. 22 Although the neural pathways underlying this response are incompletely understood, this behaviour is associated with concurrent increases in hypothalamic turnover of norepinephrine, dopamine, and serotonin. 23 The neuropeptide orexin‐A, released by neurons located in the lateral and perifornical hypothalamus, is required for fasting‐associated activity increases. Furthermore, recent mouse work demonstrated that AgRP neurons in the MBH are themselves activated during starvation and are capable of driving foraging behaviour. 24 Orexin neurons reciprocally regulate both hypothalamic AgRP neurons and catecholaminergic neurons in the locus coeruleus, establishing a brainstem‐to‐hypothalamus arousal loop that appears to mediate fasting‐associated foraging. 25 , 26 The degree of hyperactivity and foraging is balanced between fear of predation and likelihood of feeding, and these processes are influenced, in part, through amygdala circuitry. 27
Rodents will increase their locomotor activity (foraging) when calorie availability is restricted, but access to at least some nutrition is maintained. In contrast, complete removal of food causes a triphasic response in weight loss and locomotor activity in rodents. 28 , 29 , 30 The short‐lived first phase is defined by early rapid weight loss and a decline in daily activity within 24 h of fasting initiation. During a prolonged second phase, ongoing suppression of activity is associated with low rates of protein turnover, high dependence on lipid oxidation, and relatively steady body mass. Upon exhaustion of adipose depots, fasted rodents then show a profound rise in locomotor activity in the third phase, associated with rapid weight loss and protein catabolism. 31 The duration of the energy‐conserving second phase is age dependent, longest in older animals that have larger adipose depots. Similar responses were shown in other species, most notably migratory birds and emperor penguins, in which the metabolic shift from lipid to protein catabolism is a signal of expiring energy stores that switches behavioural programme from conservation to active foraging. 32 , 33 Collectively, these observations demonstrate the clear link between voluntary activity and energy balance and reveal an evolutionarily conserved mechanism whereby activity is regulated both by food availability and by long‐term energy stores.
Resting metabolic rate
Energy conservation is a key component of the adaptive response to starvation. Resting metabolic rate (RMR), the energy used to maintain body temperature, repair organs and tissues, maintain ion gradients, and support cardiorespiratory function, accounts for approximately two‐thirds of total energy expenditure. 34 Therefore, the RMR represents the greatest potential reservoir for energy conservation. Indeed, decades of research demonstrate that one of the main adaptations in humans and other species to nutrient deprivation is to reduce RMR. RMR is proportional to an animal's lean body mass, as lean tissues are far more metabolically active than adipose tissue. The suppression of RMR seen in response to even prolonged starvation exceeds that which can be explained by pure loss of lean mass, indicating that RMR depression is an active conservation strategy. This is known as ‘adaptive thermogenesis’, because heat generation is the principal component of resting energy expenditure that is modulated in response to feeding. One of the earliest reports of this process showed the basal metabolic rate (equal to the RMR upon waking, while fasted and at rest) of a man who fasted for 42 days, with intake limited to water, lemonade, or beer. Basal metabolism progressively decreased until the end of his fast, at which time, his resting energy expenditure was approximately half of that in the fed state. 35 Similarly, the Minnesota experiment challenged normal weight participants through stages of semi‐starvation, restricted refeeding, and ad libitum refeeding, demonstrating excess suppression of RMR during undernutrition. 36 Rats and mice also reduce thermogenesis response to fasting, suggesting that this is an evolutionarily conserved mechanism to preserve body mass. 37 , 38 Suppression of thermogenesis can preserve mass over a large range of caloric deficits, perhaps most recognizably in the context of failure to lose weight during dieting. 39
The RMR is largely under the control of the sympathetic nervous system (SNS). Catecholamines released into the synapse from noradrenergic nerve terminals or systemically from the adrenal medulla increase the rate of cellular metabolism and mobilize fuel stores by stimulating lipolysis in adipocytes and glycogenolysis and gluconeogenesis from the muscle and liver. 40 The SNS is responsive to nutritional status—engaged by overfeeding and suppressed by fasting. 41 This response is seen in the human studies cited above, wherein fasting decreased resting heart rate, a surrogate for decreased sympathetic tone, in addition to its effects on thermogenesis. Indeed, fasting mice and rats also exhibit decreases in heart rate and blood pressure, consistent with decreased SNS activity. 42 , 43 Fasted rats have lower levels and turnover of norepinephrine in the heart, liver, pancreas, and other sympathetically innervated tissues as compared with fed rats. 44 Although these cardiovascular effects can themselves conserve energy, the SNS has direct effects on thermogenesis mediated primarily by brown and white adipose tissue (WAT) via the β3 adrenoceptor. 45 SNS activation stimulates uncoupled oxidative respiration via the expression of uncoupling protein 1 (UCP1), leading to non‐shivering thermogenesis in brown adipose tissue (BAT). 46 Simultaneously, adrenergic input induces lipolysis in WAT, thereby providing a fuel source for BAT thermogenesis. 47 Although previously thought to only be found in infants, BAT has recently been identified as an important thermogenic tissue in adult humans, as well. 48 , 49 , 50 , 51 , 52 Thus, the decreases in RMR induced by fasting appear to be largely mediated by decreased SNS activity and the resultant restriction of thermogenesis and cardiac output.
Fuel utilization and tissue catabolism
The primary purpose of the metabolic response during fasting and starvation is to provide sufficient energy to the brain and other tissues critical for survival. During starvation, energy in the form of glucose is mobilized during early starvation, with ketone bodies serving as the primary energy source for the heart and brain in prolonged starvation. 53 If fasting proceeds beyond one day in humans, or 8–12 h in mice, hepatic stores of glycogen are rapidly depleted, and catabolism of adipose and muscle tissue serve as the major sources of energy. 54 , 55 Because fat stores are limited in their ability to generate glucose, muscle catabolism is the primary source of hepatic and renal glucose production during starvation through liberation of gluconeogenic amino acids. 56 However, proteins are not a substantial stored energy reserve, and humans evolved to preserve protein by shifting our fuel utilization from glucose to ketone bodies after just 2 days of starvation. 57 These ketone bodies are produced by the liver from lipolysis‐liberated fatty acids and significantly curtail muscle catabolism during starvation. 58 Indeed, humans preferentially catabolize fat stores over skeletal muscle mass during prolonged caloric deficit. 1 If starvation persists after fat stores are depleted, protein catabolism accelerates and can lead to severe wasting, organ failure, and ultimately death. 59 , 60 The preferential catabolism of adipose tissue is largely driven by the endocrine response to starvation. In the short term, falling blood sugar is met with the counter‐regulatory endocrine response, including release of glucocorticoids, glucagon, and growth hormone, and a concomitant decrease in insulin. 61 , 62 This stimulates both gluconeogenesis and lipolysis, engaging a catabolic programme that redistributes stored energy in the absence of food intake. 63 Although full discussion is outside of the scope of this review, a review of the major changes in circulating hormone levels is summarized in Table 2. Taken together, this strategic triaging of energy store utilization during starvation serves to reduce breakdown of proteins while providing adequate energy substrates for the brain and other tissues critical for survival (Figure 2A).
Table 2.
Overview of peripheral hormone response during starvation, protein malnutrition, and cachexia
| Starvation | Protein malnutrition | Cachexia | |
|---|---|---|---|
| Cortisol | Increased 63 | Increased 207 , 208 | Increased 96 , 209 |
| Thyroid hormone | Decreased 210 , 211 , 212 | Decreased 213 | Increased a 143 |
| Parathyroid hormone | Increased 214 | Increased 215 | Increased 138 , 216 |
| Renin–angiotensin aldosterone | Increased 217 , 218 | Increased; minimal excretion 219 | Increased 220 , 221 |
| Norepinephrine | Increased peripherally 222 ; reduced action centrally 223 | Increased peripherally; reduced action centrally 224 | Increased 221 |
| Growth hormone | Increased 61 , 62 | Increased 225 | Increased 226 |
| Insulin | Decreased 222 , 227 | Increased 228 | Decreased a 221 , 229 , 230 , 231 ; impaired secretion |
| IGF1 | Decreased 232 | Decreased 233 | Decreased 234 |
Data are compared with healthy (all human studies) or pair‐fed controls. Trends reported from human studies are italicized.
Compared with non‐cachectic cancer patients.
Figure 2.
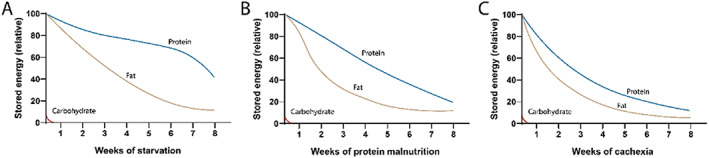
Relative rates of carbohydrate, fat, and protein catabolism during simple starvation (A), protein malnutrition (B), and cachexia (C).
Protein malnutrition
Protein malnutrition represents a special case of undernutrition, in which specific adaptations evolved to ensure adequate intake of amino acids essential for growth, reproduction, and survival. 64 In contrast to simple starvation, which is defined as caloric insufficiency, in protein malnutrition, an imbalance in amino acid content or inadequacy of one or more amino acids drives behavioural and metabolic responses designed to correct the imbalance. Protein malnutrition can refer to a broad range of protein‐deficient diets from total lack of protein content to specific amino acid shortages. For the sake of this review, we will define protein malnutrition as either overall inadequate protein intake—a low protein (LP) diet—or the dietary absence of a single essential amino acid (EAA‐deficient diet), a scenario which best exemplifies the protein‐specific homeostatic circuit. Absence or excess of single amino acids elicit powerful feeding and behavioural effects, which in some cases can exacerbate overall energy imbalance. Imbalances in amino acids are then recovered via the increased catabolism of lean tissues, which may rely upon pathways that increase RMR. This highlights a graded system in which regulation of nutritional composition preferentially drives the response programme over pure caloric content. Below, we discuss the parallel mechanisms that mediate these processes and their interactions with homeostatic systems employed during starvation.
Ingestive behaviour
Animals fed an LP diet display alterations in appetitive behaviours that vary depending on protein content. For example, rats fed a moderately LP diet, with 8–10% of energy as protein, display sustained hyperphagia, prioritizing normalizing protein levels over caloric homeostasis. 65 , 66 , 67 However, rodents consuming either very LP diet (<8% of energy in rats or <5% in mice) or EAA‐deficient diets become hypophagic, even in the context of negative energy balance. 68 , 69 When exposed to such a diet, rats will decrease meal size and increase interfeeding intervals within 20 min of meal onset, mediated by neuronal detection of the EAA imbalance. 70 , 71 Within hours, the rats then develop conditioned taste aversion to the deficient diet while also developing preference for the missing amino acid. 72 , 73 Thus, despite undernutrition, laboratory rodents will paradoxically sustain hypophagia in the presence of EAA‐deficient foods. When given access to foods containing the missing amino acid, or if the deficient EAA is injected into the brain, feeding behaviour rapidly resumes with preference shown for the nutritionally replete food. 74 Upon refeeding, rats will even prefer a protein‐free meal to an amino acid‐imbalanced chow, reinforcing the importance of amino acid composition as a driver of food preference. 75 Chronic hypophagia is therefore driven by aversion to EAA deficiency, not lack of appetite. These observations translate somewhat to human studies, as moderate restriction of dietary protein induces adaptive changes in food intake to restore adequate protein status, but people will not overeat a very LP diet to the point of protein repletion. 76 , 77 These findings led to the hypothesis of a ‘protein‐centric’ feeding paradigm, in which dietary amino acid composition is proposed to be the primary determinant of ingestive behaviour, superseding the drive for energy homeostasis.
The sensing of amino acid deficiency during protein malnutrition is complex and involves both the hypothalamus and the anterior piriform cortex (APC), a region involved in olfaction that is amongst the most primitive parts of the mammalian cortex. 78 , 79 The APC detection mechanism relies on the accumulation of uncharged transfer RNA, which activates the general amino acid control non‐derepressing kinase 2 (GCN2). Mice and drosophila lacking GCN2 do not detect or avoid EAA‐deficient diets, unless this exposure is prolonged. 78 GCN2 phosphroylates eukaryotic initiation factor 2 (EIF2A) in APC neurons, initiating a signalling cascade that functions to block general protein synthesis. 78 , 80 The net effect of this pathway is to reduce GABAergic inhibition in the APC circuit and increase glutamatergic transmission. 81 Although it remains unclear which targets of APC mediate the anorectic response, fMRI assessments in rats show rapid activation of both the ventromedial and lateral hypothalamus, two regions involved in feeding behaviours that receive APC axonal projections. 81 , 82 However, recent conflicting studies question the GCN2/EIF2A‐dependent mechanism in the APC, suggesting that neuronal sensing of amino acids remain incompletely understood. 83 A similar mechanism for EAA detection in the MBH was proposed, supported by the blunting of the anorectic response to a leucine‐deficient diet following adenoviral knockdown of GCN2 in the arcuate nucleus of mice. 84 Although appetite‐regulating neurons in the hypothalamus may directly detect EAA deficiencies, central melanocortin signalling appears to only play a minor role in the acute feeding response. Mice depleted of the MC4R have a slightly attenuated acute anorectic response to EAA‐deficient diet, but neither pharmacologic blockade nor genetic blockade impacts the chronic hypophagia induced by dietary EAA deficiency. 68 This observation illustrates the mechanistic and behavioural divergence between feeding responses—driven principally by aversion—and appetite, which is preserved.
Locomotor activity
Rodents fed EAA‐deficient diets develop rapid and sustained anorexia yet display increased foraging behaviours in effort to find foods containing the needed EAA. 81 This supports the observation that hypophagia observed during EAA‐deficient diet consumption is not reflective of a global decrease in appetitive behaviours. Similar to fasting, both LP diet and EAA deficiency significantly increase locomotor activity compared with rodents fed normal chow. 85 During the initial EAA‐deficient meal, increased activity corresponds temporally to meal termination and is characterized by digging in their food cup, suggesting that the purpose of this activity is to seek new foods. 86 Accordingly, this behaviour is rapidly extinguished upon reintroduction of the deficient EAA, in a process dependent upon normal protein synthesis in the APC. 87
As with the regulation of food intake, the regulation of foraging behaviour because of protein malnutrition is complex. The key site of signal integration appears to be orexin neurons in the lateral hypothalamus, which receive both excitatory projections from the APC and peptidergic projections from the MBH. Disinhibition of APC neurons in response to EAA deficiency then directly activates lateral hypothalamic orexin neurons, which coordinate the locomotor appetitive behaviours (reviewed in Gietzen and Aja 81 ). Karnani and colleagues demonstrate that orexin neurons are also activated by non‐EAAs, which may be increased in the context of EAA deficiency, leading to increased foraging activity. 88 These authors were further able to show that non‐EAAs at physiologic concentrations were able to overcome glucose inhibition of orexin neurons, providing a mechanistic explanation for the activation of foraging in the EAA‐deficient setting despite adequate calorie intake. 88 Collectively, it is clear that protein malnutrition induces foraging behaviours similar to that of a starving animal and, combined with a strong preference for amino acid replete food sources, serve to maximize the animal's chances of rectifying nutrient imbalances.
Resting metabolic rate
The central sensing of amino acid deprivation can regulate energy expenditure and autonomic outflow through mechanisms that are independent of other macronutrients. Dietary leucine deprivation increases thyrotropin‐releasing hormone expression in the hypothalamus, ultimately increasing energy expenditure as indicated by measures of locomotor activity, oxygen consumption, and temperature regulation. 89 This collective increase in thyrotropin‐releasing hormone observed during leucine deprivation results in SNS activation and autonomic outflow to peripheral tissues that increases RMR and lipid catabolism, as reviewed earlier. Specifically, leucine deprivation increases the expression of the β3‐adrenoceptor, Adrb3, as well as Ucp1 in BAT consistent with sympathetic activation of thermogenesis. 90 , 91 Similarly, we found that Ucp1 expression was increased in mice and rats fed with a valine‐deficient diet, and Guo and colleagues showed that both valine and isoleucine deficiency increase lipid mobilization and energy expenditure suggesting that hypermetabolism is a conserved response to amino acid imbalance. 68 , 85 Other investigators demonstrated that leucine deprivation induced the induction of Ucp1 and other markers of thermogenic activation (generally known as ‘browning’) in WAT, via a CNS pathway and activation of sympathetic outflow. 92 The evolutionary benefit of this increase in metabolic rate and metabolic reprogramming of adipose tissue in an undernourished animal is not immediately clear, but may be important to balance the energy demands of foraging, via SNS‐mediated activation of lipolysis. 90
Fuel utilization and tissue catabolism
When compared with starvation, protein malnutrition is broadly associated with an earlier onset and increased protein and fat catabolism (Figure 2B). After being fed a diet deficient in EAA, rodents quickly deplete carbohydrate stores similar to that observed during simple starvation. However, rodents catabolize muscle at a significantly higher rate than their normal chow pair‐fed counterparts, demonstrating that a distinct and independent catabolic pathway is associated with EAA deficiency. 68 Active muscle catabolism releases EAAs into the blood, thereby providing a source of diet‐limited EAAs and allowing for ongoing protein synthesis to maintain essential physiological processes. Mechanistically, this process is driven by the induction of catabolism‐inducing E3 ubiquitin ligases (MuRF1 and MAFbx) in skeletal muscle. These catabolic proteins are upregulated in the muscle of rodents fed an EAA‐deficient diet after just 3 days and remain elevated after nearly 3 weeks. 68 To a lesser extent, this increased catabolic state observed in the muscle compartment for EAA‐deficient animals is also observed in fat tissues. Specifically, rodents on an EAA‐deficient diet catabolize fat stores similarly to pair‐fed controls for the first week, but burn fat at a much higher rate after 2 weeks. 68 This catabolic programme is likely to be influenced both by increased energy expenditure described earlier, along with the actions of circulating glucocorticoids. Although the general trends of hormonal changes associated with protein malnutrition are similar to those found in starvation (Table 2), administration of a valine‐deficient diet increased plasma corticosterone compared with pair‐fed control mice and rats. 68 In response to a leucine‐deficient diet, Xia and colleagues define a novel role of p70 S6 kinase 1 (S6K1) in modulating expression of corticotropin‐releasing hormone in MC4R‐positive hypothalamic neurons. This induction of hypothalamic corticotropin‐releasing hormone expression is essential for stimulating lipolysis in response to leucine deprivation. 93 Furthermore, glucocorticoids play a well‐established role in mediating skeletal muscle catabolism via the induction of the E3 ubiquitin ligases referenced previously in multiple pathophysiologic conditions. 94 , 95 , 96 Although the dependence of muscle catabolism in response to EAA deficiency upon glucocorticoid elevation has not been directly confirmed, the associative data provide compelling evidence that glucocorticoids serve as a unifying endocrine mediator of macronutrient mobilization. Conversely, the levels of the anabolic hormones insulin and insulin‐like growth factor‐1 are decreased in rodents on an EAA‐deficient diet compared with pair‐fed animals, further shifting the metabolic balance towards catabolism. 68 In total, EAA‐deficiency results in a sustained catabolic state of peripheral tissues, liberating fat and muscle stores at a faster pace than simple starvation.
Cachexia
Cachexia is a wasting syndrome associated with a broad range of acute and chronic illnesses, including infection, heart disease, cancer, and chronic inflammatory conditions. Unlike starvation and protein malnutrition, which are dictated by environmental nutrient availability, cachexia results from internal factors and cannot be fully reversed by nutritional supplementation. Cachexia is characterized by the co‐occurrence of anorexia, lethargy, hypermetabolism, and accelerated catabolism. 97 This programme is the result of inflammatory and metabolic signals that reorient the homeostatic mechanisms employed during starvation and protein malnutrition, establishing a hierarchy of context‐specific afferent signals. That cachexia is conserved across species, and inflammatory conditions suggest that it likely evolved as an adaptive response to life‐threatening illness. Indeed, several studies demonstrate survival benefits of this catabolic state during acute infectious processes. 98 , 99 These benefits are traditionally thought to result from the redirection of valuable metabolic resources from the brain and gut to the immune response. 100 Recent work adds that this metabolic programme aids in tissue tolerance during the immune response, preventing end organ dysfunction. 98 , 99 Proinflammatory cytokines are common amongst cachectic conditions and sufficient to drive much of the metabolic physiology. Recent work has expanded the list of afferent mediators contributing to wasting and offered new insights into its aetiology. We contrast the CNS response to cachexia from those of starvation and protein malnutrition and discuss the mediators that drive these divergent programmes.
Ingestive behaviours
Because of the increase in resting energy expenditure during cachexia, an increase in energy intake would be required to offset the overall catabolic state. However, cachexia induces appetite suppression that amplifies the overall energy deficit (Figure 3). In some cases, this presents as frank anorexia, whereas in other cases, a more subtle failure to appropriately increase feeding is observed. 101 , 102 , 103 , 104 The resistance to the effects of energy debt are most notable after substantial weight is lost, when the hypothalamic feeding rheostat would be under increased pressure to reestablish homeostasis. The disinterest in feeding during cachexia is in direct opposition to what is observed in simple starvation or protein malnutrition, each of which results in behaviours that attempt to restore macronutrient homeostasis.
Figure 3.
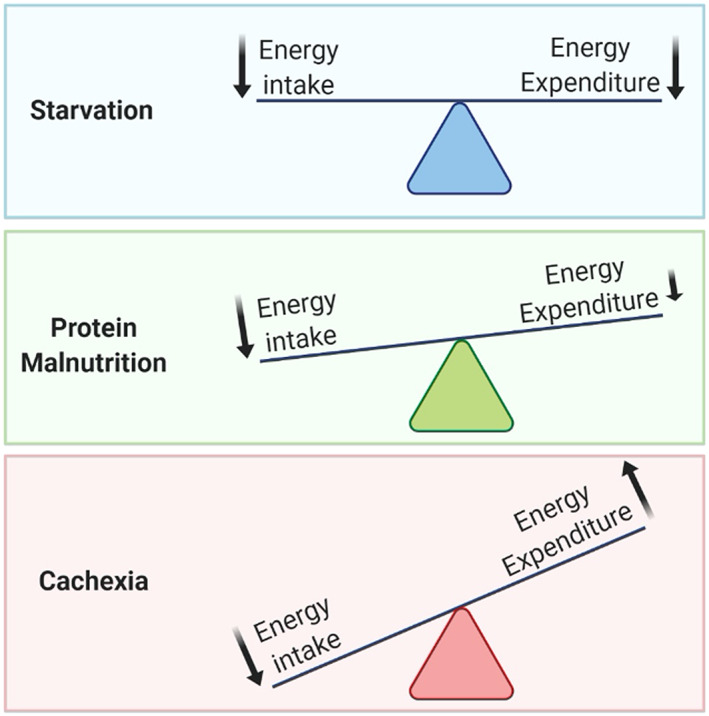
Energy intake and expenditure during starvation, protein malnutrition, and cachexia.
Multiple systemic and central factors converge to inhibit ingestive behaviours, with the hypothalamus and brainstem thought to be the major sites of signal integration. Principal amongst anorectic factors are inflammatory cytokines, including interleukin‐1β (IL‐1β), tumour necrosis factor (TNF), leukaemia inhibitory factor (LIF), Type 1 interferon (IFN), and prostaglandins. 97 , 105 Each of these molecules is independently capable of inducing anorexia when administered either peripherally or directly in the brain. Research in our laboratories and others' over the past two decades demonstrated that a principal site of action for these cytokines is in the MBH, where specialized fenestrated endothelium and localized inflammatory cells allow for the transmission and amplification of peripheral inflammatory signals (reviewed in Burfeind et al. 106 ). These cytokines then act directly or indirectly to increase signalling through the MC4R, in a fashion similar to leptin. 101 Pharmacologic or genetic ablation of MC4R signalling reverses cachexia in multiple acute and chronic laboratory rodent models of cachexia. 107 , 108 , 109 , 110 , 111 , 112
More recent work implicates two populations of neurons in the brainstem in the anorectic component of cachexia. The first consists of neurons in the PBN, located in the pons, which relays noxious stimuli from the viscera to the amygdala as a component of the threat circuit. Activation of PBN calcitonin gene‐related peptide (CGRP)‐expressing neurons potently suppresses appetite. 113 These neurons were activated in two murine models of cancer cachexia and sterile inflammation, and their chemogenetic inhibition was sufficient to reverse the anorexia and weight loss associated with each model. 113 , 114 As PBN CGRP neurons are inhibited by hypothalamic AgRP neurons and express MC4R, they may function as a downstream effector of hypothalamic‐mediated cachexia. 115 , 116 Given their role in relaying aversive signals, these neurons may also have non‐overlapping influences on cachexia–anorexia, generating redundancy in this system. A second brainstem site involved in the transmission of noxious signals is located in the area postrema, a circumventricular organ most well known for its role in nausea. A population of neurons there expresses the GDNF family receptor alpha like, the only known receptor for growth differentiation factor 15 (GDF‐15), a member of the transforming growth factor beta ligand family that suppresses food intake. 117 , 118 , 119 Elevated serum levels of GDF‐15 are found in multiple cachectic states and serum levels correlate with weight loss in prostate cancer. 99 , 120 Treatment of mice with GDF‐15 or implantation with a GDF‐15 overexpressing tumour is sufficient to induce cachexia, whereas treatment with a neutralizing antibody reversed cachexia in multiple murine cancer models. 104 , 121 , 122 Although there is some integration of these anorectic pathways, the existence of multiple disparate mediators represents a level of redundancy found in few biological systems, suggesting that suppression of food intake during illness is an essential response. Indeed, a provocative study by Wang and colleagues demonstrated that improvement in survival in mice with a Salmonella infection required fasting‐induced ketogenesis. 98 Whether decreased nutrient intake provides an adaptive advantage in the contexts of cancer or other conditions remains unclear.
Locomotor activity
Unlike starvation or protein malnutrition, cachexia is associated with a profound lethargy and decrease in both foraging and non‐foraging locomotor activity. Lethargy is amongst the first signs of sickness, often observed prior to the onset of fever in patients with microbial infections or murine models of sterile inflammation. 123 From an evolutionary perspective, this response makes sense both to reserve metabolic resources for the fight against infection and to avoid exposure to the elements or predation while in a weakened state. Cachectic rodents neither exhibit the increase in foraging behaviour seen in other states of undernutrition nor develop anticipatory activity when entrained with time‐restricted feeding paradigms. 124 As described earlier, foraging behaviour is mediated by perifornical/lateral hypothalamic orexin neurons, which receive input from the MBH, APC, and brainstem arousal centres. Orexin neuron activity is decreased in mice treated with lipopolysaccharide and sarcoma‐bearing rats, and intracerebroventricular administration of orexin can restore normal locomotor activity in these models, suggesting that the inhibition of orexin neuron activity underlies sickness‐associated lethargy. 124 This inhibition appears to be mediated by an increase in the activity of local interneurons that express neurotensin, but the link between inflammation and the activity of these neurons remains poorly described. 124 Within the MBH, AgRP neuron activity is necessary to engage in foraging behaviour, yet AgRP neuron activity and peptide release are reduced in cachectic rodents. 24 , 125 , 126 As AgRP neurons are known to share reciprocal projections with the lateral hypothalamus and thought to drive foraging via activation of orexin neurons, the loss of AgRP neuron activity may mediate the downregulation of orexin neuron activity. The aforementioned study that evaluated PBN CGRP neurons also found that their chemogenetic inhibition reversed lethargy. 114 Neurons in the lateral PBN send projections to orexin neurons that then innervate the locus coeruleus, but it remains unclear whether orexin neurons play a role in lateral PBN CGRP‐mediated lethargy or these are two parallel pathways. 127
Decreased activity in cachectic humans and rodents appears to be multifactorial, involving peripheral mechanisms, as well. Muscles from cachectic mice exhibit decreased mass, strength, and function and exhibit early fatigability. 128 , 129 Because inflammatory signalling can drive both skeletal muscle wasting and lethargy, it can be difficult to attribute the decrease in locomotor activity specifically to muscle wasting. However, transgenic mice overexpressing the transcription factor Forkhead box protein O1 (FoxO1), a driver of skeletal muscle catabolism, show reduced muscle mass and spontaneous locomotor activity, suggesting that skeletal muscle loss is sufficient to suppress activity. 130 Cachexia is further associated with metabolic changes that alter fuel mobilization and utilization. Recent work in mice bearing head and neck cancers shows that decreases in locomotor activity can occur independent of central inflammation and prior to significant muscle loss. 131 Metabolic phenotyping in these mice revealed significant changes in carbohydrate metabolism associated with lower blood glucose levels and increased skeletal muscle lactate accumulation, positing an additional contribution of metabolic exhaustion to hypoactivity in cachexia. 132
Resting metabolic rate
The regulation of RMR in cachexia is most thoroughly studied in the context of cancer, with the majority of studies demonstrating normal or increased energy expenditure in a variety of different cancer types. 133 , 134 , 135 , 136 Cancer patients frequently have reduced caloric intake and weight loss, so even ‘normal’ energy expenditure should be considered excessive in this context. RMR is related to the degree of cachexia, with elevated energy expenditure documented prior to overt weight loss, sustained through early cachexia, then declining in those most severely affected (refractory cachexia), 133 , 137 perhaps because of depletion of available metabolic substrates. Indeed, increased fat oxidation is observed in cancer patients, irrespective of weight loss, and browning of WAT is observed in murine cachexia models and in humans with cachexia. 103 , 138 Although direct action of circulating factors (e.g. parathyroid hormone related peptide) can activate thermogenesis and browning of adipose tissue in a subset of cancer types, the most robust and consistent driver of this process is chronic activation of adrenergic sympathetic inputs. A number of studies demonstrate activation of pre‐autonomic neurons in the paraventricular nucleus of the hypothalamus during both the early and late stages of cachexia, suggesting that this is a common mechanism of induction of thermogenesis in this disease. 139 , 140 Although few in number, studies that explored the impact of glucocorticoids on human adipose tissue thermogenesis demonstrated an increase in BAT activation by glucocorticoids, suggesting that this is another mechanism driving increased energy utilization during cachexia. 141 , 142 Furthermore, the hormonal changes associated with cancer cachexia are characterized by increased release of thyroid hormone as compared with non‐cachectic patients, indicating that this may further increase the RMR 143 (Table 2). Collectively, existing data argue that increased metabolic rate (or lack of compensatory metabolic response to insufficient caloric intake) is an important feature of cachexia, driven by a combination of metabolic inefficiency (‘futile’ metabolic cycles) and tissue reprogramming. However, the afferent signals driving these events, as well as the relative contribution of CNS vs. peripheral mechanisms remain poorly described.
Fuel utilization and tissue catabolism
As in starvation and protein malnutrition, cachexia is associated with a global shift from carbohydrate to lipid oxidation in both patients and rodent models of disease. 144 , 145 , 146 Levels of both glucose and lipids are frequently elevated in the blood of cachectic patients and rodents, indicating adequate substrate availability, particularly in early stages of cachexia. 147 , 148 That hyperglycaemia is common in cancer patients despite the tumour's increased glucose avidity, implies a global metabolic reprogramming favouring lipid as a substrate. The relative fuel utilization can be measured by indirect calorimetry and is expressed as the respiratory exchange ratio (RER)—the ratio between the amount of carbon dioxide generated and the amount of oxygen consumed. Multiple studies show a decrease in RER of cachectic patients and rodents, indicating a preference for lipid oxidation over glucose. 103 , 144 , 149 Evidence from models of early cachexia suggests that this transition occurs before substantial weight loss occurs. 144 Lipid oxidation appears to be elevated principally in the skeletal muscle, where excess lipid oxidation may be sufficient to drive muscle wasting. 144 , 150 However, hepatic lipid oxidation, required for ketone generation, is decreased in multiple models of cachexia, demonstrating a tissue‐dependent reprogramming. 151 , 152 This impaired that ketogenesis is hypothesized to be a driver of tissue wasting, but conflicting data exist, with dietary or pharmacologic activation of ketogenesis shown to reverse cachexia in mouse models of lung and pancreatic cancer, but not in a rat sarcoma model or human cancer patients. 151 , 153 , 154 , 155 The use of proteins as a metabolic fuel is more difficult to measure, as RER does not take them into account, and they generally constitute a small contribution to overall metabolism. Although muscle wasting is a cardinal feature of cachexia, levels of serum amino acids and urinary nitrogen excretion are largely unaltered or paradoxically decreased in cachectic patients and rodents. 156 , 157 , 158 Much of the protein mobilized from muscles during cachexia is thought to provide substrate for the hepatic acute phase response to inflammation, a metabolically costly process involving the synthesis and release of bioactive proteins involved in modifying the metabolic environment of the threatened host. Because the amino acid composition of muscle differs substantially from acute phase reactants, it is hypothesized that this drives further wasting to supply adequate levels of the limiting amino acids. 159
In comparison with both starvation and protein malnutrition, cachexia is associated with the greatest muscle and fat catabolism relative to the degree of caloric deficiency (Figure 2C). 102 , 160 Using computational models of cachexia in humans, it is estimated that lipolysis increases by up to 30–80% over baseline, 161 , 162 , 163 while reports suggest muscle catabolism may increase by 40–60%. 164 , 165 , 166 , 167 Muscle loss in cachexia owes to a combination of reduced protein synthesis and increased protein catabolism. 168 The relative influence of altered synthesis and degradation to wasting varies amongst studies, with early reports suggesting that decreased synthesis played a dominant role. 169 , 170 More recent studies clearly established that cachectic patients retain anabolic potential, with a clinical trial showing net gain in muscle mass with the ghrelin mimetic (anamorelin) in patients with cancer cachexia. 171 , 172 , 173 , 174 , 175 Amino acid supplementation in cachectic tumour‐bearing rats increased protein synthesis, yet degradation outpaced gains in protein synthesis. 176 This catabolic programme in skeletal muscle is mediated through the activation of the ubiquitin proteasome pathway and enhanced Mafbx, Murf1, and Foxo1 expression. 177 Although common to all three states of undernutrition, the ubiquitin proteasome pathway is activated to a greater degree in cachexia than in either starvation or protein malnutrition. 68 , 102 The activation of the ubiquitin proteasome pathway in cachexia reflects the influences of direct inflammatory cytokine signalling on muscle, persistently elevated glucocorticoid signalling, and disuse. 178 Multiple in vitro and preclinical studies confirm that inflammatory cytokines, including IL‐1, TNF, and IFNγ, and glucocorticoids, are independently sufficient to induce E3 ubiquitin ligase expression in skeletal muscle, thereby amplifying the catabolic effect of undernutrition. 179 , 180 , 181
Autophagy, the digestion and recycling of cellular contents by the lysosome, also contributes to muscle catabolism. This process is an important component of cellular homeostasis, allowing for the degradation of damaged organelles, toxic protein aggregates, and misfolded proteins. 182 Autophagy is increased following prolonged fasting in mice, but notably is elevated in the muscles of cachectic human mice as well, as evidenced by increased levels of autophagy mediators BNIP3 and LC3B and the autophagy‐promoting transcription factor FOXO3. 183 , 184 , 185 , 186 , 187 , 188 , 189 Similar to the ubiquitin‐proteasome pathway, autophagy can be activated in skeletal muscle by metabolic (calorie restriction), hormonal (glucocorticoid), or inflammatory (cytokine) challenges, illustrating the high degree of conservation in muscle‐intrinsic catabolic mechanisms across contexts. 185 As in skeletal muscle, cardiac wasting can be driven by both the ubiquitin proteasome pathway and increased autophagy. Few extant data support the role of increased MAFbx and MuRF‐1 in hearts from cachectic mice, however, with conflicting reports in mice with cancer cachexia. 102 , 190 , 191 Conversely, autophagy markers LC3‐II, cathepsin L, and beclin are elevated in hearts from rodents with cancer cachexia. 190 , 192 , 193 The relative roles for these pathways in cardiac wasting remain unclear and a topic of active investigation. Ultimately, the cachectic humoral milieu, characterized by increased levels of proinflammatory mediators and glucocorticoids, is able to augment physiologic activation of these catabolic pathways in skeletal and cardiac muscle beyond that of undernutrition alone.
Loss of WAT in cachexia is due to enhanced lipolysis, associated with elevated levels of circulating free fatty acids and glycerol. 194 Lipolysis is mediated by two enzymes in adipocytes—adipose triglyceride lipase (ATGL), which catalyzes the initial hydrolysis of triglycerides to diacylglycerol, and hormone sensitive lipase, which is responsible for the subsequent hydrolysis of diacylglycerol. Although hormone sensitive lipase is generally considered the main inducible driver of lipolysis, deletion of Atgl prevented lipolysis in the B16 melanoma murine model of cancer cachexia. 195 Lipolysis in cachexia is mediated by increased SNS activation and a host of humoral mediators, including TNF, IL‐6, and zinc alpha glycoprotein, each of which is commonly elevated in the serum of cachectic patients or rodents. 194 Cachexia is also associated with browning of WAT to promote thermogenesis via the expression of the UCP1. 103 , 138 In this way, cachexia modifies adipocyte biology both to induce WAT atrophy via lipolysis and to increase metabolic rate through excess energy dissipation. Fat and muscle catabolism are largely studied as independent events in cachexia; however, several murine studies demonstrate that preventing adipose wasting also reversed skeletal muscle loss. 138 , 195 The link between adipose wasting and muscle loss remains unclear but may owe to oxidative stress associated with the increase in fatty oxidation seen in cachectic muscle. 150 When compared with simple starvation and protein malnutrition, the metabolic programmes of cachexia are broadly more catabolic and energetically inefficient, leading to an increase in resting energy expenditure and depletion of metabolic reserves.
Conclusion
Throughout our evolutionary history, humans developed behavioural and biochemical strategies to cope with nutrient scarcity in the context of famine. However, starvation was not the only threat to survival associated with undernutrition, as changes in ingestive behaviours and metabolism are seen in the contexts of protein malnutrition and infection or inflammation, as well. Herein, we sought to summarize general macronutrient utilization and tissue catabolism shifts observed amongst these three metabolic states, as well as the associated deviations in neurophysiology and behaviours that serve to rectify (or propagate) nutritional imbalances. In the context of simple starvation and protein malnutrition, the metabolic and neuroendocrine responses induce changes in behaviour and metabolism that facilitate correction of the nutritional deficiencies. Throughout the spectrum of starvation and protein malnutrition, the brain receives both local neuroendocrine signals and distant neuroendocrine signals to interpret the body's overall nutritional state and modulates behaviours and motivations in attempt to balance energy needs, with the requirement to balance amino acid composition eclipsing the drive to maintain overall caloric sufficiency. This hierarchy may seem somewhat surprising and may suggest that EAAs and not simple energy equivalents are the limiting nutritional reagent for organismal survival and replication.
The constellation of metabolic and behavioural responses observed during cachexia represent a highly coordinated series of adaptations designed to survive acute insults by shifting priorities to both combat and tolerate the inflammatory challenge. As systemic infection represented the most salient existential threat to animals in the pre‐antibiotic era, the reorganization of metabolism around disease survival provides a teleological narrative for this paradoxical response to energy depletion. From an evolutionary perspective, humans rarely lived long enough to develop chronic diseases associated with cachexia. Because the metabolic alterations of cachexia, including browning of adipose tissue, skeletal muscle, and adipose catabolism, and elevated basal metabolic rate lead to a severe mismatch in energy balance, it is commonly thought that these responses become maladaptive when engaged over a prolonged period, as during chronic disease. Furthermore, the sickness behaviours of cachexia (including appetite suppression, fatigue, and debility) significantly impact patients' quality of life, stimulating efforts to develop treatments aimed specifically towards reversing cachexia. However, future research may yet show advantages to this physiology in chronic cachectic conditions.
We recognize the important contributions that cognitive, emotional, and hedonic inputs play in feeding motivation during both normal physiology and pathology. Because of the widely varying influences these psychosocial inputs play in both energy metabolism and feeding, we chose to focus solely on the brain's integration of metabolic and neuroendocrine cues during starvation, protein malnutrition, and cachexia, with a particular focus on neurological pathways that are distinct amongst these three metabolic states. Nearly all of the mechanistic data discussed in this review are derived from studies in rodents. It is clear that these responses are likely to be heavily modified by telencephalic inputs in humans, which may provide an additional level of context matching to enhance survival.
In starvation, classical neuroendocrine cues, such as gut‐derived and fat‐derived hormones, are predominantly responsible for organismal metabolic and behavioural outputs. During protein malnutrition, the direct sensing of amino acid imbalances through recently identified neuronal pathways in the APC likely play a direct role in driving peripheral tissue catabolism. The neuroscience of cachexia is defined by the production of cachexia‐promoting factors that are not regulated by nutritional stress alone, but by systemic inflammation because of the underlying disease. The CNS‐based pathways that regulate energy homeostasis during cachexia remains an area of active investigation, but a growing body of evidence demonstrates the capacity of the brain to recognize peripheral mediators of sickness, amplify these signals in circumventricular structures, and modulate energy homeostasis through appetite regulation and neuroendocrine and autonomic control of peripheral tissue metabolism. 97 , 106 , 196 Collectively, the neurophysiology of cachexia exacerbates energy losses, whereas the neurophysiology of simple starvation and protein malnutrition ultimately serve to rectify energy imbalances (Figure 3). As evolution is driven by competing pressures, these divergent responses are the result of a natural hierarchy of needs and illustrate the profound impact that disease had in shaping animal physiology. Future investigations into the metabolism and neuroscience of these metabolic states may identify distinct diverging points in our evolutionary history, unique neurobiological pathways of CNS metabolic control, and offer new insights into the physiologic plasticity needed for long‐term survival of a species.
Funding
This work is supported by the National Cancer Institute grants F30 CA254033 (B.O.), R01 CA217989 (D.L.M.), R01 CA234006 (D.L.M.), and K08 CA245188 (A.J.G.), and the Brenden‐Colson Center for Pancreatic Care (D.L.M. and A.J.G.).
Acknowledgements
This work is supported by the National Cancer Institute grants F30 CA254033 (BO), R01 CA217989 (DLM), R01 CA234006 (DLM), and K08 CA245188 (AJG), and the Brenden‐Colson Center for Pancreatic Care (DLM and AJG). The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 197
The authors have no conflict of interests to disclose.
Olson B., Marks D. L., and Grossberg A. J. (2020) Diverging metabolic programmes and behaviours during states of starvation, protein malnutrition, and cachexia, Journal of Cachexia, Sarcopenia and Muscle, 11, 1429–1446, 10.1002/jcsm.12630
References
- 1. Cahill GF Jr. Starvation in man. N Engl J Med 1970;282:668–675. [DOI] [PubMed] [Google Scholar]
- 2. Cahill GF Jr. Fuel metabolism in starvation. Annu Rev Nutr 2006;26:1–22. [DOI] [PubMed] [Google Scholar]
- 3. Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr 2003;78:361–369. [DOI] [PubMed] [Google Scholar]
- 4. Lowell BB. New neuroscience of homeostasis and drives for food, water, and salt. N Engl J Med 2019;380:459–471. [DOI] [PubMed] [Google Scholar]
- 5. Edholm OG. Energy balance in man studies carried out by the Division of Human Physiology, National Institute for Medical Research. J Hum Nutr 1977;31:413–431. [PubMed] [Google Scholar]
- 6. Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 2000;404:661–671. [DOI] [PubMed] [Google Scholar]
- 7. Lowell BB, Goodman MN. Protein sparing in skeletal muscle during prolonged starvation. Dependence on lipid fuel availability. Diabetes 1987;36:14–19. [DOI] [PubMed] [Google Scholar]
- 8. Rennie MJ, Harrison R. Effects of injury, disease, and malnutrition on protein metabolism in man. Unanswered questions. Lancet 1984;1:323–325. [DOI] [PubMed] [Google Scholar]
- 9. Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature 2006;444:854–859. [DOI] [PubMed] [Google Scholar]
- 10. Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev 2005;85:495–522. [DOI] [PubMed] [Google Scholar]
- 11. Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, et al. A role for ghrelin in the central regulation of feeding. Nature 2001;409:194–198. [DOI] [PubMed] [Google Scholar]
- 12. Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003;37:649–661. [DOI] [PubMed] [Google Scholar]
- 13. Chen SR, Chen H, Zhou JJ, Pradhan G, Sun Y, Pan HL, et al. Ghrelin receptors mediate ghrelin‐induced excitation of agouti‐related protein/neuropeptide Y but not pro‐opiomelanocortin neurons. J Neurochem 2017;142:512–520. [DOI] [PubMed] [Google Scholar]
- 14. Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene‐related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate‐treated mice. Proc Natl Acad Sci U S A 1998;95:15043–15048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short‐term starvation in healthy men. J Clin Invest 2003;111:1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pan WW, Myers MG. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci 2018;19:95–105. [DOI] [PubMed] [Google Scholar]
- 17. Flak JN, Myers MG Jr. Minireview: CNS Mechanisms of Leptin Action. Mol Endocrinol (Baltimore, Md) 2016;30:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brunstrom JM, Cheon BK. Do humans still forage in an obesogenic environment? Mechanisms and implications for weight maintenance. Physiol Behav 2018;193:261–267. [DOI] [PubMed] [Google Scholar]
- 19. Schuppli C, Graber SM, Isler K, van Schaik CP. Life history, cognition and the evolution of complex foraging niches. J Hum Evol 2016;92:91–100. [DOI] [PubMed] [Google Scholar]
- 20. Ross CT, Winterhalder B. Sit‐and‐wait versus active‐search hunting: a behavioral ecological model of optimal search mode. J Theor Biol 2015;387:76–87. [DOI] [PubMed] [Google Scholar]
- 21. Miller JR, Ament JM, Schmitz OJ. Fear on the move: predator hunting mode predicts variation in prey mortality and plasticity in prey spatial response. J Anim Ecol 2014;83:214–222. [DOI] [PubMed] [Google Scholar]
- 22. Mistlberger RE. Neurobiology of food anticipatory circadian rhythms. Physiol Behav 2011;104:535–545. [DOI] [PubMed] [Google Scholar]
- 23. Pirke KM, Broocks A, Wilckens T, Marquard R, Schweiger U. Starvation‐induced hyperactivity in the rat: the role of endocrine and neurotransmitter changes. Neurosci Biobehav Rev 1993;17:287–294. [DOI] [PubMed] [Google Scholar]
- 24. Dietrich MO, Zimmer MR, Bober J, Horvath TL. Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell 2015;160:1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horvath TL, Diano S, van den Pol AN. Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. J Neurosci 1999;19:1072–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Horvath TL, Peyron C, Diano S, Ivanov A, Aston‐Jones G, Kilduff TS, et al. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comp Neurol 1999;415:145–159. [PubMed] [Google Scholar]
- 27. Choi JS, Kim JJ. Amygdala regulates risk of predation in rats foraging in a dynamic fear environment. Proc Natl Acad Sci U S A 2010;107:21773–21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sclafani A, Rendel A. Food deprivation‐induced activity in normal and hypothalamic obese rats. Behav Biol 1978;22:244–255. [DOI] [PubMed] [Google Scholar]
- 29. Jakubczak LF. Age differences in the effects of terminal food deprivation (starvation) on activity, weight loss, and survival of rats. J Gerontol 1967;22:421–426. [PubMed] [Google Scholar]
- 30. Goodman MN, Larsen PR, Kaplan MM, Aoki TT, Young VR, Ruderman NB. Starvation in the rat. II. Effect of age and obesity on protein sparing and fuel metabolism. Am J Physiol 1980;239:E277–e86. [DOI] [PubMed] [Google Scholar]
- 31. Koubi HE, Robin JP, Dewasmes G, Le Maho Y, Frutoso J, Minaire Y. Fasting‐induced rise in locomotor activity in rats coincides with increased protein utilization. Physiol Behav 1991;50:337–343. [DOI] [PubMed] [Google Scholar]
- 32. Robin JP, Boucontet L, Chillet P, Groscolas R. Behavioral changes in fasting emperor penguins: evidence for a "refeeding signal" linked to a metabolic shift. Am J Physiol 1998;274:R746–R753. [DOI] [PubMed] [Google Scholar]
- 33. Ferretti A, Maggini I, Lupi S, Cardinale M, Fusani L. The amount of available food affects diurnal locomotor activity in migratory songbirds during stopover. Sci Rep 2019;9:19027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dulloo AG. Explaining the failures of obesity therapy: willpower attenuation, target miscalculation or metabolic compensation? Int J Obes (Lond) 2012;36:1418–1420. [DOI] [PubMed] [Google Scholar]
- 35. Kleitman N. BASAL METABOLISM IN PROLONGED FASTING IN MAN. Am J Physiol Legacy Content 1926;77:233–244. [Google Scholar]
- 36. Keys A, Brožek J, Henschel A, Mickelsen O, Taylor HL. The biology of human starvation, Vol. 2 Oxford, England: Univ. of Minnesota Press; 1950. [Google Scholar]
- 37. Overton JM, Williams TD, Chambers JB, Rashotte ME. Cardiovascular and metabolic responses to fasting and thermoneutrality are conserved in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 2001;280:R1007–R1015. [DOI] [PubMed] [Google Scholar]
- 38. Tanner JM, Kearns DT, Kim BJ, Sloan C, Jia Z, Yang T, et al. Fasting‐induced reductions in cardiovascular and metabolic variables occur sooner in obese versus lean mice. Exp Biol Med (Maywood) 2010;235:1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenbaum M, Hirsch J, Gallagher DA, Leibel RL. Long‐term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am J Clin Nutr 2008;88:906–912. [DOI] [PubMed] [Google Scholar]
- 40. Young JB, Landsberg L. Catecholamines and intermediary metabolism. Clin Endocrinol Metab 1977;6:599–631. [DOI] [PubMed] [Google Scholar]
- 41. Landsberg L, Young JB. Fasting, feeding and regulation of the sympathetic nervous system. N Engl J Med 1978;298:1295–1301. [DOI] [PubMed] [Google Scholar]
- 42. Einhorn D, Young JB, Landberg L. Hypotensive effect of fasting: possible involvement of the sympathetic nervous system and endogenous opiates. Science 1982;217:727–729. [DOI] [PubMed] [Google Scholar]
- 43. Swoap SJ. Altered leptin signaling is sufficient, but not required, for hypotension associated with caloric restriction. Am J Physiol Heart Circ Physiol 2001;281:H2473–H2479. [DOI] [PubMed] [Google Scholar]
- 44. Young JB, Landsberg L. Suppression of sympathetic nervous system during fasting. Science 1977;196:1473–1475. [DOI] [PubMed] [Google Scholar]
- 45. Cypess AM, Weiner LS, Roberts‐Toler C, Franquet Elía E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a β3‐adrenergic receptor agonist. Cell Metab 2015;21:33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oelkrug R, Polymeropoulos ET, Jastroch M. Brown adipose tissue: physiological function and evolutionary significance. J Comp Physiol B 2015;185:587–606. [DOI] [PubMed] [Google Scholar]
- 47. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med 2011;17:200–205. [DOI] [PubMed] [Google Scholar]
- 48. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold‐activated brown adipose tissue in healthy men. N Engl J Med 2009;360:1500–1508. [DOI] [PubMed] [Google Scholar]
- 49. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 2009;360:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med 2009;360:1518–1525. [DOI] [PubMed] [Google Scholar]
- 51. Saito M, Okamatsu‐Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio‐Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 2009;58:1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, et al. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J 2009;23:3113–3120. [DOI] [PubMed] [Google Scholar]
- 53. Puchalska P, Crawford PA. Multi‐dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab 2017;25:262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 1991;254:573–576. [DOI] [PubMed] [Google Scholar]
- 55. Felig P, Owen OE, Wahren J, Cahill GF Jr. Amino acid metabolism during prolonged starvation. J Clin Invest 1969;48:584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF Jr. Liver and kidney metabolism during prolonged starvation. J Clin Invest 1969;48:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Berg JM, Tymoczko JL, Stryer L. Food Intake and Starvation Induce Metabolic Changes. New York: W H Freeman; 2002. [Google Scholar]
- 58. Garber AJ, Menzel PH, Boden G, Owen OE. Hepatic ketogenesis and gluconeogenesis in humans. J Clin Invest 1974;54:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Henry CJ. Body mass index and the limits of human survival. Eur J Clin Nutr 1990;44:329–335. [PubMed] [Google Scholar]
- 60. Wang T, Hung CC, Randall DJ. The comparative physiology of food deprivation: from feast to famine. Annu Rev Physiol 2006;68:223–251. [DOI] [PubMed] [Google Scholar]
- 61. Gahete MD, Córdoba‐Chacón J, Luque RM, Kineman RD. The rise in growth hormone during starvation does not serve to maintain glucose levels or lean mass but is required for appropriate adipose tissue response in female mice. Endocrinology 2013;154:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marks V, Howorth N, Greenwood FC. Plasma growth‐hormone levels in chronic starvation in man. Nature 1965;208:686–687. [Google Scholar]
- 63. Duclos M, Corcuff JB, Roger P, Tabarin A. The dexamethasone‐suppressed corticotrophin‐releasing hormone stimulation test in anorexia nervosa. Clin Endocrinol (Oxf) 1999;51:725–731. [DOI] [PubMed] [Google Scholar]
- 64. Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell 2015;161:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest 2014;124:3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morrison CD, Xi X, White CL, Ye J, Martin RJ. Amino acids inhibit Agrp gene expression via an mTOR‐dependent mechanism. Am J Physiol Endocrinol Metab 2007;293:E165–E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. White BD, Porter MH, Martin RJ. Effects of age on the feeding response to moderately low dietary protein in rats. Physiol Behav 2000;68:673–681. [DOI] [PubMed] [Google Scholar]
- 68. Zhu X, Krasnow SM, Roth‐Carter QR, Levasseur PR, Braun TP, Grossberg AJ, et al. Hypothalamic signaling in anorexia induced by indispensable amino acid deficiency. Am J Physiol Endocrinol Metab 2012;303:E1446–E1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gietzen DW, Hao S, Anthony TG. Mechanisms of food intake repression in indispensable amino acid deficiency. Annu Rev Nutr 2007;27:63–78. [DOI] [PubMed] [Google Scholar]
- 70. Koehnle TJ, Russell MC, Gietzen DW. Rats rapidly reject diets deficient in essential amino acids. J Nutr 2003;133:2331–2335. [DOI] [PubMed] [Google Scholar]
- 71. Leung PM, Larson DM, Rogers QR. Food intake and preference of olfactory bulbectomized rats fed amino acid imbalanced or deficient diets. Physiol Behav 1972;9:553–557. [DOI] [PubMed] [Google Scholar]
- 72. Mori M, Kawada T, Ono T, Torii K. Taste preference and protein nutrition and l‐amino acid homeostasis in male Sprague‐Dawley rats. Physiol Behav 1991;49:987–995. [DOI] [PubMed] [Google Scholar]
- 73. Feurté S, Tomé D, Gietzen DW, Even PC, Nicolaïdis S, Fromentin G. Feeding patterns and meal microstructure during development of a taste aversion to a threonine devoid diet. Nutr Neurosci 2002;5:269–278. [DOI] [PubMed] [Google Scholar]
- 74. Naito‐Hoopes M, McArthur LH, Gietzen DW, Rogers QR. Learned preference and aversion for complete and isoleucine‐devoid diets in rats. Physiol Behav 1993;53:485–494. [DOI] [PubMed] [Google Scholar]
- 75. Sanahuja JC, Harper AE. Effect of amino acid imbalance on food intake and preference. Am J Physiol 1962;202:165–170. [DOI] [PubMed] [Google Scholar]
- 76. Griffioen‐Roose S, Mars M, Siebelink E, Finlayson G, Tome D, de Graaf C. Protein status elicits compensatory changes in food intake and food preferences. Am J Clin Nutr 2012;95:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Martens EA, Tan SY, Dunlop MV, Mattes RD, Westerterp‐Plantenga MS. Protein leverage effects of beef protein on energy intake in humans. Am J Clin Nutr 2014;99:1397–1406. [DOI] [PubMed] [Google Scholar]
- 78. Hao S, Sharp JW, Ross‐Inta CM, McDaniel BJ, Anthony TG, Wek RC, et al. Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science 2005;307:1776–1778. [DOI] [PubMed] [Google Scholar]
- 79. Rudell JB, Rechs AJ, Kelman TJ, Ross‐Inta CM, Hao S, Gietzen DW. The anterior piriform cortex is sufficient for detecting depletion of an indispensable amino acid, showing independent cortical sensory function. J Neurosci 2011;31:1583–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hao S, Ross‐Inta CM, Gietzen DW. The sensing of essential amino acid deficiency in the anterior piriform cortex, that requires the uncharged tRNA/GCN2 pathway, is sensitive to wortmannin but not rapamycin. Pharmacol Biochem Behav 2010;94:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gietzen DW, Aja SM. The brain's response to an essential amino acid‐deficient diet and the circuitous route to a better meal. Mol Neurobiol 2012;46:332–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yokawa T, Tabuchi E, Takezawa M, Ono T, Torii K. Recognition and neural plasticity responding to deficient nutrient intake scanned by a functional MRI in the brain of rats with l‐lysine deficiency. Obes Res 1995;3:685s–688s. [DOI] [PubMed] [Google Scholar]
- 83. Leib DE, Knight ZA. Re‐examination of dietary amino acid sensing reveals a GCN2‐independent mechanism. Cell Rep 2015;13:1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bjordal M, Arquier N, Kniazeff J, Pin JP, Léopold P. Sensing of amino acids in a dopaminergic circuitry promotes rejection of an incomplete diet in Drosophila . Cell 2014;156:510–521. [DOI] [PubMed] [Google Scholar]
- 85. Du Y, Meng Q, Zhang Q, Guo F. Isoleucine or valine deprivation stimulates fat loss via increasing energy expenditure and regulating lipid metabolism in WAT. Amino Acids 2012;43:725–734. [DOI] [PubMed] [Google Scholar]
- 86. Rozin P. Specific aversions as a component of specific hungers. J Comp Physiol Psychol 1967;64:237–242. [DOI] [PubMed] [Google Scholar]
- 87. Beverly JL 3rd, Gietzen DW, Rogers QR. Protein synthesis in the prepyriform cortex: effects on intake of an amino acid‐imbalanced diet by Sprague‐Dawley rats. J Nutr 1991;121:754–761. [DOI] [PubMed] [Google Scholar]
- 88. Karnani MM, Apergis‐Schoute J, Adamantidis A, Jensen LT, de Lecea L, Fugger L, et al. Activation of central orexin/hypocretin neurons by dietary amino acids. Neuron 2011;72:616–629. [DOI] [PubMed] [Google Scholar]
- 89. Xia T, Zhang Q, Xiao Y, Wang C, Yu J, Liu H, et al. CREB/TRH pathway in the central nervous system regulates energy expenditure in response to deprivation of an essential amino acid. Int J Obes (Lond) 2015;39:105–113. [DOI] [PubMed] [Google Scholar]
- 90. Cheng Y, Meng Q, Wang C, Li H, Huang Z, Chen S, et al. Leucine deprivation decreases fat mass by stimulation of lipolysis in white adipose tissue and upregulation of uncoupling protein 1 (UCP1) in brown adipose tissue. Diabetes 2010;59:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cheng Y, Zhang Q, Meng Q, Xia T, Huang Z, Wang C, et al. Leucine deprivation stimulates fat loss via increasing CRH expression in the hypothalamus and activating the sympathetic nervous system. Mol Endocrinol 2011;25:1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yuan F, Jiang H, Yin H, Jiang X, Jiao F, Chen S, et al. Activation of GCN2/ATF4 signals in amygdalar PKC‐δ neurons promotes WAT browning under leucine deprivation. Nat Commun 2020;11:2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Xia T, Cheng Y, Zhang Q, Xiao F, Liu B, Chen S, et al. S6K1 in the central nervous system regulates energy expenditure via MC4R/CRH pathways in response to deprivation of an essential amino acid. Diabetes 2012;61:2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, et al. Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J 2013;27:3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Braun TP, Szumowski M, Levasseur PR, Grossberg AJ, Zhu X, Agarwal A, et al. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014;9:e106489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Braun TP, Zhu X, Szumowski M, Scott GD, Grossberg AJ, Levasseur PR, et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic‐pituitary‐adrenal axis. J Exp Med 2011;208:2449–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grossberg AJ, Scarlett JM, Marks DL. Hypothalamic mechanisms in cachexia. Physiol Behav 2010;100:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 2016;166:1512–1525, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, et al. GDF15 is an inflammation‐induced central mediator of tissue tolerance. Cell 2019;178:1231–44.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 2008;9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Grossberg AJ, Scarlett JM, Zhu X, Bowe DD, Batra AK, Braun TP, et al. Arcuate nucleus proopiomelanocortin neurons mediate the acute anorectic actions of leukemia inhibitory factor via gp130. Endocrinology 2010;151:606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, et al. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 2017;8:824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Petruzzelli M, Schweiger M, Schreiber R, Campos‐Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer‐associated cachexia. Cell Metab 2014;20:433–447. [DOI] [PubMed] [Google Scholar]
- 104. Suriben R, Chen M, Higbee J, Oeffinger J, Ventura R, Li B, et al. Antibody‐mediated inhibition of GDF15‐GFRAL activity reverses cancer cachexia in mice. Nat Med 2020;26 8:1264–1270. [DOI] [PubMed] [Google Scholar]
- 105. Wang A, Luan HH, Medzhitov R. An evolutionary perspective on immunometabolism. Science 2019;363:eaar3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Burfeind KG, Michaelis KA, Marks DL. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin Cell Dev Biol 2016;54:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Marks DL, Ling N, Cone RD. Role of the central melanocortin system in cachexia. Cancer Res 2001;61:1432–1438. [PubMed] [Google Scholar]
- 108. Cheung W, Yu PX, Little BM, Cone RD, Marks DL, Mak RH. Role of leptin and melanocortin signaling in uremia‐associated cachexia. J Clin Invest 2005;115:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Marks DL, Butler AA, Turner R, Brookhart G, Cone RD. Differential role of melanocortin receptor subtypes in cachexia. Endocrinology 2003;144:1513–1523. [DOI] [PubMed] [Google Scholar]
- 110. Zhu X, Callahan MF, Gruber KA, Szumowski M, Marks DL. Melanocortin‐4 receptor antagonist TCMCB07 ameliorates cancer‐ and chronic kidney disease‐associated cachexia. J Clin Invest 2020;130:4921–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Scarlett JM, Bowe DD, Zhu X, Batra AK, Grant WF, Marks DL. Genetic and pharmacologic blockade of central melanocortin signaling attenuates cardiac cachexia in rodent models of heart failure. J Endocrinol 2010;206:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Markison S, Foster AC, Chen C, Brookhart GB, Hesse A, Hoare SR, et al. The regulation of feeding and metabolic rate and the prevention of murine cancer cachexia with a small‐molecule melanocortin‐4 receptor antagonist. Endocrinology 2005;146:2766–2773. [DOI] [PubMed] [Google Scholar]
- 113. Carter ME, Soden ME, Zweifel LS, Palmiter RD. Genetic identification of a neural circuit that suppresses appetite. Nature 2013;503:111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Campos CA, Bowen AJ, Han S, Wisse BE, Palmiter RD, Schwartz MW. Cancer‐induced anorexia and malaise are mediated by CGRP neurons in the parabrachial nucleus. Nat Neurosci 2017;20:934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Campos CA, Bowen AJ, Schwartz MW, Palmiter RD. Parabrachial CGRP neurons control meal termination. Cell Metab 2016;23:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Paues J, Mackerlova L, Blomqvist A. Expression of melanocortin‐4 receptor by rat parabrachial neurons responsive to immune and aversive stimuli. Neuroscience 2006;141:287–297. [DOI] [PubMed] [Google Scholar]
- 117. Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med 2017;23:1215–1219. [DOI] [PubMed] [Google Scholar]
- 118. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjaer SB, et al. GFRAL is the receptor for GDF15 and is required for the anti‐obesity effects of the ligand. Nat Med 2017;23:1158–1166. [DOI] [PubMed] [Google Scholar]
- 119. Hsu JY, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, et al. Non‐homeostatic body weight regulation through a brainstem‐restricted receptor for GDF15. Nature 2017;550:255–259. [DOI] [PubMed] [Google Scholar]
- 120. Tsai VW, Husaini Y, Manandhar R, Lee‐Ng KK, Zhang HP, Harriott K, et al. Anorexia/cachexia of chronic diseases: a role for the TGF‐β family cytokine MIC‐1/GDF15. J Cachexia Sarcopenia Muscle 2012;3:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, et al. MAP 3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 2016;7:467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, et al. Tumor‐induced anorexia and weight loss are mediated by the TGF‐beta superfamily cytokine MIC‐1. Nat Med 2007;13:1333–1340. [DOI] [PubMed] [Google Scholar]
- 123. Kozak W, Conn CA, Kluger MJ. Lipopolysaccharide induces fever and depresses locomotor activity in unrestrained mice. Am J Physiol 1994;266:R125–R135. [DOI] [PubMed] [Google Scholar]
- 124. Grossberg AJ, Zhu X, Leinninger GM, Levasseur PR, Braun TP, Myers MG Jr, et al. Inflammation‐induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci 2011;31:11376–11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Padilla SL, Qiu J, Soden ME, Sanz E, Nestor CC, Barker FD, et al. Agouti‐related peptide neural circuits mediate adaptive behaviors in the starved state. Nat Neurosci 2016;19:734–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Scarlett JM, Zhu X, Enriori PJ, Bowe DD, Batra AK, Levasseur PR, et al. Regulation of agouti‐related protein messenger ribonucleic acid transcription and peptide secretion by acute and chronic inflammation. Endocrinology 2008;149:4837–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Arima Y, Yokota S, Fujitani M. Lateral parabrachial neurons innervate orexin neurons projecting to brainstem arousal areas in the rat. Sci Rep 2019;9:2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Murphy KT, Chee A, Trieu J, Naim T, Lynch GS. Importance of functional and metabolic impairments in the characterization of the C‐26 murine model of cancer cachexia. Dis Model Mech 2012;5:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Voltarelli FA, Frajacomo FT, Padilha CS, Testa MTJ, Cella PS, Ribeiro DF, et al. Syngeneic B16F10 melanoma causes cachexia and impaired skeletal muscle strength and locomotor activity in mice. Front Physiol 2017;8:715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T, et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down‐regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem 2004;279:41114–41123. [DOI] [PubMed] [Google Scholar]
- 131. Grossberg AJ, Vichaya EG, Christian DL, Molkentine JM, Vermeer DW, Gross PS, et al. Tumor‐associated fatigue in cancer patients develops independently of IL1 signaling. Cancer Res 2018;78:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Grossberg AJ, Vichaya EG, Gross PS, Ford BG, Scott KA, Estrada D, et al. Interleukin 6‐independent metabolic reprogramming as a driver of cancer‐related fatigue. Brain Behav Immun 2020;88:230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hyltander A, Drott C, Körner U, Sandström R, Lundholm K. Elevated energy expenditure in cancer patients with solid tumours. Eur J Cancer 1991;27:9–15. [DOI] [PubMed] [Google Scholar]
- 134. Nixon DW, Kutner M, Heymsfield S, Foltz AT, Carty C, Seitz S, et al. Resting energy expenditure in lung and colon cancer. Metabolism 1988;37:1059–1064. [DOI] [PubMed] [Google Scholar]
- 135. Lindmark L, Bennegård K, Edén E, Ekman L, Scherstén T, Svaninger G, et al. Resting energy expenditure in malnourished patients with and without cancer. Gastroenterology 1984;87:402–408. [PubMed] [Google Scholar]
- 136. Rohm M, Zeigerer A, Machado J, Herzig S. Energy metabolism in cachexia. EMBO Rep 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cao DX, Wu GH, Zhang B, Quan YJ, Wei J, Jin H, et al. Resting energy expenditure and body composition in patients with newly detected cancer. Clin Nutr 2010;29:72–77. [DOI] [PubMed] [Google Scholar]
- 138. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature 2014;513:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Konsman JP, Blomqvist A. Forebrain patterns of c‐Fos and FosB induction during cancer‐associated anorexia‐cachexia in rat. Eur J Neurosci 2005;21:2752–2766. [DOI] [PubMed] [Google Scholar]
- 140. Scarlett JM, Jobst EE, Enriori PJ, Bowe DD, Batra AK, Grant WF, et al. Regulation of central melanocortin signaling by interleukin‐1 beta. Endocrinology 2007;148:4217–4225. [DOI] [PubMed] [Google Scholar]
- 141. Ramage LE, Akyol M, Fletcher AM, Forsythe J, Nixon M, Carter RN, et al. Glucocorticoids acutely increase brown adipose tissue activity in humans, revealing species‐specific differences in UCP‐1 regulation. Cell Metab 2016;24:130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Scotney H, Symonds ME, Law J, Budge H, Sharkey D, Manolopoulos KN. Glucocorticoids modulate human brown adipose tissue thermogenesis in vivo. Metabolism 2017;70:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tancini G, Barni S, Crispino S, Paolorossi F, Lissoni P. A study of thyroid function in cancer cachexia. Tumori 1989;75:185–188. [DOI] [PubMed] [Google Scholar]
- 144. Kliewer KL, Ke JY, Tian M, Cole RM, Andridge RR, Belury MA. Adipose tissue lipolysis and energy metabolism in early cancer cachexia in mice. Cancer Biol Ther 2015;16:886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Legaspi A, Jeevanandam M, Starnes HF Jr, Brennan MF. Whole body lipid and energy metabolism in the cancer patient. Metabolism 1987;36:958–963. [DOI] [PubMed] [Google Scholar]
- 146. Hansell DT, Davies JW, Shenkin A, Burns HJ. The oxidation of body fuel stores in cancer patients. Ann Surg 1986;204:637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bishop JS, Marks PA. Studies on carbohydrate metabolism in patients with neoplastic disease. II. Response to insulin administration. J Clin Invest 1959;38:668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lang CH, Bagby GJ, Nowotny A, Spitzer JJ. Effects of toxic and nontoxic endotoxin derivatives on glucose kinetics. Circ Shock 1985;17:301–311. [PubMed] [Google Scholar]
- 149. Dodesini AR, Benedini S, Terruzzi I, Sereni LP, Luzi L. Protein, glucose and lipid metabolism in the cancer cachexia: a preliminary report. Acta Oncol 2007;46:118–120. [DOI] [PubMed] [Google Scholar]
- 150. Fukawa T, Yan‐Jiang BC, Min‐Wen JC, Jun‐Hao ET, Huang D, Qian CN, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med 2016;22:666–671. [DOI] [PubMed] [Google Scholar]
- 151. Fearon KC, Tisdale MJ, Preston T, Plumb JA, Calman KC. Failure of systemic ketosis to control cachexia and the growth rate of the Walker 256 carcinosarcoma in rats. Br J Cancer 1985;52:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor‐induced IL‐6 reprograms host metabolism to suppress anti‐tumor immunity. Cell Metab 2016;24:672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Fearon KC, Borland W, Preston T, Tisdale MJ, Shenkin A, Calman KC. Cancer cachexia: influence of systemic ketosis on substrate levels and nitrogen metabolism. Am J Clin Nutr 1988;47:42–48. [DOI] [PubMed] [Google Scholar]
- 154. Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2014;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Goncalves MD, Hwang SK, Pauli C, Murphy CJ, Cheng Z, Hopkins BD, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci U S A 2018;115:E743–e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lorite MJ, Cariuk P, Tisdale MJ. Induction of muscle protein degradation by a tumour factor. Br J Cancer 1997;76:1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Beck SA, Tisdale MJ. Nitrogen excretion in cancer cachexia and its modification by a high fat diet in mice. Cancer Res 1989;49:3800–3804. [PubMed] [Google Scholar]
- 158. Yang QJ, Zhao JR, Hao J, Li B, Huo Y, Han YL, et al. Serum and urine metabolomics study reveals a distinct diagnostic model for cancer cachexia. J Cachexia Sarcopenia Muscle 2018;9:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Reeds PJ, Fjeld CR, Jahoor F. Do the differences between the amino acid compositions of acute‐phase and muscle proteins have a bearing on nitrogen loss in traumatic states? J Nutr 1994;124:906–910. [DOI] [PubMed] [Google Scholar]
- 160. Hall KD, Baracos VE. Computational modeling of cancer cachexia. Curr Opin Clin Nutr Metab Care 2008;11:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Jeevanandam M, Horowitz GD, Lowry SF, Brennan MF. Cancer cachexia and the rate of whole body lipolysis in man. Metabolism 1986;35:304–310. [DOI] [PubMed] [Google Scholar]
- 162. Zuijdgeest‐van Leeuwen SD, van den Berg JW, Wattimena JL, van der Gaast A, Swart GR, Wilson JH, et al. Lipolysis and lipid oxidation in weight‐losing cancer patients and healthy subjects. Metabolism 2000;49:931–936. [DOI] [PubMed] [Google Scholar]
- 163. Klein S, Wolfe RR. Whole‐body lipolysis and triglyceride‐fatty acid cycling in cachectic patients with esophageal cancer. J Clin Invest 1990;86:1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Fearon KC, Hansell DT, Preston T, Plumb JA, Davies J, Shapiro D, et al. Influence of whole body protein turnover rate on resting energy expenditure in patients with cancer. Cancer Res 1988;48:2590–2595. [PubMed] [Google Scholar]
- 165. McMillan DC, Preston T, Fearon KC, Burns HJ, Slater C, Shenkin A. Protein synthesis in cancer patients with inflammatory response: investigations with [15N]glycine. Nutrition 1994;10:232–240. [PubMed] [Google Scholar]
- 166. Melville S, McNurlan MA, Calder AG, Garlick PJ. Increased protein turnover despite normal energy metabolism and responses to feeding in patients with lung cancer. Cancer Res 1990;50:1125–1131. [PubMed] [Google Scholar]
- 167. Richards EW, Long CL, Nelson KM, Tohver OK, Pinkston JA, Navari RM, et al. Protein turnover in advanced lung cancer patients. Metabolism 1993;42:291–296. [DOI] [PubMed] [Google Scholar]
- 168. Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care 2010;13:225–229. [DOI] [PubMed] [Google Scholar]
- 169. Eley HL, Tisdale MJ. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J Biol Chem 2007;282:7087–7097. [DOI] [PubMed] [Google Scholar]
- 170. Lundholm K, Bennegård K, Edén E, Svaninger G, Emery PW, Rennie MJ. Efflux of 3‐methylhistidine from the leg in cancer patients who experience weight loss. Cancer Res 1982;42:4807–4811. [PubMed] [Google Scholar]
- 171. Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, et al. Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo‐controlled, double‐blind trials. Lancet Oncol 2015;16:108–116. [DOI] [PubMed] [Google Scholar]
- 172. Prado CM, Bekaii‐Saab T, Doyle LA, Shrestha S, Ghosh S, Baracos VE, et al. Skeletal muscle anabolism is a side effect of therapy with the MEK inhibitor: selumetinib in patients with cholangiocarcinoma. Br J Cancer 2012;106:1583–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. MacDonald AJ, Johns N, Stephens N, Greig C, Ross JA, Small AC, et al. Habitual myofibrillar protein synthesis is normal in patients with upper GI cancer cachexia. Clin Cancer Res 2015;21:1734–1740. [DOI] [PubMed] [Google Scholar]
- 174. Winter A, MacAdams J, Chevalier S. Normal protein anabolic response to hyperaminoacidemia in insulin‐resistant patients with lung cancer cachexia. Clin Nutr 2012;31:765–773. [DOI] [PubMed] [Google Scholar]
- 175. Prado CM, Sawyer MB, Ghosh S, Lieffers JR, Esfandiari N, Antoun S, et al. Central tenet of cancer cachexia therapy: do patients with advanced cancer have exploitable anabolic potential? Am J Clin Nutr 2013;98:1012–1019. [DOI] [PubMed] [Google Scholar]
- 176. Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, et al. Increased ATP‐ubiquitin‐dependent proteolysis in skeletal muscles of tumor‐bearing rats. Cancer Res 1994;54:5568–5573. [PubMed] [Google Scholar]
- 177. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 178. Tisdale MJ. Mechanisms of Cancer Cachexia. Physiol Rev 2009;89:381–410. [DOI] [PubMed] [Google Scholar]
- 179. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 180. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin‐1, a muscle‐specific F‐box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A 2001;98:14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Llovera M, Carbó N, López‐Soriano J, García‐Martínez C, Busquets S, Alvarez B, et al. Different cytokines modulate ubiquitin gene expression in rat skeletal muscle. Cancer Lett 1998;133:83–87. [DOI] [PubMed] [Google Scholar]
- 182. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mofarrahi M, Sigala I, Guo Y, Godin R, Davis EC, Petrof B, et al. Autophagy and skeletal muscles in sepsis. PLoS ONE 2012;7:e47265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hermans G, Casaer MP, Clerckx B, Güiza F, Vanhullebusch T, Derde S, et al. Effect of tolerating macronutrient deficit on the development of intensive‐care unit acquired weakness: a subanalysis of the EPaNIC trial. Lancet Respir Med 2013;1:621–629. [DOI] [PubMed] [Google Scholar]
- 185. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–1378. [DOI] [PubMed] [Google Scholar]
- 186. Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O. Autophagic‐lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr 2013;98:1485–1492. [DOI] [PubMed] [Google Scholar]
- 187. Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, Laugs F, et al. Nuclear transcription factor κ B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 2013;98:738–748. [DOI] [PubMed] [Google Scholar]
- 188. Johns N, Hatakeyama S, Stephens NA, Degen M, Degen S, Frieauff W, et al. Clinical classification of cancer cachexia: phenotypic correlates in human skeletal muscle. PLoS ONE 2014;9:e83618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Aversa Z, Pin F, Lucia S, Penna F, Verzaro R, Fazi M, et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci Rep 2016;6:30340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res 2011;71:1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Tian M, Asp ML, Nishijima Y, Belury MA. Evidence for cardiac atrophic remodeling in cancer‐induced cachexia in mice. Int J Oncol 2011;39:1321–1326. [DOI] [PubMed] [Google Scholar]
- 192. Manne ND, Lima M, Enos RT, Wehner P, Carson JA, Blough E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int J Oncol 2013;42:2134–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, et al. Prevention of liver cancer cachexia‐induced cardiac wasting and heart failure. Eur Heart J 2014;35:932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Das SK, Hoefler G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol Med 2013;19:292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer‐associated cachexia. Science 2011;333:233–238. [DOI] [PubMed] [Google Scholar]
- 196. Bodnar RJ, Pasternak GW, Mann PE, Paul D, Warren R, Donner DB. Mediation of anorexia by human recombinant tumor necrosis factor through a peripheral action in the rat. Cancer Res 1989;49:6280–6284. [PubMed] [Google Scholar]
- 197. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD. Cholecystokinin‐mediated suppression of feeding involves the brainstem melanocortin system. Nat Neurosci 2004;7:335–336. [DOI] [PubMed] [Google Scholar]
- 199. Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon‐like peptide‐1 in the central regulation of feeding. Nature 1996;379:69–72. [DOI] [PubMed] [Google Scholar]
- 200. Meeran K, O'Shea D, Edwards CM, Turton MD, Heath MM, Gunn I, et al. Repeated intracerebroventricular administration of glucagon‐like peptide‐1‐(7‐36) amide or exendin‐(9‐39) alters body weight in the rat. Endocrinology 1999;140:244–250. [DOI] [PubMed] [Google Scholar]
- 201. Halatchev IG, Cone RD. Peripheral administration of PYY(3‐36) produces conditioned taste aversion in mice. Cell Metab 2005;1:159–168. [DOI] [PubMed] [Google Scholar]
- 202. Batterham RL, Heffron H, Kapoor S, Chivers JE, Chandarana K, Herzog H, et al. Critical role for peptide YY in protein‐mediated satiation and body‐weight regulation. Cell Metab 2006;4:223–233. [DOI] [PubMed] [Google Scholar]
- 203. Makimura H, Mizuno TM, Roberts J, Silverstein J, Beasley J, Mobbs CV. Adrenalectomy reverses obese phenotype and restores hypothalamic melanocortin tone in leptin‐deficient ob/ob mice. Diabetes 2000;49:1917–1923. [DOI] [PubMed] [Google Scholar]
- 204. Drazen DL, Wortman MD, Schwartz MW, Clegg DJ, van Dijk G, Woods SC, et al. Adrenalectomy alters the sensitivity of the central nervous system melanocortin system. Diabetes 2003;52:2928–2934. [DOI] [PubMed] [Google Scholar]
- 205. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 2002;5:566–572. [DOI] [PubMed] [Google Scholar]
- 206. Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Münzberg H, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet‐sensitive obesity. J Clin Invest 2006;116:1886–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Jaya Rao KS, Srikantia SG, Gopalan C. Plasma cortisol levels in protein‐calorie malnutrition. Arch Dis Child 1968;43:365–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Bajaj JS, Khardori R, Deo MG, Bansal DD. Adrenocortical function in experimental protein malnutrition. Metabolism: clinical and experimental 1979;28:594–598. [DOI] [PubMed] [Google Scholar]
- 209. Drott C, Svaninger G, Lundholm K. Increased urinary excretion of cortisol and catecholami‐NES in malnourished cancer patients. Ann Surg 1988;208:645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Lei J, Nowbar S, Mariash CN, Ingbar DH. Thyroid hormone stimulates Na‐K‐ATPase activity and its plasma membrane insertion in rat alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2003;285:L762–L772. [DOI] [PubMed] [Google Scholar]
- 211. Williams LT, Lefkowitz RJ, Watanabe AM, Hathaway DR, Besch HR Jr. Thyroid hormone regulation of beta‐adrenergic receptor number. J Biol Chem 1977;252:2787–2789. [PubMed] [Google Scholar]
- 212. Croxson MS, Ibbertson HK. Low serum triiodothyronine (T3) and hypothyroidism in anorexia nervosa. J Clin Endocrinol Metab 1977;44:167–174. [DOI] [PubMed] [Google Scholar]
- 213. Turkay S, Kus S, Gokalp A, Baskin E, Onal A. Effects of protein energy malnutrition on circulating thyroid hormones. Indian Pediatr 1995;32:193–197. [PubMed] [Google Scholar]
- 214. Kawane T, Saikatsu S, Akeno N, Abe M, Horiuchi N. Starvation‐induced increase in the parathyroid hormone/PTH‐related protein receptor mRNA of bone and kidney in sham‐operated and thyroparathyroidectomized rats. Eur J Endocrinol 1997;137:273–280. [DOI] [PubMed] [Google Scholar]
- 215. Kerstetter JE, Svastisalee CM, Caseria DM, Mitnick ME, Insogna KL. A threshold for low‐protein‐diet‐induced elevations in parathyroid hormone. Am J Clin Nutr 2000;72:168–173. [DOI] [PubMed] [Google Scholar]
- 216. Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab 2016;23:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Garnett ES, Cohen H, Nahmias C, Viol G. The roles of carbohydrate, renin, and aldosterone in sodium retention during and after total starvation. Metabolism: clinical and experimental 1973;22:867–874. [DOI] [PubMed] [Google Scholar]
- 218. Ljutić D, Raos V, Biocić M. Aldosterone and blood pressure in obese subjects during short‐term starvation. Acta medica Croatica: casopis Hravatske akademije medicinskih znanosti 1995;49:165–168. [PubMed] [Google Scholar]
- 219. Aldosterone secretion in infantile malnutrition. Nutr Rev 1974;32:296–298. [DOI] [PubMed] [Google Scholar]
- 220. Anker SD, Sharma R. The syndrome of cardiac cachexia. Int J Cardiol 2002;85:51–66. [DOI] [PubMed] [Google Scholar]
- 221. Anker SD, Chua TP, Ponikowski P, Harrington D, Swan JW, Kox WJ, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation 1997;96:526–534. [DOI] [PubMed] [Google Scholar]
- 222. Zauner C, Schneeweiss B, Kranz A, Madl C, Ratheiser K, Kramer L, et al. Resting energy expenditure in short‐term starvation is increased as a result of an increase in serum norepinephrine. Am J Clin Nutr 2000;71:1511–1515. [DOI] [PubMed] [Google Scholar]
- 223. Gotoh M, Iguchi A, Tajima T, Ikari H, Hirooka Y. Starvation reduces norepinephrine activities in both hypothalamus and heart in rats. Brain Res 1996;706:351–354. [DOI] [PubMed] [Google Scholar]
- 224. Petry CJ, Dorling MW, Wang CL, Pawlak DB, Ozanne SE. Catecholamine levels and receptor expression in low protein rat offspring. Diabetic Med: J British Diabetic Assoc 2000;17:848–853. [DOI] [PubMed] [Google Scholar]
- 225. Günöz H, Neyzł O, Sencer E, Molvalilar S, Argun A. Growth hormone secretion in protein energy malnutrition. Acta Paediatr Scand 1981;70:521–526. [DOI] [PubMed] [Google Scholar]
- 226. Svaninger G, Isaksson O, Lundholm K. Growth hormone and experimental cancer cachexia. J Natl Cancer Inst 1987;79:1359–1365. [PubMed] [Google Scholar]
- 227. Buchanan KD, Vance JE, Williams RH. Effect of starvation on insulin and glucagon release from isolated islets of Langerhans of the rat. Metabolism: clinical and experimental 1969;18:155–162. [DOI] [PubMed] [Google Scholar]
- 228. Das BK, Ramesh J, Agarwal JK, Mishra OP, Bhatt RP. Blood sugar and serum insulin response in protein‐energy malnutrition. J Trop Pediatr 1998;44:139–141. [DOI] [PubMed] [Google Scholar]
- 229. Bennegård K, Lundgren F, Lundholm K. Mechanisms of insulin resistance in cancer associated malnutrition. Clin Physiol (Oxford, England) 1986;6:539–547. [DOI] [PubMed] [Google Scholar]
- 230. Edén E, Edström S, Bennegård K, Scherstén T, Lundholm K. Glucose flux in relation to energy expenditure in malnourished patients with and without cancer during periods of fasting and feeding. Cancer Res 1984;44:1718–1724. [PubMed] [Google Scholar]
- 231. Rofe AM, Bourgeois CS, Coyle P, Taylor A, Abdi EA. Altered insulin response to glucose in weight‐losing cancer patients. Anticancer Res 1994;14:647–650. [PubMed] [Google Scholar]
- 232. Rahmani J, Kord Varkaneh H, Clark C, Zand H, Bawadi H, Ryan PM, et al. The influence of fasting and energy restricting diets on IGF‐1 levels in humans: A systematic review and meta‐analysis. Ageing Res Rev 2019;53:100910, 10.1016/j.arr.2019.100910 [DOI] [PubMed] [Google Scholar]
- 233. Ammann P, Zacchetti G, Gasser JA, Lavet C, Rizzoli R. Protein malnutrition attenuates bone anabolic response to PTH in female rats. Endocrinology 2015;156:419–428. [DOI] [PubMed] [Google Scholar]
- 234. Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, et al. IGF‐1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2006;291:R674–R683. [DOI] [PubMed] [Google Scholar]