

Abstract
Iron regulatory protein 2 (IRP2) is a key regulator of iron homeostasis and is found to be altered in several types of human cancer. However, how IRP2 contributes to tumorigenesis remains to be elucidated. In this study, we sought to investigate the role of IRP2 in tumorigenesis and found that IRP2 promotes cell growth by repressing TAp63, a member of p53 tumor suppressor family. Specifically, we found that IRP2 overexpression decreased, whereas IRP2 deficiency increased, TAp63 expression. We also showed that the repression of TAp63 by IRP2 was independent of tumor suppressor p53. To uncover the molecular basis, we found that IRP2 stabilized TAp63 mRNA by binding to an iron response element in the 3′UTR of p63 mRNA. To determine the biological significance of this regulation, we showed that IRP2 facilitates cell proliferation, at least in part, via repressing TAp63 expression. Moreover, we found that IRP2 deficiency markedly alleviated cellular senescence in TAp63-deficient mouse embryo fibroblasts. Together, we have uncovered a novel regulation of TAp63 by IRP2 and our data suggest that IRP2 exerts its oncogenic activities at least in part by repressing TAp63 expression.
Implications:
We have revealed a novel regulation of TAp63 by IRP2 and our data suggest that IRP2 exerts its oncogenic activities, at least in part, by repressing TAp63 expression.
Introduction
Iron is essential for all organisms. It is required by variety of proteins involved in cell growth and proliferation. Notably, recent studies showed that dysregulation of iron metabolism frequently occurs in cancer cells and contributes to cancer development. At the cellular level, the uptake, storage, and usage of iron are tightly governed by iron regulatory protein 1 (IRP1) and IRP2 (also known as ACO1 and IREB2, respectively; refs. 1, 2). IRP1 and IRP2 are cytosolic RNA-binding proteins that bind to iron-responsive elements (IRE) in their target mRNAs and regulate the translation or stability of mRNAs encoding proteins involved in cellular iron homeostasis (3–7). IRP1/2 repress mRNA translation when bound to an IRE located at the 5′UTR, including the ones coding for ferritin L (FTL) and H (FTH1; ref. 8), ALAS2 (9), ferroportin (SLC40A1; ref. 10), mitochondrial aconitase ACO2 and SDH (11), and HIF2α (12). In contrast, IRP1/2 regulate the stability of their target mRNAs through interaction with IREs in the 3′UTR, including the ones coding for TFR1 (8), SLC11A2 (9), CDC14A (13), Cdc42-binding kinase alpha (CDC42BPA; ref. 14) and glycolate oxidase (Hao1; ref. 15). The biological function of IRPs in regulating cellular iron metabolism has been confirmed in mouse models. Loss of Irp2 in mouse models results in microcytic anemia, altered body iron distribution, and adult-onset progressive neurode-generation (16, 17). IRP1 deficiency was initially found to be asymptomatic in mice (18), but later found to result in age-dependent erythropoietic abnormalities and dysregulation of body iron metabolism (19, 20). Despite the well-established role of IRP1/2 in iron metabolism, very little is known about their role in tumorigenesis.
p63 belongs to the p53 tumor suppressor family, including p53, p63, and p73 (21). Because of differential promoter usage and splicing, the TP63 gene produces multiple isoforms: two N-terminal isoforms (TA and ΔN) and five C-terminal isoforms (α, β, γ, δ, ε; refs. 21, 22). All TP63 isoforms contain an identical DNA-binding domain (DBD) and an oligomerization domain and are able to transactivate some p53 target genes related to tumor suppression, such as CDKN1A (p21) and Bax (21, 23). However, some TP63 isoforms contain one or both unique domains at their C termini: sterile alpha motif and transcription-inhibitory domain, both of which can modulate target gene specificity (24–26). The biological function of TP63 has been explored in multiple studies with various mouse models. For example, loss of all TP63 isoforms leads to aberrant epithelial development (21, 27). Interestingly, mice deficient in TAp63 are tumor prone and display features of accelerated aging (28, 29). In contrast, mice deficient in ΔNp63 have developmental defects, which resemble the phenotypes of total TP63 knockout mice (30). These data suggest that TP63 has a profound role in development, tumor suppression, and aging.
Because of the critical role of TP63 in modulating various cellular processes, TP63 expression is tightly controlled, which is mainly through posttranscriptional levels (31, 32). For example, c-Ab1, IKKβ, ATM, CDK2, and p38 can regulate TP63 activity through protein phosphorylation and stability (33–36). Both ΔNp63 and mutant p53 can inactivate TAp63 in a dominant negative fashion (21, 37, 38). Our group found that TP63 can be regulated via mRNA stability by Rbm24 and Rbm38 RNA-binding proteins (39–42). We and others also found that TP63 is regulated by several miRNAs (41, 43, 44). However, whether TP63 is regulated by other RNA-binding proteins remains unclear.
Among all three p53 family members, p53 has been found to play a role in iron metabolism as well as ferroptosis, an iron-mediated cell death (45). However, it is not certain whether other p53 family members play a role in iron metabolism. In this study, we sought to determine whether TAp63 is regulated by IRP2 and the biological significance of this regulation. Indeed, we made novel observations that ectopic expression of IRP2 decreases, whereas IRP2 deficiency increases, p63 expression. We also found that IRP2 promotes cell proliferation at least in part via suppressing TAp63 expression. Furthermore, we found that loss of IRP2 inhibits TAp63 deficiency-induced cellular senescence. Together, our data suggest that IRP2 exerts its oncogenic activity via suppressing TAp63 expression and that the IRP2–p63 pathway may be explored as a cancer therapeutic strategy.
Materials and Methods
IRP2- and TAp63-mutant mouse models
Irp2+/− mice (ID: MMRRC: 030490-MU, on a C57BL/6N background) were obtained from Mutant Mouse Resource and Research Center (MMRRC) at the University of Missouri (Columbia, MO). The TAp63+/− mice on C57BL/6 background were kindly provided by Dr. E. Flores’ laboratory (46). All animals were housed in a specific pathogen-free environment at the University of California, Davis (Davis, CA). The procedures for all animal experiments were approved by the Institutional Animal Care and Use Committee at University of California, Davis (Davis, CA).
Mouse embryonic fibroblasts’ isolation
Irp2+/−;TAp63+/− mice were intercrossed to generate ~13.5-day embryos, which were then used to isolate WT, TAp63+/−, Irp2−/− and Irp2−/−;TAp63+/− MEFs as described previously (47).
Cell culture
MIA-PaCa2, HepG2, Huh7, and Hep3B cells were originally purchased from the ATCC. The cell lines and their derivatives were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Hyclone). Because these cell lines were authenticated and tested free for Mycoplasma by ATCC, no further authentication and Mycoplasma testing were performed, especially considering that these cell lines at low passages were used.
Plasmid construction and cell line generation
Cells deficient in IRP2 or p53 were generated and confirmed as described previously (48). The primers used to generate sgRNA expression vectors were listed in Supplementary Table S1. The primers used for genotyping cell lines were listed in Supplementary Table S2.
The pcDNA3–5′UTR-TAp63, pcDNA3-TAp63-CDS and pcDNA3-TAp63–3′UTR expression vectors were generated as described previously (40). pcDNA3 vector expressing GFP was generated as described previously (49). The 5′UTR-GFP and GFP-3′UTR reporters were generated by cloning p63 5′UTR or 3′UTR into pcDNA3-GFP downstream of GFP. p63 5′UTR was amplified with forward primer, 5′-AAG CTT CCC GGC TTT ATA TCT ATA TA-3′, and reverse primer, 5′-GGA TCC TTC CTT CAA CTG TCT TTG A-3′. p63 3′UTR was amplified as described previously (41). To generate pcDNA3-GFP-TAp63-ΔIRE, which lacks an IRE site (CAGUGU) at nt 2792, a two-step PCR strategy was used and pcDNA3-p63-3′UTR was used as a template. Fragment 1 was amplified with forward primer, 5′-CTC GAG GCC TCA CCA TGT GAG CTC-3′, and reverse primer, 5′-ATA AAA TAT ACC AAG AAA AGA ATA CAT-3′. Fragment 2 was amplified with forward primer, 5′-ATG TAT TCT TTT CTT GGT TAT ATT TTA T-3′, and reverse primer, 5′-TCT AGA GCA TGT CCT GGC AAA CAA AAA G-3′. The second-step PCR was performed with forward primer, 5′-CTC GAG GCC TCA CCA TGT GAG CTC-3′, and reverse primer, 5′-TCT AGA GCA TGT CCT GGC AAA CAA AAA G-3′. The resulting PCR fragment was then cloned into pcDNA3-GFP vector to generate pcDNA3-GFP-p63–3′UTR-ΔIRE.
Western blot analysis
Western blot was performed as previously described (48). Antibodies against TAp63, p130, PML, IRP2, and actin were purchased from Santa Cruz Biotechnology. Anti-mouse IRP2 antibody was kindly gifted from Professor E. Leibold (University of Utah, Salt Lake City, UT; ref. 50).
RNA isolation, RT-PCR analysis, and qRT-PCR
Total RNA isolation, cDNA synthesis, and RT-PCR measurement of p63 and actin transcripts with the primers were performed as described previously (41).
RNA-chromatin immunoprecipitation assay
RNA-chromatin immunoprecipitation (RNA-ChIP) was carried out as described previously (51). RT–PCR analysis was performed to determine the level of RNA transcripts associated with IRP2. The primers for human TFR were as follows: forward primer, 5′-ACG CCA GAC TTT GCT GAG TT-3′; reverse primer, 5′-GAG GAG CCA GGA GAG GAC TT-3′. The primers for human TAp63 were as follows: forward primer, 5′-TTA TTA CCG ATC CAC CAT GTC-3′; reverse primer, 5′-TGC GGA TAC AGT CCA TGC TA-3′. The primers used to amplify human actin were as follows: forward primer, 5′-CTG AAG TAC CCC ATC GAG CAC GGC A-3′; reverse primer, 5′-GGA TAG CAC AGC CTG GAT AGC AAC G-3′.
RNA interference
Scrambled siRNA (5′-GCA GUG UCU CCA CGU ACU AdTdT-3′), siRNAs against IRP2 (siIRP2#1:5′-GCG AUU UCC AGG CUU GCU UdTdT −3′ and #2: 5′-GCA AAC AUG UGU CCG GAA dTdT-3′), and siRNA against human TAp63 (5′-GAU GGU GCG ACA AAC AAG AdTdT-3′) were purchased from Dharmacon. For siRNA transfection, RNAiMax Lipid Reagent (Thermo Fisher Scientific) was used according to the manufacturer’s manual. The siRNAs were transfected into the cells at the final concentration of 30 nmol/L for 3 days.
Colony formation assay
Colony formation assay was performed with HepG2, Hep3B, or their derivatives (1,000 cells per well) in 6-well plates for two weeks. Fixation and staining for colonies were performed as described previously (48). The density of colonies was scanned and analyzed using Image J (52).
Statistical analysis
The data were presented as mean ± SD. Statistical significance was determined by two-tailed Student t test. Values of P < 0.05 were considered significant.
Results
Ectopic expression of IRP2 represses, whereas knockdown or knockout of IRP2 increases, TAp63 expression regardless of p53 status
To test whether IRP2 regulates TP63, IRP2 was ectopically expressed in HepG2 (carrying WT p53), Hep3B (p53-null), and MIA-PaCa2 (carrying mutant p53) cells. We showed that the levels of TAp63α protein were significantly reduced by ectopic expression of IRP2 (Fig. 1A, C, and E). We also showed that the level of TAp63 transcript was decreased by ectopic expression of IRP2 (Fig. 1B, D, and F). To confirm this, IRP2 was knocked down by two siRNAs that target different regions of IRP2 mRNA (Fig. 2A, siIRP2#1 and siIRP2#2) in Hep3B cells. We showed that the level of TAp63α protein was reduced by silencing of IRP2 (Fig. 2A). We also showed that the level of TAp63 transcript was increased by silencing of IRP2 (Fig. 2B and C). To further confirm this, we generated multiple IRP2-deficient cell lines by CRISPR/Cas9 from HepG2 (clone #35 and #57), MIA-PaCa2 (clone #8 and #46) and Huh7 (carrying mutant p53, clone #28 and #54) cells. We found that the level of TAp63 protein was much higher in IRP2−/− cells than that in isogenic control cells (Fig. 2D, G, and J). We also found that the level of TAp63 transcript was increased in IRP2−/− cells (Fig. 2E and F, H and I, K and L). These data indicate that IRP2 represses TAp63 expression regardless of p53 status.
Figure 1.
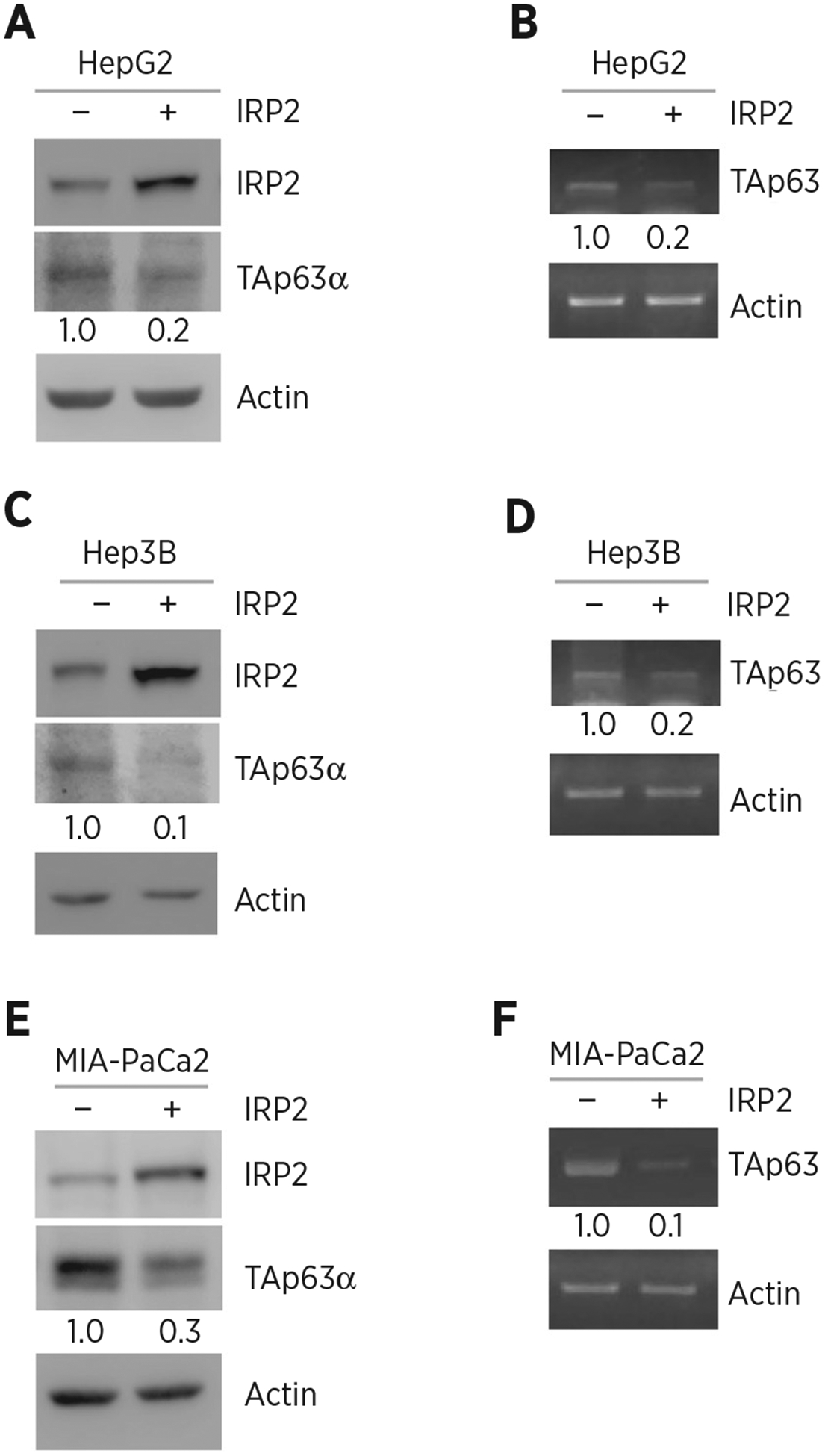
Ectopic expression of IRP2 represses TAp63 expression. A, HepG2 cells were transfected with a control vector or a vector expressing HA-tagged IRP2 for 24 hours. Cell lysates were collected and subjected to Western blot analysis with various antibodies as indicated. The level of proteins was normalized to that of actin, and the relative fold change is shown below each lane. B, The experiments were performed as in A except that the levels of TAp63 and actin transcripts were measured. The level of transcripts was normalized to that of actin, and the relative fold change is shown below each lane. C and D, The experiments were performed as in A and B except that Hep3B cells were used. E and F, The experiments were performed as in A and B except that MIA-PaCa2 cells were used.
Figure 2.
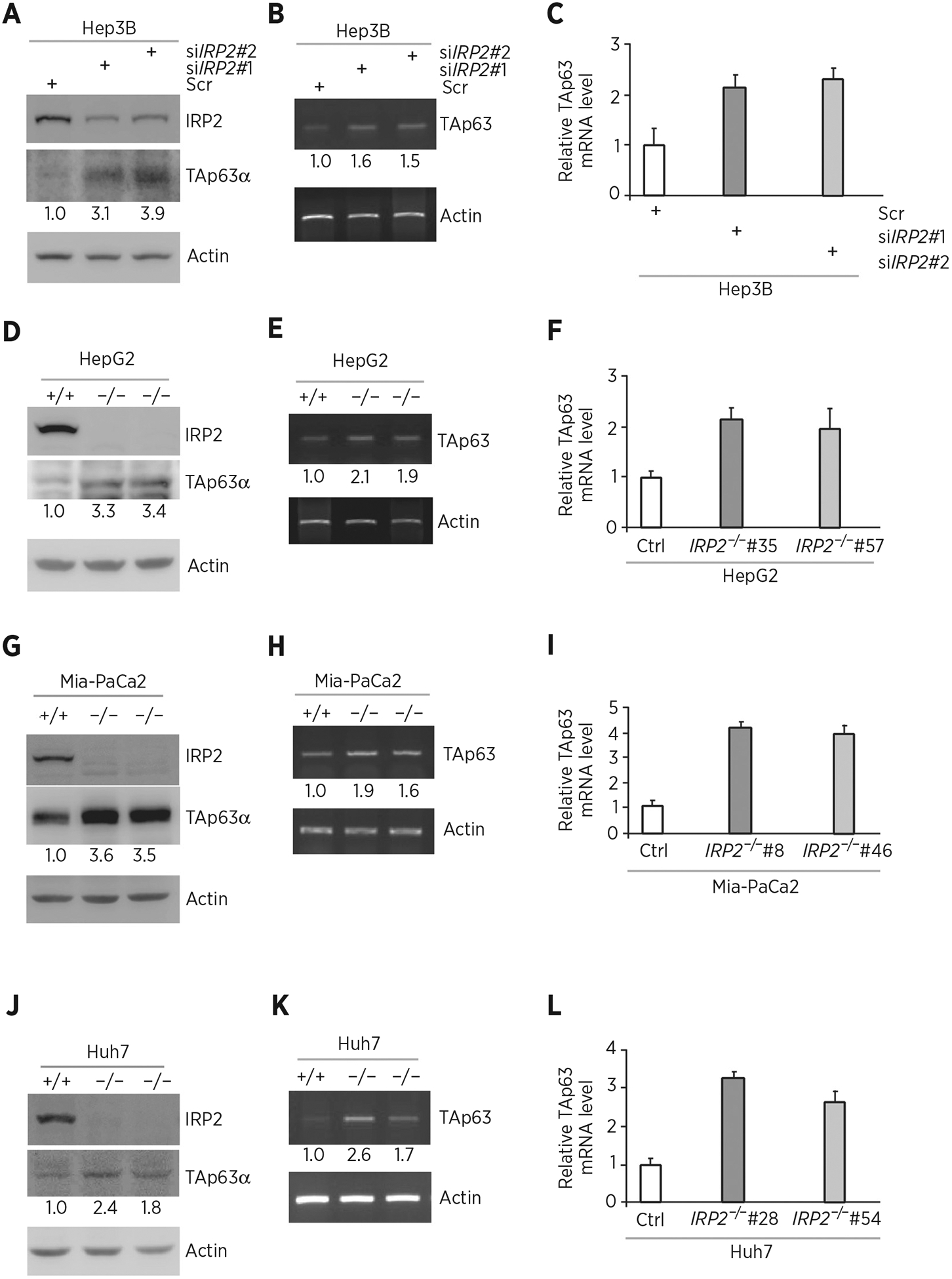
Knockdown or knockout of IRP2 increases p63 expression. A, Hep3B cells were transfected with scrambled siRNA (Scr) or siRNA against IRP2 for 72 hours. Cell lysates were collected and subjected to Western blot analysis with various antibodies as indicated. The level of proteins was normalized to that of actin, and the relative fold change is shown below each lane. B, The levels of TAp63 and actin transcripts were measured in Hep3B cells transfected with scrambled siRNA or siRNA against IRP2 for 72 hours. The level of transcripts was normalized to that of actin, and the relative fold change is shown below each lane. C, Quantitative RT-PCR was performed to measure the levels of TAp63 transcripts in Hep3B cells transfected with scrambled siRNA or siRNA against IRP2 for 72 hours. D, Western blots were prepared with lysates from isogenic control or IRP2−/− HepG2 cells, and then probed with antibodies against IRP2, TAp63α, and actin, respectively. The level of proteins was normalized to that of actin, and the relative fold change is shown below each lane. E, The levels of TAp63 and actin transcripts were measured in isogenic control or IRP2−/− HepG2 cells. The level of transcripts was normalized to that of actin, and the relative fold change is shown below each lane. F, Quantitative RT-PCR was performed to measure the levels of TAp63 transcripts in isogenic control or IRP2−/− HepG2 cells. G–I, The experiments were performed as in D–F except that isogenic control or IRP2−/− MIA-PaCa2 cells were used. J–L, The experiments were performed as in D–F except that isogenic control or IRP2−/− Huh7 cells were used.
IRP2 regulates p63 expression via mRNA stability
Because IRP2 regulates p63 expression by altering the level of TP63 transcript, we next determined whether TP63 mRNA stability is regulated by IRP2. To test this, isogenic control and IRP2−/− MIA-PaCa2 cells were treated with 5,6-dichlorobenzimidazole-β-D-ribofuranoside (DRB; 100 μmol/L) to inhibit de novo RNA synthesis. We found that knockout of IRP2 increased the half-life of TAp63 mRNA from approximately 2.55 hours in isogenic control cells to approximately 3.58 hours in IRP2−/− cells (Fig. 3A). As a RNA-binding protein, we hypothesized that IRP2 might bind to TP63 mRNA and then regulate p63 expression. To test this, RNA-ChIP assay was performed and showed that p63 transcript was detected in IRP2-immunocomplexes from isogenic control cells, but little if any from IRP2-KO cells (Fig. 3B, compare lanes 5 and 6). We also showed that IRP2 bound to TFR mRNA, which is known to be recognized by IRP2 but not actin mRNA (Fig. 3B, compare lanes 5 and 6).
Figure 3.
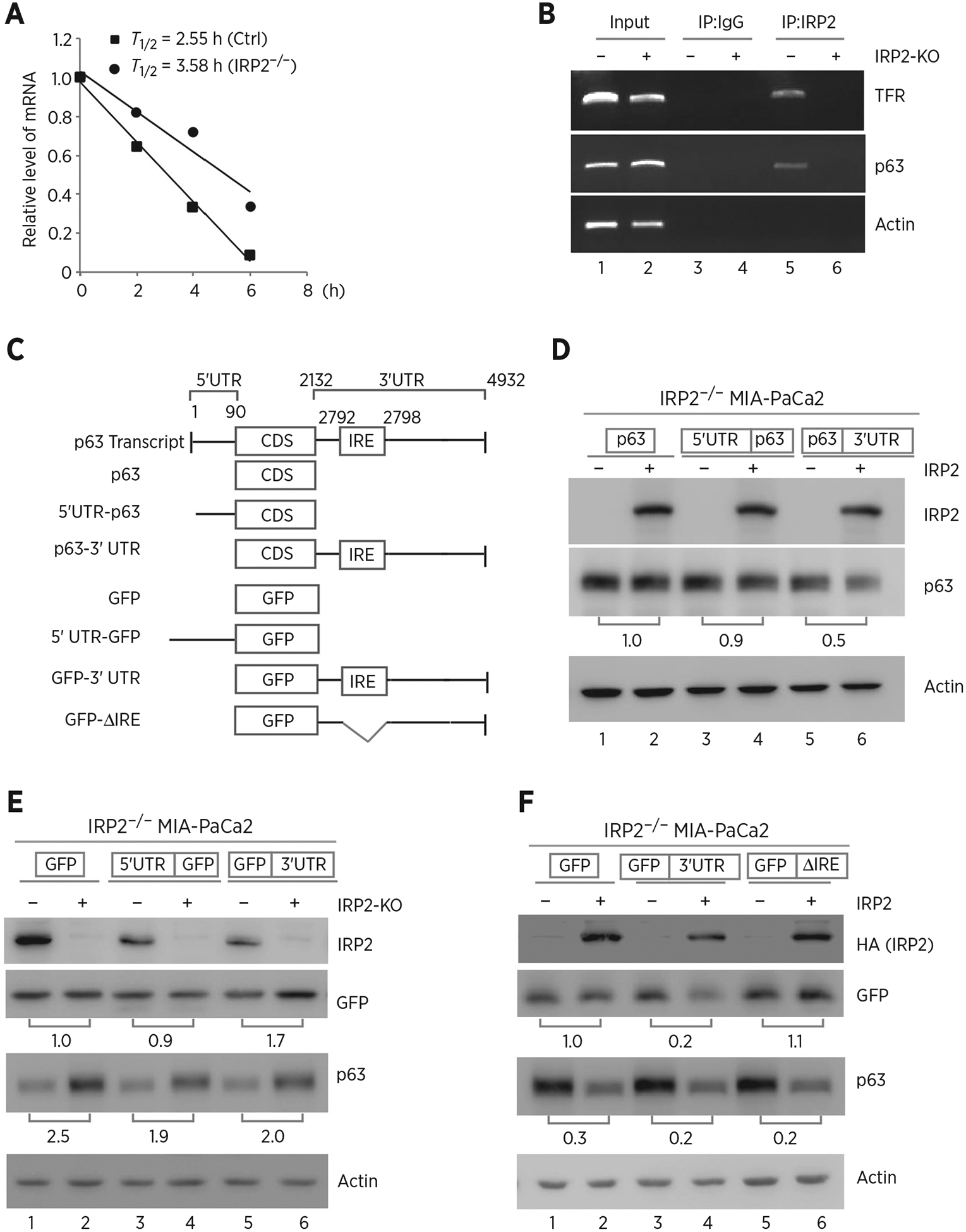
IRP2 regulates TP63 mRNA stability. A, The half-life of TP63 mRNA was determined in isogenic control or IRP2−/− MIA-PaCa2 cells. The level of TAp63 transcript was measured by qRT-PCR in isogenic control or IRP2−/− MIA-PaCa2 cells treated with 5,6- dichlorobenzimidazole-β-D-ribofuranoside (DRB; 100 μmol/L) for various times. mRNA was normalized by level of actin mRNA from triplicate samples and presented as mean ± SD. B, RNA-ChIP was performed with DNAase I–treated extracts from isogenic control (−) or IRP2−/− (+) MIA-PaCa2 cells with control IgG or anti-IRP2. Total RNAs were purified from immunocomplexes and subjected to RT-PCR to measure the levels of IRP2, p63, TFR and actin mRNAs. C, Schematic presentation of TAp63 transcript and TAp63 reporters that carry TAp63 coding region alone, or together with TAp63 5′ or 3′UTR. Also shown below is schematic presentation of GFP reporters that carry GFP coding region alone, or together with TAp63 5′UTR, 3′UTR, or a mutant 3′UTR with deletion of the putative IRE region. D, IRP2−/− MIA-PaCa2 cells were transfected with control pcDNA3 (−) or a vector expressing HA-IRP2 (+) along with a reporter that contains TAp63 coding region alone or together with TAp63 5′ or 3′UTR. Twenty-four hours posttransfection, cell lysates were collected and subjected to Western blot analysis to detect IRP2, TAp63 and actin. The level of proteins was normalized to that of actin, and the relative fold change is shown below each pair. E, Isogenic control (−) and IRP2-KO (+) MIA-PaCa2 cells were transfected with a reporter that contains GFP coding region alone or together with TAp63 5′ or 3′UTR. Twenty-four hours posttransfection, cell lysates were collected and subjected to Western blot analysis to detect IRP2, GFP, TAp63, and actin. The levels of GFP and TAp63 proteins were normalized to that of actin, and the relative fold change is shown below each pair. F, A putative IRE in TP63 3′UTR is recognized by and responsive to IRP2. IRP2−/− MIA-PaCa2 cells were transfected with control pcDNA3 (−) or a vector expressing HA-IRP2 (+) along with a GFP reporter as listed in C. Twenty-four hours posttransfection, cell lysates were collected and subjected to Western blot analysis to detect IRP2, GFP, TAp63, and actin proteins. The levels of GFP and TAp63 proteins were normalized to that of actin, and the relative fold change is shown below each pair.
Next, several reporters were generated to determine whether a region in TP63 mRNA can be recognized by and responsive to IRP2 (Fig. 3C). We found that ectopic expression of IRP2 decreased the level of p63 protein upon cotransfection with reporter expressing p63 CDS plus its 3′UTR in IRP2−/− MIA-PaCa2 cells (Fig. 3D, compare lanes 5 and 6). However, IRP2 could not alter p63 expression upon cotransfection with reporters expressing p63 CDS alone or together with p63 5′UTR (Fig. 3D, compare lane 1 and 3 with 2 and 4, respectively). To verify this, GFP expression vectors that carry GFP alone or together with 5′ or 3′UTR of p63 mRNA was generated (Fig. 3C). Consistently, we observed that loss of IRP2 led to an increased GFP expression upon transfection with reporter expressing GFP CDS plus p63 3′UTR in MIA-PaCa2 cells (Fig. 3E, lanes 5 and 6). However, loss of IRP2 could not alter GFP expression upon transfection with reporters expressing GFP CDS alone or together with p63 5′UTR reporter (Fig. 3E, lanes 1–4). As an internal control, we showed that endogenous p63 expression in MIA-PaCa2 cells was increased by IRP2-KO regardless of the overexpression of GFP reporters (Fig. 3E). Because p63 3′UTR is responsive to IRP2 (Fig. 3D and E), we searched for a conserved IRE consensus sequence consist of six-membered (CAGUGX) loop that can be recognized by IRP2 (53, 54). Interestingly, we located a putative IRE (CAGUGU) at nt 2792 in p63 3′UTR (Fig. 3C). We thus generated a GFP reporter harboring p63 3′UTR but lacks of such a putative IRE (Fig. 3C). We showed that IRP2 inhibited GFP expression upon transfection with a reporter that contains an intact p63 3′UTR in IRP2−/− MIA-PaCa2 cells (Fig. 3F, compare lanes 3 and 4). In contrast, IRP2 had no effect on GFP expression upon cotransfection with a reporter expressing IRE-deleted p63 3′UTR (Fig. 3F, compare lanes 5 and 6). As an internal control, we showed that endogenous p63 expression was reduced by ectopic expression of IRP2 regardless of the transfection with a GFP reporter in IRP2−/− cells (Fig. 3F).
Ectopic expression of IRP2 promotes, whereas knockdown of IRP2 inhibits, cell growth, at least in part, via repressing TAp63
Previously, we demonstrated that IRP2 represses p53 expression via mRNA translation (47, 48). As p53 is a potent suppressor of cell growth, it is necessary to examine the effect of IRP2-mediated TAp63 expression on cell growth in the absence of p53. In this regard, p53−/− HepG2 cell lines were generated by using CRISPR/Cas9. Next, p53−/− HepG2 cells were transiently transfected with an IRP2 expression vector along with a control vector or a vector expressing TAp63 (Fig. 4A). We found that upon ectopic expression of IRP2, the levels of ectopic and endogenous TAp63 protein were decreased (Fig. 4A, compare lane 2 with 1, lane 4 with 3, respectively), consistent with the above observation in Fig. 1. Next, colony formation assay was performed and found that ectopic IRP2 enhanced, whereas ectopic TAp63 inhibited, colony formation in p53−/− HepG2 cells (Fig. 4B, compare the 1st column with the 2nd and 3rd column, respectively). Interestingly, we found that the reduced colony numbers by ectopic TAp63 can be increased by ectopic IRP2, suggesting that IRP2 facilitates cell growth via repressing TAp63 expression (Fig. 4B, compare the 4th column with the 1st and 2nd columns, respectively). Similar results were observed in p53−/− Hep3B cell (Fig. 4C and D).
Figure 4.
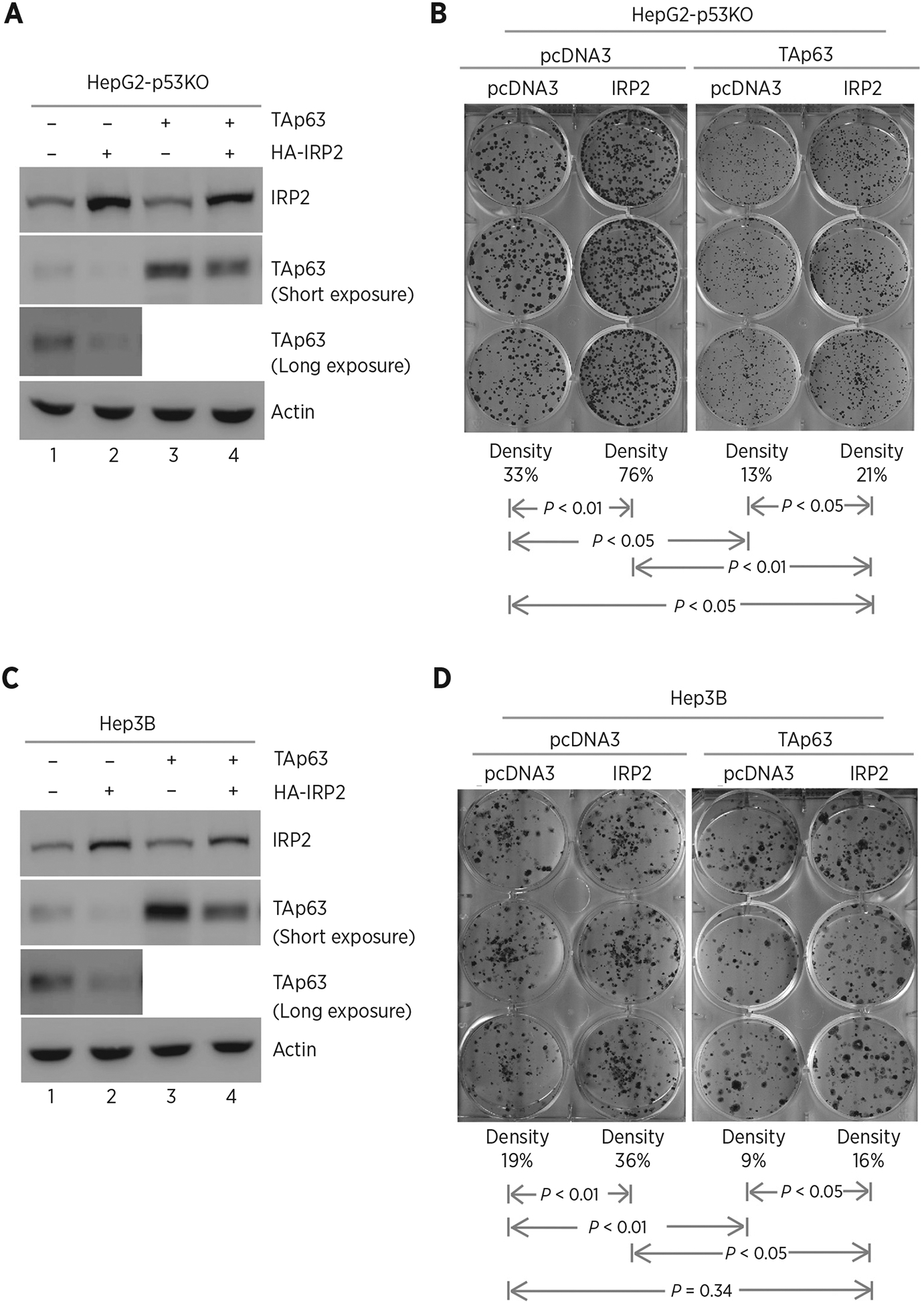
Ectopic expression of IRP2 promotes cell growth via TAp63. A, p53−/− HepG2 cells were transfected with control pcDNA3 or a vector expressing HA-tagged IRP2 along with a control vector or a vector expressing TAp63. Twenty-four hours posttransfection, cell lysates were collected and subjected to Western blot analysis to detect IRP2, TAp63, and actin proteins. B, Colony formation assay was performed with p53−/− HepG2 cells transfected with control pcDNA3 or a vector expressing HA-tagged IRP2, followed by cotransfection with a control vector or a vector expressing TAp63. The relative density for colonies was showed below each image. C and D, The experiments were performed as in A and B except that p53−/− Hep3B cells were used.
To further confirm that IRP2 promotes cell growth via repressing TAp63, p53−/− HepG2 cells were transiently transfected with a scrambled siRNA or a siRNA against IRP2, TAp63, or both. We showed that the level of TAp63 protein was increased by silencing of IRP2 in p53−/− HepG2 cells (Fig. 5A, compare lanes 2 and 4 with 1 and 3, respectively), consistent with the above observation in Fig. 2. However, the level of IRP2 was not altered by silencing of TAp63 (Fig. 5A, compare lanes 3 and 4 with 1 and 2, respectively). Next, we performed colony formation assay and showed that silencing of IRP2 decreased the number of colonies in p53−/− HepG2 (Fig. 5B, compare the 1st with the 2nd column). In contrast, the number of colonies was increased by silencing of TAp63 (Fig. 5B, compare the 3rd with the 1st column). Most importantly, the decreased colony formation by silencing of IRP2 was increased by silencing of TAp63, reaching the level in control cells (Fig. 5B, compare the 4th column with the 1st and 2nd columns, respectively). Similar results were observed in p53−/− Hep3B cells (Fig. 5C and D). Together, these observations suggest that IRP2 is required for cell growth, at least in part, via repressing TAp63 expression.
Figure 5.
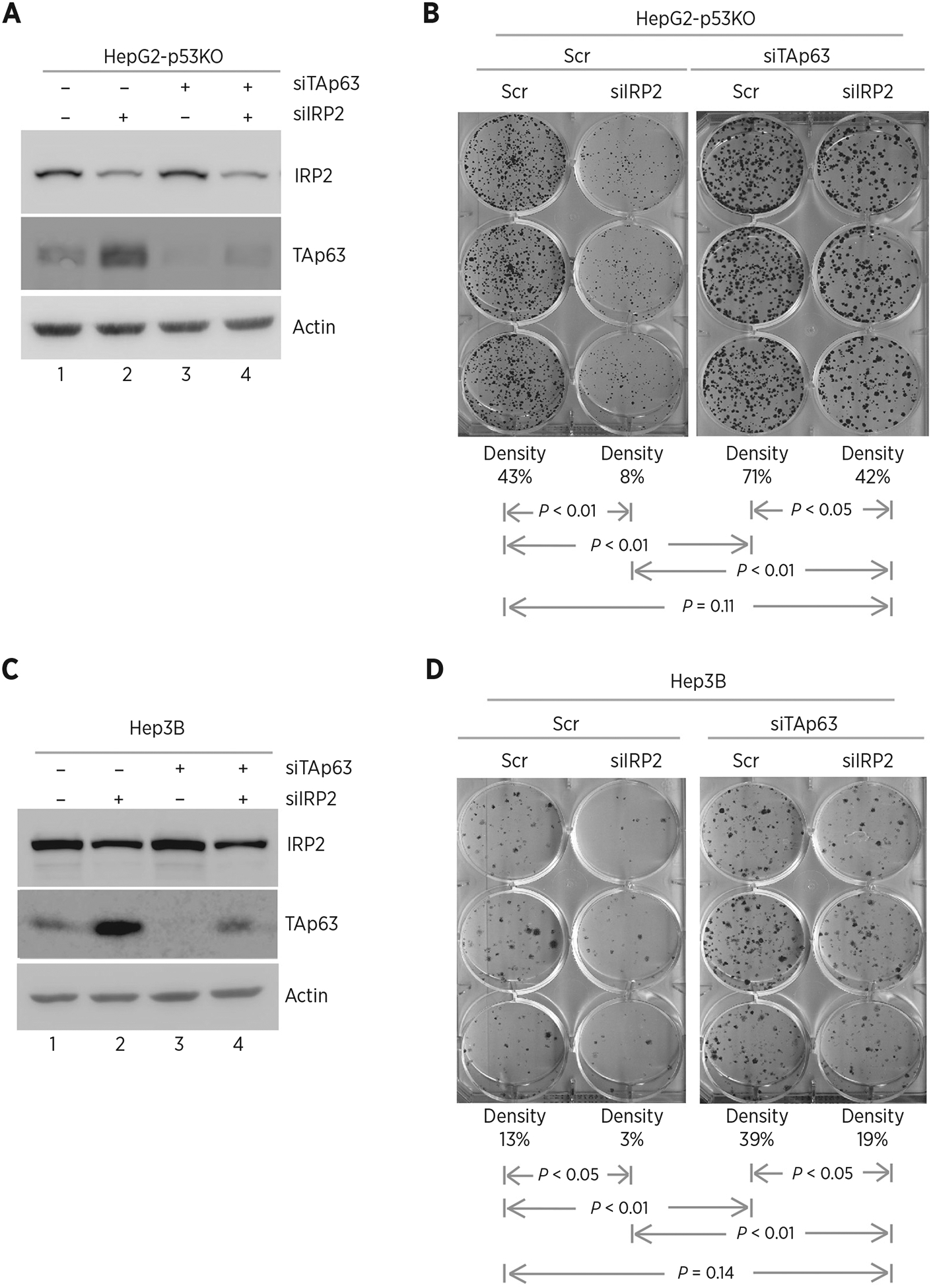
Knockdown of IRP2 inhibits cell growth via TAp63. A, p53−/− HepG2 cells were transfected with scrambled siRNA or siRNA against IRP2, followed by cotransfection with scrambled siRNA or siRNA against TAp63 for 72 hours. Cell lysates were collected and subjected to Western blot analysis with various antibodies as indicated. B, Colony formation assay was performed with p53−/− HepG2 cells transfected with scrambled siRNA or siRNA against IRP2, followed by cotransfection with scramble siRNA or siRNA against TAp63 for 72 hours. The relative density for colonies was showed below each image. C and D, The experiments were performed as in A and B except that p53−/− Hep3B cells were used.
Loss of IRP2 inhibits cellular senescence induced by TAp63 deficiency
To further explore the biological significance of IRP2-mediated TAp63 repression, we generated a cohort of WT, TAp63+/−, Irp2−/− and Irp2−/−;TAp63+/− MEFs. As expected, the level of IRP2 protein was not detectable in Irp2−/− and Irp2−/−;TAp63+/− MEFs (Fig. 6A, compare lanes 3 and 4 with 1 and 2) and loss of Irp2 resulted in an increase in TAp63 transcript in Irp2−/− MEFs (Fig. 6B, compare lanes 1 and 3). We also found that the level of TAp63 transcript in TAp63+/− MEFs was restored to near WT level by loss of Irp2 (Fig. 6B, compare lane 4 with lanes 1 and 2, respectively). Next, SA-β-gal assay was performed and we found that the percentage of SA-β-gal–positive cells was markedly increased in TAp63+/− MEFs (63%) as compared with that in WT MEFs (21%; Fig. 6C), consistent with the previous reports (29, 39). However, loss of Irp2 alone had little effect on cellular senescence in Irp2−/− MEFs (Fig. 6C, the 3rd column). Importantly, loss of Irp2 markedly reduced the number of SA-β–positive cells from 63% in TAp63+/− MEFs to 23% in Irp2−/−;TAp63+/− MEFs (P < 0.05; Fig. 6C). Furthermore, we analyzed the expression of p130 and PML, both of which are known to play a role in triggering the senescence process (55, 56). We found that the levels of p130 and PML were highly increased in TAp63+/− MEFs (Fig. 6D, compare lane 2 with 1), consistent with previous reports (57, 58). In contrast, the levels of p130 and PML increased in TAp63+/− MEFs were reduced to near WT levels in Irp2−/−;TAp63+/− MEFs (Fig. 6D, compare lane 4 with lanes 1 and 2, respectively). Together, these data indicate that loss of Irp2 restores TAp63 expression in TAp63+/− MEFs and consequently inhibits cellular senescence induced by loss of TAp63.
Figure 6.
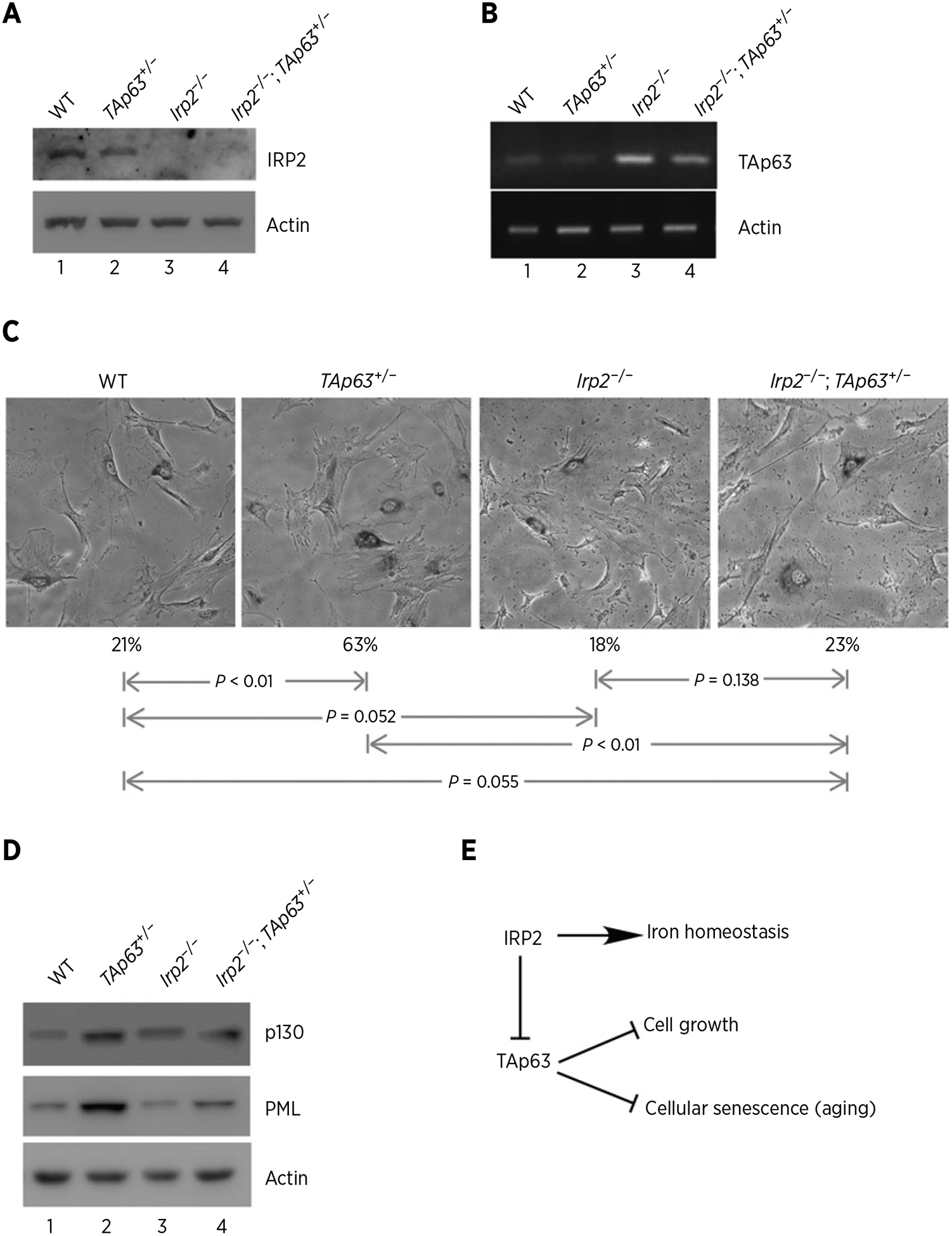
Loss of IRP2 inhibits cellular senescence induced by TAp63 deficiency. A, Western blots were prepared using extracts from WT, TAp63+/−, Irp2−/−, and Irp2−/−;TAp63+/− littermate MEFs. The blots were probed with antibodies against Irp2 and actin. B, The levels of p63 and actin transcripts were measured in WT, TAp63+/−, Irp2−/−, and Irp2−/−;TAp63+/− MEFs. C, Representative images of SA-β-Gal–stained WT, TAp63+/−, Irp2−/−, and Irp2−/−;TAp63+/− MEFs. Quantification of the percentage of SA-β-Gal–positive cells was shown below each image. D, Western blots were prepared using extracts from WT, TAp63+/−, Irp2−/−, and Irp2−/−;TAp63+/− MEFs. The blots were probed with antibodies against p130, PML and actin, respectively. E, A model of how IRP2 modulates cell growth and cellular senescence via TAp63.
Discussion
Recent studies indicate that iron metabolism plays a critical role in contributing tumorigenesis. However, very little is known about the role of IRP2 in tumorigenesis despite its well-studied role in modulating iron homeostasis. In this study, we examined whether IRP2 regulates expression of TAp63, a member of p53 tumor suppressor family. We found that IRP2 represses TAp63 expression regardless of p53 status (Figs. 1 and 2), suggesting that TAp63 expression is regulated directly by IRP2 in a p53-independent manner. In addition, we found that IRP2 stabilizes TAp63 mRNA stability via a putative IRE in p63 3′UTR (Fig. 3). We would like to note that p63 3′UTR is also recognized by and responsive to Rbm38 and Rbm24, both of which regulate p63 expression via mRNA stability (39–42). These data indicate that TP63 mRNA stability is uniquely regulated by multiple RNA-binding proteins through its 3′UTR. Thus, further studies are warranted to determine whether these RNA-binding proteins can coordinate to regulate p63 expression.
In our study, we found that ectopic expression of IRP2 suppresses TAp63 expression and subsequently, promotes cell growth, the latter of which can be blocked by ectopic expression of TAp63 (Fig. 4). In contrast, loss of IRP2 increases TAp63 expression and subsequently, inhibits cell growth, the latter of which can be blocked by knockdown of TAp63 (Fig. 5). These data suggest that IRP2 promotes cell growth, at least in part, via repressing TAp63 expression. A hypothesis is summarized in Fig. 6E. Nevertheless, several questions remain to be addressed. First, in vivo studies are needed to determine whether IRP2-mediated TAp63 repression is capable of promoting tumor formation. Second, as IRP2 functions as a key regulator of iron homeostasis, it would be interesting to determine whether IRP2-mediated TAp63 repression plays a role in iron metabolism, such as modulating the liable iron pool. Third, due to the usage of two different promoters, TP63 is expressed as two isoforms, TAp63 and ΔNp63, with opposing functions. As we showed that IRP2 stabilizes TAp63 mRNA (Fig. 3), it is possible that IRP2 stabilizes ΔNp63 mRNA due to the same 3′UTR. Thus, further studies are needed to examine the regulation of ΔNp63 by IRP2 and whether this regulation contributes to tumorigenesis.
It is known that cellular senescence is a tumor-suppressing mechanism but also leads to accelerated aging (59, 60). In our study, we showed that loss of IRP2 alleviates cellular senescence mediated by TAp63 deficiency (Fig. 6). Notably, several reports showed that dysfunctional iron homeostasis is associated with aging (61, 62). In addition, TAp63-deficient mice are prone to premature aging. Thus, it would be important to examine whether the IRP2–TAp63 axis plays a role in aging by altering iron metabolism.
In conclusion, our study revealed a novel regulation of TAp63 by IRP2 and our data indicate that IRP2 exerts its oncogenic activity at least partly via suppressing TAp63 expression. Given that TAp63 is capable of suppressing tumorigenesis (63), these observations prompt us to speculate that targeting IRP2 may be used to restore TAp63 expression, which can be explored as a cancer therapeutic strategy.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH grants CA224433-01 and CA195828 and Center for Companion Animal Health (CCAH) Faculty Grants 2017-26-F, School of Veterinary Medicine, University of California, Davis.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Gray NK, Hentze MW. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J 1994; 13:3882–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci 2004;1012:1–13. [DOI] [PubMed] [Google Scholar]
- 3.Hentze MW, Seuanez HN, O’Brien SJ, Harford JB, Klausner RD. Chromosomal localization of nucleic acid-binding proteins by affinity mapping: assignment of the IRE-binding protein gene to human chromosome 9. Nucleic Acids Res 1989;17:6103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouault TA, Tang CK, Kaptain S, Burgess WH, Haile DJ, Samaniego F, et al. Cloning of the cDNA encoding an RNA regulatory protein–the human iron-responsive element-binding protein. Proc Natl Acad Sci U S A 1990;87:7958–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell 1993;72:19–28. [DOI] [PubMed] [Google Scholar]
- 6.Leibold EA, Guo B. Iron-dependent regulation of ferritin and transferrin receptor expression by the iron-responsive element binding protein. Annu Rev Nutr 1992;12:345–68. [DOI] [PubMed] [Google Scholar]
- 7.Theil EC. The IRE (iron regulatory element) family: structures which regulate mRNA translation or stability. Biofactors 1993;4:87–93. [PubMed] [Google Scholar]
- 8.Hentze MW, Rouault TA, Caughman SW, Dancis A, Harford JB, Klausner RD. A cis-acting element is necessary and sufficient for translational regulation of human ferritin expression in response to iron. Proc Natl Acad Sci U S A 1987;84:6730–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dandekar T, Stripecke R, Gray NK, Goossen B, Constable A, Johansson HE, et al. Identification of a novel iron-responsive element in murine and human erythroid delta-aminolevulinic acid synthase mRNA. EMBO J 1991;10:1903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000;5:299–309. [DOI] [PubMed] [Google Scholar]
- 11.Gray NK, Pantopoulos K, Dandekar T, Ackrell BA, Hentze MW. Translational regulation of mammalian and Drosophila citric acid cycle enzymes via iron-responsive elements. Proc Natl Acad Sci U S A 1996;93:4925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat Struct Mol Biol 2007;14:420–6. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stolte J, et al. Iron regulation and the cell cycle: identification of an iron-responsive element in the 3′-untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. J Biol Chem 2006;281:22865–74. [DOI] [PubMed] [Google Scholar]
- 14.Cmejla R, Petrak J, Cmejlova J. A novel iron responsive element in the 3′UTR of human MRCKalpha. Biochem Biophys Res Commun 2006;341:158–66. [DOI] [PubMed] [Google Scholar]
- 15.Kohler SA, Menotti E, Kuhn LC. Molecular cloning of mouse glycolate oxidase. High evolutionary conservation and presence of an iron-responsive element-like sequence in the mRNA. J Biol Chem 1999;274:2401–7. [DOI] [PubMed] [Google Scholar]
- 16.Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA. Microcytic anemia, erythropoietic protoporphyria, and neuro-degeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005;106:1084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, et al. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005;106:2580–9. [DOI] [PubMed] [Google Scholar]
- 18.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J 2004;23:386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh MC, Zhang DL, Jeong SY, Kovtunovych G, Ollivierre-Wilson H, Noguchi A, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2alpha. Cell Metab 2013;17:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson SA, Nizzi CP, Chang YI, Deck KM, Schmidt PJ, Galy B, et al. The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab 2013;17:282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998;2:305–16. [DOI] [PubMed] [Google Scholar]
- 22.Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisa E, Pesole G, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res 2009;37:6092–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004;2:371–86. [PubMed] [Google Scholar]
- 24.De Laurenzi V, Melino G. Evolution of functions within the p53/p63/p73 family. Ann N Y Acad Sci 2000;926:90–100. [DOI] [PubMed] [Google Scholar]
- 25.Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci 2003;28:663–70. [DOI] [PubMed] [Google Scholar]
- 26.Yang A, McKeon F. P63 and P73: P53 mimics, menaces and more. Nat Rev Mol Cell Biol 2000;1:199–207. [DOI] [PubMed] [Google Scholar]
- 27.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999;398: 708–13. [DOI] [PubMed] [Google Scholar]
- 28.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010;467:986–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 2009;5:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 2012;139:772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong SR, Wu H, Wang B, Abuetabh Y, Sergi C, Leng RP. The regulation of tumor suppressor p63 by the ubiquitin-proteasome system. Int J Mol Sci 2016;17;2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci U S A 2006;103:12753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 2009;15:1179–85. [DOI] [PubMed] [Google Scholar]
- 34.Hildesheim J, Belova GI, Tyner SD, Zhou X, Vardanian L, Fornace AJ Jr. Gadd45a regulates matrix metalloproteinases by suppressing DeltaNp63alpha and beta-catenin via p38 MAP kinase and APC complex activation. Oncogene 2004;23:1829–37. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, et al. ATM kinase is a master switch for the Delta Np63 alpha phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008;7:2846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacPartlin M, Zeng SX, Lu H. Phosphorylation and stabilization of TAp63-gamma by IkappaB kinase-beta. J Biol Chem 2008;283:15754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009;137:87–98. [DOI] [PubMed] [Google Scholar]
- 38.Stindt MH, Muller PA, Ludwig RL, Kehrloesser S, Dotsch V, Vousden KH. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015; 34:4300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Y, Xu E, Zhang J, Chen M, Flores E, Chen X. The Rbm38-p63 feedback loop is critical for tumor suppression and longevity. Oncogene 2018;37:2863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Jun Cho S, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci U S A 2010;107:9614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Feng X, Sun W, Zhang J, Chen X. Serine 195 phosphorylation in the RNA-binding protein Rbm38 increases p63 expression by modulating Rbm38’s interaction with the Ago2-miR203 complex. J Biol Chem 2019; 294:2449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu E, Zhang J, Zhang M, Jiang Y, Cho SJ, Chen X. RNA-binding protein RBM24 regulates p63 expression via mRNA stability. Mol Cancer Res 2014;12:359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lena AM, Shalom-Feuerstein R, Rivetti di Val Cervo P, Aberdam D, Knight RA, Melino G, et al. miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ 2008;15:1187–95. [DOI] [PubMed] [Google Scholar]
- 44.Mehrazarin S, Chen W, Oh JE, Liu ZX, Kang KL, Yi JK, et al. The p63 gene is regulated by grainyhead-like 2 (GRHL2) through reciprocal feedback and determines the epithelial phenotype in human keratinocytes. J Biol Chem 2015;290:19999–20008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Chen X. p53 tumor suppressor and iron homeostasis. FEBS J 2019;286:620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su X, Gi YJ, Chakravarti D, Chan IL, Zhang A, Xia X, et al. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab 2012;16: 511–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Qian Y, Zhang J, Yan W, Jung YS, Chen M, et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev 2017;31:1243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Feng X, Zhang J, Chen M, Huang E, Chen X. Iron regulatory protein 2 is a suppressor of mutant p53 in tumorigenesis. Oncogene 2019;38:6256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang M, Zhang J, Chen X, Cho SJ, Chen X. Glycogen synthase kinase 3 promotes p53 mRNA translation via phosphorylation of RNPC1. Genes Dev 2013;27:2246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo B, Yu Y, Leibold EA. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J Biol Chem 1994;269:24252–60. [PubMed] [Google Scholar]
- 51.Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiriksdottir E, Langel U, et al. Immunoprecipitation of mRNA-protein complexes. Nat Protoc 2006;1:577–80. [DOI] [PubMed] [Google Scholar]
- 52.Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 2014;9:e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butt J, Kim HY, Basilion JP, Cohen S, Iwai K, Philpott CC, et al. Differences in the RNA binding sites of iron regulatory proteins and potential target diversity. Proc Natl Acad Sci U S A 1996;93:4345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henderson BR, Menotti E, Kuhn LC. Iron regulatory proteins 1 and 2 bind distinct sets of RNA target sequences. J Biol Chem 1996;271:4900–8. [DOI] [PubMed] [Google Scholar]
- 55.Helmbold H, Deppert W, Bohn W. Regulation of cellular senescence by Rb2/p130. Oncogene 2006;25:5257–62. [DOI] [PubMed] [Google Scholar]
- 56.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000;406:207–10. [DOI] [PubMed] [Google Scholar]
- 57.Bernassola F, Oberst A, Melino G, Pandolfi PP. The promyelocytic leukaemia protein tumour suppressor functions as a transcriptional regulator of p63. Oncogene 2005;24:6982–6. [DOI] [PubMed] [Google Scholar]
- 58.McDade SS, Patel D, McCance DJ. p63 maintains keratinocyte proliferative capacity through regulation of Skp2-p130 levels. J Cell Sci 2011; 124:1635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Narita M, Lowe SW. Senescence comes of age. Nat Med 2005;11:920–2. [DOI] [PubMed] [Google Scholar]
- 60.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005;436:660–5. [DOI] [PubMed] [Google Scholar]
- 61.Xu J, Jia Z, Knutson MD, Leeuwenburgh C. Impaired iron status in aging research. Int J Mol Sci 2012;13:2368–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wawer AA, Jennings A, Fairweather-Tait SJ. Iron status in the elderly: a review of recent evidence. Mech Ageing Dev 2018;175:55–73. [DOI] [PubMed] [Google Scholar]
- 63.Westfall MD, Pietenpol JA. p63: Molecular complexity in development and cancer. Carcinogenesis 2004;25:857–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.