

Abstract
High-throughput RNA sequencing unveiled the complexity of transcriptome and significantly increased the records of long noncoding RNAs (lncRNAs), which were reported to participate in a variety of biological processes. Identification of lncRNAs is a key step in lncRNA analysis, and a bunch of bioinformatics tools have been developed for this purpose in recent years. While these tools allow us to identify lncRNA more efficiently and accurately, they may produce inconsistent results, making selection a confusing issue. We compared the performance of 41 analysis models based on 14 software packages and different data sets, including high-quality data and low-quality data from 33 species. In addition, computational efficiency, robustness, and joint prediction of the models were explored. As a practical guidance, key points for lncRNA identification under different situations were summarized. In this investigation, no one of these models could be superior to others under all test conditions. The performance of a model relied to a great extent on the source of transcripts and the quality of assemblies. As general references, FEELnc_all_cl, CPC, and CPAT_mouse work well in most species while COME, CNCI, and lncScore are good choices for model organisms. Since these tools are sensitive to different factors such as the species involved and the quality of assembly, researchers must carefully select the appropriate tool based on the actual data. Alternatively, our test suggests that joint prediction could behave better than any single model if proper models were chosen. All scripts/data used in this research can be accessed at http://bioinfo.ihb.ac.cn/elit.
Keywords: long noncoding RNA identification, tools comparison, simulated and biological data sets, joint prediction, non-model species
INTRODUCTION
Long noncoding RNAs (lncRNAs) are transcripts with little or no coding ability and longer than 200 nt (Mercer et al. 2009). As a class of important biomacromolecules in eukaryotes, lncRNAs were reported to participate in the regulation of gene expression, cell differentiation, cancer progress, and many other biological processes (Fatica and Bozzoni 2014; Yang et al. 2014). Benefiting from the advances in RNA sequencing (RNA-seq) and computational techniques, a large number of novel lncRNAs have been identified and their entries in databases are accumulating rapidly (Pauli et al. 2012; Iyer et al. 2015; Maracaja-Coutinho et al. 2019). While the GENCODE database has recorded 15,512 lncRNAs (Harrow et al. 2012), the ENCODE project predicted that 62%–75% of the human chromosome sequences could be transcribed, most of which are noncoding sequences (Djebali et al. 2012). Iyer claimed that there are 58,648 lncRNA genes in the human genome, accounting for 68% of expressed genes (Iyer et al. 2015), which are more than 2 times of protein-coding genes (21,313). In fact, the NONCODE website has collected up to 144,134 human lncRNA genes (Zhao et al. 2016b), which are approximately as many as 6–7 times of the protein-coding genes.
While lncRNA shares many features with mRNA (e.g., both can be multiexonic and be polyadenylated [Kung et al. 2013]), its roles in organisms are completely different from those of mRNA, and research approaches for them differ as well (Rinn and Chang 2012; Yan et al. 2012; Cech and Steitz 2014; Holoch and Moazed 2015; Liu et al. 2015; Kashi et al. 2016). Therefore, the discrimination of the two RNA types is the first thing in research, and the development of effective lncRNA recognition methods becomes a basic issue in lncRNA research.
Despite the discovery of bifunctional RNAs that function as both mRNA and lncRNA (Nam et al. 2016; Williamson et al. 2017; Ransohoff et al. 2018), the essential difference between mRNA and lncRNA lies in the protein-coding ability in the overwhelming majority of cases. Thus, lncRNA recognition becomes a problem of classifying coding capabilities. Open reading frame (ORF) is a straightforward feature for discrimination because transcripts with longer ORFs are more likely to encode proteins. However, the transcripts assembled using popular RNA-seq technologies are pretty poor in the integrity of ORFs, which obstructs this simple classification (Uszczynska-Ratajczak et al. 2018). Therefore, other features independent of the integrity of transcripts, such as MLCDS (the most-like coding domain sequence) and k-mers, were applied in lncRNA identification. In addition, lncRNA shows a distinct difference from mRNA in terms of sequence conservation, which can be measured from multiple sequence alignments (Corona-Gomez et al. 2020). In brief, the exons of lncRNAs are far less conserved than the exons of mRNAs (Ulitsky 2016; Hon et al. 2017). Thus, sequence conservation is widely used in lncRNA identification as well.
With the increase of researches on lncRNA, a number of tools for lncRNA identification have been developed recently. Most classification solutions are based on machine learning using well-characterized mRNA/lncRNA data sets. Logistic regression and support vector machines are popular training algorithms, while deep learning and random forests based on the decision tree models are frequently applied as well. Data from GENCODE and RefSeq are often used as the golden standard for training sets (Sun et al. 2013b; Li et al. 2014; Hu et al. 2017; Kang et al. 2017; Wucher et al. 2017). There is no doubt that human and mouse are the two most studied species. Public databases have accumulated huge omics data from human and mouse individuals, including genome sequences, RNA-seq data, ChIP-seq data, multigenome alignments, etc. More importantly, many of these data were manually validated and fairly reliable (Harrow et al. 2014; Lagarde et al. 2016; The UniProt Consortium 2017). Therefore, these data are most commonly used to build prediction models within vertebrates. Some other model species, such as zebrafish and Arabidopsis, are also considered as training species. Among the dozens of lncRNA–mRNA classification software tools developed until now, tools such as CNCI (Sun et al. 2013b) and CPAT (Wang et al. 2013) are applicable to multiple species, while other tools using species-specific prebuilt models are only applicable to certain species.
Different methods inevitably lead to more or less inconsistent results, which makes selection a confusing problem. Although some reviews have explained in detail the principles and algorithms for lncRNA identification (Guo et al. 2016; Housman and Ulitsky 2016), and some comparisons have been made at the time of the release of software tools, we still lack a comprehensive assessment of all of these methods. This paper aims to provide such a comparison as a guidance to selecting appropriate tools for research. RNAcode (Washietl et al. 2011), PhyloCSF (Lin et al. 2011), COME (Hu et al. 2017), and iSeeRNA (Sun et al. 2013a) are prediction tools based on sequence conservation. According to the comparisons made at the time of publication, the last two tools were superior to the first two not only in accuracy but also in usability. Since we aim to give practical recommendations for users, only COME and iSeeRNA were evaluated as representatives of these sequence-conservation-based tools. In this study, we compared 14 popular software packages using three high-standard databases/data sets, including two golden-standard data sets that are frequently used for model training and one transcriptome assembled from more than 7000 RNA-seq samples. Then the robustness of the software packages was tested on the transfrags or other imperfect transcripts through simulation data. We also compared their performance on data from different biological species and explored the joint use of these tools. The transcripts assembled from two real RNA-seq data were used to compare their performance under various data quality. The time efficiency was discussed last.
RESULTS
For representative and reliable evaluations, we carefully selected 14 popular or newly published software packages covering most of the statistical models and analysis algorithms commonly used in lncRNA recognition (Table 1; Kong et al. 2007; Mistry et al. 2013; Sun et al. 2013a,b, 2015; Wang et al. 2013; Li et al. 2014; Zhao et al. 2016a; Hu et al. 2017; Kang et al. 2017; Schneider et al. 2017; Singh et al. 2017; Wucher et al. 2017; Negri et al. 2019).
TABLE 1.
Software for lncRNA identification

A total of 41 analytic models (see Materials and Methods) based on the 14 lncRNA recognition software were evaluated using various data sets, including high-quality data and data with sequencing errors (Table 2). For most software tools, lncRNA recognition can be based entirely on sequence, except for COME, iSeeRNA, lncRScan-SVM, and lncScore, which require genome annotation files in GTF format. By using different quality transcripts from multiple species, this benchmark testing is designed to provide solutions for a variety of application scenarios. As the results showed, it makes sense to adopt a prior model combination based on a specific application scenario.
TABLE 2.
Data used for evaluation

Best performance on golden-standard data
The golden-standard data sets (golden_human and golden_mouse) were applied to obtain the best performance for most software tools except PLncPRO and RNAplonc, which were specifically trained for plants. Unfortunately, just as the golden-standard data, the mRNAs and lncRNAs of the training data for CNCI, PLEK, and CPAT are from RefSeq and GENCODE, respectively, while the training data of lncScore, lncRScan-SVM, and COME are from GENCODE as well. The intersection of testing data and training data will undoubtedly give these tools additional benefits, we must be aware of this when evaluating tool performance.
Potentials in prediction
For human data, there are four prediction models with AUC values greater than 0.99, and nine models with AUC values between 0.95 and 0.99 in representative model sets (Supplemental Fig. S1; Supplemental Table S1). The corresponding numbers for mouse data are 3 and 10 (Supplemental Fig. S2; Supplemental Table S1). The performance of the tools on the two species is comparable since the difference of AUC values is very small (≤0.02) for most classifiers except PLEK, whose AUC value ranks second in human or 24th in mouse (Supplemental Table S1). In general, CPC and FEELnc_all_cl are the two best models for both human and mouse data, while RNAplonc_guess and CNCI_ve are the two worst in both data sets (Fig. 1A; Supplemental Figs. S1 and S2). The poor performance of RNAplonc_guess can be ascribed to its training data from plants.
FIGURE 1.
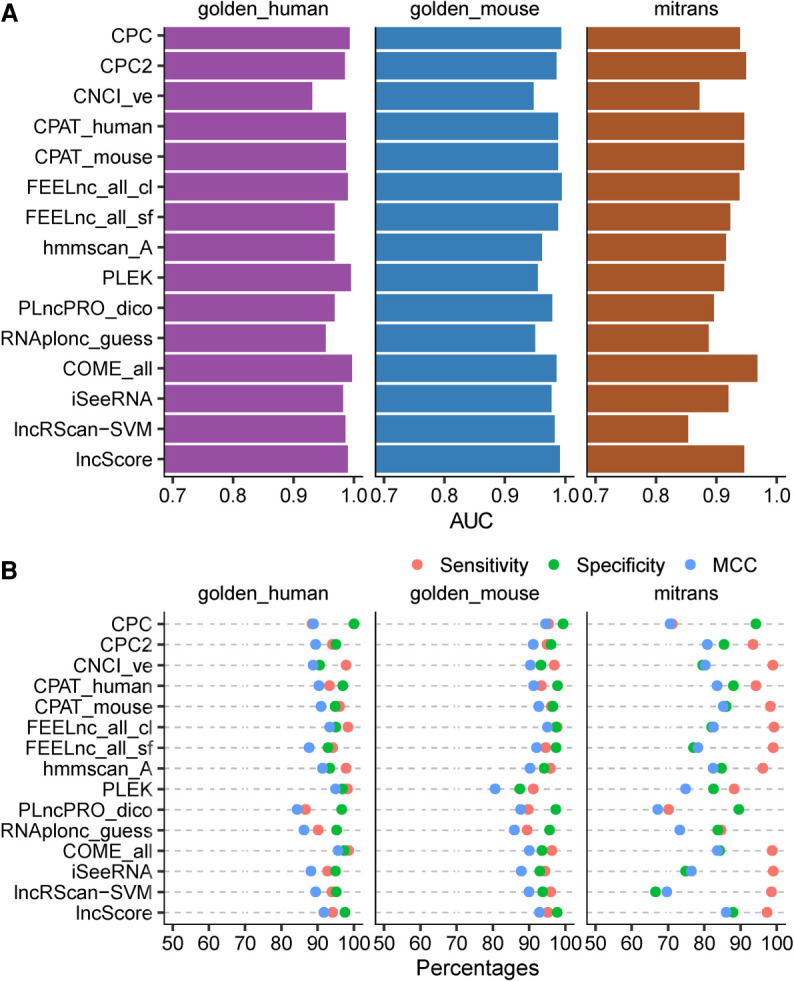
The performance of representative models on golden_human, golden_mouse, and mitrans. The representative models are a subset of the models that have been tested (see Materials and Methods). (A) AUC value of ROC curve for each model on golden_human (purple), golden_mouse (blue), and mitrans (brown). (B) Sensitivity (red), Specificity (green), and Matthews correlation coefficient (MCC, blue) values for each model on the testing data. Sensitivity indicates the proportion of true classified positive samples (lncRNAs) on the total input positive samples (in percentage). Specificity indicates the proportion of true classified negative samples (mRNAs) on the total input negative samples (in percentage). MCC indicates the overall performance ranges from −100 to 100 (in percentage), where 100 implies a perfect prediction, 0 implies a random prediction, and −100 implies a totally wrong prediction.
The ROC curve of CNCI showed a slower rise, indicating that CNCI has a false positive rate higher than other tools when the classification threshold is strict (Supplemental Figs. S4, S5). Such a result implies that it is impracticable for CNCI to achieve higher Specificity (SPE) by setting tighter thresholds and sacrificing Sensitivity (SEN). Surprisingly, longdist's performance is far worse than the official description (Supplemental Figs. S7, S8). Therefore, it was excluded from this comparison and some subsequent analyses.
As gene-structure-dependent methods, COME, lncRScan-SVM, lncScore and iSeeRNA, are capable of utilizing additional annotation information for prediction, they were expected to make better prediction. However, these four tools did not show obvious superiority over others (Fig. 1A; Supplemental Figs. S1, S2).
FEELnc can train prediction models in two different modes (“cl” and “sf”). While both mRNAs and lncRNAs have to be supplied for model training in the cl mode, the sf mode can generate lncRNAs from shuffled mRNAs. As the result of our testing, the cl mode performed reasonably much better than the sf mode (Supplemental Figs. S10, S11). This fact indicates that for some sequence features, the shuffled mRNAs are not comparable to the real lncRNAs.
Prediction accuracy
The performance of a software tool depends not only on its algorithm, but also on the thresholds used in analysis. Considering the fact that default values are most often adopted by users, the performance of prediction tools at their default thresholds was compared. In general, default values are set to achieve the highest accuracy (ACC), which is determined by all positive and negative cases with equal weights. Although CNCI_ve ranked the second lowest according to the AUC values on the two test data, its ACC and Matthews correlation coefficient (MCC) values rank in the middle (Fig. 1A,B; Supplemental Tables S1, S2). This suggests that CNCI_ve has carefully chosen a threshold to achieve better results in practice. On the contrary, ACC/MCC of CPC was far worse than its AUC in human data. Considering its outstanding SPE and positive predictive value (PPV), we can conclude that CPC sacrificed its SEN and negative predictive value (NPV).
On the human data, ACC/MCC did not differ significantly between COME_all and PLEK, and the both models were superior to other models (α = 0.05. Fig. 1B; Supplemental Fig. S16; Supplemental Table S3). On the mouse data, FEELnc_all_cl had ACC/MCC significantly better than other models except CPC (α = 0.05. Fig. 1B; Supplemental Fig. S16; Supplemental Table S3). Although the best classifiers in the two data sets were different, the rank patterns of theses models are similar.
Based on higher accuracy predictions (ACC > 70%) on the human and mouse data, FEELnc_ff_cl and FEELnc_all_cl had better SEN and NPV, and CPC showed better SPE and PPV (Supplemental Fig. S16; Supplemental Table S2).
Transcripts assembled from a large number of samples
MiTranscriptome (mitrans) is a well-assembled human transcriptome database containing a large number of mRNA and lncRNA records (Iyer et al. 2015). Although most of these lncRNA sequences have not been experimentally validated, their reliability was ensured by strict thresholds used in the assembly of transcript sequences, and the accuracy of classification was promised by excluding transcripts of uncertain coding potential (TUCP). Since it has not been taken as a training set by any of the models in this study, mitrans is an ideal data set for testing. Of all the software tools in this study, CPAT and hmmscan were used for the construction of the mitrans database, but different thresholds were applied to the two software in our models. For most of the models being tested, it is pretty fair to use mitrans in general. Some models performed poorly with mitrans (ACC < 70%), and these low ACC models (LACCMs) might be excluded from some of the comparisons below.
Comparing with golden_human, the AUC of mitrans decreased in all models including hmmscan which is not based on machine-learning (Fig. 1A; Supplemental Figs. S1, S3; Supplemental Table S1). This fact reflected the difference in the inclusion criteria between the two databases. COME had the smallest AUC drop and ranked first in both data sets. Although CPC2 also declined in terms of AUC, its ranking increased by seven, indicating that its robustness is superior to other tools. PLEK and lncRScan-SVM have the largest drop in AUC, which means they may have the problem of overfitting.
For mitrans data, lncScore and CPAT_mouse were the best models, which had ACC significantly better than any other model (α = 0.05) (Fig. 1B; Supplemental Fig. S16; Supplemental Tables S2 and S3). COME_all, CPAT_human, COME_seq, FEELnc_ all_cl and hmmscan came in second, and their ACC exceeded 0.9, which were significantly better than the other models except FEELnc_ms_cl and FEELnc_hm_cl. Compared with golden_human, mitrans led to declines of ACC in all models. If LACCMs were not considered, FEELnc_hm_sf, FEELnc_zf_sf and lncScore had the smallest ACC drop, reflecting the fact that these tools are more robust for transcripts assembled from high-throughput sequencing data.
Excluding LACCMs, four models (CPAT_fly, FEELnc_ff_cl, FEELnc_ff_sf and FEELnc_all_cl) had the best SEN and NPV (Fig. 1B; Supplemental Fig. S16; Supplemental Table S2). The three FEELnc-based models even performed better in mitrans than in golden_human. Considering that the average SEN/NPV of these models were comparable between in mitrans and in golden_human, the four models were very robust to the imperfect transcripts (Supplemental Table S4). These transcripts are not as good as those manually curated, but they are much more reliable than the transcripts assembled in conventional RNA-seq analysis.
CPC, hmmscan_both, PLncPRO_dico and lncScore showed high SPE and PPV, where hmmscan_both had the best SPE, and CPC had the best PPV (Fig. 1B; Supplemental Fig. S16; Supplemental Table S2). For all models except LACCMs, despite the decreases of the SPE and PPV, the test with mitrans resulted in a rank similar to that with golden_human.
Erroneous transcripts
In RNA-seq data analysis, many of the challenges come from erroneously assembled transcripts that cannot be corrected at the state of the art. In some cases, slightly misassembled transcripts have little adverse effect on subsequent data analysis. To test the robustness of the models on erroneous transcripts, we used known transcript sequences as templates to generate simulated sequencing data and assembled them through a popular pipeline (see Materials and Methods). The models were assessed by comparing the classification between the new transcript and its corresponding template. If the model gives the same classification, the prediction is considered “good” regardless of the true classification. The corresponding relationships between these assembled transcripts and the original transcripts were classified by gffcompare and each was assigned a single-character class code (Pertea and Pertea 2020), among which we named “=,” “c,” and “j” as “CRE,” “CRC,” and “CRJ” in order. In detail, CRE means a transcript newly assembled shares all introns with its template transcript, while the edge of the first and the last exon may be different; CRC means that a template entirely contains its assembled counterpart, which is known as transfrag; CRJ means that a transcript shares at least one splicing site with its template, indicating a wrong assembly in our analysis. The three types of correspondences were studied further to determine the consistency of the prediction (Fig. 2A).
FIGURE 2.

The performance of the representative models on the transcripts assembled from the simulated data. Based on the golden-standard sequence sets (golden_human and golden_mouse), simulated Illumina-sequencing data sets were generated by Polyester with five sequencing depths: 30× (which means that, on average, 30 reads were simulated from each transcript), 100×, 300×, 600×, and 1200×. (A) The relationship between assembled transcripts and its templates. The CRE transcript shares an identical intron chain with its template; the CRC transcript is covered by its template; and the CRJ transcript shares at least one splicing site with its template. (B) Box plots for the MCC of all testing models on the simulated data of different sequencing depths. (C) Performance of representative models on human CRC transcripts. (D) Performance of representative models on human CRJ transcripts.
GTF can greatly affect the number of different types of transcripts assembled. If there is no GTF guidance, more CRC, less CRE, and a comparable number of CRJ transcripts were generated (Supplemental Fig. S17). To test the robustness, we mainly discussed the performance on transcripts assembled without GTF guidance.
As expected, most models performed better for CRE transcripts according to MCC in our test (Fig. 2B). As the depth increased, the models worked better for CRC transcripts, but such a correlation was not observed for CRE or CRJ transcripts.
While most of the models performed well and are similar for CRE transcripts, CPAT_human, CPAT_mouse, COME_all, and hmmscan_A were slightly better (Supplemental Figs. S18 and S19; Supplemental Table S5). The two models using FEELnc_wm had poor SEN and MCC.
Generally, CRJ transcripts assembled from sequencing data are considered to be potential alternative splicing transcripts, but we can see that they were actually misassembled in our study because all templates were known in the simulation (Trapnell et al. 2010). The models behaved very differently on these transcripts (Fig. 2D; Supplemental Fig. S20; Supplemental Table S5). CPC had the best MCC when testing with mouse data; CPC, CPAT_fly, and CPAT_mouse presented good SEN; CPC, hmmscan_A, and hmmscan_both showed good SPE. However, very few intersections showed in the results from human data in which FEELnc_all_cl was best in MCC; FEELnc_all_cl and CNCI_ve showed good SEN; hmmscan_A, CPC, and FEELnc_all_cl presented good SPE.
When classifying CRC transcripts with human data, CPAT_zebrafish and CPC2 had good SEN, and hmmscan_A, hmmscan_both and CPC had good SPE (Fig. 2C). For mouse data, FEELnc_wm_sf, FEELnc_wm_cl, and CPAT_mouse showed better SEN, while COME_all, COME_seq, hmmscan_A, and hmmscan_both showed better SPE (Supplemental Fig. S21; Supplemental Table S5). Furthermore, almost all tools had SPEs worse than SENs, especially in less sequencing depth. This fact indicates that the tools tend to predict incomplete sequences as lncRNAs. However, COME, hmmscan_A, and hmmscan_both were the exceptions, because they are less dependent on the integrity of CDS for prediction of transcript coding ability. K-mer based PLEK is another tool that is independent of CDS and surprisingly performed poorly.
In general, the performance of each model varied significantly across different types of erroneous transcripts. Although no single model showed excellent stability in all situations, each of COME_all, CPC, CNCI_ve, and COME_seq has a sum of ranks less than 300, that is, on average, the performance ranks in the top 10. Considering the extensive existence of assembly errors and sequence diversity across different species (Supplemental Table S6), these models are commendable.
Joint prediction
Joint prediction strategies are often used to improve the reliability of lncRNA predictions (Qiu et al. 2016; Wang et al. 2016, 2019; Chen et al. 2017; Rolland et al. 2019). Obviously, applying more thresholds from different models can improve the SPE and the PPV in prediction, but it also leads to an increasing of false negative predictions, and thus a decrease in overall ACC/MCC.
A rough principle and a vote principle were used in our analysis (see Materials and Methods). When the rough principle was applied, ACC and MCC decreased as the increasing number of the models used for analysis. For the voting method, ACC and MCC increased slightly before reaching a plateau, on which the mean was equal to or slightly higher than the best single model (Fig. 3A; Supplemental Figs. S22 and S23). When all 41 analytic models were used in the joint prediction, ACC and MCC decreased significantly for both strategies; obviously the comprehensive result was hindered by some worse models, such as those based on londist. This result suggests that when doing a joint prediction, choosing a proper set of models is essential for better performance.
FIGURE 3.

Summary of joint predictions. (A) The MCC of joint predictions with different number of models on the golden_human data. The combination methods (“rough” and “voting”) were described in the “Materials and Methods” section. (B) Comparison of optimal joint predictions and optimal single models on three data sets (golden_human, golden_mouse, and mitrans). The optimal model is defined as one joint/single prediction performed best in terms of any metric of Sensitivity, Specificity, positive predictive value (PPV), negative predictive value (NPV), Accuracy, and MCC on any data set. The names of joint prediction follow this rule: (combination method, v for vote and r for rough)_(the number of models was used in combination)_(descriptions, where characters represent data sets: h for golden_human, m for golden_mouse, and i for mitrans; numbers 1–6 represent Sensitivity, Specificity, PPV, NPV, Accuracy, and MCC, respectively). For example, r_15_m23_i23 means 15 models were combined in the joint prediction following the rough rule, and this prediction showed the best Specificity and PPV on golden_mouse data as well as on mitrans data. Particularly, r_2_h_m_i is the abbreviation of r_2_h14_m1456_i14, which was a two-model joint prediction that has best Sensitivity and NPV on golden_human; Sensitivity, NPV, Accuracy, and MCC on golden_mouse; Sensitivity and NPV on mitrans. The complete list of models used in combination was recorded in Supplemental Table S15.
By significantly sacrificing other indicators, the rough principle could increase SPE and PPV to nearly 100% in some cases (Fig. 3B; Supplemental Fig. S28; Supplemental Table S7). For instance, r3_h23 (joint prediction in rough principle using three models: CPAT_human, CPC, and PLEK) and r15_m23_i23 (rough-principle joint prediction of 15 models: CNCI_ve, COME_all, CPAT_human, CPAT_mouse, CPC2, CPC, FEELnc_all_cl, FEELnc_all_sf, hmmscan_A, iSeeRNA, lncRScan-SVM, lncScore, PLEK, PLncPRO_dico, and RNAplonc_guess) showed poor SEN and exhibited SPE or PPV slightly higher than those in CPC. Specifically, when applied to mitrans data, r15_m23_i23 achieved 4% improvement in SPE by allowing SEN nearly 30% lower than that of CPC.
The key point of the voting method is to weigh lncRNA and mRNA equally and to weigh all models equally as well. Assuming that the prediction errors are random and independent, the voting method can promote the predictions by following the decision of the majority. The voting method did significantly improve the prediction in our test, where v5_m6 (voting-principle joint prediction of five models: CPC2, CPC, FEELnc_all_cl, hmmscan_A, and PLEK), v5_m5 (voting models: CPAT_human, CPC, FEELnc_all_cl, hmmscan_A, and PLEK), and v5_i56 (voting models: CNCI_ve, CPAT_human, CPC, hmmscan_A, and PLEK) had a better MCC and ACC than any single model (P ≤ 0.05; see Fig. 3B; Supplemental Fig. S28; Supplemental Tables S7, S8). When using golden_human and golden_mouse data sets, the improvements of the voting method were very limited, and for the mitrans data, the voting method could get obviously better results. Because of the neutrality of the voting strategy, the best SPE obtained by the voting method was worse than the best one by the rough method, and was only close to the best of the single models (Supplemental Fig. S28; Supplemental Table S7).
To see whether the increase of models in a combination brings better outcomes, the performance of a nine-model combination, a 15-model combination, and a 41-model combination were compared (Supplemental Figs. S22–S27). The nine-model combination worked best, the 15-model combination was slightly worse, and the 41-model combination was the worst, with all metrics falling, except that the SPE was slightly better in the rough method. The voting strategy in the nine-model combination statistically outperformed any single model in mitrans data (Supplemental Table S8), but did not show an advantage on the golden_human and golden_mouse data sets (Supplemental Fig. S29; Supplemental Table S7).
Every possible combination of the nine models selected for joint prediction was compared with the single tools with one or more best metrics (Fig 3B; Supplemental Fig. S28; Supplemental Table S7). v_3_h56 (voting models: CPC, FEELnc_all_cl, and PLEK), v_5_m6 (CPC2, CPC, FEELnc_all_cl, hmmscan_A, and PLEK), and v_5_i56 (CNCI_ve, CPAT_human, CPC, hmmscan_A, and PLEK) performed best in golden_human, golden_mouse and mitrans, respectively. However, v_3_h56 and v_5_m6 behaved ordinarily in mitrans, where lncScore as a single prediction tool worked even better than v_3_h56 statistically. Such results might imply impracticality to find a superior combination working best for all data sets.
In order to find the models that play more important roles, we inspected the frequency of the models in 17 well performed joint predictions, the top 10% according to MCC. For predictions using the vote principle, CPC and hmmscan_A are the two most frequent models among three testing data sets (Supplemental Fig. S30); they play roles in at least 15 of the 17 well-performed predictions. Hmmscan_A and FEELnc_all_cl are the most frequent models for predictions using the rough method, with frequencies no more than 11. Interestingly, CPC, hmmscan_A, and FEELnc_all_cl were not the best three models on the three data sets (see the section “Best performance on golden-standard data”). We also investigated the occurrence of concurrent set—a set of models coexisting in multiple joint predictions. No concurrent models were found in more than seven joint predictions using the rough principle on any of the three data sets. In predictions using the vote principle, CPC-hmmscan_A is the most frequent two-model concurrent set across three data sets (Supplemental Fig. S31), while each of CPC−hmmscan_A−PLEK and CPC−FEELnc_all_cl−hmmscan_A has 10 or more occurrences with two data sets (Supplemental Fig. S32).
In summary, the principle for joint prediction showed dramatic impacts on results. Nearly all metrics benefited from the vote method, while only SPE and PPV benefited from the rough method. Meanwhile, the reliability of results heavily depends on how many models and which models are used for prediction. Although no model combination is found to be superior in all data sets, the two models, CPC and hmmscan_A, exist or even coexist in many well-performed combinations.
Performance on different species
Considering the variability of the lncRNA sequence, the biological source of training sets, and the potential bias of training algorithm, a model might show discrepancy across different organisms and outperform others on the data of some species. Such preferences were investigated using SEN and SPE, which are robust on unbalanced data, and some test samples in this section are unbalanced.
Because CNCI is a standard tool used to screen transcripts for the NONCODE database, it outperformed other models in NONCODE data as expected (Fig. 4; Supplemental Table S9). For the lncRNAs in RefSeq (RefSeq-SEN), models behaved drastically differently (Supplemental Fig. S33), with CPAT and FEELnc performing best. Longdist had a relatively high SEN and an extremely low SPE, showing a strong tendency to give lncRNA predictions (Supplemental Table S16).
FIGURE 4.

Performance on multiple species (in percentage). The tree on the left depicts the taxonomy of the species involved, regardless of the evolutionary distance. The sampling procedure for core species (labeled with *) and peripheral species is described in the “Materials and Methods” section. For each species, this plot displays the optimal model with regard to the four metrics: Sensitivity on Ensembl, NONCODE, RefSeq, and Specificity on RefSeq. The best prediction for multiple-model software is shown.
The accuracy of a prediction is highly dependent on the relationship between the species for training and for prediction. Generally, the results of most models followed a relationship rule, that is, the closer the relationship, the more accurate the prediction (Supplemental Figs. S34–S36). In this test, most species (as many as 23) were mammals. Generally, the models performed inconsistently across these mammalian species, although they are thought to be genetically close (Fig. 4; Supplemental Table S9). However, when predicting with the mRNAs from RefSeq, a relatively high species consistency was observed in these models, among which CPC performed best, hmmscan_both and CPAT_human were the second. In addition, FEELnc_all_cl and CNCI_ve showed the best SEN when testing with three databases: Ensembl, NONCODE, and RefSeq. As a summary, FEELnc_all_cl, CPC, and hmmscan_A were the best three models in mammalian.
FEELnc_all_cl performed best for birds (represented by Gallus gallus), followed by hmmscan_both, CPAT_mouse, and CNCI_ve (Supplemental Figs. S34–S36; Supplemental Table S16). The best software for reptiles (represented by Anolis carolinensis) were hmmscan, CPC, and CPAT. For fishes, CPC2, CPC, and CPAT_mouse worked relatively well. The two worm-based models of FEELnc_wm worked best for worms as expected, but performed poorly in other species. FEELnc_ff_cl and CPAT_fly, the two models trained with the fly data set, performed best for fly reasonably. Particularly, FEELnc_all_cl was close to the best model for the fly and worms.
Among the four plants tested, Zea mays is a monocot, while Solanum tuberosum, Brassica napus, and Arabidopsis thaliana are eudicots. FEELnc_ab performed well for Arabidopsis thaliana, but just average for other plants, and poorly for Zea mays, the only monocot (Supplemental Figs. S34–S36; Supplemental Table S16). CPAT_fly and CPAT_mouse were good in SEN, and FEELnc_ab_cl showed good SPE. In general, CPAT_fly, FEELnc_all_cl, CPAT_mouse, and CPC2 were relatively well-performing models. Unexpectedly, CNCI_pl, the plants model of CNCI, worked relatively poorly.
In summary, FEELnc_all_cl, CPC, and CPAT_mouse showed relatively broad adaptabilities, although no single model could perform well in a variety of species.
Transcripts assembled from real sequencing data
Since the real classifications of transcripts that assembled from real data were unknown, the metrics for evaluation used in former sections were not available here. Despite that most transcripts were predicted as lncRNA in rainbow trout or mRNA in seahorse, generally the prediction tendencies of most models remained the same in the two data sets (Fig. 5A). PLncPRO-based models were likely to predict transcripts as mRNA, while models PLEK, CPC2, and hmmscan_B tended to predict transcripts as lncRNA. Various FEELnc-based models showed different predictive biases. While the two models of FEELnc_wm were most likely to predict transcripts as lncRNA, FEELnc_hm_sf and FEELnc_ms_sf were the two models most likely to give mRNA predictions in all models. The FEELnc models trained in the “cl” mode produced more lncRNA predictions than those in the “sf” mode, indicating that the species and the quality of training sets had a significant impact on the prediction of FEELnc.
FIGURE 5.

Performance on real data and time efficiency. (A) Prediction bias. This figure shows the counts of positive (red) and negative (blue) results predicted by each model in addition to the longdist-based models. (B) The relationship between Accuracy and the count of models that give consensus predictions. Representative models were used in this analysis. (C) The counts of positive and negative disagreed-predictions on rainbow trout (left) and seahorse (right), respectively. The transcripts that were reported positive by one model while negative by other representative models were regarded as positive disagreed-predictions and vice versa. (D) Average time consumption of each software tool for predicting 5000 transcripts on golden_human, golden_mouse, and mitrans. The error bar shows the 95% confidence interval of Gaussian distribution.
Most models showed a high level of similarity in the pairwise agreement analysis between models (Supplemental Figs. S37 and S38). When applied to the rainbow trout data set, FEELnc_hm_sf and FEELnc_ms_sf differed greatly from other models. For the seahorse data set, FEELnc_wm_sf and FEELnc_wm_cl showed more differences with other models, and their performance was similar to hmmscan_B, which tended to predict transcripts as lncRNA.
The relationship between ACC and the number of consensus models indicated that, in general, the more models supported, the more reliable the prediction (Fig. 5B). Hence, we treated the predictions supported by only one model as not credible. From the rainbow trout data (Fig. 5C), FEELnc_all_sf produced the most such predictions, most of which were lncRNA. Hmmscan_A and PLEK hold the second-most unique predictions. With the seahorse data (Fig. 5C), PLEK made the most unique predictions. Hmmscan_A and PLncPRO_dico ranked second. On both data sets, CPAT_mouse and FEELnc_all_cl output the least unique predictions. While we cannot say that the tools producing fewer unique predictions are better, it is understandable that the tools with more unique predictions are worse.
Time efficiency
We recorded the time for each model to classify 5000 transcripts in golden_human, golden_mouse, and mitrans (see Materials and Methods; Fig. 5D; Supplemental Table S10). PLncPRO, CNCI, CPC, and hmmscan were the most time-consuming software and required more than 20,000 sec, especially PLncPRO, which took 150,873 sec to complete the classification task. As a computationally intensive task and an indispensable key step in prediction, BLAST alignment greatly slowed down CPC and PLncPRO. For CNCI, the MLCDS/MCSS finding step based on dynamic programming may limit its speed. Fortunately, CPC, CNCI, and PLncPRO can be used in parallel computing, which may shorten waiting time greatly.
The average time of FEELnc was 3599.89 sec, which included the training step that had already been done for the other models. Hence, FEELnc would be more effective if the prebuilt models were available. In a classic whole-transcriptomic analysis, more than 100,000 transcripts will be classified. Therefore, only the efficient tools should be considered. Hmmscan can complete this work in 100 CPU hours, and the software slower than hmmscan should support multithreads to be practical.
DISCUSSION
Open reading frame (ORF) is a widely used feature that is highly dependent on the integrity of sequence and is sensitive to indels. Hexamer usage bias (or ANT in CNCI) is another important feature for sequence analysis. While CPAT treats the in-frame hexamer as a classification feature, CNCI and lncScore use hexamer to extract MLCDS/MCSS, which can serve to some extent as ORFs for fragment sequences. The utilization of sequence conservation can promote prediction performance and bring other limitations. CPC and RNAcode obtain sequence conservation through a time-consuming alignment step (Washietl et al. 2011), while iSeeRNA, lncRNAScan, and COME assess sequence conservation through the phastCons score, which is based on a specific version of the genome and therefore loses versatility (Siepel et al. 2005). In particular, PLEK attempts to solve the prediction problem for incomplete transcripts by choosing only k-mer frequencies (1-mer to 5-mer) as features and obtained relatively good results.
For human, mouse, or other well-established model organisms with high-quality transcripts in manually curated public databases (Lagarde et al. 2016), most software worked well and achieved ACC over 90% under appropriate models (see tests on the golden data). However, transcripts assembled from high-throughput sequencing data are usually incomplete and erroneous. In such a case, COME, which performed well on mitrans and simulation data, is a good choice, followed by CNCI, lncScore, and FEELnc_all_cl.
The lack of high-quality training data sets is a common problem for non-model organisms; sometimes we have to predict with the models trained by other well-studied species. Considering the incompleteness of the transcript structure of non-model organisms under the state-of-the-art assembly technology, we recommend FEELnc_all_cl, CPAT_mouse, and CNCI_ve, where CNCI_ve performed better for transfrags, and FEELnc_all_cl and CPAT_mouse performed better for most species. Unfortunately, there were no perfect tools in our tests that can perform well in various states. Firstly, it is challenging to find a set of generic features, the efficiency of which varies from species to species in prediction (Ventola et al. 2017). Secondly, the performance of tools varied even in closely related species in our tests, indicating a higher heterogeneity in the lncRNA collections. Obviously, more work is necessary to develop a series of generic criteria for collecting sequences as standard lncRNA sets.
A number of studies have applied joint prediction for large-scale lncRNA identification (Zhang et al. 2014; Qiu et al. 2016; Wang et al. 2016, 2019; Chen et al. 2017; Rolland et al. 2019). Based on our results, researchers need to carefully select a combination of models, only well-performed models should be considered for joint analysis, and it is not wise to simply use all available models. In fact, benefits were very limited when the number of tools exceeded three. Although no perfect combination was found in our tests, it is feasible to achieve better performance by joint prediction. According to the principles of ensemble learning, excellent models trained with divergent features, such as HMMER with CPC (Zhang et al. 2014), or HMMER with CPAT (Wang et al. 2013), are good combinations. Since the overall optimal model was not found, we summarized our findings and made suggestions for different analysis scenarios (Table 3). These suggestions were based on the rank of the models according to their SEN and SPE values (Supplemental Table S16).
TABLE 3.
Suggestions for choosing models in different situations

Some deep-learning-based tools, for example, lncRNAnet, lncADeep, and lncFinder, were tested for lncRNA identification (Amin et al. 2019). Since the data sets used in these tests were from GENCODE, the same source of the lncRNAs in our golden data, the SEN values (i.e., the “recall” values used by Amin et al.) can be directly compared to those in our test. The highest SEN obtained by lncADeep ranks very high in our test, but the ACC by lncADeep is poor overall. While the performance of lncRNAnet reduces with transcript length, some models in our test have similar behaviors, for example, the FEELnc-based models trained with data from Arabidopsis or worm, and all longdist-based models (Supplemental Figs. S39 and S40). However, we cannot come to a conclusion for correlation in our cases because the sequence length distribution is not the only potential cause that may have impacts on the models’ performance. For instance, although PLEK did not perform as well as golden_human on the mitrans data set, which has a longer average sequence length (Supplemental Fig. S41), its prediction accuracy showed length independence within both data sets (Fig. 1B; Supplemental Figs. S39 and S40). This shows that the length correlation is not universal, and it may be associated with specific software tools, or may be related to other factors, especially the differences in data set quality.
Longdist-based models showed very low SPE and much higher SEN on our golden data sets. This fact implies that longdist have wrongly classified most mRNAs as lncRNAs. Considering longdist performed very well on the mRNA data (the file “pct.fa”) provided by itself (Supplemental Table S11), we transformed our test data to the format pct.fa to exclude potential technical issues: Capitalize all sequence letters and keep 60 letters per line. However, the results showed no difference. So it is likely that longdist has a problem of overfitting.
Because of the diversity of species and transcript quality, it is very common that a software tool performs differently under various situations. The prediction model of PLEK was trained with human data and performed very poorly on golden_mouse comparing to golden_human. The underlying cause might be the k-mer, which is used as the classification feature by PLEK and its distribution varies across species (Chor et al. 2009; Han and Cho 2019). CNCI_pl was originally trained with plants data; however, it performed relatively poorly on our plants data. Interestingly, although CPAT_mouse was trained with mouse data, it worked well on a broad range of species. It is not easy to find the reason behind these behaviors; what we are trying to do is to unveil the advantages and limitations of available tools and to provide a reference for researchers to choose proper tools.
Because a broad range of species were involved in this research, the quality of data from different sources may vary drastically and lead to bias in analysis, we tried to keep the congruity in quality, source, and other aspects of the data for different species when we chose the sources of data sets for testing. So, the testing data sets were downloaded from databases covering a broad spectrum of species. That is why some high-quality databases were not used in this analysis, for example, the two databases specifically for plants, CANTATAdb and GreeNC (Paytuvi Gallart et al. 2016; Szczesniak et al. 2016). With the accumulation of high-quality lncRNA data, it can be expected that the software performance can be more accurately detected for a certain class of species in the future.
With the discovery of the bifunctional RNAs that can serve as lncRNA and can encode small peptides as well (Nam et al. 2016; Williamson et al. 2017; van Heesch et al. 2019), the boundary of lncRNA and mRNA becomes fuzzy. One source of bifunctional RNA is the lncRNA-encoding peptides. A ribosome profiling analysis showed the proportion of lncRNAs interacting with ribosomes is 39.17% in human and 48.16% in mouse (Zeng et al. 2018). Since the original definition of lncRNA mainly relies on coding ability, whether we should refer to “translatable lncRNA” as bifunctional RNA or mRNA depends on whether the sequence can actually serve as lncRNA or not. Another source of bifunctional RNA is the mRNA playing functional roles as lncRNA. Jin-Wu Nam has listed 15 mRNAs that can participate in translation, transcription, and scaffolding (Nam et al. 2016). To the best of our knowledge, there is still a lack of a large-scale analysis to estimate the number of bifunctional RNAs with definite coding ability and noncoding functions. But one thing is certain, the lack of coding ability does not appear to be the essential characteristic of lncRNA. Instead, a more intrinsic standard seems to be the functional capability before being translated. Since the functional elements of lncRNA have not been fully uncovered, even the function of most lncRNAs has not been verified, more exploration is needed to obtain better characteristics of lncRNA, especially bifunctional RNA.
MATERIALS AND METHODS
Software
Prediction models
A total of 14 software packages were evaluated. Because of the limitation of the strategy used in software development, four software tools (COME, iSeeRNA, lncRScan-SVM, and lncScore) in this study can only make within-species predictions (Table 1); that is, the training data and the testing data should come from the same organism species. The other 10 tools can perform cross-species predictions. In order to make predictions, different data sets and training strategies were applied to train software tools, resulting in 41 different prediction models. A model name starts with the software name, and is optionally followed by a character string for some other information, typically the species or the source of the training data and distinct technical handling details in prediction (Table 4).
TABLE 4.
Models for lncRNA identification

Hmmscan, a subprogram of the HMMER software package (Mistry et al. 2013), was tested with reference to Iyer's work (Iyer et al. 2015). Among all 14 tested tools, hmmscan is the only one not based on machine learning. In application, each transcript was translated into six protein sequences based on all potential reading frames and searched against the Pfam database using hmmscan. The classification was based on arbitrary cut-offs. More specifically, transcripts having one or more mRNA-specific domains (e < 0.0001) are negative (mRNA), otherwise positive (lncRNA). The name “hmmscan_A” was used for the analytic model with Pfam-A as the only database. Similarly, “hmmscan_B” was based on the Pfam-B database and “hmmscan_both” was based on these two databases. HMMER (version 3.1b2), Pfam-A (version 30) and Pfam-B (version 27.0) were used in this study.
Occasionally some predictions by RNAplonc do not have a classification tag. We have made it clear by communicating with the author: The software txCdsPredict is packaged in RNAplonc to extract ORFs from sequences. When an ORF is not given by txCdsPredict, RNAplonc will skip the transcript due to a lack of information for prediction (pers. comm.). Considering the fact that lncRNAs usually have very short ORFs or do not have ORFs, it is perhaps appropriate to regard these transcripts as lncRNAs. Hence, these nonlabeled results were processed in two ways in our test: (i) simply discarded (RNAplonc_cut); and (ii) labeled lncRNA (RNAplonc_guess).
In our test, FEELnc is the only one software tool published without any prebuilt model. Hence, we trained 14 models for FEELnc with the seven sets of data published in the original paper of CPC2. There are two training data sets used in the original paper for FEELnc; however, a simple comparison showed that the models trained by those data are weaker than ours, so we did not use them in this study. FEELnc has two strategies to use a training data set. In short, the first is to use both the coding sequences and the noncoding sequences provided by the user, and the other is to use only the coding sequences while the noncoding sequences are generated by the shuffling method of FEELnc itself.
Representative models
One or two analytical models that performed best on the gold standard data set were selected as the representative models for each software package except longdist due to its poor performance; the total of representative models is 15 (Table 4). For the purpose of conciseness, some plots only showed representative models, although all available models were tested throughout our study.
Every possible combination of models will be tested in joint prediction. Because the number of combinations increases drastically with the number of models, the representative model set is still too big for such a test. Therefore, nine models were selected as a core set, and only those capable of cross-species prediction were considered.
Data sets
LncRNA identification requires sequence data in FASTA or GTF format. Most sequence data in this study were downloaded directly from public databases; other data were assembled using data generated from in silico simulation or actual sequencing.
The general procedures for sequence processing
Most of the data were downloaded from public databases, including GENCODE, RefSeq, Ensembl, NONCODE, etc. (Supplemental Table S12). All sequences were pooled together after a quality control procedure, which simply discarded any sequence shorter than 200 bp or containing characters other than “ATCG.” The test data were sampled from the sequence pool in different ways (Supplemental Fig. S43).
From GENCODE, we downloaded all FASTA files and GTF files labeled “Long noncoding RNA.” A transform called LiftOver (Hinrichs et al. 2006) was then run to convert the GTF files from human genome version hg38 to version hg19 to meet the requirements of hg19-based tools (e.g., COME, iSeeRNA). For the sake of data quality, a record from RefSeq was used only when its name starts with “NM” and the source field is labeled with “BestRefSeq” in the GTF file.
Golden-standard sequences
Based on the richness and reliability of the data, human and mouse sequences from GENCODE and RefSeq were used as the golden standard for evaluating tool performance. For each species, the positive data set consists of 5000 sequences randomly sampled from GENCODE, while the 5000 negative sequences were sampled from RefSeq. The golden-standard data sets for human and mouse were named golden_human and golden_mouse, respectively.
Transcripts assembled from a large number of samples
The positive data are 5000 lncRNA sequences and the negative data are 5000 mRNA sequences, both of which were randomly sampled from the downloaded MiTranscriptome (mitrans) data.
Sequences of different species
In order to investigate the performance of the tools on different species, we collected mRNA sequences prefixed with “NM” from RefSeq, as well as noncoding RNA sequences from RefSeq, Ensembl, and NONCODE (Supplemental Table S13). There are 33 species with 50 or more entries in Ensembl, Ensembl_plants, and RefSeq. According to the phylogenetic relationships, they fall into two categories: 18 representative core species and 15 peripheral species. The taxonomy of species was based on NCBI's Taxonomy database, and the phylogenetic tree was plotted by ete3, a handy web tool (http://etetoolkit.org/treeview/) (Huerta-Cepas et al. 2016). From each database, we randomly selected up to 2000 sequences for each core species and 500 for each peripheral species. If the sequences in a database were insufficient for sampling, the entire collection was used.
Simulated data
Based on the golden-standard sequence sets (golden_human and golden_mouse), simulated Illumina sequencing data sets were generated by Polyester with five sequencing depths: 30× (which means that average 30 reads were simulated from each transcript), 100×, 300×, 600×, and 1200× (Frazee et al. 2015). The simulated data were then assembled into transcripts in two ways via an HISAT2 and StringTie: with or without genome annotation files (GTFs) (Pertea et al. 2016). The performance of tools on the assembled sequences was compared to those on the golden-standard sequences.
Real RNA-seq data
Real sequencing data of two non-model fishes (seahorse and rainbow trout) were used for testing. A poor sequencing was represented by the rainbow trout data set, which was a single-end sequencing with a lower mapping rate (Supplemental Table S14). In contrast, the seahorse data set was pair-end sequenced with a higher mapping rate, representing a high-quality sequencing. The real RNA-seq data for rainbow trout and seahorse were downloaded from the SRA website and subsequently assembled to transcript sequences following the same procedure as for the simulated data. Tens of thousands of sequences were randomly sampled for succedent testing.
Joint prediction
Mitrans and the golden data were used to investigate the effects of joint predictions. Joint prediction was achieved by combining predictions from different models. Briefly, a set of models were separately applied to a sample sequence to obtain a list of predictions that determined the classification of the transcript sequence by voting or using a rough principle.
In the vote scheme, a given transcript is classified as mRNA or lncRNA, whichever is more supported by predictions. In particular, lncRNA is in preference to mRNA in case of a tie. If operated according to a rough principle, as long as it is classified as mRNA by any model, the transcript is mRNA, otherwise, it is lncRNA.
From the 15 representative models, nine well-performed ones were chosen as a core model set for joint prediction (Table 4), and any possible combination of the nine models was investigated. In addition, the joint prediction was also tested using all the representative models or all 41 models.
A joint prediction can be named using an expression starting with a letter (“v” for vote and “r” for rough), an underscore character, and the number of models used in this prediction, then followed by one or more best-performance results, each of which consists of an underscore, a letter for data set (“h” for golden_human, “m” for golden_mouse, and “I” for mitrans), and one or more digits for the best metrics resulting from this joint prediction (1–6 represent SEN, SPE, PPV, NPV, ACC, and MCC, respectively). For example, “r_2_h14_m1456_i14” is a two-model joint prediction using the rough rule, and it resulted in the best SEN and NPV on golden_human, best SEN/NPV/ACC/MCC on golden_mouse, and best SEN/NPV on mitrans. In particular, this name can be abbreviated as “r_2_h_m_i” because it does not cause ambiguity.
Metrics for performance evaluation
The performance of prediction models was evaluated using six indicators: (i) sensitivity (SEN). The fraction of true positive predictions on all positive test samples; (ii) specificity (SPE), the fraction of true negative predictions on all negative test samples; (iii) positive predictive value (PPV), the fraction of true positive predictions on all positive predictions; (iv) negative predictive value (NPV), the fraction of true negative predictions on all negative predictions; (v) accuracy (ACC), the fraction of total true predictions on all test samples; and (vi) Matthews correlation coefficient (MCC) (Matthews 1975), an indicator used in machine learning as a measure of the quality of prediction, MCC is more robust to unbalanced test samples comparing with ACC. All computational formulas are listed below.
Abbreviations in these equations are FN for false negative, FP for false positive, TN for true negative, and TP for true positive. The relationships among metrics are also illustrated in Figure 6.
FIGURE 6.
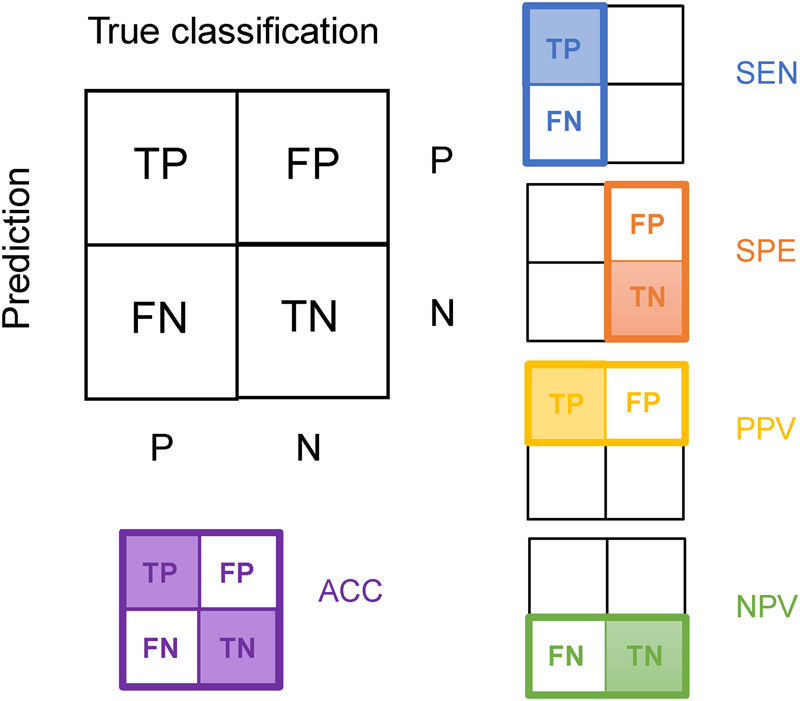
Metrics used to evaluate prediction results (except MCC). The value is calculated by dividing the predicted count in the shadowy box by the predicted count in the whole rectangle (or square) with a colored border.
In balanced samples, SEN and NPV performed similarly, as did SPE and PPV. Hence, only one indicator of each pair was discussed in most subsequent analyses. Two other measurements, the receiver operating characteristic (ROC) curve and the area under the curve (AUC) value, were also occasionally used. Typically, a classifier can form a probability value or other number, which will be compared with a given threshold to determine the classification. Therefore, most of the indices, including ACC, SEN, and SPE, are actually calculated at certain thresholds. However, the ROC curve and its corresponding AUC value may reflect the expected generalization ability of a classifier at different thresholds or under general circumstances.
Venn diagram was drawn through an R package, VennDiagram. Some in-house scripts have been written for analysis and are available to be provided in a query. Programs ran with R (version 3.3.0) and Python (version 2.7.9) under a Linux environment (CentOS 7).
Time efficiency
Time consumptions were measured by the “user CPU time,” an indicator reported by the Linux system's “time” command. The golden-standard data sets and mitrans data were used to access the time efficiencies of the tools.
Statistics
The McNemar test was used for assessment of discrepancy between two prediction models.
H0: There is no significant difference in the prediction accuracy between the two models being compared.
H1: There is a significant difference in the prediction accuracy between the two models being compared.
The Bonferroni method and the false discovery rate (FDR) were used for correction of the P-values in multiple tests (α = 0.05).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mr. Xia Lei for language guidance in manuscript preparation (Institute of Hydrobiology, Chinese Academy of Science). This work was supported by the Wuhan Branch, Supercomputing Center, Chinese Academy of Sciences, China. This work was supported by the National Natural Science Foundation of China (Grant No. 31571275), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA08020201), and the National High-Technology Research and Development Program (863 Program, Grant No. 2011AA100403).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.074724.120.
REFERENCES
- Amin N, McGrath A, Chen Y-PP. 2019. Evaluation of deep learning in non-coding RNA classification. Nat Mach Intell 1: 246–256. 10.1038/s42256-019-0051-2 [DOI] [Google Scholar]
- Cech TR, Steitz JA. 2014. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157: 77–94. 10.1016/j.cell.2014.03.008 [DOI] [PubMed] [Google Scholar]
- Chen CK, Yu CP, Li SC, Wu SM, Lu MJ, Chen YH, Chen DR, Ng CS, Ting CT, Li WH. 2017. Identification and evolutionary analysis of long non-coding RNAs in zebra finch. BMC Genomics 18: 117 10.1186/s12864-017-3506-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chor B, Horn D, Goldman N, Levy Y, Massingham T. 2009. Genomic DNA k-mer spectra: models and modalities. Genome Biol 10: R108 10.1186/gb-2009-10-10-r108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona-Gomez JA, Garcia-Lopez IJ, Stadler PF, Fernandez-Valverde SL. 2020. Splicing conservation signals in plant long noncoding RNAs. RNA 26: 784–793. 10.1261/rna.074393.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. 2014. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15: 7–21. 10.1038/nrg3606 [DOI] [PubMed] [Google Scholar]
- Frazee AC, Jaffe AE, Langmead B, Leek JT. 2015. Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics 31: 2778–2784. 10.1093/bioinformatics/btv272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Gao L, Wang Y, Chiu DK, Wang T, Deng Y. 2016. Advances in long noncoding RNAs: identification, structure prediction and function annotation. Brief Funct Genomics 15: 38–46. 10.1093/bfgp/elv022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GB, Cho DH. 2019. Genome classification improvements based on k-mer intervals in sequences. Genomics 111: 1574–1582. 10.1016/j.ygeno.2018.11.001 [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774. 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow JL, Steward CA, Frankish A, Gilbert JG, Gonzalez JM, Loveland JE, Mudge J, Sheppard D, Thomas M, Trevanion S, et al. 2014. The Vertebrate Genome Annotation browser 10 years on. Nucleic Acids Res 42: D771–D779. 10.1093/nar/gkt1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34: D590–D598. 10.1093/nar/gkj144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D. 2015. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16: 71–84. 10.1038/nrg3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, et al. 2017. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543: 199–204. 10.1038/nature21374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housman G, Ulitsky I. 2016. Methods for distinguishing between protein-coding and long noncoding RNAs and the elusive biological purpose of translation of long noncoding RNAs. Biochim Biophys Acta 1859: 31–40. 10.1016/j.bbagrm.2015.07.017 [DOI] [PubMed] [Google Scholar]
- Hu L, Xu Z, Hu B, Lu ZJ. 2017. COME: a robust coding potential calculation tool for lncRNA identification and characterization based on multiple features. Nucleic Acids Res 45: e2 10.1093/nar/gkw798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Serra F, Bork P. 2016. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 33: 1635–1638. 10.1093/molbev/msw046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. 2015. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47: 199–208. 10.1038/ng.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Yang DC, Kong L, Hou M, Meng YQ, Wei L, Gao G. 2017. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res 45: W12–W16. 10.1093/nar/gkx428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashi K, Henderson L, Bonetti A, Carninci P. 2016. Discovery and functional analysis of lncRNAs: Methodologies to investigate an uncharacterized transcriptome. Biochim Biophys Acta 1859: 3–15. 10.1016/j.bbagrm.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. 2007. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35: W345–W349. 10.1093/nar/gkm391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JT, Colognori D, Lee JT. 2013. Long noncoding RNAs: past, present, and future. Genetics 193: 651–669. 10.1534/genetics.112.146704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde J, Uszczynska-Ratajczak B, Santoyo-Lopez J, Gonzalez JM, Tapanari E, Mudge JM, Steward CA, Wilming L, Tanzer A, Howald C, et al. 2016. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat Commun 7: 12339 10.1038/ncomms12339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Zhang J, Zhou Z. 2014. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics 15: 311 10.1186/1471-2105-15-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, Kellis M. 2011. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27: i275–i282. 10.1093/bioinformatics/btr209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hao L, Li D, Zhu L, Hu S. 2015. Long non-coding RNAs and their biological roles in plants. Genomics Proteomics Bioinformatics 13: 137–147. 10.1016/j.gpb.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maracaja-Coutinho V, Paschoal AR, Caris-Maldonado JC, Borges PV, Ferreira AJ, Durham AM. 2019. Noncoding RNAs databases: current status and trends. In Computational biology of non-coding RNA: methods and protocols (ed. Lai X, Gupta SK, Vera J), pp. 251–285. Springer, New York. [DOI] [PubMed] [Google Scholar]
- Matthews BW. 1975. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 405: 442–451. 10.1016/0005-2795(75)90109-9 [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. 2009. Long non-coding RNAs: insights into functions. Nat Rev Genet 10: 155–160. 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41: e121 10.1093/nar/gkt263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JW, Choi SW, You BH. 2016. Incredible RNA: dual functions of coding and noncoding. Mol Cells 39: 367–374. 10.14348/molcells.2016.0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri T, Alves WAL, Bugatti PH, Saito PTM, Domingues DS, Paschoal AR. 2019. Pattern recognition analysis on long noncoding RNAs: a tool for prediction in plants. Brief Bioinformatics 20: 682–689. 10.1093/bib/bby034 [DOI] [PubMed] [Google Scholar]
- Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, et al. 2012. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22: 577–591. 10.1101/gr.133009.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paytuvi Gallart A, Hermoso Pulido A, Anzar Martinez de Lagran I, Sanseverino W, Aiese Cigliano R. 2016. GREENC: a Wiki-based database of plant lncRNAs. Nucleic Acids Res 44: D1161–D1166. 10.1093/nar/gkv1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Pertea M. 2020. GFF Utilities: GffRead and GffCompare [version 1; peer review: 3 approved]. F1000Res 9: 304 10.12688/f1000research.23297.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650 10.1038/nprot.2016.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Ren Z, Yan J. 2016. Identification and functional analysis of long non-coding RNAs in human and mouse early embryos based on single-cell transcriptome data. Oncotarget 7: 61215–61228. 10.18632/oncotarget.11304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff JD, Wei Y, Khavari PA. 2018. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 19: 143–157. 10.1038/nrm.2017.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. 2012. Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145–166. 10.1146/annurev-biochem-051410-092902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland AD, Evrard B, Darde TA, Le Beguec C, Le Bras Y, Bensalah K, Lavoue S, Jost B, Primig M, Dejucq-Rainsford N, et al. 2019. RNA profiling of human testicular cells identifies syntenic lncRNAs associated with spermatogenesis. Hum Reprod 34: 1278–1290. 10.1093/humrep/dez063 [DOI] [PubMed] [Google Scholar]
- Schneider HW, Raiol T, Brigido MM, Walter M, Stadler PF. 2017. A support vector machine based method to distinguish long non-coding RNAs from protein coding transcripts. BMC Genomics 18: 804 10.1186/s12864-017-4178-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 12: 47–56. 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh U, Khemka N, Rajkumar MS, Garg R, Jain M. 2017. PLncPRO for prediction of long non-coding RNAs (lncRNAs) in plants and its application for discovery of abiotic stress-responsive lncRNAs in rice and chickpea. Nucleic Acids Res 45: e183 10.1093/nar/gkx866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Chen X, Jiang P, Song X, Wang H, Sun H. 2013a. iSeeRNA identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genomics 14: S7 10.1186/1471-2164-14-S2-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, Liu Y, Chen R, Zhao Y. 2013b. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41: e166 10.1093/nar/gkt646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liu H, Zhang L, Meng J. 2015. lncRScan-SVM: a tool for predicting long non-coding RNAs using support vector machine. PLoS ONE 10: e0139654 10.1371/journal.pone.0139654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesniak MW, Rosikiewicz W, Makalowska I. 2016. CANTATAdb: a collection of plant long non-coding RNAs. Plant Cell Physiol 57: e8 10.1093/pcp/pcv201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. 2017. UniProt: the universal protein knowledgebase. Nucleic Acids Res 45: D158–D169. 10.1093/nar/gkw1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I. 2016. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat Rev Genet 17: 601–614. 10.1038/nrg.2016.85 [DOI] [PubMed] [Google Scholar]
- Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigo R, Johnson R. 2018. Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet 19: 535–548. 10.1038/s41576-018-0017-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL, et al. 2019. The translational landscape of the human heart. Cell 178: 242–260 e229. 10.1016/j.cell.2019.05.010 [DOI] [PubMed] [Google Scholar]
- Ventola GM, Noviello TM, D'Aniello S, Spagnuolo A, Ceccarelli M, Cerulo L. 2017. Identification of long non-coding transcripts with feature selection: a comparative study. BMC Bioinformatics 18: 187 10.1186/s12859-017-1594-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. 2013. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res 41: e74 10.1093/nar/gkt006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xue S, Liu X, Liu H, Hu T, Qiu X, Zhang J, Lei M. 2016. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci Rep 6: 20238 10.1038/srep20238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ji N, Chen Z, Wu C, Sun Z, Yu W, Hu F, Huang M, Zhang M. 2019. Next generation sequencing for long non-coding RNAs profile for CD4+ T cells in the mouse model of acute asthma. Front Genet 10: 545 10.3389/fgene.2019.00545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Findeiss S, Muller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N. 2011. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA 17: 578–594. 10.1261/rna.2536111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L, Saponaro M, Boeing S, East P, Mitter R, Kantidakis T, Kelly GP, Lobley A, Walker J, Spencer-Dene B, et al. 2017. UV irradiation induces a non-coding RNA that functionally opposes the protein encoded by the same gene. Cell 168: 843–855 e813. 10.1016/j.cell.2017.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucher V, Legeai F, Hedan B, Rizk G, Lagoutte L, Leeb T, Jagannathan V, Cadieu E, David A, Lohi H, et al. 2017. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res 45: e57 10.1093/nar/gkw1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Wang ZH, Guo JT. 2012. The research strategies for probing the function of long noncoding RNAs. Genomics 99: 76–80. 10.1016/j.ygeno.2011.12.002 [DOI] [PubMed] [Google Scholar]
- Yang G, Lu X, Yuan L. 2014. LncRNA: a link between RNA and cancer. Biochim Biophys Acta 1839: 1097–1109. 10.1016/j.bbagrm.2014.08.012 [DOI] [PubMed] [Google Scholar]
- Zeng C, Fukunaga T, Hamada M. 2018. Identification and analysis of ribosome-associated lncRNAs using ribosome profiling data. BMC Genomics 19: 414 10.1186/s12864-018-4765-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Huang K, Luo Y, Li S. 2014. Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genomics 15: 845 10.1186/1471-2164-15-845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Song X, Wang K. 2016a. lncScore: alignment-free identification of long noncoding RNA from assembled novel transcripts. Sci Rep 6: 34838 10.1038/srep34838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, Li Z, Bu D, Sun N, Zhang MQ, et al. 2016b. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res 44: D203–D208. 10.1093/nar/gkv1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.