

Abstract
A recent crystal structure of a ribosome complex undergoing partial translocation in the absence of elongation factor EF-G showed disruption of codon–anticodon pairing and slippage of the reading frame by −1, directly implicating EF-G in preservation of the translational reading frame. Among mutations identified in a random screen for dominant-lethal mutations of EF-G were a cluster of six that map to the tip of domain IV, which has been shown to contact the codon–anticodon duplex in trapped translocation intermediates. In vitro synthesis of a full-length protein using these mutant EF-Gs revealed dramatically increased −1 frameshifting, providing new evidence for a role for domain IV of EF-G in maintaining the reading frame. These mutations also caused decreased rates of mRNA translocation and rotational movement of the head and body domains of the 30S ribosomal subunit during translocation. Our results are in general agreement with recent findings from Rodnina and coworkers based on in vitro translation of an oligopeptide using EF-Gs containing mutations at two positions in domain IV, who found an inverse correlation between the degree of frameshifting and rates of translocation. Four of our six mutations are substitutions at positions that interact with the translocating tRNA, in each case contacting the RNA backbone of the anticodon loop. We suggest that EF-G helps to preserve the translational reading frame by preventing uncoupled movement of the tRNA through these contacts; a further possibility is that these interactions may stabilize a conformation of the anticodon that favors base-pairing with its codon.
Keywords: EF-G, domain IV, frameshifting, ribosome, translocation
INTRODUCTION
Errors in maintenance of the translational reading frame are much more dangerous than missense errors. While most proteins can tolerate substitutions at many different positions (Kurland 1992), shifting of the reading frame not only results in incorporation of a continuous series of incorrect amino acids, but soon leads to premature termination at an out-of-frame stop codon. The resulting incomplete polypeptides are often toxic, caused by dominant-lethal effects (Drummond and Wilke 2008). The importance of maintaining the reading frame is reflected in the high fidelity of translocation, with frameshifting error rates less than 10−5 (Kurland 1992). In spite of decades of study, the mechanisms by which the translational reading frame is preserved during the coupled translocation of the mRNA and tRNAs are still only poorly understood. We know that tRNAs can be translocated in the absence of mRNA in an elongation factor EF-G-catalyzed reaction (Tnalina et al. 1982; Yusupova et al. 1986), showing that the translocation mechanism indeed acts on the tRNA; however, there is no convincing evidence that the mRNA can be actively translocated in the absence of tRNA. This suggests that the mRNA is moved only passively, by virtue of its base-pairing to tRNA. Coupled movement of mRNA and A-tRNA would thus appear to rely strongly, if not completely, on maintaining correct codon–anticodon pairing during translocation. Although these weak triplet duplexes are stabilized by their interactions with the ribosome, they immediately become vulnerable to disruption as they move out of their binding sites during translocation. How, then, is mRNA movement strictly coupled to tRNA movement?
A clue to the mechanism of reading-frame maintenance comes from cryo-EM and X-ray structures of ribosome-EF-G complexes trapped in various states of translocation. One key structure is that of the chimeric hybrid-state intermediate, in which the head domain of the 30S subunit is rotated by ∼20°, and the A- and P-site tRNAs are bound in ap/P (or ap/ap) and pe/E states, respectively (Ramrath et al. 2013; Zhou et al. 2014). In this state, the anticodon stem–loop (ASL) of the A-tRNA is held approximately between the A site of the head domain and the P site of the body domain of the 30S subunit, while the ASL of the P-tRNA is positioned between the P site of the head domain and the E site of the body domain, hence the term “chimeric.” Their acceptor ends have moved fully into the 50S P site (or in the ap/ap state, shared between the 50S A and P sites) and the 50S E site, respectively. In these structures, the tip of the long, flexible domain IV of EF-G contacts the codon–anticodon duplex (Ramrath et al. 2013; Zhou et al. 2014). An X-ray structure of a posttranslocation EF-G-ribosome complex (Gao et al. 2009) and a cryo-EM structure of a pretranslocation complex (Brilot et al. 2013) both found domain IV in contact with their respective P-site and A/P-state codon–anticodon duplexes. Taken together, these three structures suggest that domain IV maintains contact with the duplex during its trajectory from the A site to the P site, consistent with a smFRET study that directly observed rearrangement of domain IV in pre and posttranslocation complexes in solution (Salsi et al. 2014b). Finally, in a recent crystal structure of a ribosome complex containing tRNAs that translocated spontaneously into chimeric hybrid states in the absence of EF-G, codon–anticodon pairing was disrupted, resulting in a −1 shift of the reading frame, providing direct evidence for a role for EF-G in coupling of mRNA and tRNA movement (Zhou et al. 2019). Interestingly, the A-tRNA had moved further in this complex than in the corresponding EF-G-containing complex, suggesting that EF-G may actually restrict movement of the tRNA during translocation.
In an effort to further understand the role of EF-G in translocation, we have undertaken a global screen for dominant-lethal mutations in EF-G of E. coli (Nelson C, Leung CS, Noller HF, et al., in prep.). Among these, we identified a cluster of six dominant-lethal mutations mapping to the tip of domain IV (Fig. 1). Prompted by the structural evidence described above, we tested whether any of these mutant forms of EF-G influence the translational reading frame. Using a system based on in vitro translation of a full-length protein containing a “slippery sequence,” we find that the presence of each of these six mutant EF-Gs greatly stimulates shifting into the −1 reading frame in our in vitro system. All but one of these mutations also confer reduced rates of mRNA translocation and rotation of the head and body domains of the 30S ribosomal subunit. Our frameshifting results are in agreement with recent studies by Peng et al. (2019) showing −1 frameshifting during in vitro translation of an oligopeptide with mutant forms of EF-G carrying several mutations at two positions in domain IV. In the structure of the chimeric hybrid-state intermediate (Zhou et al. 2014), all of the contacts between domain IV and the codon–anticodon duplex are formed with ribose or phosphate moieties in the RNA backbone of the anticodon loop. We propose that these contacts between domain IV and the anticodon loop help to preserve the translational reading frame by preventing uncoupled movement of the tRNA during translocation; a further possibility is that these contacts may also help to stabilize an anticodon conformation that favors base-pairing with its codon.
FIGURE 1.
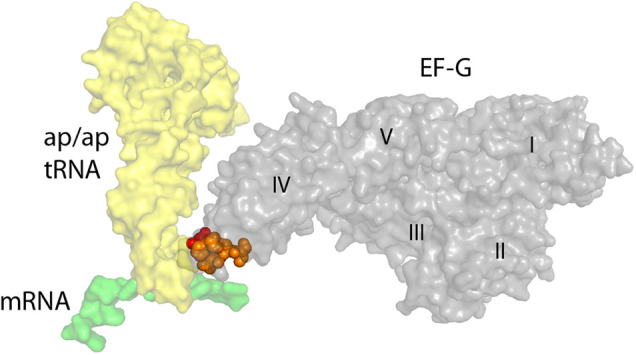
Domain IV of EF-G contacts the codon–anticodon duplex during translocation. In the crystal structure of a trapped chimeric hybrid-state translocation intermediate (Zhou et al. 2014), the tip of domain IV of EF-G contacts the RNA backbones of the anticodon loop of the ap/ap tRNA (yellow) and the codon of the mRNA (green) at their point of convergence. The positions of dominant-lethal mutations in loop I (red) and loop II (orange) of domain IV are indicated.
RESULTS
EF-G mutants were isolated from an unbiased global screen, using random PCR mutagenesis (Materials and Methods), from which dominant-lethal mutations were mapped to all five structural domains of EF-G (Nelson C, Leung CS, Noller HF, et al., in prep.). Here, we focus on a cluster of mutations in loops I (positions 507–513) and II (positions 579–589) at the tip of domain IV (Fig. 1). These include mutant Q507H in loop I and H583R, D586V, S587Y, S587P, and S588P in loop II (Table 1), all of which are highly conserved, with the exception of S588. In addition, Q507D, which was found by Peng et al. (2019) to induce strong frameshifting, was created by directed mutagenesis and included in our studies. The numbering of EF-G residues refers to EF-G from the fusA gene of E. coli throughout, excluding its amino-terminal methionine residue.
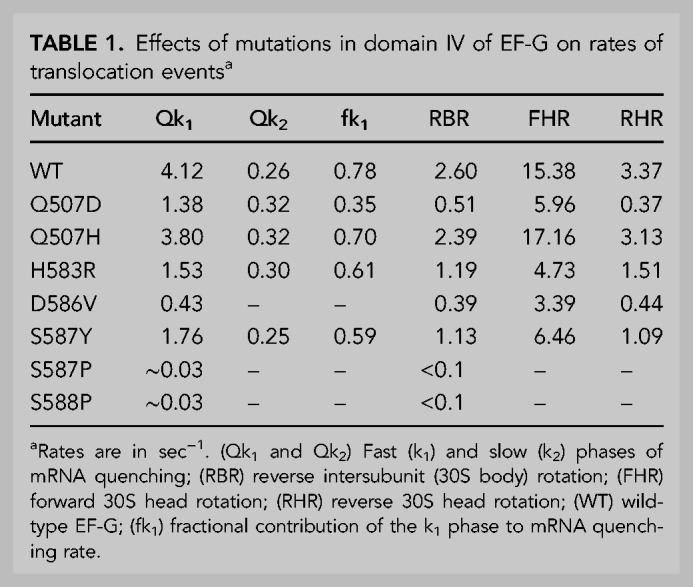
TABLE 1.
Effects of mutations in domain IV of EF-G on rates of translocation eventsa
In vitro translocation activities of domain IV mutants
The seven domain IV mutant EF-Gs were tested for their ability to undergo multiple rounds of translocation using a toe-printing assay (Fig. 2; Hartz et al. 1988; Fredrick and Noller 2003). Pretranslocation complexes bound to an mRNA coding for MVVV were prepared by binding N-Ac-Met-tRNAMet to the P site, followed by introducing excess Val-tRNA·EF-Tu·GTP ternary complex and EF-G·GTP. A DNA primer annealed to the 3′ end of the mRNA was then extended by reverse transcriptase. The register of the ribosome on the mRNA can be determined from the length of the resulting extended DNA. All seven mutants were capable of translocating through all 3 Val codons during the 5 min incubation (followed by an additional 5 min during primer extension; see Materials and Methods) (Fig. 2). EF-G mutant H91L, a dominant-lethal mutation at a position known to be critical for GTP hydrolysis (Cunha et al. 2013; Holtkamp et al. 2014; Salsi et al. 2014a), which was included as a negative control, showed only a single round of translocation, as expected (Fig. 2).
FIGURE 2.
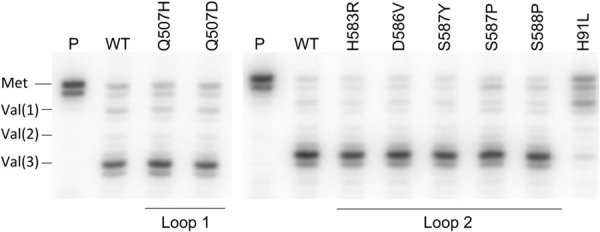
All domain IV mutants catalyze multiple rounds of translocation. Toeprint analysis of translocation by domain IV mutant EF-Gs. (P) Pretranslocation complex; (WT) wild-type EF-G. Positions of reverse transcriptase stops indicate register of mRNA with (Met), P site occupied by N-Ac-Met-tRNAMet; [Val(1), Val(2), (Val(3)], translocation through 1, 2, or 3 consecutive Val codons. The domain I EF-G mutant H91L, which is defective in GTPase activity (Cunha et al. 2013; Holtkamp et al. 2014; Salsi et al. 2014a) is included as a negative control.
Rates of mRNA translocation were measured by a fluorescence quenching assay using a mRNA with pyrene attached to position +9, which is quenched by contact with the ribosome upon translocation by one codon (Studer et al. 2003). At t = 0, EF-G·GTP was rapidly mixed with a pretranslocation complex containing N-Ac-Met-Val-tRNAVal in the ribosomal A site and tRNAMet in the P site in a stopped-flow fluorimeter. Two of the mutants, S587P and S588P, have severe rate defects, more than 100-fold slower than wild-type EF-G (Table 1; Fig. 3A,B). The other mutants show moderate defects, having rates three- to 10-fold down from wild-type, except for Q507H, whose kinetics are nearly indistinguishable from those of wild-type (Fig. 3B). Interestingly, although nearly all of the mutants show mRNA quenching rate behaviors that are clearly biphasic, as has consistently been reported for wild-type EF-G (Peske et al. 2004; Shi et al. 2009; Munro et al. 2010; Ermolenko and Noller 2011), D586V can be fitted well to a pure single-exponential curve (Fig. 3A). The quenching curves are dominated by the fast phase (Qk1), except for Q507D, where Qk2 is predominant (Table 1). Due to their very low activities, the rates of mRNA quenching by S587P and S588P were measured manually (Materials and Methods) (Fig. 3C,D). Thus, all mutant EF-Gs can support multiple rounds of translocation but show a considerable range of rate defects.
FIGURE 3.

Domain IV mutations cause mRNA translocation defects. A fluorescence quenching assay (Studer et al. 2003) was used to measure mRNA translocation complex containing a 3′-pyrene-labeled mRNA. (A,B) A pretranslocation complex was rapidly mixed with mutant forms of EF-G·GTP and quenching of fluorescence of the pyrene label was measured in a stopped-flow fluorimeter. Data were fit to double-exponential curves (Table 1). (C,D) Rates of mRNA quenching for EF-G mutants (C) S587P and (D) S588P were measured manually, due to their low translocation rates. Data could be fit to single-exponential curves.
Domain IV mutants were then tested for their ability to carry out synthesis of the full-length 27 kDa ribosomal protein S2, which contains seven internal methionines, in an in vitro translation reaction using [35S]-methionine to label the polypeptide products. In initial experiments, each mutant EF-G was added in an approximately twofold molar excess over the wild-type EF-G present in the S100 extract in the in vitro system (Supplemental Fig. S1). The amount of full-length protein synthesized by mutant EF-Gs in the presence of wild-type EF-G was comparable to the amount synthesized by wild-type EF-G alone, except for the S588P mutant, whose presence caused a strong dominant inhibitory effect (Fig. 4A). We next asked if the domain IV mutants can support synthesis of a full-length protein in the absence of wild-type EF-G. To do this, we constructed a strain in which the genomic copy of wild-type EF-G was replaced with EF-G containing an amino-terminal 6-His sequence. We then prepared S100 extract from the strain, removing the 6-His-EF-G with Ni++ resin (Materials and Methods). In the absence of wild-type EF-G, all mutant EF-Gs had robust activity, catalyzing synthesis of full-length protein in amounts comparable to that of wild-type EF-G, with the exception of S587P and S588P, which were unable to produce full-length protein above background amounts (Fig. 4B). In addition to the full-length S2 protein, a shorter ∼23 kDa product is consistently produced at a reduced level in both S100 extracts (Fig. 4). The 23 kDa product is likely the result of translation of a ∼100-nt 3′-truncated version of the S2 mRNA. We observed that incubation of the S2 mRNA with S100 extract results in an RNA product whose size is consistent with the loss of ∼100 nt from the mRNA caused by a nuclease activity present in the extract (Supplemental Fig. S2). This conclusion is supported by quantification of these products across all mutants in independent experiments, which shows that the relative proportions of the full-length and 23 kDa products remain constant for a given mutant across multiple experiments, independent of the amount of full-length S2 protein synthesized (Supplemental Table SI).
FIGURE 4.

In vitro translation of a full-length protein by domain IV mutant EF-Gs. A mRNA coding for ribosomal protein S2 was translated in vitro by 70S ribosomes in an E. coli system (Ali et al. 2006) using mutant forms of EF-G with S100 extract (A) containing or (B) lacking endogenous wild-type EF-G. (WT) Addition of wild-type EF-G; (No EF-G) wild-type EF-G not added; (S2) position of full-length protein S2; (23 kD) a translation product likely made from a 3'-truncated mRNA. All mutant EF-Gs except S587P and S588P are capable of catalyzing synthesis of full-length protein.
Rates of rotation of 30S subunit body and head domains
Rates of intersubunit (30S body domain) rotation, which is coupled to movement of tRNAs from their classical states to hybrid states (Ermolenko et al. 2007), were measured by FRET changes using doubly labeled ribosomes containing a Cy5–S6 acceptor on the 30S subunit and a Cy3–L9 donor on the 50S subunit, in a stopped-flow fluorimeter (Ermolenko et al. 2007). Starting with a pretranslocation complex in the rotated, hybrid state containing mRNA, N-Ac-Met-Val-tRNAVal in the ribosomal A site and tRNAMet in the P site, we followed reverse intersubunit rotation to the nonrotated classical state by an increase in FRET efficiency upon rapid mixing with EF-G·GTP (Fig. 5A,B; Ermolenko et al. 2007). Reverse intersubunit rotation rates for the mutant EF-Gs paralleled the order of their rates of mRNA translocation: Q507H has virtually no defect, but the rates for H583R and S587Y are approximately twofold down, Q507D and D586V slower, and S587P and S588P barely detectable (Table 1; Fig. 5A,B). All intersubunit rotation kinetics followed single-exponential behavior.
FIGURE 5.

Domain IV mutations affect rates of intersubunit and 30S head rotation. (A,B) Reverse intersubunit rotation during translocation was measured by FRET using doubly labeled 70S ribosomes containing a Cy3 donor on 50S protein L9 and a Cy5 acceptor on 30S protein S6 in a stopped-flow fluorimeter (Ermolenko and Noller 2011). Data were fit to single-exponential curves. (C,D) Forward and reverse rotation of the head domain of the 30S subunit during translocation was measured by FRET using 70S ribosomes formed from doubly labeled 30S subunits containing an Alexa488 donor on protein S12 in the 30S body domain and an Alexa568 acceptor on protein S19 in the 30S domain (Guo and Noller 2012), in a stopped-flow fluorimeter. Data were fit to double-exponential curves corresponding to initial forward head rotation (downward curves) followed by reverse head rotation (upward curves). (WT) Wild-type EF-G.
Forward and reverse rotation of the 30S subunit head domain were also measured using a FRET-based assay, with protein S12 in the 30S body domain labeled with the donor Alexa 488 and protein S19 in the head domain with the acceptor Alexa 568 (Guo and Noller 2012). All mutants except Q507D showed forward and reverse head rotation rates that paralleled their respective rates of 30S body rotation, including barely detectable rates for S587P and S588P (Table 1; Fig. 5C,D). The rates of mRNA quenching were most similar to the rates of reverse 30S head rotation, in keeping with our previous conclusion that mRNA quenching is the result of contact by the 30S head domain with the 3′-pyrene fluor during its return to the nonrotated state (Guo and Noller 2012). An apparent anomaly is seen for Q507D, whose rate of reverse head rotation is several-fold slower than its rate of mRNA quenching. However, the mRNA quenching kinetics for Q507D, unlike the other EF-G mutants and wild-type EF-G, are dominated by the slow phase (Qk2) (Table 1). Its reverse head rotation rate (0.37 sec−1) in fact bears similarity to the slow phase of its mRNA quenching kinetics (0.32 sec−1), suggesting that Q507D has an interesting atypical defect involving reverse head rotation (Table 1). The overall relative rates of translocation-related events for most of the mutant EF-Gs are generally in the order of forward head rotation > reverse head rotation ≈ mRNA quenching > reverse body rotation, as previously reported for wild-type EF-G (Guo and Noller 2012).
Although the activities of S587P and S588P mutants were only barely detectable in our stopped-flow kinetic measurements (Fig. 5), they were nevertheless fully capable of supporting multiple rounds of translocation in a toe-printing assay, likely due to the longer incubation times in the latter assay (Fig. 2). To further clarify this point, we retested S588P in a toe-printing time-course experiment at room temperature, quenching the translocation reaction with viomycin at different time points prior to primer extension (Supplemental Fig. S3; Fredrick and Noller 2003). It can be seen that an extent of translocation comparable to that of wild-type EF-G at 30 sec is only reached by S588P after 4 min (Supplemental Fig. S3).
Mutations in domain IV cause increased levels of −1 frameshifting
We based our frameshifting assay on the dnaX gene, which contains three elements that promote −1 frameshifting: an internal Shine–Dalgarno (SD) sequence, a slippery sequence, and a downstream 11 bp hairpin (Tsuchihashi and Brown 1992; Larsen et al. 1994). We excluded the downstream hairpin from our construct, but introduced the internal SD (AGGGAG) and the slippery sequence (AA AAA AAG) into the S2 protein mRNA sequence at positions 365–370 and 381–388, respectively (see Materials and Methods). This construct is predicted to stimulate frameshifting to the −1 reading frame, which would result in translation termination at an out-of-frame UGA stop codon at position 417, creating a truncated 16 kDa polypeptide product. In vitro translation of this modified mRNA with wild-type EF-G resulted in synthesis of the predicted 16 kDa frameshift product at 23% frameshifting efficiency (Table 2; Fig. 6A). This is consistent with results that show ∼9%–27% frameshifting with dna X in the absence of downstream secondary structure (Tsuchihashi and Kornberg 1990; Larsen et al. 1994, 1997; Chen et al. 2014; Kim and Tinoco 2017). All seven domain IV mutant EF-Gs, even in the presence of endogenous wild-type EF-G in our S100 extract (Supplemental Fig. S1), increased the synthesis of frameshifted product to 50%–67%. This corresponds to a 3.4- to 6.8-fold increase in the relative abundance of frameshifted product to zero-frame product over that of wild-type EF-G, with H583R, S587Y, and S588P showing the highest rates of frameshifting (Fig. 6A,B). This result demonstrates the dominant properties of these mutant forms of EF-G.
TABLE 2.
Stimulation of −1 frameshifting by mutations in domain IV of EF-Ga

FIGURE 6.

Stimulation of −1 frameshifting by domain IV mutants. SDS gels showing in vitro translation of [35S]-labeled ribosomal protein S2 from a mRNA containing a “slippery sequence” with domain IV mutants using S100 extract (A) containing wild-type EF-G and (C) lacking wild-type EF-G. (B,D) Histograms showing frameshifting efficiencies for A and C, respectively, plotted as the ratio of frameshifted product to 0-frame product. (S2) Full-length S2; (23 kDa) carboxy-terminal truncated S2 product; (FS) −1 frameshift product; (WT) wild-type EF-G. Error bars represent standard errors of the mean.
We next measured frameshifting in the absence of endogenous wild-type EF-G. For the mutants that show robust translational activity (Q507D, Q507H, H583R, D586V, and S587Y), the relative abundance of frameshifted product induced by the mutant EF-Gs increased to 3.7- to 7.2-fold over that of wild-type EF-G (Table 2; Fig. 6C,D). The most dramatic increase was seen for Q507D, suggesting that this mutant does not compete as well with wild-type EF-G (Table 2). For mutants S587P and S588P, there is a clear increase in the amount of frameshifted protein product compared to S100 extract alone (Fig. 6D), but we could not quantify the level of frameshifting for these mutants because they do not produce a full-length protein (Fig. 4B). The increase in frameshifting caused by the domain IV mutants (55%–70% vs. 24% for wild-type EF-G) is comparable to frameshift stimulation by the downstream secondary structure element in the dna X system that is absent in our mRNA construct (Tsuchihashi and Kornberg 1990; Tsuchihashi and Brown 1992; Larsen et al. 1994, 1997; Chen et al. 2014; Kim and Tinoco 2017). Frameshifting efficiencies showed a roughly inverse correlation with rates of translocation, intersubunit rotation and 30S head rotation (Fig. 7).
FIGURE 7.

Rates of translocation events versus frameshifting efficiencies. The rates of (A) reverse 30S head rotation and (B) mRNA quenching are roughly correlated inversely with frameshifting efficiencies. The outlier in (B) Q507D, whose rate of reverse head rotation is unusually slow compared to its rate of mRNA quenching (Table 1), suggests that reverse head rotation is more closely correlated with frameshifting than intersubunit rotation or mRNA quenching. Error bars indicate standard errors of the mean.
In order to confirm that these mutations are indeed causing −1 frameshifting, we created mRNAs with UGA to GGA read-through mutations in the −1 and +1 frames at the first out-of-frame stop codon following the slippery sequence (Fig. 8A). The resulting read-throughs would be predicted to create products with a ∼3 kDa increase in size over the frameshift product from the nonmutated mRNA. When the mutated mRNAs were tested, only the mRNA containing the UGA to GGA substitution in the −1 reading frame generated a product of the predicted size, along with disappearance of the original frameshift product (Fig. 8B). This result was observed for translation with both wild-type EF-G and all seven domain IV mutants.
FIGURE 8.

Domain IV mutants stimulate frameshifting into the −1 reading frame. (A) Schematic showing (no RT) the S2 mRNA containing a slippery sequence at positions 381–388 showing the positions of +1 and −1 out-of-frame stop codons. (−1 RT) A mRNA designed to create read-through of the −1 frame UGA stop codon at position 417, which was replaced by a GGA sense codon. (+1 RT) A +1 read-through mRNA in which the out-of-frame +1 UGA stop codon at position 404 was replaced by a GGA sense codon. Frameshifting events can be assigned to the −1 or +1 reading frames according to whether a frameshift product of increased size appears when translating the −1 RT or +1 RT mRNA. (B) SDS gel showing results of translation through the +1 read-through (+1 RT), −1 read-through (−1 RT), and no-read-through (No RT) mRNAs. Products indicated are (S2), full-length S2 protein; (23 kD) 23 kD carboxy-terminal truncated EF-G product; (−1 RT) read-through product in the −1 frame; (−1 FS) frameshift product in the −1 frame; (−2 FS) product of 2 −1 frameshifting events. Appearance of the 16.6 kDa product with the −1 RT mRNA confirms that the 19.5 kDa band is indeed the product of −1 frameshifting.
For mutants with the highest degrees of frameshifting, an additional band is seen corresponding to the predicted size for a +1 frameshift product (Fig. 8). The intensity of this band is greatest in the presence of EF-G mutants with the highest degrees of frameshifting and is absent in the translation products from the +1 bypass mRNA (Fig. 8B). Given that the slippery sequence is compatible with a −2 frameshift, but does not allow cognate tRNA-mRNA pairing with a +1 frameshift, we infer that this product is likely generated from a −2 slip caused by two −1 slips, rather than an authentic +1 frameshifting event.
DISCUSSION
Our study was prompted by a recent crystal structure of a ribosome-tRNA-mRNA complex that had undergone spontaneous partial translocation in the absence of EF-G or antibiotics (Zhou et al. 2019). The resulting translocation intermediate is similar to a previous EF-G-containing chimeric hybrid-state complex that had been trapped with fusidic acid (Zhou et al. 2014), providing an opportunity to compare in detail the influence of EF-G on the movements of tRNA and mRNA during translocation. In both complexes, the tRNAs were trapped in intermediate states between their pre- and posttranslocation positions. The anticodon ends of the A-site tRNAs had both moved into chimeric a/p states, positioned between A site features of the 30S head domain and P site features of the 30S body. Most unexpected was the finding that, in the absence of EF-G, base-pairing of the A-tRNA codon–anticodon duplex was disrupted, and the anticodon end of the tRNA had actually moved further than in the corresponding EF-G-containing complex. Examination of the relative positions of the codon and anticodon in the EF-G-deficient complex showed that the anticodon register had slipped by one position, into the −1 reading frame, providing direct evidence that EF-G plays a role in maintaining the reading frame. Meanwhile, in a screen for dominant-lethal mutations in EF-G (Nelson C, Leung CS, Noller HF, et al., in prep.), we identified a cluster of six mutations that map to the tip of domain IV, which was found to contact the codon–anticodon duplex in the trapped chimeric hybrid-state intermediate (Ramrath et al. 2013; Zhou et al. 2014), a pretranslocation complex (Brilot et al. 2013), and a posttranslocation complex (Gao et al. 2009). Our results show that all six mutations in domain IV cause an increase in −1 frameshifting as well as a range of defects in translocation itself. These mutations have escaped detection in previous searches for mutations in the genomic copy of the fusA gene (Dahlfors and Kurland 1990; Hou et al. 1994), possibly because of their dominant-lethal phenotypes.
Domain IV of EF-G has long been implicated in catalysis of translocation (Rodnina et al. 1997; Martemyanov and Gudkov 1999; Savelsbergh et al. 2000; Gao et al. 2009; Khade and Joseph 2011; Ramrath et al. 2013; Liu et al. 2014; Peng et al. 2019). Indeed, for the seven mutant forms of EF-G tested in our studies, all of them, with the exception of Q507H, conferred diminished rates of translocation-associated processes, including defects in mRNA translocation, intersubunit rotation and rotation of the head domain of the 30S subunit (Table 1). Their rates of translocation-associated processes were in the order WT ≈ Q507H > H583R ≈ S587Y > Q507D > D586V >>> S587P ≈ S588P. It has been shown that complete deletion of domain IV results in a ∼1000-fold decrease in the rate of translocation without impairing binding of EF-G to the ribosome, single-round GTP hydrolysis, or Pi release (Rodnina et al. 1997; Martemyanov and Gudkov 1999). Site-directed mutations at four positions at the tip of domain IV have been found in several previous studies to cause decreased rates of translocation, including defects for H583K, H583R (Savelsbergh et al. 2000); Q507L, H583K, S588P, and E589A (Liu et al. 2014); and Q507D, Q507E, and H583K (Peng et al. 2019). Three of these four positions were found among the mutations from our dominant-lethal screen (Q507H, H583R, and S588P), which also identified mutations (S587Y, S587P, and D586V) at two additional positions.
While it is clear that these mutations in domain IV confer translocation defects, what are their effects on frameshifting? We used an in vitro assay to monitor stimulation of frameshifting, based on translation of full-length ribosomal protein S2 using a mRNA containing a “slippery sequence” with an upstream Shine–Dalgarno-like sequence. To exclude the possible effects of downstream structured mRNA elements, which are known to strongly increase frameshifting efficiency (Atkins et al. 2016; Choi et al. 2020), our construct lacks any such downstream hairpin or pseudoknot elements (Materials and Methods). All six of the domain IV mutants identified in our screen, plus an additional Q507D mutant (Peng et al. 2019) dramatically stimulate frameshifting into the −1 reading frame by approximately four- to sevenfold over that of wild-type EF-G (Table 2; Fig. 6). Even experiments done with mutant EF-Gs in the presence of wild-type EF-G showed strong increases in frameshifting (Table 2; Fig. 6A,B). Thus, the observed stimulation of −1 frameshifting is dominant in vitro, although we cannot distinguish whether or not this frameshifting defect is responsible for the observed in vivo dominant-lethal phenotypes of the domain IV mutations. While our studies were in progress, Rodnina and coworkers (Peng et al. 2019) reported stimulation of frameshifting by their mutations at positions 507 in loop I and 583 in loop II, using an assay based on in vitro translation of oligopeptides through three different slippery sequences, analyzed by incorporation of radioactively labeled amino acids. All of their mutations conferred −1 frameshifting, with efficiencies ranging from 30% to 80%, in good overall agreement with our findings, in spite of the very different methodologies used in the two studies. In addition to residues Q507 and H583, we find that mutations at D586, S587, and S588 in loop II also stimulate frameshifting (Table 2).
How do mutations in domain IV promote frameshifting? Structures of complexes representing three different states of the translocation process (Gao et al. 2009; Brilot et al. 2013; Zhou et al. 2014) suggest that domain IV remains in contact with the codon–anticodon duplex throughout its trajectory between the A and P sites. Contacts made by domain IV in the chimeric hybrid state (Fig. 9A–D; Zhou et al. 2014) overlap, but are not identical with those seen in the posttranslocation state (Fig. 9E; Gao et al. 2009), indicating some shifting of their positions. In the pretranslocation state (Brilot et al. 2013), the corresponding segments of the domain IV and tRNA backbones are similarly juxtaposed, although the 7.6 Å cryo-EM structure is not of sufficient resolution to conclude whether any of the same contacts occur.
FIGURE 9.

Contacts between the tip of domain IV of EF-G and the RNA backbones of the codon–anticodon duplex. The crystal structure of a trapped chimeric hybrid-state translocation intermediate (Zhou et al. 2014) shows that (A) Gln507 and Tyr514 in loop I (red) form H-bonds with phosphate 37 in the anticodon loop; Gly 509 and Thr 508 are within Van der Waals contact distance of riboses 36 and 37. (B) His583 and Asp586 at the tip of loop II form H-bonds with the 2′-OH of ribose 35 and phosphate 37 in the anticodon loop. (C) Gly510 in loop I makes the sole Van der Waals contact between domain IV and the mRNA backbone, at U20 of the GUA Val codon. (D) Schematic representations of domain IV interactions with the codon–anticodon duplex in the chimeric hybrid state complex (Zhou et al. 2014) and posttranslocation complex (Gao et al. 2009). Mutations at Gln507, His583, and Asp586 were found to confer severe frameshifting and translocation phenotypes (Tables 1, 2). In both crystal structures, all contacts with domain IV are with ribose and phosphate backbone moieties of the tRNA and mRNA. Contacts observed in the two structures are overlapping, but not identical; interactions with Thr508, Gly509, His583 are preserved between the two translocational states. Most of the contacts are with the backbone of the anticodon loop, centered around nucleotides that interact with the first and second codon positions. Note that both crystal structures were obtained from T. thermophilus, although we use E. coli numbering here; all residues are identical between the two species, except for Thr508, which is replaced by Ser508 in E. coli EF-G.
We propose that contact between domain IV and the RNA backbone of the anticodon loop helps to preserve the reading frame by restraining the tRNA from uncoupled movement during translocation. Frameshifting efficiency would then be expected to be increased by mutations in the tip of domain IV that disrupt these interactions. Q507 (loop I), H583 and D586 (loop II), which have strong frameshifting phenotypes, all contact the backbone of the anticodon loop of the transiting A-site tRNA in the chimeric-hybrid state (Fig. 9; Zhou et al. 2014). Although S587P and S588P show frameshifting effects, neither S587 nor S588 actually contact the translocating tRNA or mRNA. However, proline substitutions at these positions likely cause misfolding of the loop II region, creating both translocation and frameshifting defects. The S587Y mutation does not affect translocation as dramatically as the S587P mutation, but strongly stimulates frameshifting, presumably because the bulky tyrosine side chain interferes with normal interaction of domain IV with the anticodon loop. Interestingly, contacts between domain IV and the anticodon loop are centered around nucleotides that interact with the crucial first and second codon positions, suggesting the further possibility that these interactions may stabilize a conformation of the anticodon that favors base-pairing with its codon.
Rodnina and coworkers (Peng et al. 2019) have pointed out that the increased frameshifting efficiencies of domain IV EF-G mutants appear to be correlated with slow rates of translocation-associated processes. Our findings are in general agreement with this (Fig. 7); for example, S588P, which is extremely slow at translocation, shows one of the strongest frameshifting efficiencies. Peng et al. (2019) have proposed that during fast translocation, the ribosome remains committed to the 0 frame before it can slip into the −1 frame, whereas long pauses can allow equilibrium that favors the −1 frame, depending on the mRNA sequence. However, a counterexample from our results is Q507H, which has virtually wild-type translocation rates (Table 1), yet causes a strong increase in frameshifting efficiency (Table 2). This result shows that increased frameshifting cannot be explained solely by the effects of slow translocation.
How would slow translocation promote frameshifting? Structures (Ramrath et al. 2013; Zhou et al. 2014, 2019) suggest that the chimeric hybrid state is the state most vulnerable to reading frame disruption, so any mutation that prolongs this state would likely induce higher levels of frameshifting. This idea is supported by the Q507D mutant, which has an unusually strong defect in its rate of reverse 30S head rotation relative to forward head rotation (Table 1). This implies that Q507D, which is one of the most efficient frameshifters of the mutants that show robust translational activity, must spend more time in the rotated, chimeric-hybrid state. The P-site tRNA ASL, bound tightly to the 30S head, moves with forward head rotation toward the 30S E site, whereas the A-site tRNA, lacking strong contact with the head, can move freely into the space vacated by the transiting P-tRNA, which can result in disruption of its codon–anticodon interaction, as seen in the absence of EF-G (Zhou et al. 2019). Thus, the more time spent in this state, especially with a weakened domain IV contact, the more likely a frameshift event will occur. Peng et al. (2019) also observed a significantly delayed reverse head rotation for the Q507D mutant, although they report a several 100-fold defect, whereas we observe a ninefold rate decrease compared to wild-type EF-G. One difference between the two studies is that Peng et al. monitored head rotation via fluorescence quenching between probes on S13 in the 30S head and L33 on the 50S subunit, thus requiring deconvolution of the rates of 30S head and body rotation, whereas our FRET probes, on S12 in the 30S body and S19 in the 30S head, allow direct measurement of forward and reverse head rotation. Nevertheless, our findings are in agreement in that for Q507D, reverse head rotation is the most defective step. Quite independently, the strong stimulatory effects of downstream secondary structure elements on frameshifting have been attributed to inhibition of reverse head rotation (Caliskan et al. 2014; Yan et al. 2015), providing further support for this possibility.
Interestingly, early searches for EF-G mutations affecting frameshifting identified mutants that decrease the rate of frameshifting. One of these, a G502D mutation (Hou et al. 1994), which introduces a negatively charged side chain into loop I, is consistent with a role in reading frame maintenance for domain IV. It is less obvious how another mutation, Q121R (Dahlfors and Kurland 1990), would affect frameshifting, raising the possibility of a connection between the GTPase and reading-frame maintenance functions of EF-G.
The effects of domain IV mutations on translocation rates are not well understood. It has been proposed that domain IV is involved in initiating translocation by disrupting contacts between the codon–anticodon duplex and the decoding center, which is believed to be the rate-limiting step of translocation (Gao et al. 2009; Khade and Joseph 2011; Ramrath et al. 2013; Liu et al. 2014). Our results, together with the aforementioned previous mutational studies, implicate the two conserved loops I and II at the tip of domain IV in catalysis. Initial contact between domain IV and the pretranslocation complex must somehow trigger release of the codon–anticodon duplex from the 30S A site, possibly by inducing a conformational change in the decoding center around nucleotides G530, A1492, and A1493 of 16S rRNA. Our sole view of EF-G bound to a pretranslocation complex is a 7.6 Å resolution cryo-EM structure, which reveals the positions of the protein and RNA backbones of EF-G, the tRNAs, mRNA, ribosomal proteins and rRNA (Brilot et al. 2013). This structure shows loop I of domain IV within contact distance of the anticodon loop of the A/P tRNA; together with the chimeric hybrid-state and posttranslocation structures (Gao et al. 2009; Ramrath et al. 2013; Zhou et al. 2014), this observation shows that loop I remains in contact with the tRNA anticodon loop during its entire excursion from the A site to the P site. Most interesting is that loop II of domain IV is within contact distance of the 530 loop of 16S rRNA around positions 517 and 530 (Brilot et al. 2013) (PDB 4V7D), raising the intriguing possibility that contact between loop II and the 530 loop might trigger release of the codon–anticodon duplex from the 30S A site. Earlier studies showing that mutations at positions 517 and 529 of 16S rRNA confer frameshifting phenotypes (O'Connor et al. 1992, 1997; Santer et al. 1995) could be explained by weakening of the contacts between loop II and the 530 loop, and are consistent with the finding that many of the same EF-G mutations that increase frameshifting also cause defects in translocation. It should be noted that these mutants also have miscoding phenotypes, so their increased frameshifting could derive from effects on near-cognate tRNA binding, in addition to any potential effects on EF-G function. Merging both the translocation and reading-frame functions in domain IV would ensure that upon release of the codon–anticodon duplex from the decoding site, movement of the tRNA is immediately restrained to prevent its uncoupled translocation.
MATERIALS AND METHODS
Screening for dominant-lethal mutations in EF-G and purification of mutant EF-G proteins
PCR mutagenesis (Cadwell and Joyce 1992) was used to generate random mutations in the E. coli MRE600 gene coding for EF-G, modified to contain a carboxy-terminal 6-His tag, which was then ligated into the expression vector pBAD-18 (Guzman et al. 1995). To screen for dominant-lethal mutants, DH10B transformants were replica-plated on plates containing 0.5 mM arabinose or 0.2% glucose, to turn expression on or off, respectively. Transformants that grew on glucose but not on arabinose were scored as dominant-lethal candidates, as will be described in detail elsewhere (Nelson C, Leung CS, Noller HF, et al., in prep.). Following DNA sequencing, candidate mutants were recreated by site-directed mutagenesis (Kunkel 1985) and retested for their resulting phenotypes. Mutant EF-G proteins were expressed in strain DH10B by induction with 2 mM arabinose for 3 h at 37°C, and purified using Ni-NTA resin (Qiagen) as described (Guo and Noller 2012).
Materials
Tight-couple 70S ribosomes were purified as described (Lancaster et al. 2002). IF1, IF2, and IF3 were prepared as described (Lancaster and Noller 2005), as was wild-type EF-G (Ramrath et al. 2013). To prepare in vitro-transcribed tRNAMet, the gene from E. coli strain MRE600 was cloned into plasmid pRZ (Walker et al. 2003), which is designed to produce RNA transcripts with homogeneous 3′ ends via catalytic HDV ribozyme cleavage of the transcript. Following transcription and gel purification, 2′,3′-cyclic phosphate was removed from the 3′ end of tRNAMet by incubation with T4 polynucleotide kinase (NEB) (Schurer et al. 2002). Aminoacylation (Moazed and Noller 1989) of transcribed tRNAMet was shown to be >95% as monitored by acid gel electrophoresis (Varshney et al. 1991). fMet-tRNAfMet (Sigma), Val-tRNAVal1 (Subriden) and NAcMet-tRNAMet (transcribed) were prepared as described (Moazed and Noller 1989; Lancaster and Noller 2005).
Toeprint analysis
mRNA MVVV_100 (5′GGAAAGGAAAUAAAAAUGGUAGUAGUAGAUAGAAAAUAAUAGAAGAAUCGGAUAAGAGAACACAGGAUCCAGCUGGCGUAAUAGCGAAGAGGCCCGCACC), coding for MVVV, was preannealed to 5′-[32P]-labeled DNA primer (5′GGTGCGGGCCTCTTCGC), then used to form P-site tRNA complexes containing 0.4 µM 70S ribosomes, 0.8 µM N-Ac-Met-tRNA, and 1.2 µM mRNA MVVV_100/primer, in 25 mM Tris·HCl (pH 7.5), 100 mM NH4Cl, 15 mM MgCl2 and 1 mM DTT, that was incubated for 20 min at 37°C. Ternary complex was formed with 7.5 µM EF-Tu, 1.5 µM Val-tRNAVal1, and 2 mM GTP in 25 mM Tris·HCl (pH 7.5), 100 mM NH4Cl and 1 mM DTT and incubated for 5 min at 37°C. Translocation was initiated by adding 6 pmol ternary complex and 10 pmol EF-G to 2 pmol of P-site complex in a final condition of 25 mM Tris·HCl (pH 7.5), 100 mM NH4Cl, 7.5 mM MgCl2, 1 mM DTT, and 1 mM GTP, in a total volume of 10 µL. After 5 min incubation at 37°C, primer extension was initiated by adding 1 µL of extension mix containing 0.55 mM of each dNTP and 0.05 µL of AMV reverse transcriptase (Seikagaku) in the same buffer, and incubation was continued for 5 min at 37°C. Reactions were ethanol precipitated, run on an 8 M urea, 7.5% polyacrylamide gel and autoradiographed. For the toe-printing time-course reactions (Fredrick and Noller 2003), the P-site complex was cooled to room temperature prior to addition of ternary complex and EF-G, and incubation was at room temperature. To stop translocation at each time point, 9 µL of the reaction were added to 1 µL of 10 mM viomycin. Primer extension was initiated and incubated at 37°C as above.
Genomic replacement of wild-type EF-G with 6His-EF-G
The DNA sequence containing 475 nt upstream and 1000 nt downstream from the amino terminus of the fusA gene was PCR-amplified from E. coli MRE600 genomic DNA, then cloned into the Bam HI site of plasmid pKC. The synthetic oligonucleotide 5′GCGATGGGTGTTGTACGAGCGTGGTGGTGGTGGTGGTGCATTTGTTTCCTCGTTTATC was used to add a 6-His sequence to the amino terminus of the EF-G gene (Kunkel 1985). The DNA was then subcloned into the Bam HI site of plasmid pKO3 (Link et al. 1997). Strain 6His_EF-G was generated by genomic replacement of the EF-G gene with 6-His-EF-G in E. coli strain CSH142 as described (Link et al. 1997). The mutation was confirmed by sequencing genomic DNA that was PCR-amplified using primers flanking the cloned region.
Preparation of S100 extracts
S100 extract was prepared from 10 g of E. coli MRE600 cells, or 4 L of strain 6His_EFG, and purified over DEAE resin according to previously published protocols (Traub et al. 1981). To deplete EF-G, S100 extract from strain 6His_EF-G was mixed with 1 mL Ni-NTA agarose resin (Qiagen; 50% slurry) for 1 h at 4°C. The mixture was packed in a column and the flow-through was collected and filtered through a 0.8 µ syringe filter (Corning). The absence of EF-G in the flow-through was confirmed by SDS gel electrophoresis of the S100 extract before and after the column, with purified EF-G used as a marker.
Construction of frameshift mRNAs
Site-directed mutagenesis (Kunkel 1985) of pET24b::S2 (Culver and Noller 1999) was used to generate frameshifting mRNAs by modifying the S2 gene to obtain an internal Shine–Dalgarno sequence (AGGGAG) at positions 365–370, and a slippery sequence (AAAAAAG) at positions 381–388 (Tsuchihashi and Brown 1992; Farabaugh 1996; Tinoco et al. 2013), as shown in Table 3. In addition, the plus-one frame stop codon (TGA) at positions 404–406 and the minus-one stop codon (TGA) at positions 417–419 were mutated to GGA in +1RT and −1RT, respectively, to create read-through of the first out-of-frame stop codons. The mRNAs were transcribed from plasmid linearized with Xho I (NEB).
TABLE 3.
mRNA constructs

In vitro translation
Initiation complexes were formed with a 1:2:3:3 molar ratio of 70S ribosomes (0.5–1.0 µM), fMet-tRNAfMet, mRNA, and initiation factors IF1, IF2, and IF3 in a buffer containing 50 mM Tris·HCl (pH 7.5), 60 mM NH4Cl, 8 mM MgCl2, 2 mM DTT, 1 mM GTP, and incubated at 37°C for 30 min. Total E. coli MRE600 tRNA (Roche) was aminoacylated with 1.0–1.3 µL S100 per 0.1 A260 Unit of tRNA in the same buffer with the addition of 1 mM ATP, 0.2 mM amino acids minus methionine, 6.75 µM methionine, and 0.07 to 0.14 µM [35S]-methionine (1175 Ci/mMol, PerkinElmer) at 37°C for 15 min. Both reactions were added to a translation mix containing 100 pmol EF-Tu and 37.5 pmol wild-type or mutant EF-G per pmol of ribosomes. The final combined mixture contained 0.2 mM total amino acids (except for methionine), 1 mM ATP and GTP, 5 mM phosphoenolpyruvate, 100 nM ribosomes, 0.0075 A260 units/µL tRNA, and 0.75-1.0 µL of S100 per pmol of ribosomes. The combined mixture was incubated for 30 min at 37°C. After incubation, the mixture was analyzed on a 15% (29:1 polyacrylamide:bis) gel and visualized by autoradiography.
Quantification of frameshifting
The amounts of 0, −1, and −2-frame product produced from in vitro translation were quantified using ImageLab (BioRad). Background subtraction was adjusted to isolate individual peaks corresponding to protein products from the three reading frames and the peaks were integrated and normalized according to the number of [35S]-methionines in the predicted sequence of each product: S2 full-length (S2FL) = 7 Met; 23 kDa (23 kDa) = 7 Met; −1 frameshift (−1 FS) = 5 Met; and −2 frameshift (−2 FS) = 5 Met. Percent frameshifting (%FS) was calculated as (−1 FS + −2 FS)/(−1 FS + −2 FS + S2FL + 23 kDa) × 100. Ratio of frameshifted product (FS/0-F) was calculated as (−1 FS + −2 FS)/(S2 FL + 23 kDa). FS/0-F was normalized to wild-type EF-G by dividing the FS/0-F ratio of each mutant by the EF-G wild-type FS/0-F ratio to determine the fold-increase in the frameshift ratio for each mutant.
Fluorescent labeling of ribosomal proteins
Ribosomal proteins S6–D41C and L9–N11C were labeled with Cy5 and Cy3 (Amersham GE) respectively, and S12–K108C and S19–Q56C were labeled with Alexa488 and Alexa568 (Invitrogen), respectively, essentially as described previously (Cornish et al. 2008; Guo and Noller 2012), except that excess dye was removed from labeled S6 and L9 by size exclusion chromatography on a Superdex 75 column (Pharmacia Biotech), in a buffer containing 1 M NH4Cl, 6 mM βME, and 20 mM Tris-HCl (pH 7.5). The same procedure was followed for S12 and S19, except that the buffer was supplemented with 0.0025% Nikkol and 1 M urea.
Preparation of doubly labeled ribosomes
A total of 30S subunits lacking S6, and 50S subunits lacking L9, were isolated from S6 deletion (Keio collection; CGSC #10995), and L9 deletion (Lieberman et al. 2000) strains, respectively, essentially as described (Moazed and Noller 1989; Hickerson et al. 2005), and reconstituted with S6–D41C–Cy5 and L9–N11C–Cy3 as described for L9 (Ermolenko et al. 2007) with the following modifications. Subunits were incubated with a 2.5 molar excess of protein for 1 h in 200 mM NH4Cl, 20 mM MgCl2, 5 mM βME, and 25 mM Tris·HCl (pH 7.5) prior to reassociating and isolating as 70S subunits as described (Hickerson et al. 2005). Doubly labeled S12–Alx488/S19–Alx568 70S ribosomes were reconstituted as described (Guo and Noller 2012) and stored in 20 mM Tris·HCl (pH 7.5), 100 mM NH4Cl, 15 mM MgCl2, 5 mM βME, and 0.01% Nikkol.
Stopped-flow mRNA quenching and intersubunit and 30S head rotation kinetics
Fluorescence changes due to mRNA quenching, intersubunit rotation, and 30S head rotation were measured using an Applied Photophysics SX-20 stopped-flow apparatus by rapidly mixing pretranslocation complex with EF-G·GTP, as described (Guo and Noller 2012). All stopped-flow experiments were conducted in 100 mM NH4Cl, 10 mM MgCl2, 1 mM DTT, 0.01% Nikkol, and 25 mM Tris·HCl (pH 7.5) at 22°–23°C, with final concentrations of 37.5 nM 70S ribosomes, 375 nM EF-G, and 0.5 mM GTP after mixing. Pretranslocation complexes contained 3′-pyrene labeled mv24 mRNA (or unlabeled mv39 mRNA), deacylated elongator tRNAMet bound to the P site and N-acetyl-Met-Val-tRNAVal bound to the A site. For intersubunit rotation experiments, Cy3 was excited at 550 nm and emission from Cy5 was collected using a 645 nm long-pass filter. For 30S head rotation experiments, fluorescence readings were collected as described previously (Guo and Noller 2012), with the exception that Alexa488 was excited at 496 nm. Rates were obtained by fitting individual mRNA quenching and head rotation traces to double exponentials and intersubunit rotation traces to single exponentials using a standard R package nlsLM, which is the R interface to the Levenberg-Marquardt Nonlinear Least-Squares algorithm included in package minpack.lm (http://CRAN.R-project.org/package=minpack.lm). Kinetic values from traces that were collected after the effects of mixing within the stopped-flow had dissipated and had exponential fits that converged were averaged.
Manual recording of mRNA quenching rates
For the two mutants S587P and S588P, manual measurements of mRNA fluorescence quenching were done using a Cary Varian Eclipse Fluorescence Spectrophotometer with excitation at 343 nm. Pretranslocation complexes and EF-G·GTP mixes were formed essentially as described above except that the concentrations of 70S ribosomes and EF-G were 800 nM and 8 pmol/µL, respectively. After combining pretranslocation complexes with EF-G·GTP, fluorescence emission at 380 nm was recorded over 5 min at 22°C. Final concentrations were 400 nM 70S ribosomes, 4 µM EF-G, 0.5 mM GTP, 100 mM NH4Cl, 10 mM MgCl2, 1 mM DTT, 0.01% Nikkol, and 25 mM Tris·HCl (pH 7.5). Data for both mutants could be fit to a single exponential.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by MIRA grant no. R35-GM118156 from the National Institutes of Health (NIH). We thank John Paul Donohue for outstanding computational support.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.077339.120.
REFERENCES
- Ali IK, Lancaster L, Feinberg J, Joseph S, Noller HF. 2006. Deletion of a conserved, central ribosomal intersubunit RNA bridge. Mol Cell 23: 865–874. 10.1016/j.molcel.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Atkins JF, Loughran G, Bhatt PR, Firth AE, Baranov PV. 2016. Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res 44: 7007–7078. 10.1093/nar/gkw530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brilot AF, Korostelev AA, Ermolenko DN, Grigorieff N. 2013. Structure of the ribosome with elongation factor G trapped in the pretranslocation state. Proc Natl Acad Sci 110: 20994–20999. 10.1073/pnas.1311423110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. 1992. Randomization of genes by PCR mutagenesis. PCR Methods Appl 2: 28–33. 10.1101/gr.2.1.28 [DOI] [PubMed] [Google Scholar]
- Caliskan N, Katunin VI, Belardinelli R, Peske F, Rodnina MV. 2014. Programmed −1 frameshifting by kinetic partitioning during impeded translocation. Cell 157: 1619–1631. 10.1016/j.cell.2014.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Petrov A, Johansson M, Tsai A, O'Leary SE, Puglisi JD. 2014. Dynamic pathways of −1 translational frameshifting. Nature 512: 328–332. 10.1038/nature13428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, O'Loughlin S, Atkins JF, Puglisi JD. 2020. The energy landscape of −1 ribosomal frameshifting. Sci Adv 6: eaax6969 10.1126/sciadv.aax6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish PV, Ermolenko DN, Noller HF, Ha T. 2008. Spontaneous intersubunit rotation in single ribosomes. Mol Cell 30: 578–588. 10.1016/j.molcel.2008.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver GM, Noller HF. 1999. Efficient reconstitution of functional Escherichia coli 30S ribosomal subunits from a complete set of recombinant small subunit ribosomal proteins. RNA 5: 832–843. 10.1017/S1355838299990714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha CE, Belardinelli R, Peske F, Holtkamp W, Wintermeyer W, Rodnina MV. 2013. Dual use of GTP hydrolysis by elongation factor G on the ribosome. Translation (Austin) 1: e24315 10.4161/trla.24315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlfors AA, Kurland CG. 1990. Stoichiometry of elongation factor G function in translation. J Mol Biol 216: 311–314. 10.1016/S0022-2836(05)80322-5 [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352. 10.1016/j.cell.2008.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolenko DN, Noller HF. 2011. mRNA translocation occurs during the second step of ribosomal intersubunit rotation. Nat Struct Mol Biol 18: 457–462. 10.1038/nsmb.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolenko DN, Majumdar ZK, Hickerson RP, Spiegel PC, Clegg RM, Noller HF. 2007. Observation of intersubunit movement of the ribosome in solution using FRET. J Mol Biol 370: 530–540. 10.1016/j.jmb.2007.04.042 [DOI] [PubMed] [Google Scholar]
- Farabaugh PJ. 1996. Programmed translational frameshifting. Annu Rev Genet 30: 507–528. 10.1146/annurev.genet.30.1.507 [DOI] [PubMed] [Google Scholar]
- Fredrick K, Noller HF. 2003. Catalysis of ribosomal translocation by sparsomycin. Science 300: 1159–1162. 10.1126/science.1084571 [DOI] [PubMed] [Google Scholar]
- Gao YG, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, Ramakrishnan V. 2009. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science 326: 694–699. 10.1126/science.1179709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Noller HF. 2012. Rotation of the head of the 30S ribosomal subunit during mRNA translocation. Proc Natl Acad Sci 109: 20391–20394. 10.1073/pnas.1218999109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121–4130. 10.1128/JB.177.14.4121-4130.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz D, McPheeters DS, Traut R, Gold L. 1988. Extension inhibition analysis of translation initiation complexes. Methods Enzymol 164: 419–425. 10.1016/S0076-6879(88)64058-4 [DOI] [PubMed] [Google Scholar]
- Hickerson R, Majumdar ZK, Baucom A, Clegg RM, Noller HF. 2005. Measurement of internal movements within the 30 S ribosomal subunit using Forster resonance energy transfer. J Mol Biol 354: 459–472. 10.1016/j.jmb.2005.09.010 [DOI] [PubMed] [Google Scholar]
- Holtkamp W, Cunha CE, Peske F, Konevega AL, Wintermeyer W, Rodnina MV. 2014. GTP hydrolysis by EF-G synchronizes tRNA movement on small and large ribosomal subunits. EMBO J 33: 1073–1085. 10.1002/embj.201387465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Yaskowiak ES, March PE. 1994. Carboxyl-terminal amino acid residues in elongation factor G essential for ribosome association and translocation. J Bacteriol 176: 7038–7044. 10.1128/JB.176.22.7038-7044.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khade PK, Joseph S. 2011. Messenger RNA interactions in the decoding center control the rate of translocation. Nat Struct Mol Biol 18: 1300–1302. 10.1038/nsmb.2140 [DOI] [PubMed] [Google Scholar]
- Kim HK, Tinoco I Jr. 2017. EF-G catalyzed translocation dynamics in the presence of ribosomal frameshifting stimulatory signals. Nucleic Acids Res 45: 2865–2874. 10.1093/nar/gkw1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci 82: 488–492. 10.1073/pnas.82.2.488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland CG. 1992. Translational accuracy and the fitness of bacteria. Annu Rev Genet 26: 29–50. 10.1146/annurev.ge.26.120192.000333 [DOI] [PubMed] [Google Scholar]
- Lancaster L, Noller HF. 2005. Involvement of 16S rRNA nucleotides G1338 and A1339 in discrimination of initiator tRNA. Mol Cell 20: 623–632. 10.1016/j.molcel.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Lancaster L, Kiel M, Kaji A, Noller H. 2002. Orientation of ribosome recycling factor in the ribosome from directed hydroxyl radical probing. Cell 111: 129–140. 10.1016/S0092-8674(02)00938-8 [DOI] [PubMed] [Google Scholar]
- Larsen B, Wills NM, Gesteland RF, Atkins JF. 1994. rRNA-mRNA base pairing stimulates a programmed −1 ribosomal frameshift. J Bacteriol 176: 6842–6851. 10.1128/JB.176.22.6842-6851.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen B, Gesteland RF, Atkins JF. 1997. Structural probing and mutagenic analysis of the stem–loop required for Escherichia coli DNAX ribosomal frameshifting: programmed efficiency of 50%. J Mol Biol 271: 47–60. 10.1006/jmbi.1997.1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman KR, Firpo MA, Herr AJ, Nguyenle T, Atkins JF, Gesteland RF, Noller HF. 2000. The 23S rRNA environment of ribosomal protein L9 in the 50S ribosomal subunit. J Mol Biol 297: 1129–1143. 10.1006/jmbi.2000.3621 [DOI] [PubMed] [Google Scholar]
- Link AJ, Phillips D, Church GM. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol 179: 6228–6237. 10.1128/JB.179.20.6228-6237.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Song G, Zhang D, Li Z, Lyu Z, Dong J, Achenbach J, Gong W, Zhao XS, Nierhaus KH, et al. 2014. EF-G catalyzes tRNA translocation by disrupting interactions between decoding center and codon–anticodon duplex. Nat Struct Mol Biol 21: 817–824. 10.1038/nsmb.2869 [DOI] [PubMed] [Google Scholar]
- Martemyanov KA, Gudkov AT. 1999. Domain IV of elongation factor G from Thermus thermophilus is strictly required for translocation. FEBS Lett 452: 155–159. 10.1016/S0014-5793(99)00635-3 [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF. 1989. Interaction of tRNA with 23S rRNA in the ribosomal A, P, and E sites. Cell 57: 585–597. 10.1016/0092-8674(89)90128-1 [DOI] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung CS, Cate JH, Sanbonmatsu KY, Blanchard SC. 2010. Spontaneous formation of the unlocked state of the ribosome is a multistep process. Proc Natl Acad Sci 107: 709–714. 10.1073/pnas.0908597107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor M, Goringer HU, Dahlberg AE. 1992. A ribosomal ambiguity mutation in the 530 loop of E. coli 16S rRNA. Nucl Acids Res 20: 4221–4227. 10.1093/nar/20.16.4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor M, Thomas CL, Zimmermann RA, Dahlberg AE. 1997. Decoding fidelity at the ribosomal A and P sites: influence of mutations in three different regions of the decoding domain in 16S rRNA. Nucl Acids Res 25: 1185–1193. 10.1093/nar/25.6.1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng BZ, Bock LV, Belardinelli R, Peske F, Grubmuller H, Rodnina MV. 2019. Active role of elongation factor G in maintaining the mRNA reading frame during translation. Sci Adv 5: eaax8030 10.1126/sciadv.aax8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peske F, Savelsbergh A, Katunin VI, Rodnina MV, Wintermeyer W. 2004. Conformational changes of the small ribosomal subunit during elongation factor G-dependent tRNA-mRNA translocation. J Mol Biol 343: 1183–1194. 10.1016/j.jmb.2004.08.097 [DOI] [PubMed] [Google Scholar]
- Ramrath DJ, Lancaster L, Sprink T, Mielke T, Loerke J, Noller HF, Spahn CM. 2013. Visualization of two transfer RNAs trapped in transit during elongation factor G-mediated translocation. Proc Natl Acad Sci 110: 20964–20969. 10.1073/pnas.1320387110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Savelsbergh A, Katunin VI, Wintermeyer W. 1997. Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature 385: 37–41. 10.1038/385037a0 [DOI] [PubMed] [Google Scholar]
- Salsi E, Farah E, Dann J, Ermolenko DN. 2014a. Following movement of domain IV of elongation factor G during ribosomal translocation. Proc Natl Acad Sci 111: 15060–15065. 10.1073/pnas.1410873111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsi E, Farah E, Netter Z, Dann J, Ermolenko DN. 2014b. Movement of elongation factor G between compact and extended conformations. J Mol Biol 427: 454–467. 10.1016/j.jmb.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer UV, Cekleniak J, Kansil S, Santer M, O'Connor M, Dahlberg AE. 1995. A mutation at the universally conserved position 529 in Escherichia coli 16S rRNA creates a functional but highly error prone ribosome. RNA 1: 89–94. [PMC free article] [PubMed] [Google Scholar]
- Savelsbergh A, Matassova NB, Rodnina MV, Wintermeyer W. 2000. Role of domains 4 and 5 in elongation factor G functions on the ribosome. J Mol Biol 300: 951–961. 10.1006/jmbi.2000.3886 [DOI] [PubMed] [Google Scholar]
- Schurer H, Lang K, Schuster J, Morl M. 2002. A universal method to produce in vitro transcripts with homogeneous 3′ ends. Nucleic Acids Res 30: e56 10.1093/nar/gnf055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Chiu K, Ghosh S, Joseph S. 2009. Bases in 16S rRNA important for subunit association, tRNA binding, and translocation. Biochemistry 48: 6772–6782. 10.1021/bi900472a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer SM, Feinberg JS, Joseph S. 2003. Rapid kinetic analysis of EF-G-dependent mRNA translocation in the ribosome. J Mol Biol 327: 369–381. 10.1016/S0022-2836(03)00146-3 [DOI] [PubMed] [Google Scholar]
- Tinoco I Jr, Kim HK, Yan S. 2013. Frameshifting dynamics. Biopolymers 99: 1147–1166. 10.1002/bip.22293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tnalina G, Belitsina NV, Spirin AS. 1982. [Template-free polypeptide synthesis from aminoacyl-tRNA in Escherichia coli ribosomes]. Dokl Akad Nauk SSSR 266: 741–745. [PubMed] [Google Scholar]
- Traub P, Mizushima S, Lowry CV, Nomura M. 1981. Reconstitution of ribosomes from subribosomal components. In RNA and protein synthesis (ed. Moldave K), pp. 521–539. Academic Press, New York. [Google Scholar]
- Tsuchihashi Z, Brown PO. 1992. Sequence requirements for efficient translational frameshifting in the Escherichia coli dnaX gene and the role of an unstable interaction between tRNALys and an AAG lysine codon. Genes Dev 6: 511–519. 10.1101/gad.6.3.511 [DOI] [PubMed] [Google Scholar]
- Tsuchihashi Z, Kornberg A. 1990. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc Natl Acad Sci 87: 2516–2520. 10.1073/pnas.87.7.2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, Lee CP, RajBhandary UL. 1991. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem 266: 24712–24718. [PubMed] [Google Scholar]
- Walker SC, Avis JM, Conn GL. 2003. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res 31: e82 10.1093/nar/gng082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Wen JD, Bustamante C, Tinoco I Jr. 2015. Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160: 870–881. 10.1016/j.cell.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupova GZ, Belitsina NV, Spirin AS. 1986. Template-free ribosomal synthesis of polypeptides from aminoacyl-tRNA. Polyphenylalanine synthesis from phenylalanyl-tRNALys. FEBS Lett 206: 142–146. 10.1016/0014-5793(86)81356-4 [DOI] [PubMed] [Google Scholar]
- Zhou J, Lancaster L, Donohue JP, Noller HF. 2014. How the ribosome hands the A-site tRNA to the P site during EF-G-catalyzed translocation. Science 345: 1188–1191. 10.1126/science.1255030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lancaster L, Donohue JP, Noller HF. 2019. Spontaneous ribosomal translocation of mRNA and tRNAs into a chimeric hybrid state. Proc Natl Acad Sci 116: 7813–7818. 10.1073/pnas.1901310116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.