

Abstract
We improve multiphoton structured illumination microscopy using a nonlinear guide-star to determine optical aberrations and a deformable mirror to correct them. We demonstrate our method on bead phantoms, cells in collagen gels, nematode larvae and embryos, Drosophila brain, and zebrafish embryos. Peak intensity is increased (up to 40-fold) and resolution recovered (to 176 ± 10 nm laterally, 729 ± 39 nm axially) at depths ~250 μm from the coverslip surface.
Two photon laser scanning microscopy (2PM) is well suited to imaging in samples tens to hundreds of microns thick, enabling excellent background rejection due to its long wavelength and nonlinear excitation. The performance of 2PM can be improved by applying methods that overcome the effect of optical aberrations by first measuring distortions in the wavefront and then adaptively correcting them, thereby recovering resolution, signal, and contrast that would otherwise be lost. Such adaptive optics (AO) approaches have improved pharyngeal imaging in C. elegans1, imaging of blood flow and the developing olfactory bulb within larval zebrafish2, centrosome imaging in Drosophila embryos3, and morphological and functional imaging within the mouse cortex4,5. Nevertheless, even if AO is implemented efficiently in 2PM, spatial resolution remains limited by diffraction to ~0.3 μm laterally and ~0.8 μm axially6.
Resolution in point-scanning microscopes can be enhanced to ~140 nm laterally and ~350-400 nm axially by combining information from excitation and emission point-spread functions (PSFs)7–13. Such structured illumination microscopy (SIM) methods can be implemented ‘instantaneously’, i.e. as rapidly as the point-scanning microscopes upon which they are based14,15. We reasoned that combining direct wavefront sensing AO6 with two photon instant SIM15,16 would complement both approaches, as aberrations limit the resolution of any microscope and most AO has been applied to diffraction-limited microscopy.
We began by modifying our 2P-ISIM for aberration correction, using direct wavefront sensing6 (Supplementary Fig. 1, Methods). Since two photon excitation produces a nonlinear guide-star1 (localized point source within the sample), we measured the averaged wavefront created during image acquisition by scanning our guide-star over the sample and descanning the fluorescent emission onto a Shack-Hartmann wavefront sensor (SHS). This procedure averages out fine structure at each scan point17, producing a more accurate estimate of the mean aberration than would be obtained using a single, stationary guide-star6. Once this is determined, a compensatory wavefront is applied to a deformable mirror (DM), correcting aberrations in both excitation and emission arms of the microscope. Both SHS and DM are easily integrated into the underlying 2P-ISIM optical path (Supplementary Fig. 1). A super-resolution image is then obtained by scanning the corrected excitation through the imaging area, simultaneously rescanning the resulting emission (doubling the distance between adjacent scan points) before collection with a camera, and deconvolving the result15.
We benchmarked AO-2P ISIM performance by measuring the PSF in increasingly aberrated environments (Supplementary Fig. 2). First, we estimated the diffraction-limited resolution of our microscope by measuring the apparent size of 100 nm yellow-green fluorescent beads at the coverslip surface (lateral full width at half maximum, FWHM: 260 ± 22 nm; axial FWHM 611 ± 32 nm, N = 30 beads) after correcting system aberrations. As expected, after rescanning and deconvolution we obtained the previously reported resolution improvement15 (Supplementary Fig. 3, lateral FWHM 140 ± 20 nm, axial FWHM 390 ± 43 nm, 30 beads).
Images of fluorescent beads deposited on the interior surface of a curved glass coverslip were contaminated by substantial astigmatism and coma (Supplementary Figs. 2, 4) that masked any resolution improvement possible by rescanning and exhibited a ~40-fold reduction in peak intensity relative to unaberrated beads. Measuring the aberrated wavefront and correcting it with the deformable mirror restored PSFs to near-optimal size after rescanning (lateral FWHM 247 ± 23 nm, axial FWHM 655 ± 72 nm, N = 10 beads, 100 nm diameter), enabling deconvolution and subsequent super-resolution imaging (lateral FWHM 160 ± 31 nm, axial FWHM 419 ± 62 nm, N = 10 beads).
To simulate a strong spherical aberration due to refractive index mismatch, we embedded 100 nm fluorescent beads within polyacrylamide (refractive index 1.452), and imaged the beads at increasing distance from the coverslip (Supplementary Fig. 2). As expected, before correction, PSFs were broadened laterally and especially axially due to the increasingly severe aberration (Supplementary Fig. 5). Measurement and subsequent reduction of the aberration improved peak signal levels up to ~5-fold (Supplementary Fig. 5) and spatial resolution (Supplementary Table 1) throughout the 250 μm thick imaging volume (close to the 280 μm working distance of our objective lens).
We next applied AO 2P-ISIM to biological samples, imaging Alexa Fluor 488 phalloidin-labeled actin in fixed 3rd instar larval Drosophila brain sections (Fig. 1, Supplementary Fig. 6, Supplementary Video 1). Inspection of the average wavefront up to ~65 μm from the coverslip revealed that the dominant aberration was spherical (Supplementary Fig. 6), and that it progressively worsened with depth. AO correction at each plane (Supplementary Fig. 6) substantially improved imaging resolution and contrast (Fig. 1a, b). After deconvolution (Fig. 1c, d), we achieved sub-250 nm lateral resolution (compared to ~500 nm without AO) and ~500 nm axial resolution (equivalent to the axial step-size, improved from ~2 μm without AO). These improvements enabled the visualization of fine, actin-enclosed structures that were otherwise masked due to diffraction and aberrations in axial views, and better resolved the dense actin network in lateral views.
Fig. 1. AO correction based on direct wavefront sensing improves spatial resolution of 2P-ISIM in biological samples, as revealed in labeled Drosophila third instar larval brain.
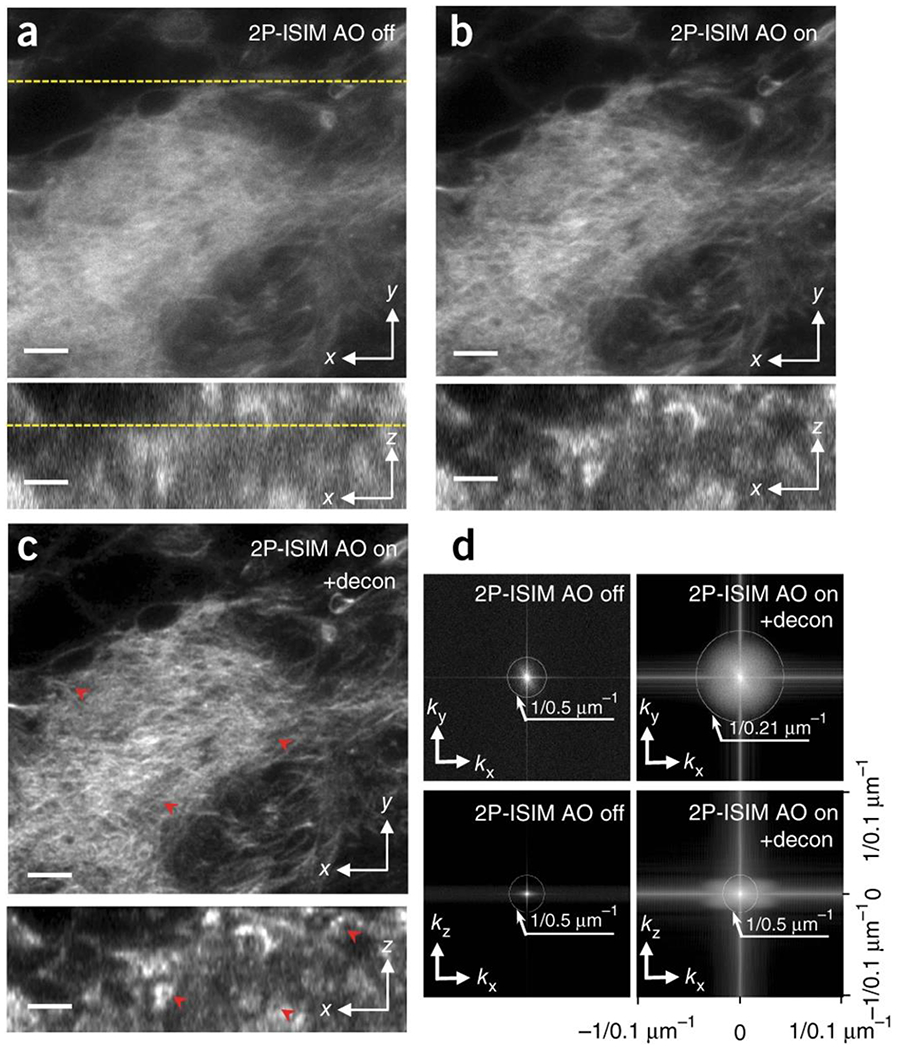
Lateral (top) and axial (bottom) 2P ISIM images of Alexa Fluor 488 phalloidin labeled actin in fixed larval brain lobe, shown without (a) and with (b) adaptive correction, and after subsequent deconvolution (c). The lateral slice is taken 35 μm from the surface of the brain lobe; correspondence with axial slice is indicated with yellow dotted line. Progressive improvements in spatial resolution and contrast are evident in a-c (see also red arrowheads), and further quantified in d), where lateral (top row) and axial (bottom row) MTFs are shown. Note that axial resolution is limited to the step size used when acquiring stacks (0.5 μm for this dataset). See also Supplementary Fig. 6, Supplementary Video 1. Scale bars: 5 μm.
In brightly fluorescent (Fig. 1) or autofluorescent (Supplementary Fig. 7) biological samples, an average wavefront is measured at each plane by diverting fluorescence derived from the entire image scan to the SHS prior to aberration correction, rescanning, and super-resolution image formation. For other samples, such as live, transgenic nematode larvae expressing fluorescent protein in neural structures, the wavefront associated with individual neurons within a single plane (Supplementary Fig. 8) was still helpful in improving the overall image volume. In another case, GFP-labeled histones in nematode embryos proved sufficiently bright for wavefront measurement at the medial image plane. Using the same correction for all planes, we performed 4D (volumetric time-lapse) imaging over 60 time-points, capturing cellular divisions over one hour (~220-280 minutes post fertilization) without photobleaching or obvious phototoxicity (Supplementary Video 2).
However, the majority of biological specimens examined did not inherently provide enough signal for direct wavefront sensing. Here, we added bright fluorescent labels near or coincident with the imaging field. For example, we imaged microtubule bundles in fixed primary mouse endothelial cells embedded in collagen matrices using fluorescent beads added to the collagen matrix to correct aberrations to improve resolution and contrast up to 230 μm from the coverslip (Supplementary Fig. 9). In another case, we improved imaging of rhodamine-phalloidin labeled actin in fixed primary mouse endothelial cells embedded in a collagen gel, 100-150 μm from the coverslip surface (Fig. 2, Supplementary Video 3). Lateral (Fig. 2a, c) and axial (Fig. 2b, d) views were sharpened due to the AO-based reduction in spherical aberration (Fig. 2n), enabling spatial resolution similar to at the coverslip surface (Fig. 2m). We obtained high signal-to-noise images (Fig. 2e, f) of actin bundles (Fig. 2g–j), resolving bundles laterally to ~190 nm (Fig. 2k) and axially to ~400 nm (Fig. 2l). Lamellipodial and filopodial structures (Fig. 2c), actin bundles at the cell cortex, and lateral lamellipodia with apparent actin meshwork (Fig. 2e, red arrows) were clearly resolved. Dim cortical actin structures were also visible (Fig. 2c, yellow arrows), resembling small star-like structures which have been observed in cells cultured on two dimensional substrates18.
Fig. 2. Fiducial-based AO correction enables super-resolution imaging at depths exceeding 100 μm, as revealed by cytoskeletal imaging in fixed cells embedded in collagen matrices.
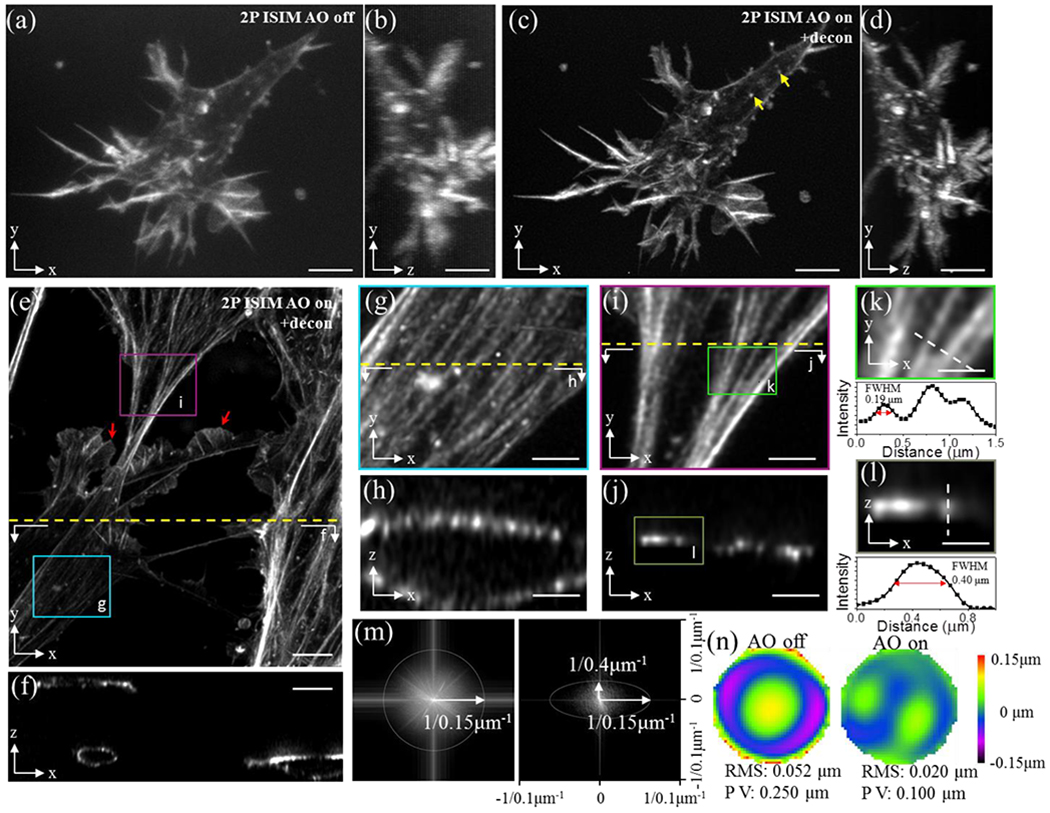
a) Rhodamine-phalloidin stained actin in a primary mouse endothelial cell, embedded in collagen matrix and imaged with 2P ISIM 150 μm from surface of coverslip. Lateral (a) and axial (b) views are shown. c, d) Images as in a, b but after AO correction using a 1 μm fluorescent bead fiducial as a guide-star. Additional cells 100 μm from the coverslip are also shown (lateral view, e) and axial view f). Higher magnification views of boxed regions in e) are shown in g, i along with corresponding axial views h, j. Resolution is estimated from actin bundles, laterally in k and axially in l (higher magnification views of boxed region in i, j respectively), and further quantified in MTFs in m (lateral, left; axial, right). Wavefronts before (left) and after AO correction (right), corresponding to sample in e) are also shown (n). Note that (a-e, g, i) display maximum intensity projections; (f, h, j, l) are single plane cross sections as indicated by dotted lines in (e, g, i) and boxed region in j; and k is a single lateral plane at boxed region indicated in i). Yellow arrows in c) indicate cortical actin structures; red arrows in e) indicate lamellipodial structures. See also Supplementary Video 3. Scale bars: 5 μm (a-f), 2 μm (g-j), 1 μm (k, l).
If placing fiducial beads precisely at the imaging site is either impossible or inconvenient, direct wavefront sensing is facilitated by injecting or expressing a bright, spectrally distinct fluorophore within the imaging vicinity5. We adapted this approach for AO-2P ISIM, demonstrating its application in vivo within embryonic zebrafish (Fig. 3, Supplementary Video 4). By soaking GFP-microtubule labeled larvae in the bright CellTracker Orange dye we enabled wavefront measurement at each plane throughout the 50 x 50 x 70 μm3 volume (Fig. 3a) by diverting both dim GFP- and the much brighter CellTracker Orange fluorescence to the SHS. After wavefront measurement and AO correction we imaged the GFP signal (Fig. 3h). Comparing images before (Fig. 3b, d, f) and after AO correction and deconvolution (Fig. 3c, e, g) reveal obvious improvements in resolution and signal, sufficient to resolve a complex network of smaller microtubules within the developing eye in addition to the larger and brighter structures observed without 2P ISIM AO. Improvements after AO are even more pronounced in deeper slices, where areas with otherwise weak and diffuse signal show a dense network of thin microtubule fibers (Fig. 3f, g). We used the same approach to improve 2P ISIM imaging of somites in live embryonic zebrafish (Supplementary Fig. 10), periodic phalloidin-labeled actin striae in gastrocnemius muscle of excised mouse leg (Supplementary Fig. 11), and presumed gut granules19 in live nematode larvae (Supplementary Fig. 12).
Fig.3. Dye-based AO correction improves 2P ISIM imaging of GFP-labeled microtubules in 38-40 hpf-old embryonic zebrafish lens in vivo.
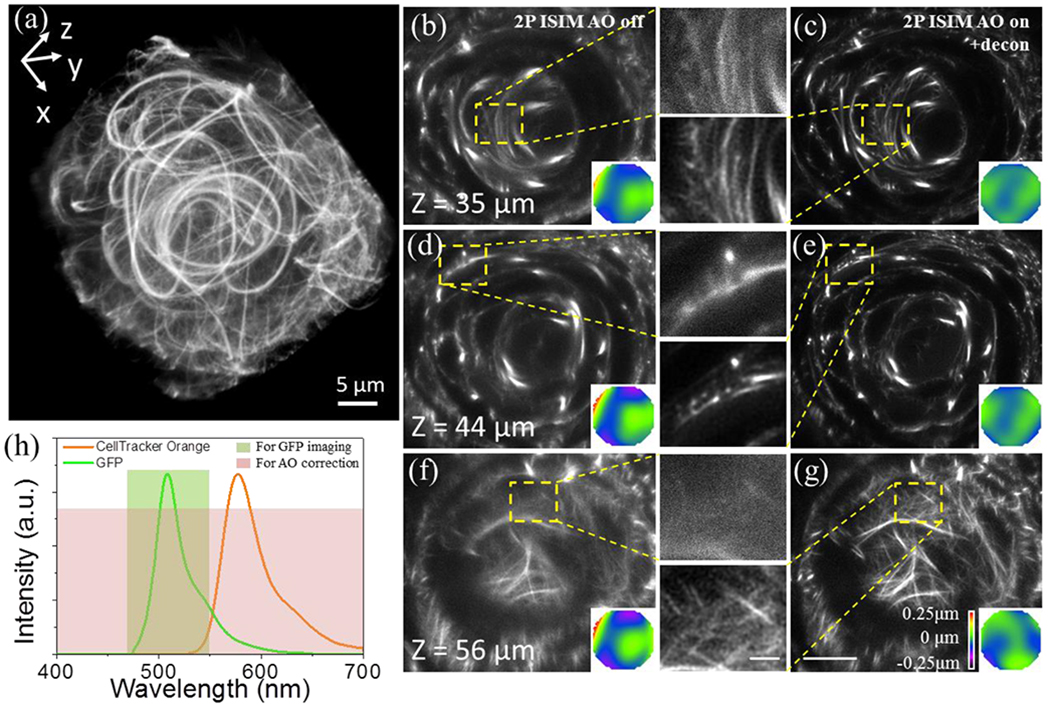
a) Overview rendering of AO-corrected and deconvolved 2P-ISIM volume. Selected slices at indicated axial depth are shown before (b, d, f) and after AO correction and deconvolution (c, e, g), along with wavefront maps (inset) and higher magnification views of yellow dashed rectangular regions. h) Spectra of GFP and CellTracker Orange dye, indicating spectral regions used for AO correction and imaging. See also Supplementary Fig. 10, Supplementary Video 4. Scale bars in b-g: 10 μm (zoomed out views), 2 μm (insets).
AO has been used to improve localization microscopy in cellular samples up to 10 μm thick20, stimulated emission depletion (STED) microscopy through 10-25 μm of tissue21, and SIM through 35 μm of tissue22. Although the resolution in AO 2P ISIM is less than these studies, it enables super-resolution imaging up to 250 μm from the coverslip, considerably greater than previous approaches. Furthermore, we demonstrate live, aberration-corrected volumetric and 4D super-resolution imaging (Fig. 3, Supplementary Figs. 8, 10, 12, Supplementary Videos 2, 4) over hundreds of imaging planes, tens of time points, and with second level temporal resolution – currently impractical with either localization or STED microscopy.
We note several caveats to the current method. First, direct wavefront sensing and SIM require image formation (on the SHS for wavefront sensing, and the detection camera for resolution enhancement). In highly scattering samples, conventional 2PM with point-based detection outperforms our method because contamination from scattered emission is minimized as no image is formed. Second, our design uses a single DM to correct both excitation-side- and emission-side aberrations, simplifying the instrument. This choice assumes that aberrations are similar at both near infrared excitation and bluer fluorescence emission. Chromatic or other aberrations that differentially affect the two imaging paths would benefit from separate aberration correction. Since our ability to correct aberrations was critically dependent on sample brightness, our relatively insensitive SHS was a practical bottleneck. Using a more sensitive SHS – e.g. with an EM-CCD6 – would improve the speed of wavefront sensing, especially in dim samples. Evaluating the aberrations ‘indirectly’ from the image itself23–26 (removing the need for a SHS, additional labels, simplifying instrument design, and reducing the overall cost of the setup) is an interesting alternative to direct wavefront sensing, and may be more light efficient and faster than our current approach.
Finally, our system uses a single high numerical aperture objective for point-scanning illumination and detection. Compared to light sheet microscopy, AO 2P ISIM is fundamentally slower and less dose-efficient but offers better spatial resolution, optical sectioning, and (arguably) easier sample preparation. For transparent samples close to the coverslip (where linear excitation provides sufficient depth penetration), the speed and gentleness of the current approach could be enhanced without compromising spatial resolution if single-photon structured illumination is used for imaging, especially if performed in a parallelized, ‘instantaneous’ implementation14.
Online Methods
Two photon excitation instant SIM /adaptive optics microscope
Our microscope is based upon a previously reported design15 that uses a 2D galvanometric scanner for excitation and a matched 2D emission side galvanometric scanner for rescanning, thereby directly providing super-resolution images that are captured on a widefield detector (an EM-CCD, Andor, DU-855K-CS0-#VP, back-thinned, 1004 pixels x 1002 pixels, 8 μm x 8 μm). The optical components, layout, and microscope base in this study are identical to the previous setup, with two important additions: a Shack Hartmann sensor (SHS, Imagine Optic, HA-7364, HASO3-First, 40 X 32 microlenses) for direct wavefront sensing and a deformable mirror (DM, Imagine Optic, 52 actuators, +/− 50 μm stroke, 15 mm pupil) for aberration correction (Supplementary Fig. 1).
To measure the wavefront, we diverted the descanned emission fluorescence to the SHS using a flip mirror (Thorlabs, GVS211), reimaging the emission scanner (and thus the back focal plane of our 1.2 NA water objective) to the SHS with a pair of achromatic lenses (L8 and L9, f = 250 mm and f = 100 mm, Thorlabs, AC508-250-A-ML and AC254-100-A-ML) placed in a 4f imaging configuration. Emission filters (F2, Semrock, Supplementary Table 2) placed after L9 rejected excitation and isolated fluorescence before collection on the SHS.
To control aberrations, we imaged the back focal plane of our objective lens onto a DM with lens pair L5 and L6 (Thorlabs, both AC508-200-A-ML, both f = 200 mm), placed in a 4f configuration. Lens pair L3 and L4 (Thorlabs, both AC508-250-A-ML, both f = 250 mm, also in a 4f imaging configuration) ensured that the DM was optically conjugate to the excitation scanner. We matched the diameter of the excitation beam to the diameter of the emission beam at the DM with a 5-fold beam expander (L1 and L2, Thorlabs, AC254-040-B-ML and AC254-200-B-ML, f = 40 mm and f = 200 mm, with pinhole, Edmund Optics, 36-392, 100 μm diameter placed at the co-focal point between the lenses to spatially filter the beam) and iris (placed after L2 for fine adjustment of beam diameter), thereby allowing aberration correction in both excitation and emission paths with the same DM setting.
Once the appropriate correction was applied to the DM, emission fluorescence was diverted away from the wavefront sensor by the same flip mirror mentioned above, and imaged onto the EM-CCD via lens L7 (Thorlabs, AC508-250-A-ML, f = 250 mm), filtering out excitation light with filter FI (Semrock, Supplementary Table 2). In some experiments an additional filter was used (Semrock, Supplementary Table 2) in order to further isolate fluorescence in a particular spectral band.
Data acquisition, wavefront correction, and deconvolution
For all imaging, the acquisition time was set to 0.5 s / frame. In most experiments, 5-10 frames were averaged to improve SNR. The imaging field of view spanned 1004 * 48 nm x 1002 * 48 nm = 48.2 μm x 48.1 μm. The axial step size used for acquiring stacks and further acquisition parameters (excitation wavelength, fluorescence filter, total axial extent of stack) are provided in Supplementary Table 2. The electron multiplication gain of the EM-CCD was set to 200 for all measurements.
For wavefront correction, the image exposure on the SHS was set to 0.5 s to 1.5 s depending on the sample brightness. The excitation laser power measured post objective varied: ~5 mW for fluorescent beads, ~30 mW for cells and live samples (nematodes, zebrafish embryos and algae), and ~90 mW for fixed, thick tissue (Drosophila brain lobe and mouse leg muscle).
For datasets of fluorescent beads and cells in gels, we used Richardson-Lucy deconvolution, as previously described14. We assumed Gaussian PSFs with lateral and axial FWHMs derived from 100 nm fluorescent beads (Supplementary Table 1), and ran each deconvolution for 20 iterations.
For time-lapse volumetric AO 2P-ISIM embryo data, we used Richardson-Lucy deconvolution, but implemented the algorithm in the Fourier domain with a graphics processing unit (GPU) card27 to speed processing. We modeled the system PSF as the product of the two-photon excitation PSF and wide field emission PSF, i.e., PSFSYSTEM = PSFEXC X PSFEXC X PSFEMISSION, where PSFEXC and PSFEMISSION were simulated with PSF Generator (ImageJ plugin, http://bigwww.epfl.ch/algorithms/psfgenerator/) using the ‘Born and Wolf’ model with appropriate numerical aperture (1.2 NA), refractive index (1.33) and wavelength (930 nm for PSFEXC, and 550 nm for PSFEMISSION). The resulting system PSF has FWHM of 220 nm, corresponding well to the measured case (Supplementary Table 1). The number of iterations was set to 60.
For other biological samples, datasets were deconvolved with Huygens deconvolution software (Scientific Volume Imaging), which provided better de-noising capability, perhaps due to regularization. We used the following settings: (1) modeling a theoretical PSF based on the microscopy parameters: ‘confocal’ PSF type, 48 nm pixel dimensions, 1.2 NA objective, water immersion, and 1.33 index of refraction; (2) using classic maximum likelihood estimation (CMLE) as the restoration method; (3) setting the quality stop criterion to 0.1 and the targeted signal-to-noise ratio to 20; and (4) setting 20-30 iterations for convergence.
Setting up, characterizing, and using the AO Loop
The DM, SHS, and relay optics between them form the heart of the AO system (Supplementary Fig. 1). Both DM and SHS are controlled through the software CASAO, provided by the vendor. For measuring and correcting aberrations in biological samples, we also wrote a master control program in LabVIEW (National Instruments) for communications between the piezo stage and CASAO software development kit (SDK), enabling plane-by-plane AO correction during image volume collection. AO correction includes three steps. First, we establish an interaction matrix relating the amplitude of the Zernike wavefront modes and the voltages applied to the DM. Second, the wavefront is measured with the SHS and the necessary corrective voltages to the DM are computed. Third, we apply the corrective voltages to the DM, canceling or reducing aberrations. We provide more detail on these steps below:
Determining the interaction matrix.
A 1 μm diameter fluorescent bead (Molecular Probes, F13081) deposited on a glass coverslip was excited by a 488 nm laser (Spectra-Physics, Excelsior-488-200c-CDRH) and the transmitted fluorescence signal isolated with a 500 nm longpass filter (Semrock, FF01-500/LP-25) was detected by the SHS. An iris was placed in an imaging plane to selectively image only the wavefront of a single bead. Second, +/− 0.2 V was applied to each actuator successively to push or pull on the surface of the deformable mirror, and the consequent change in wavefront was recorded by the SHS. We obtained a total of 104 wavefront shapes corresponding to the push/pull of all 52 actuators of the DM. Third, each measured wavefront shape was decomposed into the amplitudes of 36 Zernike modes (up to 7th order). Using the function ‘IM-CM’ provided in CASAO, the above decomposition and voltages were used to compute the interaction matrix. Each wavefront was collected in ~500 ms. The entire process took ~1 minute, but only needed to be performed once during instrument setup.
Measuring the system wavefront and correcting system aberrations.
A single layer of 100 nm fluorescent beads was deposited on a coverslip, and this sample immersed in water and used to measure system aberrations (i.e. those aberrations inherent to the optical setup). After exciting the beads with 2P illumination, the descanned fluorescence signal from the beads (i.e., the nonlinear guide-star) was detected by the SHS (exposure time set to 0.5 s) and decomposed into Zernike modes. Using the interaction matrix, the DM was then set to cancel the system aberrations. This DM setting was recorded and subsequently used as a basis for future AO correction on biological samples.
Measuring the distorted wavefront and correcting aberrations in biological specimens.
We used two types of AO correction: plane-by-plane (every plane in the sample is corrected) or fiducial-based (a fiducial at a particular location in the sample is used for correction and the resulting correction applied throughout the image volume). Differences between these modes of correction are the area over which the measurement and correction are derived (the entire imaging field vs. a selected subregion covering the fiducial) and the frequency of measurement/correction (a unique measurement/correction for each plane vs. a single measurement in one plane and the same correction applied throughout the entire volume). The general protocol is similar in either case: i) set the deformable mirror to correct system aberrations and set the flip mirror to divert the fluorescence emission to the SHS (Supplementary Fig.1). ii) Scan the focal point across a defined area, descan the fluorescence with the emission-side scanner, and record the descanned fluorescence signal with the SHS. The typical exposure time on the SHS for biological specimens varied between 0.5 s – 1.5 s depending on the sample brightness. iii) Decompose the wavefront into Zernike modes, and use the interaction matrix to compute the corrective voltages that, when applied to the DM, will set the 4th to 36th Zernike modes to zero. Steps ii to iii were often repeated (and sometimes 3 iterations were used) to achieve better correction. iv) Add these voltages to the base state of the mirror and shift the flip mirror so that the fluorescence will now be collected by the EM-CCD. v) Finally, form the 2P-ISIM image by scanning the excitation focus across the imaging area again, now rescanning the emission for super-resolution.
Preparation of bead samples
We employed three bead samples: a monolayer of fluorescent beads on a coverslip surface for assessing system resolution, a monolayer of fluorescent beads on a curved tube surface for verifying our ability to correct significant coma/astigmatism, and fluorescent beads embedded in a 3D polyacrylamide gel that allowed us to produce and correct substantial spherical aberration.
To prepare a bead monolayer on a conventional coverslip, 24 × 50 mm #1.5 coverslips (VWR, #48393241) were coated with 100 mg/ml poly-L lysine (Sigma, P8920) and ~50 μL of 100 nm diameter yellow–green fluorescent beads (Invitrogen, F8803, 1:1000 dilution in water) were deposited on the coated coverslips. After several minutes, the coverslips were gently washed with water to remove excess, unbound beads. During imaging, the fluorescent beads were immersed in water by dripping ~500 μL deionized water on the beads.
To prepare bead monolayers on a curved surface, a monolayer with 100 nm diameter beads (Invitrogen, F8803) or 200 nm diameter beads (Invitrogen, F8811) was deposited on a 24 × 50 mm #1.5 coverslip as described above. Next, a 0.9 mm diameter glass tube (Microcells, CAT. # 8100-100) was sandwiched between a glass slide and the coverslip, and pressure applied until the coverslip buckled, thereby forming a curved surface with radius ~270 mm.
Beads in polyacrylamide were prepared by suspending 100 μL of 100 nm diameter yellow–green fluorescent beads (Invitrogen, F8803, 1:100 dilution) and 25 μL of 1 μm diameter yellow-green fluorescent beads (Molecular Probes, F13081,1:1000 dilution) together into a solution containing 125 μL Bis : acrylamide (30 : 0.8), 3 μL 10% ammonium persulfate (APS) and 0.5 μL TEMED. The solution was mixed, vortexed vigorously, and then deposited on a #1.5 glass-bottomed dish (Matek, P35G-1.5-14-C). The gel was allowed to solidify for an hour and imaged immediately thereafter. The assumed refractive index was 1.452.
Preparation of Algae
Zygnema (Carolina Biological Supply, 152695) were sandwiched between a coverslip and a glass slide. To avoid overcrowding of algae, two pieces of plastic plate with thickness ~500 μm were inserted between the coverslip and glass slide, along the two borders of coverslip. Algae were kept moist during imaging by immersing them in their original buffer (alga-Gro freshwater).
Preparation of larval and embryonic nematodes
C. elegans were grown at 20°C on NGM media plates containing E. coli OP50, picked at the larval stage, immobilized in 50 mM levamisole and mounted on an agarose pad sandwiched between a slide and coverslip (VWR, #48393241, #1.5 thickness). Neuronal imaging was performed on strain DCR4315 [olaEx2537 [pncs-1::GFP; punc-122::mCH]] and gut granule imaging on strain BV24 [Itls44 [ple-1p-mCherry::PH(PLC1delta1) + unc-119(+)]; zuls178 [(his-72 1 kb::HIS-72::GFP); unc-119(+)] V]. The latter strain was incubated overnight on NGM plates containing a 1:1 solution of CellTracker Orange and E. coli OP50. Nematode embryos (strain BV24) were prepared for imaging as previously described15, except that #1.5 coverslips were used instead of well chambers.
Preparation of endothelial cells in gels
Primary mouse endothelial cells were cultured and stained as previously described28, with minor modifications. C57bl/6 mouse aorta sections were embedded in 2.5 mg/mL bovine collagen I (Corning, Bedford, MA) in DMEM (ThermoFisher), which was supplemented with 1 μm fluorescent microspheres (F13081, Molecular Probes, Life Technologies, OR), in MatTek dishes (MatTek, Ashland, MA). Gels were covered with EGM-2 endothelial growth medium (Lonza, Houston TX) and maintained at 37°C for 4 days. Gels were fixed in 4% paraformaldehyde in cytoskeleton buffer (CB; 20 mM PIPES pH 6.9, 145 mM KCl, 3 mM MgCl2, 2 mM EGTA) at 25°C for 40 minutes. Cells in gels were permeabilized in the presence of 100 nM Alexa-488-phalloidin (ThermoFisher) in CB supplemented with 0.5% Triton X-100 for 2 hours at 25°C. Gels were rinsed 3 times in CB with 0.1% Tween and one time in CB buffer alone. For microtubule staining, 0.5% glutaraldehyde in CB buffer was used as fixative. Autofluorescence was quenched with 1 mg/mL sodium borohydride in CB, and following CB+Tween washes, gels were blocked with blocking buffer (CB supplemented with 3% bovine serum albumin and 0.1% Tween) for 1 hour at 25°C or 16 hours at 4°C. Gels were incubated with anti-tubulin antibody (1:300, DM1A, AbCam) in blocking buffer for 2-4 hours, followed by three washes with CB buffer, and then Alexa 488-anti-mouse antibody (1:300, ThermoFisher) in blocking buffer for 2 hours at 4°C, followed by three washes in CB. Animals were maintained and euthanized according to guidelines approved by National Heart, Lung and Blood Institute Institutional Animal Care and Use Committee
Preparation of Drosophila brain lobe
Drosophila melanogaster (strain Canton-S) brain lobes were dissected from third instar larvae and fixed in 4% paraformaldehyde/PBS for 20 mins at room temperature. After several washes with 0.1% Triton X-100/PBS the brain lobes were incubated in Alexa Fluor 488 Phalloidin (Cat# A12379, ThermoFisher Scientific) for 2 hours at room temperature. The samples were washed in 0.1% Triton X-100/PBS and mounted in ProLong (Cat# P36934, ProLong Gold Antifade Mountant, ThermoFisher Scientific).
Preparation and staining of live zebrafish embryos
Tg (XIEef1a1:dclk2–GFPio008) Danio rerio embryos were generated by natural spawning and raised at 28.5°C according to standard procedures29 in egg water (0.06g/L Instant Ocean, Doctors Foster and Smith Cat #CD-116528) supplemented with 0.003% N-phenylthiourea to prevent pigment formation (Sigma, Cat. # P7629). Dechorionated 32 or 40hpf old embryos were soaked in a solution of 1.1 μg/mL CellTracker Orange CMRA (ThermoFisher Scientific, Cat. # C34551) in egg water for one hour at room temperature in the dark. Following staining, embryos were washed in egg water twice for 5 minutes and then anesthetized in 600 μM MS-222 (Sigma, Cat. # E10521) diluted in egg water. Embryos were then mounted for imaging in 1% low melting point agarose (NuSieve GTG, Cambrex BioScience, Cat. # 50080). Zebrafish experiments were approved by the NIH intramural animal care and use committee (protocol number 15-039).
Preparation of mouse leg muscle
Four-week-old A/J mice (Jackson Laboratory) were sacrificed according to the procedures in NIH protocol ASP 14-018. The hind limbs were then separated at the femur/hip joint with the membrane and skin removed. Limbs were placed in a glass-bottomed #1.5 dish (MatTek Corportation) submerged in DMEM media (Gibco). Legs were fixed for 30 minutes at 37°C with 4% paraformaldehyde (Electron Microscopy Sciences). Legs were washed twice with phosphate buffered saline (PBS), washed with 10 mM CellTracker Green CMFDA (ThermoFisher Scientific, Cat. # C7025) in DMEM and 12 units of rhodamine phalloidin (ThermoFisher Scientific, Cat. # R415) and incubated at room temperature for two hours prior to imaging.
Estimating spatial resolution
100 nm diameter yellow–green fluorescent beads were used to characterize system resolution at the coverslip surface. The excitation wavelength was set to 900 nm, the axial interval between successive imaging planes set to 100 nm, and the pixel size in each imaging plane 48 nm for 2P-ISIM operation and 96 nm for conventional (diffraction-limited) imaging. The full width at half maximum (FWHM) values of beads were computed using a custom-written MATLAB script that fits a Gaussian function to vertical and horizontal lines centered on the brightest point in each single bead image. To suppress background, we measured the average background on a coverslip area devoid of beads and subtracted this average from each bead measurement.
In biological samples, we used the modulation transfer function (MTF) to estimate spatial resolution. In any single imaging plane or axial reslice, the MTF was noisy or poorly defined due to sparsity of the sample, so we pooled multiple planes for MTF measurement: first, we created an isotropic 3D image stack by axially interpolating the measured image data using the Scale function (Interpolation: Bicubic) in ImageJ. Second, we computed the 2D Fourier transform at different axial depths over an interval of 2 μm or at different lateral distances spanning an interval of ~4 μm. The FFT function in ImageJ was used for calculation. Finally, the Maximum Intensity Projection function in ImageJ was applied to produce the lateral and axial MTFs used in analysis and in figures.
Photobleaching/Phototoxicity Assessment
At the power levels we used, we observed minor or no photobleaching in any of our live samples (for the nematode dataset presented in Supplementary Video 2, fluorescence actually increased during the hour long time course). In live zebrafish samples (Fig. 3), fish showed no sign of morphological damage after imaging, and we confirmed that the embryonic heart continued to beat 24 hours after imaging. In live nematode embryos (Supplementary Video 2), we counted 108 nuclei at the onset of imaging and 194 nuclei after 60 minutes. The change in cell number over time is consistent with the known, highly stereotyped timing of divisions in C. elegans and further supports our assertion that photodamage was minimal during imaging.
Supplementary Material
Acknowledgements
We thank P. Clemenceau, G. Clouvel, J. Legrand, A. Jasaitis, and the entire Imagine Optic team for many helpful discussions about adaptive optics and the use of their DM and SHS. We thank J. Tam (NEI) for useful discussions about AO, H. Eden (NIBIB) for helpful comments on the manuscript, and X. Chen (NIDDK) for help in preparing polyacrylamide gel samples. Support for this work was provided by the Intramural Research Programs of the National Institute of Biomedical Imaging and Bioengineering, the National Institute of Heart, Lung, and Blood, the Eunice Kennedy Shriver National Institute of Child Health and Human Development; the National Key Basic Research (973) Program of China grant (2015CB755502); the National Natural Science Foundation of China grant (81471702); the Shenzhen Science and Technology Innovation Committee grant (KQCX20120816155352228), and NIH R01MH107238 to DBA. The authors declare no competing interests.
Footnotes
Publisher's Disclaimer: Disclaimer:
The NIH, its officials, and employees do not recommend or endorse any company, product, or service.
Code availability
Deconvolution scripts are provided as Supplementary Software. Labview control vis are available from the corresponding author upon request.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reference
- 1.Aviles-Espinosa R et al. Measurement and correction of in vivo sample aberrations employing a nonlinear guide-star in two-photon excitation fluorescence microscopy. Biomed Opt Express 2, 3135–3149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rueckel M, Mack-Bucher JA & Denk W Adaptive wavefront correction in two-photon microscopy using coherence-gated wavefront sensing. Proc Natl Acad Sci U S A 103, 17137–17142 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tao X et al. Live imaging using adaptive optics with fluorescent protein guide-stars. Optics Express 20, 15969–15982 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji N, Sato TR & Betzig E Characterization and adaptive optical correction of aberrations during in vivo imaging in the mouse cortex. PNAS 109, 22–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K et al. Direct wavefront sensing for high-resolution in vivo imaging in scattering tissue. Nature Communications 6, 7276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K et al. Rapid adaptive optical recovery of optimal resolution over large volumes. Nat Methods 11, 625–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.York AG et al. Resolution Doubling in Live, Multicellular Organisms via Multifocal Structured Illumination Microscopy. Nature Methods 9, 749–754 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingaramo M, York AG, Wawrzusin P, Milberg O, Hong A, Weigert R, Shroff H, Patterson GH Two-photon excitation improves multifocal structured illumination microscopy in thick scattering tissue. PNAS 111, 5254–5259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Luca GM et al. Re-scan confocal microscopy: scanning twice for better resolution. Biomed Opt Express 4, 2644–2656 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheppard CJR Super-resolution in Confocal Imaging. Optik 80, 53–54 (1988). [Google Scholar]
- 11.Muller CB & Enderlein J Image Scanning Microscopy. Physical Review Letters 104, 198101 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Schulz O et al. Resolution doubling in fluorescence microscopy with confocal spinning-disk image scanning microscopy. PNAS 110, 21000–21005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth S, Sheppard CJR, Wicker K & Heintzmann R Optical photon reassigment microscopy (OPRA). Optical Nanoscopy 2, 1–6 (2013). [Google Scholar]
- 14.York AG et al. Instant super-resolution imaging in live cells and embryos via analog image processing. Nat Methods 10, 1122–1126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winter PW et al. Two-photon instant structured illumination microscopy improves the depth penetration of super-resolution imaging in thick scattering samples. Optica 1, 181–191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winter PW et al. Incoherent structured illumination improves optical sectioning and contrast in multiphoton super-resolution microscopy. Optics Express 23, 5327–5334 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofer H, Artal P, Singer B, Aragon JL & Williams DR Dynamics of the eye’s wave aberration. J. Opt. Soc. Am. A 18, 497–506 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Luo W, Lieu ZZ, Manser E, Bershadsky AD & Sheetz MP Formin DAAM1 Organizes Actin Filaments in the Cytoplasmic Nodal Actin Network. PLoS One 11, e0163915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clokey GV & Jacobson LA The autofluorescent “lipofuscin granules” in the intestinal cells of Caenorhabditis elegans are secondary lysosomes. Mech Ageing Dev. 35, 79–94 (1986). [DOI] [PubMed] [Google Scholar]
- 20.Huang F et al. Ultra-High Resolution 3D Imaging of Whole Cells. Cell 166, 1028–1040. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gould TJ, Burke D, Bewersdorf J & Booth MJ Adaptive optics enables 3D STED microscopy in aberrating specimens. Optics Express 20, 20998–21009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas B, Wolstenholme A, Chaudhari SN, Kipreos ET & Kner P Enhancing resolution through thick tissue with structured illumination and adaptive optics. Journal of Biomedical Optics 20, 026006 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Gonsalves RA Phase retrieval and diversity in adaptive optics. Optical Engineering 21, 829–832 (1982). [Google Scholar]
- 24.Paxman RG, Schulz TJ & Fienup JR Joint estimation of object and aberrations by using phase diversity. J. Opt. Soc. Am. A 9, 1072–1085 (1992). [Google Scholar]
- 25.Kner P Phase diversity for three-dimensional imaging. J. Opt. Soc. Am. A 30, 1980–1987 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Debarre D et al. Image-based adaptive optics for two-photon microscopy. Opt Lett. 34, 2495–2497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y et al. Simultaneous multiview capture and fusion improves spatial resolution in wide-field and light-sheet microscopy. Optica 3, 897–910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer RS, Gardel ML, Ma X, Adelstein RS & Waterman CM Local cortical tension by myosin II guides 3D endothelial cell branching. Curr Biol. 19, 260–265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B & Schilling TF Stages of embryonic development of the zebrafish. Dev Dyn. 203, 253–310 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.