

Abstract
Clinical and neuropathological staging of Alzheimer disease (AD) neurodegeneration and neuronal loss dynamics is the baseline for identification of treatment targets and timing. The aim of this study of 14 brain regions in 25 subjects diagnosed with AD and 13 age-matched control subjects was to establish the pattern of neurodegeneration, and the severity and rate of neuronal loss in mild cognitive impairment/mild AD (Functional Assessment Staging [FAST] test 3–4), moderate to moderately severe AD (FAST 5–6), and severe AD (FAST 7). The study revealed (1) the most severe neuronal loss in FAST 3–4; (2) the highest rate of neuronal loss in FAST 5–6, to the “critical” point limiting further increase in neuronal loss; (3) progression of neurofibrillary degeneration, but decline of neuronal loss to a floor level in FAST 7; and (4) structure-specific rate of neuronal loss caused by neurofibrillary degeneration and a large pool of neuronal loss caused by other mechanisms. This study defines a range and speed of progression of AD pathology and functional decline that might potentially be prevented by the arrest of neuronal loss, both related and unrelated to neurofibrillary degeneration, during the 9-year duration of mild cognitive impairment/mild AD.
Keywords: Alzheimer disease, Clinicopathological staging, GDS/FAST, Neurofibrillary degeneration, Neuronal loss
INTRODUCTION
Alzheimer disease (AD) affects approximately 36 million people. Available treatments have had limited or no effect on the course of the disease, and the projected number of people with AD dementia may grow to approximately 115 million in 2050 (1).
Brain β-amyloidosis with amyloid plaques, amyloid angiopathy with extracellular deposition of fibrillar Aβ, and neurofibrillary degeneration resulting in accumulation of fibrillar hyperphosphorylated tau in intraneuronal neurofibrillary tangles (NFTs) are essential pathological processes of AD (2) and targets for prevention and treatments (3).
The primary goal of neuropathological studies in AD has been to identify the type, topography, and severity of changes in the memory system responsible for memory decline and dementia. Most of these studies concentrated on the entorhinal cortex (EC), especially the second layer of the stellate neurons, which is affected early by neurofibrillary degeneration and loses neurons faster than other brain structures, and the hippocampal formation, with sector 1 of the cornu Ammonis (CA1) the most consistently affected. These studies revealed that loss of neurons in the EC results in disruption of the perforant pathway, leading to functional impairment of entorhinal-hippocampal connectivity and structural isolation of the hippocampus in the late stages of AD (4, 5). The study of correlations between Braak and Braak (6, 7) staging of topographic expansion of neurofibrillary degeneration and neuronal loss revealed that neuronal loss in CA sectors is Braak stage-dependent and that cumulative neuronal loss in CA1 increases from 33% in Braak stage IV to 51% in stage V (8). Consistency of the pattern of pathology in the memory system has been documented by several morphometric studies of different populations examined with different methods but revealing similar neuronal loss in the second layer of the EC (ECII) (9–13).
This study was designed to expand the survey of neuropathological changes in the memory system, including the EC, 4 sectors of the CA, and the subiculum by characterizing AD pathology in functionally different networks and structures, including the amygdala, thalamus, magnocellular basal complex, substantia nigra, caudate nucleus, cerebellar dentate nucleus, and Purkinje cells.
Clinicopathological staging of neurodegeneration and neuronal loss is critical for identification of targets and selection of the optimal time for successful prevention and treatment of AD pathology. The aim of this study of 14 brain regions and their cytoarchitectural subdivisions in the brains of 25 subjects diagnosed with clinical decline ranging from mild cognitive impairment (MCI) to severe dementia and 13 age-matched control subjects without records of cognitive decline was to establish correlations between duration of clinical stages of AD defined by Functional Assessment Staging (FAST) (14, 15) and morphometric estimates of neuropathological changes. Establishment of the duration of each of the 16 stages and substages of FAST provides a framework for estimating the dynamics of neuropathological changes corresponding to the clinical staging of 19 years, 9 months’ duration of clinically observable AD. Neuropathological staging is focused on the number of neurons, percentage of neurons lost, rate of neuronal loss per million neurons in each structure per day, and volume loss. These evaluations are paralleled with estimates of the number and percentage of neurons with NFTs, amyloid load (%), and volume (mm3) of amyloid deposits.
The next aim of this study was to determine brain region- and stage of AD-specific rates of neuronal loss related and unrelated to neurofibrillary degeneration. Several morphometric studies revealed that neuronal loss exceeds neurofibrillary degeneration (11, 16, 17). This finding suggests that AD neuronal loss might be a cumulative product of neuronal death caused by neurofibrillary degeneration and other mechanisms. The high ratio of neuronal loss caused by other mechanisms estimated in this study suggests that identification of these mechanisms and prevention/reduction of this pool of neuronal loss may have a significant preventive and/or therapeutic effect in AD.
This study of staging of brain degeneration and neuronal death reveals that brain structure- and stage of AD-specific dynamics of neuronal loss are the most consistent correlates of the duration of AD and predictors of structural and functional decline. Staging shows that the largest percentage of neurons is lost in MCI/mild AD (FAST 3 and 4) and suggests that treatments applied during combined MCI (7 years duration) and mild AD (2 years duration) have the potential to disrupt the course of AD early and to preserve high function and quality of life. This stage is followed by acceleration of degeneration and neuronal loss, reaching a ceiling level in moderate and moderately severe AD (/FAST stages 5 and 6a–e). The acceleration associated clinically with the loss of range of function during the 4-year duration of FAST 5–6 is a transition to severe AD. The result of reaching the maximum level of neuronal loss in moderate/moderately severe AD is the decline of neuronal loss to a floor level in severe AD (FAST stages 7a–f), the decline of amyloid load due to the reduction of the number of neurons and a pool of neuronal origin Aβ, and the reduction of the number of NFTs due to the progression of neuronal loss. Established FAST-based clinicopathological staging predicts the dynamics of neuropathological and clinical progression of AD and may be helpful in designing targets and timing of treatment.
MATERIALS AND METHODS
Materials
The neuropathology in the brains of 25 subjects diagnosed with clinical decline ranging from MCI to severe AD dementia and with neurodegeneration matching criteria for an AD diagnosis was compared to changes in 13 control subjects without records of cognitive decline or dementia (Table 1). The age at death ranged from 74 to 97 years in the AD group and from 69 to 102 years in the control group. The difference between the mean age in the AD (82.3 ± 1.3 years) and control (81.7 ± 2.4 years) groups was not significant. Females constituted 52% of the AD and 38% of the control groups.
TABLE 1.
Clinical and Postmortem Characteristics of Control and AD Subjects Including FAST Clinical Staging of Functional Deterioration and Braak Staging of Neurofibrillary Degeneration and β-Amyloidosis
| Number Group | Case # | Sex | Age of Onset | Age (Years) | Cause of Death | PMI | Brain Weight | FAST | Duration (Days) | NFT Tau1 | Amyloid 4G8 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C | 459-96 | F | — | 69 | Pulmonary edema | 48 | 1320 | — | — | II | A |
| 2 | C | 129-98 | M | — | 72 | Myocardial infarct | 3 | 1200 | — | — | IV | C |
| 3 | C | 456-96 | M | — | 73 | Heart failure | 48 | 1200 | — | — | III | A |
| 4 | C | 199-96 | M | — | 77 | Abdominal hemorrhage | 19 | 1270 | — | — | II | A |
| 5 | C | 402-00 | F | — | 78 | Lymphoma | 4 | 980 | — | — | III | A |
| 6 | C | 115-00 | M | — | 80 | Lymphoma | 7 | 1120 | — | — | III | C |
| 7 | C | 113-00 | M | — | 82 | Bronchopneumonia | 7 | 1140 | — | — | IV | C |
| 8 | C | 361-99 | M | — | 83 | Heart failure | 7 | 1200 | — | — | II | A |
| 9 | C | 114-00 | F | — | 85 | Coronary artery disease | 6 | 1060 | — | — | II | A |
| 10 | C | 191-02 | M | — | 85 | Heart failure | 27 | 1080 | — | — | III | 0 |
| 11 | C | 362-99 | F | — | 87 | Brest cancer | 23 | 1120 | — | — | II | B |
| 12 | C | 293-00 | M | — | 90 | Pneumonia | 6 | 1120 | — | — | III | A |
| 13 | C | 449-02 | F | — | 102 | Not established | 10 | 984 | — | — | III | A |
| Mean ± SE | 81.8 ± 2.4 | 16.5 ± 4.4 | 1138 ± 27.8 | |||||||||
| 1 | AD | 452-02 | M | 70.5 | 74 | Lymphoma, pneumonia | 93 | 1340 | 3 | 1278 | III | C |
| 2 | AD | 189-02 | F | 93.5 | 97 | Congestive heart failure | 54 | 1120 | 3 | 1278 | IV | C |
| 3 | AD | 382-01 | M | 86.5 | 90 | Myocardial infarct | 10 | 1260 | 3 | 1278 | III | B |
| 4 | AD | M8-04 | F | 91.5 | 95 | Overdose of Nembutal | 7 | 1038 | 3 | 1278 | IV | C |
| 5 | AD | 360-99 | F | 73.0 | 81 | Congestive heart failure | 24 | 1060 | 4 | 2920 | III | C |
| 6 | AD | 375-98 | M | 78.0 | 86 | Pneumonia | 46 | 1300 | 4 | 2920 | III | A |
| 7 | AD | M15-03 | M | 85.0 | 93 | Heart failure | 8 | 1316 | 4 | 2920 | V | C |
| 8 | AD | 29-99 | F | 65.3 | 75 | Heart failure | 28 | 1040 | 5 | 3559 | VI | C |
| 9 | AD | 451-02 | F | 73.3 | 83 | Adenocarcinoma, pneumonia | 20 | 1080 | 5 | 3559 | III | A |
| 10 | AD | M6-04 | M | 66.3 | 76 | Pneumonia | 9 | 1302 | 5 | 3559 | IV | C |
| 11 | AD | M7-04 | M | 70.3 | 80 | Esophageal carcinoma | 25 | 1200 | 5 | 3559 | V | C |
| 12 | AD | 41-97 | M | 61.5 | 73 | Pneumonia | 12 | 1360 | 6C | 4213 | V | C |
| 13 | AD | 513-02 | F | 70.5 | 82 | Pneumonia | 31 | 980 | 6C | 4213 | V | C |
| 14 | AD | 178-99 | F | 64.5 | 77 | Pancreatic carcinoma | 17 | 940 | 6E | 4563 | VI | C |
| 15 | AD | 376-98 | F | 19.5 | 82 | Bronchopneumonia | 31 | 950 | 6E | 4563 | V | C |
| 16 | AD | 39-97 | F | 69.5 | 82 | Lung carcinoma | 30 | 1080 | 6E | 4563 | VI | C |
| 17 | AD | 211-98 | F | 71.5 | 84 | Heart failure | 23 | 980 | 6E | 4563 | VI | C |
| 18 | AD | 250-99 | M | 64.6 | 78 | Pneumonia | 12 | 1100 | 7A | 4898 | VI | C |
| 19 | AD | 514-02 | F | 62.4 | 77 | Pulmonary art. embolism | 24 | 1028 | 7B | 5354 | VI | C |
| 20 | AD | 188-02 | F | 70.5 | 86 | Pneumonia | 9 | 1000 | 7C | 5810 | VI | C |
| 21 | AD | 252-99 | M | 67.1 | 84 | Pneumonia | 13 | 1140 | 7D | 6176 | VI | C |
| 22 | AD | 190-02 | F | 66.1 | 83 | Pneumonia | 23 | 960 | 7D | 6176 | VI | C |
| 23 | AD | 251-99 | M | 62.9 | 81 | Aspiration pneumonia | 11 | 700 | 7E | 6632 | VI | C |
| 24 | AD | 83-02 | M | 64.6 | 84 | Bronchopneumonia | 16 | 1010 | 7F | 7088 | VI | C |
| 25 | AD | 1-02 | M | 55.6 | 75 | Pneumonia | 20 | 840 | 7F | 7088 | VI | C |
| Mean ± SE | 82.3 ± 1.3 | 23.8 ± 3.7 | 1085 ± 32.7 | |||||||||
AD, Alzheimer disease; NFT, neurofibrillary tangle; PMI, postmortem interval in hours (h); estimated age of AD onset (based on FAST staging).
In AD cohort 21 subjects were white and 1 was black. In 2 cases data were not available. One subject was of Hispanic origin, 20 subjects were non-Hispanic, and in 3 cases data about ethnicity were missing. In AD cohort education of 2 individuals was limited to 8 and 9 years, whereas other subjects’ education was in range from 12 to 18 years (mean 15 years). In 11 cases per 22 with available records (50%), AD was diagnosed among close family members. Among 10 cases with available results of apolipoprotein E genotyping, 1 was diagnosed with APOE 2/4, 4 with 3/3, another 4 with 3/4, and 1 with 4/4 APOE genotype. APOE genotyping was performed by the National Centralized Repository for Aging and Dementia Related Disorders, Indiana University School of Medicine, Indianapolis, IN.
Selection of the examined cohort was based on clinical inclusion criteria to ensure similar number of cases representing each of 7 FAST stages of AD, including 4 subjects with FAST3, 3 with FAST4, 4 with FAST5, 6 representing FAST 6c and 6e, and 8 subjects representing FAST 7 substages a, b, c, d, e, and f. Cases were selected to preserve a similar ratio of females and males in FAST subgroups.
To define neuronal and volume loss, neurofibrillary degeneration and β-amyloidosis matching FAST-based clinical staging of functional decline in AD cohort, the exclusion criteria were applied to reduce risk of distortion of “pure” AD clinicopathological correlations with non-AD pathology. Subjects with double clinical diagnosis of Alzheimer’s and Parkinson’s disease, and brain vasculopathy including stroke and hemorrhage were excluded. The result of postmortem neuropathological examination was exclusion of 6 cases with vasculopathies, including cases diagnosed with signs of hippocampal sclerosis and microinfarcts, and reduction of the number of accepted cases to 25.
Pneumonia/bronchopneumonia was a common cause of death in AD (56%) but was much less common in the control cohort (15%). Infarct, heart failure, or coronary artery disease was a cause of death in 24% of AD and 38% of control subjects. The prevalence of cancer was comparable in the AD and control groups (20% and 23%, respectively). Pulmonary edema and intraabdominal hemorrhage were the causes of death in 2 other control subjects. One AD subject was diagnosed with pulmonary artery embolism, and in 1 case the cause of death was an overdose of Nembutal.
The average postmortem interval (period between death and autopsy) was comparable in the control (16.5 hours) and AD (23.8 hours) cohorts. Details of methods for tissue retrieval, processing, and staining were published in a previous report characterizing neuronal reserve in a control group 25–102 years of age (18). Briefly, brains were removed by using standard protocols, weighed, and divided with a midsagittal cut of the corpus callosum, brainstem, and cerebellum. The brain hemisphere designated for freezing was used for tissue sampling for neuropathological diagnosis. The hemisphere assigned for morphometric study of the global pattern of AD pathology was fixed in 10% buffered formalin for at least 6 weeks and cut coronally into 1-cm-thick slabs by using a macrotome that provides uniform slab thickness and a uniform plane of coronal cuts. Results of a morphometric study of 13 control cases, age-matched to the 25 AD subjects, were used as a reference in this study.
The mean brain weight in the control group was 1138 ± 27.8 g, and brain weight correlated with age (r = −0.69; p < 0.01). Mean brain weight in the AD subject group was insignificantly less (1085 g), and the decline did not correlate significantly with age (r = 0.05); however, the correlation with the duration of AD was significant (r = −0.63; p < 0.001). The study was performed by using coded brain tissue samples and demographics. The brains of AD subjects and neuropathological diagnoses were obtained from the Alzheimer’s Disease Research Center at New York University Medical Centers. Anonymized results of the application of FAST staging were provided by the Clinical Core (Barry Reisberg), NYU Alzheimer’s Disease Center, New York University Langone Medical Center. In each case, diagnostic tissue samples were used for neuropathological evaluation, and the second hemisphere was preserved for morphometric studies. The protocols of this study were approved by the institutional review board of the New York State Institute for Basic Research in Developmental Disabilities.
Research Design to Establish Neuropathological Staging in MCI/Mild AD, Moderate and Moderately Severe AD, and Severe AD
Functional decline in the AD cohort was estimated with the FAST procedure (14) (Table 1). FAST staging was applied to estimate both the clinical course of functional deterioration and to determine duration of each stage and substage of AD. In contrast to the FAST, the Mini-Mental State Examination (19), the Clinical Dementia Rating (CDR) (20), and other staging methods utilized in AD assessment reveal a floor effect with missing standards for assessment of functional deterioration during severe AD, whereas FAST stage 7 identifies the chronological order of functional loss in 6 substages (a–f) in the last 7 years of AD (21). Application of FAST estimates for the duration of 6 clinical substages of AD was a critical factor in attempting to detect dynamics of degeneration, neuronal loss, and functional deterioration in severe AD.
This study was designed to monitor the progression of changes in 25 cases of AD in comparison with 13 age-matched control cases and to define dynamics of pathological changes in 3 major periods of AD, including the 7 years of MCI and the 2 years of mild AD (FAST stages 3–4, corresponding to 45% of AD duration); 3 years 11 months of moderate and moderately severe AD (FAST 5–6; 20% of AD duration); and 7 years of severe AD (FAST 7a–f; 35% of AD duration). This merger of FAST stages increased subgroup sizes to 7 in MCI/mild AD, 10 in moderate and moderately severe AD, and 8 in severe AD, permitting more robust statistical analyses. Supplementary DataTable S1 includes the duration of FAST stage and the cumulative duration applied for estimation of the dynamics of neuronal loss, including the number of neurons lost in structure or cytoarchitectural subdivision per day and the number of neurons lost per million neurons per day. This index allows one to compare the rate of neuronal loss in structures with a broad range of differences in the number of neurons.
Tissue Processing, Embedding, Sectioning, Staining, and Immunostaining
One-centimeter-thick coronal slabs of each brain hemisphere were dehydrated in ascending concentrations of ethyl alcohol, infiltrated with polyethylene glycol 400 (PEG; Sigma, St. Louis, MO) for 6 days, and with PEG 1000 for the next 6 days, embedded in fresh PEG 1000, cut serially into 50-µm-thick sections, and stored with section identifiers in 70% ethyl alcohol (18). Free-floating serial sections were stained with cresyl violet and mounted with Acrytol (Sigma). The library of serial cresyl violet-stained hemispheric sections with a known distance between sections was used for delineation of regions of interest, measurement of structure/cytoarchitectural subdivision area, and estimation of structure and cytoarchitectonic subdivision (nucleus, layer or sector) volume and number of neurons. To estimate the number of neurons with NFTs, serial sections were treated with alkaline phosphatase (Sigma, type VII-L, 400 µg/mL in PBS, pH7.4; 0.01% H2O2) and immunostained with mouse monoclonal antibody Tau-1 diluted 1:100 000 (22). Tau-1 immunostaining enables identification of neurons in all stages of progression of neurofibrillary degeneration, including pretangles and early, mature, and end-stage tangles (23). Degraded extracellular residues of NFTs (ghost tangles) were not included in NFT counts. To evaluate amyloid load, serial sections were pretreated with 88% formic acid and immunolabeled with mAb 4G8, which recognizes an epitope between amino acids 17–24 of the Aβ peptide (24). To estimate relationships between neuronal loss and amyloid deposits, all amyloid deposits immunodetected with mAb 4G8 were estimated as percentage of structure volume (amyloid load), including exclusively diffuse deposits in the cerebellar molecular layer and mainly fibrillar plaques in the EC, hippocampal formation and subcortical structures. Amyloid load was not estimated in islands of stellate neurons in the second layer of the EC due to poor distinction of borders of neuronal islands and amyloid in immunostained sections with weak counterstaining with hematoxylin.
To estimate potential contribution of different proteinopathies to neurodegeneration, neuronal loss and functional decline, the prevalence and severity of TDP-43 neurodegeneration and Lewy bodies were determined in the amygdala in AD subjects. The numerical density and percentage of neurons with TDP-43-positive cytoplasmic inclusions were estimated in the amygdala in 50-µm-thick sections immunostained with rabbit polyclonal antibody TDP-43 (1:500; Catalog nr 10782-2-AP; 1:500; ProteinTech, Rosemont, IL). The prevalence (percentage of AD cases positive for Lewy bodies) and severity (numerical density and percentage of neurons with Lewy bodies) were determined. Free-floating 50-µm-thick sections were immunostained with mouse monoclonal antibody 4B12 detecting α-synuclein (1:500; MA1-90346, Thermo Fisher Scientific, Rockford, IL). Numerical density (n/mm3) and percentage of neurons with TDP-43-positive cytoplasmic inclusions and neurons with Lewy bodies (cresyl violet-stained neurons—100%) were estimated using an ×40 objective lens. On average 183 sampling sites per case were examined to detect TDP-43 pathology and 146 sampling sites to detect Lewy bodies. The same test area (150 × 150 µm) and sampling grid (600 × 600 µm) were applied for estimation of both degenerative processes.
Structures Examined, Morphometric Parameters, Delineation of Brain Structures, and Their Cytoarchitectural Subdivisions
Unbiased stereology was applied to estimate the volume, number of neurons, number, and percentage of neurons with NFTs, and amyloid load in 14 brain regions and their cytoarchitectural subdivisions. Delineation of anatomical boundaries and region of interest cytoarchitectural subdivisions was described in detail in our previous report (18). This study included all layers of the EC and the islands of the stellate neurons in ECII, delineated using Amaral and Insausti criteria (25); the pyramidal layer in 4 sectors of the Ammon’s horn (CA1–4); and the subiculum proper, delineated according to Lorente de No (26), Rosene and Van Hoesen (27), and Duvernoy (28). Four nuclei—basal, accessory basal, lateral, and central—were examined in the amygdala. They were delineated according to Schumann and Amaral criteria (29). In the thalamus, all subdivisions were examined. In the caudate nucleus, estimates of the number of neurons were limited to small neurons. Fearnley and Lees criteria (30, 31) were applied to delineate the substantia nigra pars reticulata and compacta and all subdivisions of the dorsal and ventral tier of nuclei. Four nuclei (CH1–4) were examined in the magnocellular basal complex (32). Morphometry of the cerebellum included volume of the molecular and granular layer to estimate the total number of Purkinje cells in the borderline between these 2 layers. A cerebellar study included the dentate nucleus. All estimates are limited to 1 brain hemisphere examined.
Stereological and Statistical Analysis
To maintain compatibility with the results of our study of aging and neuronal reserve (18), similar standards of stereological analysis were applied in both studies (Supplementary DataTable S2). The volume of brain structures and the number of neurons with and without NFTs were estimated with unbiased stereological methods (33) by using a workstation consisting of an Axiopthot II light microscope (Carl Zeiss, Gottingen, Germany) with Plan-NEOFLUAR objectives 1.25×, 2.5×, 20×, and 40×; a specimen stage with a 3-axis, computer-controlled stepping system (Ludl Electronics, Hawthorne, NY); a CCD color camera CH9000 (MicroBrightfield Bioscience, Williston, VT); and stereology software (Stereo Investigator, MicroBrightfield Bioscience).
The mean number of equidistant sections examined per case varied from 8 for EC and amygdala to 18 for dentate nucleus (on average 11 sections per structure). To maintain low coefficient of error, the mean number of counting spaces was 364 and mean number of counted neurons 206 per case/per structure.
Comparisons of neuronal counts, structural volumes, etc., between groups were analyzed in t-tests adjusted for unequal variances when required. Pearson correlations are unadjusted. Significance levels are not adjusted for multiple comparisons. Analyses were performed by using version 16 of Stata (34).
RESULTS
Link Between Short Survival of Older AD Subjects, Longer Survival of Younger Individuals, and the Stage of AD at Death
Distribution of estimated onset of MCI in 25 nonselected consecutive AD cases examined postmortem shows that the age at onset varied in range from 55.6 to 93.5 years of life. Age-associated enhanced mortality during this 37.9-year interval results in shortening of survival time from 15.5 years in the youngest cases to only 6.3 years in the oldest subjects (Fig. 1). The reversed ratio between prevalence of younger subjects who died in late stage of AD (FAST stages 6e and 7a–f) and prevalence of older subjects who died in early stages of AD (FAST stages 3 or 4) appears to be the cause of a lack of correlation between age and severity of pathology in the examined AD cohort.
FIGURE 1.

Enhanced age-associated mortality in the eighth and ninth decades results in short survival, associated with the prevalence of early stages of Alzheimer disease (AD) in the oldest subgroup. The graphs illustrate distribution of age at onset of mild cognitive impairment (MCI) in a random sample of 25 AD subjects (A), their age at death (B), and Functional Assessment Staging (FAST) stage at death (C). Subject data are presented in the order of increasing age at onset of AD and are supported with linear trendlines. The time of survival of the 7 youngest cases was on average 15.5 years, and their diagnosis at death was in FAST stage 6e to 7f, while the 7 oldest subjects, who developed MCI late, survived on average only 6.3 years and at the time of death were diagnosed at FAST stages 3 or 4. This reversed pattern may contribute to the lack of correlations between pathology and age in a postmortem study of this cohort of sporadic AD.
Multiregional Staging of Cumulative Volume Loss in AD
Brain atrophy is an effect of the region-specific rate of neuronal and volume loss during AD. In MCI/mild AD (FAST stages 3–4), volume changes determined in comparison to the control group were insignificant in most examined structures, exceptions being the 22.3% loss of volume in the CA3 sector (p < 0.05) and 22.2% (p < 0.02) loss in the thalamus (Table 2; Fig. 2A). In subjects diagnosed with moderate and moderately severe AD (FAST 5–6), a significant volume loss was found in 7 of 15 examined regions, including all layers of the EC (20.0%), islands of stellate neurons in ECII (55.2%), CA1 (29.5%), CA2 (14.0%), CA3 (21.1%), subiculum (33.1%), and thalamus (25.1%). In severe AD (FAST 7a–f), cumulative volume loss increased significantly in the entire EC to 26.8%, ECII to 84.0%, CA1 to 50.5%, subiculum to 54.2%, amygdala to 25.5%, thalamus to 34.9%, and magnocellular basal complex to 29.4%. The progression of atrophy in 13 brain regions is reflected in the 22.2% average cumulative volume loss in MCI/mild AD, 28.3% loss in moderate/moderately severe AD, and 43.6% volume loss in severe AD (Fig. 2B).
TABLE 2.
Estimated Volume (mm3; ±SE) of 14 Structures and Cytoarchitectural Subdivision in Control Subjects 69–102 Years of Age and Subjects Diagnosed With AD FAST Stages 3–4, 5–6, and 7
| Structure/Subdivision | Control | AD FAST 3–4 (n = 7) |
AD FAST 5–6 (n = 10) |
AD FAST 7a–f (n = 8) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 69–102 Years | Vol (mm3) | Vol Loss % | p < | Vol (mm3) | Vol Loss % | p< | Vol (mm3) | Vol Loss % | p < | |
|
Entorhinal c. (all layers) |
531 ± 24 | 504 ± 50 | 5.1 | ns | 425 ± 33 | 20.0 | 0.02 | 389 ± 31 | 26.8 | 0.002 |
| Entorhinal c. (Islands) | 29 ± 1 | 24 ± 3 | 17.3 | ns | 13 ± 2 | 55.2 | 0.001 | 5 ± 1 | 84.2 | 0.0001 |
| CA1 | 389 ± 21 | 387 ± 38 | 0.4 | ns | 274 ± 21 | 29.5 | 0.002 | 192 ± 25 | 50.5 | 0.0001 |
| CA2 | 39 ± 2 | 33 ± 3 | 15.4 | ns | 34 ± 2 | 14.0 | 0.05 | 34 ± 2 | 12.9 | ns |
| CA3 | 45 ± 3 | 35 ± 4 | 22.3 | 0.05 | 35 ± 2 | 21.1 | 0.02 | 39 ± 3 | 13.3 | ns |
| CA4 | 100 ± 6 | 94 ± 8 | 5.4 | ns | 97 ± 8 | 2.2 | ns | 102 ± 6 | 0 | ns |
| Subiculum | 180 ± 12 | 157 ± 8 | 12.9 | ns | 121 ± 7 | 33.1 | 0.005 | 83 ± 7 | 54.2 | 0.001 |
| Amygdala | 420 ± 20 | 421 ± 45 | 0 | ns | 368 ± 21 | 12.6 | ns | 313 ± 20 | 25.5 | 0.0018 |
| Thalamus | 2640 ± 130 | 2053 ± 151 | 22.2 | 0.02 | 1978 ± 86 | 25.1 | 0.005 | 1718 ± 126 | 34.9 | 0.0001 |
| Substantia nigra | 82 ± 6 | 96 ± 6 | 0 | ns | 79 ± 5 | 3.7 | ns | 80 ± 3 | 2.5 | ns |
|
Magnocellular basal complex |
120 ± 5 | 125 ± 6 | 0 | ns | 102 ± 7 | 14.6 | ns | 85 ± 10 | 29.4 | 0.003 |
| Caudate nucleus | 1929 ± 71 | 2140 ± 128 | 0 | ns | 1891 ± 97 | 2.0 | ns | 1748 ± 153 | 8.4 | ns |
|
Cerebellum: mol+gran cell layer |
23 747 ± 1438 | 23 457 ± 3086 | 1.3 | ns | 24 121 ± 2651 | 0 | ns | 24 712 ± 973 | 0 | ns |
| Dentate nucleus | 271 ± 15 | 246 ± 33 | 9.2 | ns | 226 ± 17 | 16.7 | ns | 221 ± 24 | 18.5 | ns |
The cumulative volume loss was calculated as the difference between volume in 69–102 years of age control group and volume in each of 3 stages of AD.
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; FAST, Functional Assessment Staging; ns, no statistically significant volume (vol) change.
FIGURE 2.

Staging of volume loss in 13 structures in the brain of Alzheimer disease subjects (graph A). Volume loss (%) estimated in Functional Assessment Staging Test (FAST) stage 7 in comparison to 69- to 102-year-old controls shows regional differences in magnitude of atrophy, ranging from a low percentage in CA4 and substantia nigra to approximately 50.5% in the CA1 and 54.2% in the subiculum, and more than 84.2% in the entorhinal cortex islands (Table 4). The estimated average volume loss in structures with significant volume loss (graph B) shows a rather uniform progress of atrophy, with volume reduction by 22.2% in FAST stages 3–4, 28.3% in FAST stages 5–6, and 43.6% in FAST stage 7.
Neuronal Loss
To expand the understanding of brain atrophy in AD, the dynamic nature of neuronal loss was analyzed, including changes during MCI/mild AD, moderate-moderately severe AD, and severe AD (FAST 3–4, FAST 5–6, and FAST 7) in the number of neurons in 14 brain structures/cytoarchitectural subdivisions (Fig. 3), estimates of the percentage of neurons lost in each of these 3 AD stage groups (Fig. 4), and estimates of the rate of neuronal loss (number of neurons lost per million neurons per day) in each stage (Fig. 5, Tables 3–5).
FIGURE 3.
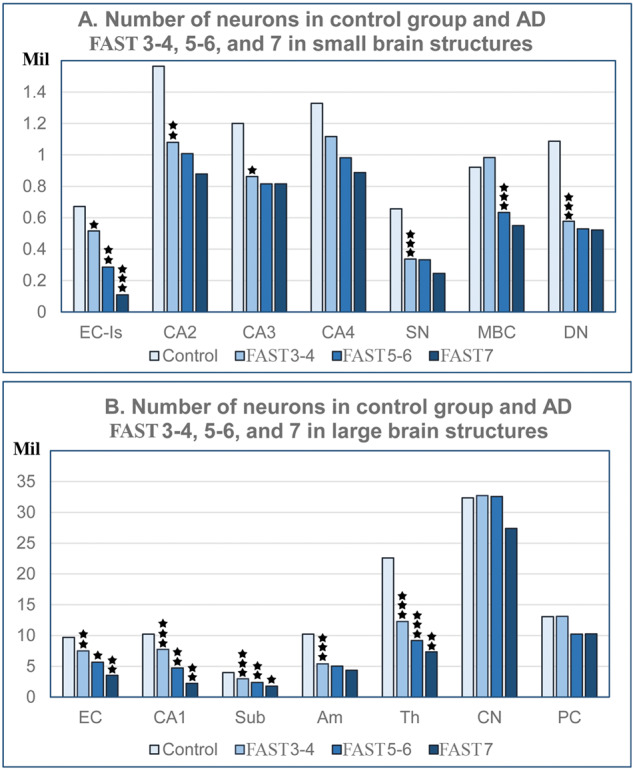
Regional differences in the number of neurons (millions) in 69- to 102-year-old nondemented control subjects and Alzheimer disease subjects diagnosed with FAST stages 3–4, 5–6, and 7. Graphs show the decline in the number of neurons in small structures with less than 1.6 million neurons (graph A) and large structures with 1.6 to approximately 32 million neurons (graph B). The pattern of neuronal loss is similar regardless of the region and the size of the neuronal population, with 2 exceptions—loss of Purkinje cells in FAST stages 5–6, and loss of small neurons in the caudate nucleus limited to FAST stage 7. EC, all layers of the entorhinal cortex; EC-Is, islands of the stellate neurons in the second layer of the EC; Cornu Ammonis, CA1, 2, 3, 4; Sub, subiculum; Am, amygdala; Th, thalamus; SN, substantia nigra; MBC, magnocellular basal complex; DN, dentate nucleus.
FIGURE 4.

Graphs A–C show differences between a high and rather uniform neuronal loss in FAST stages 3–4, and a less severe and strikingly different one in individual structures in FAST stages 5–6 and 7. Neuronal loss was calculated separately for each of the 3 clinical stages as a percentage of the number of neurons in 69- to 102-year-old control subjects. Graph D shows cumulative neuronal loss (%) during the entire course of Alzheimer disease (AD). The structure-specific percentage of neuronal loss during the 19 years and 9 months of AD ranges from 83.8% in the entorhinal cortex islands of stellate neurons to 32.1% in CA3. Graph E demonstrates the highest (31.0%) neuronal loss in FAST stages 3–4 and the decline of neuronal loss to 15.5% in FAST stages 5–6 and to 13.8% in FAST stage 7.
FIGURE 5.

Estimates of the rate of neuronal loss reveal strikingly different dynamics of neuronal loss in FAST stages 3–4 (graph A), FAST stages 5–6 (graph B), and FAST stage 7 (graph C). In the entorhinal cortex (EC), islands of stellate neurons in EC, CA1, subiculum, amygdala, and thalamus, the rate of neuronal loss in FAST stages 3–4 is rather uniform and moderate, whereas in FAST stages 5–6, the rate of neuronal loss is almost doubled, but in FAST stage 7, it drops to the lowest level. The general pattern of the rate of neuronal loss is reflected in an mean rate of neuronal loss (graph D) and cumulative neuronal loss (graph E) that is moderate in FAST stages 3–4, reaches a maximum level in FAST stages 5–6, and drops to a floor level in FAST stage 7.
TABLE 3.
Number of Neurons in Control Subjects 69–102 Years of Age and AD Subjects FAST Stages 3–4
| Structure/Subdivision | Mean Number of Neurons ± SE |
Neuronal Loss in FAST Stages 3–4 |
|||||
|---|---|---|---|---|---|---|---|
| Control 69–102 Years | AD Subjects FAST Stages 3–4 | Number | p < | Loss (%) | Loss/Day | Loss/Million Neurons/Day | |
|
Entorhinal c. All layers |
9 695 433 ± 361 074 | 7 477 533 ± 588 796 | 2 217 900 | 0.004 | 22.9 | 1119 | 115 |
| Entorhinal c. Islands | 672 074 ± 33 076 | 516 637 ± 53 472 | 155 438 | 0.020 | 23.1 | 78 | 117 |
| CA1 | 10 234 672 ± 303 305 | 7 755 632 ± 604 472 | 2 479 041 | 0.001 | 24.2 | 1251 | 122 |
| CA2 | 1 565 802 ± 64 358 | 1 080 495 ± 119 308 | 485 307 | 0.001 | 31.0 | 245 | 156 |
| CA3 | 1 201 826 ± 80 594 | 862 857 ± 89 455 | 338 970 | 0.015 | 28.2 | 171 | 142 |
| CA4 | 1 328 125 ± 39 640 | 1 116 072 ± 111 167 | 212 054 | ns | — | — | — |
| Subiculum | 3 964 418 ± 174 762 | 2 955 600 ± 117 177 | 1 008 818 | 0.0007 | 25.4 | 509 | 128 |
| Amygdala | 10 226 726 ± 525 740 | 5 383 510 ± 502 552 | 4 843 216 | 0.0001 | 47.4 | 2444 | 239 |
| Thalamus | 22 598 640 ± 1 156 514 | 12 268 846 ± 460 388 | 10 329 795 | 0.0001 | 45.7 | 5213 | 231 |
| Substantia nigra | 657 485 ± 59 449 | 337 728 ± 19 725 | 319 757 | 0.0001 | 48.6 | 161 | 245 |
|
Magno-cellular basal complex |
922 362 ± 52 143 | 983 291 ± 41 654 | 0 | ns | — | — | — |
| Caudate nucleus | 32 346 117 ± 2 177 370 | 32 722 049 ± 2 900 707 | 0 | ns | — | — | — |
| Purkinje cells | 13 068 122 ± 603 632 | 13 109 496 ± 803 390 | 0 | ns | — | — | — |
| Dentate nucleus | 1 087 720 ± 42 823 | 578 193 ± 45 776 | 509 527 | 0.0001 | 46.8 | 257 | 236 |
Estimated neuronal loss in FAST stages 3–4. Percentage of neuronal loss estimated in comparison to control subjects. The rate of neuronal loss estimated as number of lost neurons per day and per million neurons/day in subjects diagnosed in FAST stages 3–4.
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; FAST, Functional Assessment Staging; ns, no statistically significant neuronal loss.
TABLE 4.
Number of Neurons in AD Subjects FAST Stages 3–4 and 5–6 and Estimated Loss of Neurons in FAST Stages 5–6
| Structure/Subdivision | Mean Number of Neurons ± SE |
Neuronal Loss in FAST Stages 5–6 |
|||||
|---|---|---|---|---|---|---|---|
| AD Subjects FAST 3–4 | AD Subjects FAST 5–6 | Number | p < | Loss (%) | Loss/Day | Loss/Million Neurons/Day | |
| Entorhinal c. (All layers) | 7 477 533 ± 588 796 | 5 659 375 ± 558 432 | 1 818 158 | 0.047 | 18.7 | 2255 | 302 |
| Entorhinal c. (Islands) | 516 637 ± 53 472 | 285 111 ± 29 133 | 231 526 | 0.002 | 34.4 | 287 | 556 |
| CA1 | 7 755 632 ± 604 472 | 4 729 351 ± 560 656 | 3 026 281 | 0.003 | 29.5 | 3753 | 484 |
| CA2 | 1 080 495 ± 119 308 | 1 008 257 ± 89 234 | 72 238 | ns | 4.6 | — | — |
| CA3 | 862 857 ± 89 455 | 816 355 ± 49 942 | 46 502 | ns | 3.9 | — | — |
| CA4 | 1 116 072 ± 111 167 | 981 907 ± 71 632 | 134 165 | ns | — | — | — |
| Subiculum | 2 955 600 ± 117 177 | 2 350 660 ± 116 370 | 604 940 | 0.003 | 15.2 | 750 | 254 |
| Amygdala | 5 383 510 ± 502 552 | 5 013 979 ± 305 068 | 369 531 | ns | 3.6 | 458 | 85 |
| Thalamus | 12 268 846 ± 460 388 | 9 186 080 ± 419 578 | 3 082 766 | 0.0002 | 13.6 | 3823 | 312 |
| Substantia nigra | 337 728 ± 19 725 | 333 152 ± 24 338 | 4576 | ns | — | — | — |
|
Magno-cellular basal complex |
983 291 ± 41 654 | 633 343 ± 47 129 | 349 948 | 0.0001 | 35.6 | 434 | 441 |
| Caudate nucleus | 32 722 049 ± 2 900 707 | 32 558 855 ± 1 880 536 | 0 | (ns) | — | — | — |
| Purkinje cells | 13 109 496 ± 803 390 | 10 217 518 ± 1 346 464 | 2 891 978 | (ns) | — | — | — |
| Dentate nucleus | 578 193 ± 45 776 | 529 793 ± 35 930 | 48 400 | (ns) | — | — | — |
Percentage of neuronal loss estimated in comparison to control group. The rate of neuronal loss estimated as number of lost neurons per day and number/million neurons/day in FAST stages 5–6.
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; FAST, Functional Assessment Staging; ns, no statistically significant neuronal loss.
TABLE 5.
Number of Neurons in AD Subjects FAST Stages 5–6 and 7 and Estimated Neuronal Loss in FAST Stage 7
| Structure/Subdivision | Mean Number of Neurons ± SE |
Neuronal Loss in FAST 7 |
|||||
|---|---|---|---|---|---|---|---|
| AD Subjects FAST 5-6 |
AD Subjects FAST 7 | Number | p < | Loss % | Loss/Day | Loss/Million Neurons/Day | |
|
Entorhinal c. (All layers) |
5 659 375 ± 558 432 | 3 545 875 ± 429 688 | 2 113 500 | 0.010 | 21.7 | 334 | 61 |
| Entorhinal c. (Islands) | 285 111 ± 29 133 | 109 499 ± 23 877 | 175 612 | 0.0004 | 26.1 | 29 | 100 |
| CA1 | 4 729 351 ± 560 656 | 2 236 355 ± 503 577 | 2 492 996 | 0.005 | 24.3 | 405 | 86 |
| CA2 | 1 008 257 ± 89 234 | 879 922 ± 74 304 | 128 335 | ns | 8.2 | — | — |
| CA3 | 816 355 ± 49 942 | 816 611 ± 79 005 | 0 | ns | — | — | — |
| CA4 | 981 907 ± 71 632 | 888 282 ± 92 711 | 93 625 | ns | — | — | — |
| Subiculum | 2 350 660 ± 116 370 | 1 762 900 ± 178 799 | 587 760 | 0.011 | 14.8 | 96 | 41 |
| Amygdala | 5 013 979 ± 305 068 | 4 348 692 ± 279 369 | 665 287 | ns | 6.5 | — | — |
| Thalamus | 9 186 080 ± 419 578 | 7 329 607 ± 335 924 | 1 856 473 | 0.004 | 8.2 | 302 | 33 |
| Substantia nigra | 333 152 ± 24 338 | 245 924 ± 33 436 | 87 228 | ns | — | — | — |
|
Magno-cellular basal complex |
633 343 ± 47 129 | 549 995 ± 63 304 | 83 348 | ns | — | — | — |
| Caudate nucleus | 32 558 855 ± 1 880 536 | 27 376 562 ± 2 645 305 | 5 182 293 | ns | — | — | — |
| Purkinje cells | 10 217 518 ± 1 346 464 | 10 267 816 ± 648 072 | 0 | ns | — | — | — |
| Dentate nucleus | 529 793 ± 35 930 | 522 343 ± 40 897 | 7450 | ns | — | — | — |
Percentage of neuronal loss estimated in comparison to control group. The rate of neuronal loss estimated as number of lost neurons per day and number/million neurons/day in FAST stage 7.
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; FAST, Functional Assessment Staging; ns, no statistically significant neuronal loss.
Region-specific decline in the number of neurons in the 3 stages of AD was estimated as the difference in comparison to age-matched control subjects from 69 to 102 years of age. The pattern of staging and duration of AD-related neuronal loss is similar in small structures (less than 1.6 million neurons) and large structures (up to 40 million neurons) with an accentuated, more prominent decline in MCI/mild AD.
Stage-Specific Percentage of Neuronal Loss
Structure- and stage-specific neuronal loss in FAST stages 3–4 was estimated as the difference between the number of neurons in the control group and FAST stages 3–4; in FAST stages 5–6, as the difference between FAST stages 3–4 and FAST stages 5–6; and in FAST stage 7, as the difference between FAST stages 5–6 and FAST stage 7 (Tables 3–5).
Neuronal loss in FAST stages 3–4 is characterized by a rather high and uniform (22.9%–31.0%) loss of neurons in the EC and hippocampal formation subdivisions, but much more severe neuronal loss in the amygdala (47.4%) and thalamus (45.7%) (Fig. 4A). The pattern of neuronal loss changes in FAST stages 5–6 and FAST stage 7, with a shift toward an extremely broad range of regional differences, a generally lower percentage of neuronal loss with an especially low percentage of neuronal loss in FAST stage 7 in the amygdala (6.5%), thalamus and CA2 (8.2% each), and undetectable neuronal loss in CA3 (Fig. 4B, C).
Estimation of cumulative neuronal loss during the 19 years of AD duration (Fig. 4D) reveals that neuronal loss varies from 32.1% in CA3 to 83.8% in the islands of the stellate neurons in the EC. This 2.6-fold difference reflects structure-specific patterns of neuronal loss resulting in disorganization of neuronal interactions within each structure, between connected structures, and within neuronal circuits. An example of the disruption in the neuronal network within 1 anatomical subdivision is a 78% loss of neurons in CA1 but only a 43.8% loss in CA2 and 32.1% loss in CA3.
An estimation of the average neuronal loss in each of the 3 stages of AD reveals that the percentage of neuronal loss in MCI/moderate AD (FAST stages 3–4) (31% on average) is approximately twice that in moderate/moderately severe AD (FAST 5–6; 15.5%) and severe AD (FAST stage 7; 13.8%) (Fig. 4E).
Stage-Specific Speed of Neuronal Loss in AD
The rate of neuronal loss calculated per million neurons is an index of neuronal loss independent of the number of neurons in an individual brain structure and stage duration (Tables 3–5). Time-dependent estimates of neuronal loss per day detect dynamics of neuronal loss in different structures and stages of AD. The estimation of the rate of neuronal loss was limited to 6 structures with significant neuronal loss in each stage of AD. In FAST stages 3–4, the loss ranges from 115 to 128 neurons/million/day in the EC, including islands of stellate neurons, CA1, and subiculum, but the rate of neuronal loss is almost doubled in the amygdala (239 neurons/million/day) and thalamus (231 neurons/million/day) (Fig. 5A). The characteristic modification of the dynamics of neuronal loss in FAST stages 5–6 is a significant nonuniform reduction in the rate of neuronal loss to 85 neurons/million/day in the amygdala, but an almost 5-fold acceleration of neuronal loss (to 556 neurons/million/day) in the islands in the EC, a 3.9-fold increase (to 484 neurons/million/day) in CA1, and a more than 2-fold increase of the rate of neuronal loss in all layers in the EC (to 302 neurons/million/day) and subiculum (to 254 neurons/million/day) (Fig. 5B). The opposite direction of changes characterizes FAST stage 7, with a several-fold reduction in the rate of neuronal loss in all 6 regions, reflecting a terminal exhaustion of the neuronal pool (Fig. 5C).
Average and cumulative rates of neuronal loss demonstrate a general pattern of stage- and structure-specific modification of the dynamics of neuronal loss (Fig. 5D, E). The estimated average rate of neuronal loss increases from 159 neurons/million/day in FAST stages 3–4 to 332 neurons/million/day (ceiling level) in FAST stages 5–6 and decreases to 64 neurons/million/day in FAST stage 7. The cumulative rate of neuronal loss estimated in all 6 structures increases from 952 in FAST stages 3–4 to 1993 neurons/million/day in FAST stages 5–6 but decreases 6.2-fold to 321 neurons/million/day in FAST stage 7.
Neurofibrillary Degeneration
To define brain region- and stage of AD-specific relationships between neurofibrillary degeneration and neuronal loss, the number and percentage of neurons with NFTs were estimated (Table 6). Among 12 regions examined, only cerebellar Purkinje cells were free of tau pathology. In all 11 NFT-positive structures, the percentage of neurons with NFTs increases with the FAST stage but in a structure-specific pattern (Fig. 6A). Global staging of neurofibrillary degeneration is characterized by an increase in the average percentage of neurons with NFTs, from 7% in the control group to 13% in FAST stages 3–4, 24% in FAST stages 5–6, and 31% in FAST stage 7 (Fig. 6B). The broad range of differences in the percentage of neurons with NFTs reflects a brain region- and neuron type-specific susceptibility to abnormal tau hyperphosphorylation and neurofibrillary degeneration. A several-fold-higher percentage of neurons with NFTs in the CA1 than in CA2, CA3, and CA4 indicates that even within the same structure, the susceptibility of neurons to neurofibrillary degeneration is region-specific. Region- and stage-dependent differences in the dynamics and time of neurofibrillary degeneration and the loss of neurons with NFTs are illustrated by structure-specific loss of NFTs in FAST stage 7 in CA1 (−49%), islands in the second layer of the EC (−47.3%), subiculum (−32.3%), CA4 (−14.4%), and all layers of the EC (−10.3%), but a further increase in the percentage of NFTs, by 27% in the amygdala and 61.6% in the thalamus (Fig. 6C, D).
TABLE 6.
Brain Region and Neuron Type Specific Number and Percentage of Neurons With Neurofibrillary Tangles in Control Subjects and in AD FAST Stages 3–4, 5–6, and 7
| Structure/Subdivision | Control (69–102 Years) |
AD FAST 3–4 |
AD FAST 5–6 |
AD FAST 7 |
||||
|---|---|---|---|---|---|---|---|---|
| Number (SE) | % | Number (SE) | % | Number (SE) | % | Number (SE) | % | |
| Entorhinal c. (all layers) | 1 137 400 (171 955) | 11.6 | 1 787 782 (141 764) | 24.4 | 1 808 946 (246 456) | 32.9 | 1 571 175 (260 081) | 43.9 |
| Entorhinal c. (islands) | 137 072 (18 454) | 21.3 | 166 243 (20 450) | 33.5 | 160 112 (26 559) | 58.01 | 89 441 (19 683) | 84.9 |
| CA1 | 1 267 690 (201 925) | 12.2 | 2 281 756 (593 078) | 30.3 | 2 615 099 (271 304) | 58.1 | 1 331 511 (316 440) | 55.1 |
| CA2 | 71 278 (15 349) | 4.4 | 106 926 (30 223) | 9.9 | 159 046 (35 648) | 15.5 | 157 116 (32 558) | 17.9 |
| CA3 | 24 600 (4757) | 2.0 | 63 284 (20 779) | 8.8 | 91 962 (23 027) | 11.2 | 100 331 (16 295) | 12.5 |
| CA4 | 60 525 (10 525) | 4.5 | 129 633 (35 220) | 11.2 | 182 610 (38 616) | 18.3 | 156 339 (32 189) | 21.0 |
| Subiculum | 216 669 (43 838) | 5.4 | 332 050 (95 041) | 11.2 | 496 519 (32 591) | 21.5 | 336 878 (30 208) | 19.5 |
| Amygdala | 904 717 (262 576) | 8.9 | 644 262 (143 074) | 12.7 | 1 404 371 (233 755) | 28.0 | 1 784 112 (253 702) | 40.2 |
| Thalamus | 13 613 (9855) | 0.1 | 201 492 (84 178) | 1.6 | 1 120 090 (168 885) | 12.3 | 1 810 456 (177 997) | 24.5 |
| Substantia nigra | 2980 (2094) | 0.5 | 8038 (1700) | 2.4 | 18 834 (3566) | 5.9 | 18 613 (2931) | 7.9 |
| Magno-cellular basal complex | 32 033 (10 756) | 3.4 | 60 287 (14 650) | 6.2 | 62 099 (14 840) | 10.4 | 58 063 (17 403) | 9.9 |
| Caudate nucleus |
3616 (3616) |
0.01 | 291 796 (197 443) | 0.9 | 140 130 (49 330) | 0.4 | 348 780 (123 775) | 1.4 |
| Purkinje cell | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dentate nucleus | 0 | 0.0 |
1104 (823) |
0.2 |
3645 (1142) |
0.7 |
3492 (1610) |
0.7 |
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; FAST, Functional Assessment Staging.
FIGURE 6.

Staging of neurofibrillary degeneration. The percentage of neurons with neurofibrillary tangles (NFTs) ranging from 0 in Purkinje cells (not shown) to 81.2% in the second layer of the entorhinal cortex (EC) in severe Alzheimer disease, illustrates a broad spectrum of regional neuronal susceptibility to neurofibrillary degeneration. Regional differences are accentuated by a several-fold-higher percentage of neurons with NFTs in the CA1 than in 3 other segments of the cornu Ammonis (graph A). Stage-specific rate of neurofibrillary degeneration is reflected in an increase in the mean percentage of neurons with NFTs from 7% in the control group to 13% in FAST stages 3–4, 24% in FAST stages 5–6, and 31% in FAST stage 7 (graph B). Opposite directions of neurofibrillary degeneration in FAST stage 7a–f are documented by regional losses of neurons with NFTs in CA1, EC islands, subiculum, CA4, and the entire EC (graph C) and a simultaneous increase in the percentage of neurons with NFTs in the amygdala and thalamus (graph D).
Stage-Specific Rate of Neuronal Loss Dependent and Independent of Neurofibrillary Degeneration
To estimate the contribution of neurofibrillary degeneration to neuronal loss in AD, the relationships between the number of neurons with NFTs and the number of lost neurons were determined in FAST stages 3–4, 5–6, and 7 (Fig. 7). The causative link between neurofibrillary degeneration and neuron death is reflected in a similar total number of neurons with NFTs and total number of lost neurons in each of the 3 FAST stage groupings of AD in structures affected early and severely by neurofibrillary degeneration, including the entire EC and the islands of stellate neurons in ECII, hippocampal formation sector CA1, and the subiculum. However, the pattern of neurofibrillary degeneration and neuronal loss in the amygdala and thalamus is strikingly different. Limited neurofibrillary degeneration is paralleled with a 3.8 times larger number of lost neurons in the amygdala and a 51 times larger number of lost neurons than the number of NFTs in the thalamus, with a restriction of this pattern to MCI and mild AD (FAST stages 3–4). Another pattern of region- and stage-related differences between neurofibrillary degeneration and other causes of neuronal loss is detected in the caudate nucleus, with a 15 times more prominent neuronal loss than neurofibrillary degeneration observed selectively in FAST stage 7, and a severe (2.89 million) loss of Purkinje cells free of neurofibrillary degeneration and limited to FAST stages 5–6. These patterns indicate that neuronal loss is driven by neurofibrillary degeneration in structures affected in the earliest stage of AD, whereas in other structures, including the amygdala, thalamus, and caudate nucleus, non-NFT pathology appears to be a major contributor to neuronal loss, with the extreme example of severe loss of Purkinje cells free of neurofibrillary degeneration.
FIGURE 7.

Estimates of neuronal loss related and unrelated to neurofibrillary degeneration. In structures affected with neurofibrillary degeneration in the earliest stage of Alzheimer disease studied, the number of neurons with neurofibrillary tangles and the number of lost neurons are almost identical (entorhinal cortex [EC]—all layers) or are similar, but with a more prominent number of lost neurons (ECII, CA1). Contribution of nonneurofibrillary degeneration to neuronal loss increases in the subiculum by 59%. Prominent enhancement of neuronal loss not related to neurofibrillary degeneration affects the amygdala and thalamus in FAST stages 3–4, caudate nucleus in FAST stage 7, and Purkinje cells in FAST stages 5–6.
Amyloid Load (%) and Volume of Amyloid Deposits (mm3)
Estimation of the average amyloid load in 13 brain structures showed an increase of amyloid load from 0.9% in 69- to 102-year-old control subjects to 2.1% in MCI/mild AD, and a plateau with 3.8% and 3.6% load in moderate and severe AD, respectively (Fig. 8). However, the type and volume of amyloid deposits vary in a broad range in the examined regions. Diffuse nonfibrillar Aβ deposits occupy a relatively high percentage of caudate nucleus volume in control subjects (2.55%) as well as in MCI/mild AD and moderate AD (6.73% and 11.0%, respectively). The cerebellar cortex, free of NFTs in Purkinje and granular cells, is free of plaques in control subjects and in MCI/mild AD, but diffuse deposits occupy 5.69% of the molecular layer volume in FAST stages 5–6 and 3.57% in FAST stage 7. In AD, the cerebellar dentate nucleus is positive for NFTs but negative for Aβ.
FIGURE 8.

Staging of amyloid load (graph A). Amyloid load in the caudate nucleus is relatively high in control subjects (2.55%) and increases faster than in other structures to 6.7% in FAST stages 3–4 and 10.9% in FAST stages 5–6. In FAST stage 7, amyloid load decreases in several structures, including the entorhinal cortex, amygdala, caudate nucleus, and the molecular layer of the cerebellar cortex. The cerebellar dentate nucleus, positive for neurofibrillary tangles, is amyloid- negative during the entire disease. In 13 brain structures, the mean amyloid load (graph B) increases from 0.9% in control subjects to 2.1% in FAST stages 3–4 and reaches a plateau with 3.8% in FAST stages 5–6 and 3.6% in FAST stage 7.
In contrast to relative estimates of amyloid load (%), the volume of amyloid deposits is a direct measure of the amount (mm3) of amyloid in structures with volume modified by AD-related atrophy (Table 7). The trajectory of changes in amyloid volume is brain structure-specific. In 6 of 14 structures examined, the volume of amyloid increases to a top level in subjects diagnosed with FAST stages 5–6, but decreases in FAST stage 7 in several structures, including the EC (from 28.8 to17.8 mm3); CA1 (from 11.6 to 8.4 mm3); subiculum (from 5.0 to 2.9 mm3); and amygdala (with a decline from 27.3 to 15.5 mm3). This pattern is also observed in structures with diffuse amyloid deposits, including the caudate nucleus (with a decline from 215.1 to 162.5 mm3) and the molecular layer of the cerebellar cortex (from 546.3 to 328.8 mm3). In other structures, the decline of amyloid volume in late AD was not detectable or marginal.
TABLE 7.
Brain Region-Specific Patterns of β-Amyloid Deposition (Amyloid Volume, mm3, Amyloid Load, %) in Aging and AD
| Control 69–102 Years |
AD FAST Stages 3–4 |
AD FAST Stages 5–6 |
AD FAST Stage 7 |
|||||
|---|---|---|---|---|---|---|---|---|
| Structure/ Subdivision | Volume mm3 | Load % | Volume mm3 | Load % | Volume mm3 | Load % | Volume mm3 | Load % |
| Entorhinal c. (All layers) | 10.66 | 1.83 | 13.10 | 2.83 | 28.83 | 6.09 | 17.86 | 4.79 |
| CA1 | 5.55 | 1.35 | 10.37 | 2.69 | 11.66 | 4.34 | 8.43 | 5.16 |
| CA2 | 0.00 | 0.00 | 0.10 | 0.26 | 0.20 | 0.52 | 0.37 | 1.32 |
| CA3 | 0.03 | 0.06 | 0.03 | 0.12 | 0.26 | 0.69 | 0.60 | 1.79 |
| CA4 | 0.26 | 0.26 | 0.35 | 0.35 | 1.23 | 1.29 | 2.90 | 3.23 |
| Subiculum | 3.15 | 1.53 | 5.08 | 3.22 | 5.04 | 4.23 | 2.94 | 3.74 |
| Amygdala | 6.56 | 1.38 | 18.84 | 4.57 | 27.35 | 7.50 | 15.55 | 4.88 |
| Thalamus | 37.99 | 1.38 | 26.58 | 1.31 | 62.32 | 2.92 | 58.62 | 3.04 |
|
Substantia nigra |
0.02 | 0.03 | 0.16 | 0.16 | 0.09 | 0.12 | 0.30 | 0.36 |
| Magno-cellular basal complex | 2.20 | 1.85 | 6.98 | 5.67 | 5.84 | 6.20 | 5.26 | 5.99 |
| Caudate nucleus | 47.63 | 2.55 | 139.15 | 6.73 | 215.11 | 11.00 | 162.5 | 9.3 |
|
Cerebellum-molecular layer |
0.00 | 0.00 | 0.00 | 0.00 | 546.36 | 5.69 | 328.83 | 3.57 |
|
Dentate nucleus |
0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Estimation of amyloid volume in the second layer of the entorhinal cortex was postponed because measurements of amyloid were not reliable in small islands of stellate neurons.
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; FAST, Functional Assessment Staging.
Prevalence and Severity of TDP-43 and Lewy Bodies Pathology in the Amygdala in AD Subjects
Among 24 examined AD cases 9 subjects (37.5%) were TDP-43-positive. Prevalence increased from 1 case in FAST 3 and 0 cases in FAST 4–5, to 8/14 (68.7%) in FAST 6 and 7. Mean percentage of TDP-43-positive neurons was 0.1 in FAST 3–4, 0.9% in FAST 5–6, and 2.3% in FAST 7. Among 22 examined subjects 15 (68%) were positive for Lewy bodies. The percentage of Lewy body-positive neurons increased from 0.06% in FAST 3–4, to 2.0% in FAST 5–6 but declined to 0.2% in FAST 7. The study revealed strong correlation between number of neurons and NFTs but no significant correlation between number of neurons and number of TDP-43- or Lewy body-positive neurons, suggesting limitations of TDP-43 degeneration and Lewy body contribution to neuronal loss in examined cohort.
Correlations Between Pathology and AD Duration in the Absence of a Correlation With Age
In the examined AD cohort, brain weight correlates with AD duration (r = −0.63; p < 0.001) as well as with Braak and Braak staging of brain neurofibrillary degeneration (Spearman rho = −0.57; p < 0.003) (6). Table 8 shows Pearson correlations for 7 structures with a consistent pattern of changes of structure volume, number of neurons, and number of neurons with NFTs. The correlations reveal a uniform pattern of significant associations between AD duration and the volume decline of all layers of the EC, ECII, CA1, subiculum, thalamus, amygdala, and magnocellular basal complex. Strong correlations (p < 0.001) were detected between AD duration and decline in the number of neurons in all these structures except the amygdala. The link between neuronal loss and decline of structure volume is reflected in a strong correlation (p < 0.001) between number of neurons and volume in all 7 regions.
TABLE 8.
Correlations Between AD Duration and Brain Structure Volume, Number of Neurons, and Number of Neurons With NFTs; Structure Volume/Number of Neurons; Number of Neurons and Number and Percentage of Neurons With NFTs
| Structure | AD Duration | AD Duration | Structure Volume | AD Duration | Neuron Number | Neuron Number |
|---|---|---|---|---|---|---|
| Structure Volume | Neuron Number | Neuron Number | NFT % | NFT Number | NFT% | |
| EC—all layers | −0.52* | −0.80*** | 0.78*** | 0.64** | 0.46* | −0.58** |
| EC—second layer | −0.85*** | −0.89*** | 0.97*** | 0.79*** | 0.57** | −0.79*** |
| CA1 | −0.72*** | −0.84*** | 0.93*** | 0.36 | 0.47* | −0.42* |
| Subiculum | −0.84*** | −0.83*** | 0.86*** | 0.52** | 0.15 | −0.48* |
| Thalamus | −0.51** | −0.87*** | 0.55*** | 0.84*** | 0.64*** | −0.76*** |
| Amygdala | −0.60** | −0.39 | 0.74*** | 0.71*** | 0.01 | −0.28 |
| Magnocellular basal complex | −0.61** | −0.77*** | 0.83*** | 0.17 | 0.13 | −0.29 |
AD, Alzheimer disease; CA1, sector 1 of the cornu Ammonis; NFT, neurofibrillary tangle.
p < 0.05;
p < 0.01;
p < 0.001.
A significant correlation between duration of AD and the percentage of neurons with NFTs reflects progression of neurofibrillary degeneration in the entire EC, and especially in ECII, subiculum, thalamus, and amygdala. Contribution of neurofibrillary degeneration to neuronal loss is reflected in a correlation between the number of NFTs and the number of neurons in the entire EC and in ECII, CA1, and the thalamus. A similar correlation is detected between the percentage of neurons with NFTs and the number of neurons in the EC, CA1, subiculum, and thalamus.
In an AD cohort with different ages of disease onset, estimates of AD-related changes, including reduction in brain weight, decline in the volume of 15 examined structures, reduction in the number of neurons, increase in the number and percentage of neurons with NFTs, and increase in amyloid load, do not correlate with age (not shown).
There is no correlation between age and amyloid load except for a weak correlation between age and amyloid load in the CA3, CA4, and substantia nigra. A weak marginal correlation was detected between AD duration and amyloid load in these 3 structures. Amyloid load does not correlate with the volume of 14 structures, the number of neurons in 15 structures, or the number of neurons with NFTs in 11 structures (not shown).
Overview of Neuropathological and Morphometric Staging of AD
A panel of micrographs of sections stained with cresyl violet and immunostained with mAb Tau-1 illustrates striking morphological differences between control subjects’ ECII, CA1, and amygdala and neuronal degeneration and loss in AD subjects diagnosed with FAST 4, 6c, and 7f (Fig. 9A). Cresyl violet staining shows not only loss of neurons but also loss of structure-specific cytoarchitecture and demonstrates abnormal size and shape of surviving neurons. Tau-1 immunostaining shows onset of neurofibrillary degeneration in control subjects, severe neurofibrillary degeneration in FAST 3–4 and 6, and reduction of NFTs in FAST 7f with exhausted neuronal population during 19 years of AD duration.
FIGURE 9.

(A) Cresyl violet (CV)-stained sections provide overview of neuronal loss, deterioration of cytoarchitecture specific for islands of the stellate neurons in the second layer of the entorhinal cortex (EC II), CA1 pyramidal layer and lateral nucleus of the amygdala reflecting AD pathology in FAST 4, 6c, and 7f compared to control subjects. Neuronal loss and neurofibrillary degeneration are associated with reduction of neuronal soma size and neuron deformation common in FAST 6c and 7f. Immunostaining of adjacent sections with mAb Tau1 demonstrates onset of neurodegeneration with sporadically detected NFTs in 3 examined regions in control subject, massive neurofibrillary degeneration with high and comparable number of NFT-positive neurons and neurofibrillary threads in FAST 4 and 6c, but striking decline in the number of neurons, NFTs and neurofibrillary threads in final stage of AD (FAST 7f). (B) Coronal sections of the body of the hippocampal formation immunostained with mouse monoclonal antibody Tau-1 reveal that neurofibrillary degeneration affects the subiculum, CA1 and 2 sector, less CA3 sector and much less the CA4 sector in mild AD (FAST 3 stage) and that pathology reaches the highest level in moderate AD (FAST 5) in all segments including CA3 and CA4. In a very advanced stage of severe AD (FAST 7d), striking reduction of immunoreactivity reflects severe loss of neurons with NFTs in the subiculum and all sectors of the cornu Ammonis associated with prominent atrophy of all hippocampal subdivisions affected by neurofibrillary degeneration.
Three low-magnification micrographs of the hippocampus body characterize pattern of progression of pathology in mild, moderate, and severe AD (Fig. 9B). They reflect results of morphometric studies showing a surprisingly advanced neurofibrillary degeneration in mild AD (FAST 3), progression of neurofibrillary degeneration to a ceiling level in moderate AD (FAST 5), and exhaustion of neuronal population paralleled with a striking reduction of the number of neurons with NFTs and severe atrophy in all sectors of the cornu Ammonis and subicular complex in severe AD (FAST7d). The panel of box plots outlines main features of clinicopathological staging of AD in subjects diagnosed with MCI/mild AD (FAST 3–4), moderate/moderately severe AD (FAST 5–6), and severe AD (FAST 7a–f) compared to age-matched control subjects (Fig. 10). The estimates of the decline of structure volume and number of neurons, and increase of the percentage of volume loss and increase of the percentage of neurons with NFTs reflect the range of structural deterioration in the selected 5 structures, including the EC, CA1, subiculum, amygdala, and thalamus, in each stage of AD. Staging reveals surprisingly severe decline in the number of neurons and the highest percentage of neuronal loss as an integral feature of MCI/mild AD. The ceiling level of neuronal loss in moderate/moderately severe AD, especially prominent in the amygdala and the thalamus, reflects a critical point of exhaustion of the pool of neurons susceptible to AD degeneration and death, resulting in a decline in the rate of neuronal loss to the lowest level in FAST 7. Both neuropathological and morphometric staging indicate that the most effective treatment and prevention of progression might be expected in the earliest stages of AD (MCI/mild AD).
FIGURE 10.

The panel of box plots characterizes structure and FAST stage-specific patterns of volume loss, decline in the number of neurons, percentage of neuronal loss and percentage of neurons with NFTs in the entorhinal cortex (EC), CA1, subiculum (Sub), amygdala (Amyg), and thalamus (Th). Strikingly severe reduction of the number of neurons and high percentage of neuronal loss, especially in the subiculum, amygdala, and thalamus, distinguishes MCI/mild AD (FAST 3–4). The rate of neuronal loss decreases in the amygdala and thalamus in FAST 5–6 and 7. A floor level in the number of neurons and arrest of further neuronal loss is typical for FAST 7. The marker of exhaustion of neuronal population in late AD is a reduction of neuronal loss as well as a reduction of the number of neurons with NFTs in the entorhinal cortex, CA1, and subiculum, and reduced progression of neurofibrillary degeneration in other structures in FAST 7. In box plots, the median value is marked with a horizontal line within the box. The upper and lower boundaries of each box represent the interquartile range (IQR), whereas the whisker above and below the box marks the maximum and minimum values unless any data point lies more than 1.5 times the IQR above the 75th percentile or below the 25th percentile. A few records above the 75th or below the 25th percentile (outliers) are marked with dots.
DISCUSSION
No Correlation Between Sporadic AD Pathology and Age
This study reveals correlations between AD duration and structure volume, percentage and rate of neuronal loss, and neurofibrillary degeneration in the memory system, subcortical structures, and cerebellum, but no correlation between AD pathology and age. Figure 1 suggests that the absence of correlations of pathological changes with age in these nonselected consecutive 25 cases of sporadic AD is determined by age at onset of AD and the affected subject survival time/AD stage at death. The age at onset ranges from 55.6 to 93.5 years of age. A mortality rate growing with age results in reduction of survival from an average 15.5 years for the youngest subjects with AD to an average of 6.3 years in the oldest AD subjects. Brookmeyer et al (35) reported a median life span ranging from 7 to 10 years for those diagnosed in their 60s or 70s, while the median life span was 3 years or less for individuals diagnosed at more than 90 years of age. A reversed pattern, with the longest survival and the most advanced stage of AD at death in younger individuals, and the shortest duration of AD and early stages of AD in the oldest individuals, explains the lack of correlation between age and pathology, including neuronal loss. However, the pattern of pathology in sporadic AD is fundamentally different from that in a Down syndrome (DS) cohort, with onset of β-amyloidosis and neurofibrillary degeneration appearing in all trisomic subjects at around 40 years of age (36, 37), diagnosis of dementia at a mean age of 55 years (38), and an average lifespan of 55–60 years (39, 40). The relationship between age and AD in the DS cohort is supported by postmortem quantitative studies demonstrating the correlation between age and pathology, including neuronal loss (41). Absence of a correlation between age and severity of AD pathology detected in this sporadic AD cohort appears to be associated with the prevalence of advanced stages of AD in younger AD subjects with longer survival time and a combination of late onset of AD, short survival time, and death in earlier stages of AD in the oldest AD group.
MCI/Mild AD as a Target for Successful Treatment
Elderly individuals with signs of MCI are at a high risk of developing dementia (42, 43). MCI has been defined to distinguish the abnormal clinical state prodromal to dementia (44) and to identify patients destined to develop AD as candidates for early intervention to slow or stop the progression of pathology and functional deterioration (45).
Previous postmortem neuropathological studies of MCI cases revealed changes typical for AD, including neurofibrillary degeneration and amyloid plaques. In patients with MCI, the number of neuritic plaques was significantly elevated in 4 neocortical regions and in the amygdala compared to controls, whereas the number of NFTs was increased in the parietal cortex and in 4 ventromedial temporal lobe structures (46). Morphometric studies revealed significant region-specific neurodegeneration and neuronal loss in MCI. A Gomez-Isla et al (9) study limited to the EC revealed no changes in the number of neurons in 60- to 90-year-old cognitively normal subjects (CDR = 0), but a 32% reduction of the number of neurons in the EC and a 60% loss of neurons in ECII in the brains of subjects with MCI (CDR = 0.5). von Gunten et al revealed 1.9% neuronal loss in the EC and 26% in CA1 sector in a questionable dementia stage (CDR = 0.5) (17).
Our study expands the definition of MCI/mild AD pathology with evidence of significant neuronal loss in the EC (22.9%), CA1 (24.2%), subiculum (25.4%), amygdala (47.4%), thalamus (45.7%), substantia nigra (48.6%), and dentate nucleus (46.8%). This pattern indicates that MCI/mild AD-related neuronal loss is not a continuum of age-associated changes, but instead a multiregional AD pathology with significant neurodegeneration/neuronal loss and functional deficits. The detected MCI/mild AD-associated higher rate of neuronal loss/million/day in the CA1, subiculum, and thalamus compared to FAST stages 7a–f shows that in this early stage of AD with relatively limited functional decline, the speed of neuronal loss is already higher than in the overtly symptomatic severe AD subjects.
Multiregional neuronal loss is evidence of the advanced neurodegeneration/neuronal loss in progress during 9 years of MCI/mild AD that may contribute to the functional deficits detected in clinical studies. A Kluger et al study (47) of 87 subjects with MCI at baseline revealed a 67.8% decline to dementia at a 4-year follow-up, corresponding to a 17.8% per-year rate of decline to dementia. MCI revealed, in addition to classical cognitive deficits, distinctive complaints of cognitive impairment (48), behavioral disturbances (49), alterations of neurological reflexes (50), emerging motor, balance, and coordination deficits (51), deficits in activities of daily life (52), and hippocampal region atrophy (53).
The changes detected in clinical, neuropsychological, functional, and neuroimaging profiles of patients with MCI and significant multiregional neurodegeneration and neuronal loss reflect a transition of some aging subjects to AD. Treatments applied during MCI may be able to stop pathology while disease is in an early transitional stage (54). The combination of the detected pattern of neuronal loss and functional deficits in FAST stages 3–4 rationalizes the strategies of a majority of clinical trials focused on prevention of AD pathology and treatment of people diagnosed with MCI and mild AD (3).
Link Between Loss of Neurons in the Memory System and Dementia in AD
Many early morphometric studies concentrated on detection of the type, topography, and severity of changes explaining memory loss in AD. Usually they were limited to structures characterizing the earliest Alzheimer-type pathology, including EC and ECII, and the hippocampal formation or CA1. In spite of the different cohorts examined and methods of tissue processing and quantitative examination, several studies revealed a comparable estimated percentage of lost neurons in the CA1: 68% [13], 86% [10], 75% [12], 60% [11], and 78.2% cumulative neuronal loss in severe AD in our study. Neuronal loss in ECII was estimated as 90% [9] and 83.8% in this study.
A von Gunten et al study (17) of ECII using CDR revealed 1.9% neuronal loss in CDR 0.5 (questionable dementia), 46.8% cumulative neuronal loss detected in CDR 1, 52.6% in CDR 2, and 91.6% in CDR 3. Cumulative neuronal loss in the CA1 sector was estimated as 26% in CDR 0.5, 33% in CDR 1, 35% in CDR 2, and 75% in CDR 3. In our study, the estimated cumulative neuronal loss in ECII was 83.8%, and in the CA1 78.2%.
Distinct Dynamics of Neuronal Loss in Different Clinical Stages of AD
In contrast to studies of the cumulative effects of AD neurodegeneration in unspecified stages, or the cumulative effect detected in the brain of subjects diagnosed with different clinical stages of AD, part of this study was designed to estimate neuronal degeneration and the percentage and rate of neuronal loss separately in each of 3 major stages of AD, including FAST 3–4, 5–6, and 7. Pathology was characterized in 14 brain regions to test the hypothesis that dynamics of neuronal loss are stage-specific and that patterns of changes are structure-specific. Two parameters were applied to estimate different aspects of neuronal loss: the percentage of lost neurons in each of 3 stages of AD and the rate of neuronal loss per million neurons per day. While the percentage of neuronal loss depends on the duration of the stage, the rate of neuronal loss approximates the speed of neuronal loss regardless of the size of the neuronal population in small or large structures and is independent of the duration of the stage of AD.
The average neuronal loss (neurons/million/day) in all layers of the EC, island of stellate neurons in ECII, sector CA1, subiculum, amygdala, and thalamus increases 9.2 times from 15 in control subjects 69–102 years of age to 139 neurons in subjects diagnosed with MCI/mild AD (FAST stages 3–4). A further 3.9 times increase in the rate of neuronal loss in moderately severe AD (FAST 5–6) to 545 neurons reflects the ceiling level and critical point of neuronal death in AD. The reduction in the rate of neuronal loss in severe AD (FAST 7a–f) to 195 neurons/million/day appears to be a result of exceeding a “critical” point of neuronal death corresponding to exhaustion of the neuronal population to a level when further increase of the rate of neuronal loss is impossible.
Survival Time of Neurons With NFTs
Predictions that a neuron with neurofibrillary degeneration survives approximately 20 years and that NFTs may not be obligatory for death of CA1 neurons are challenged by estimates of the number of neurons with NFTs and the number of neurons lost in each of the 3 groups of FAST stages of AD (55). Staging of neurofibrillary degeneration and neuronal loss reveals that the number of neurons with NFTs and the number of lost neurons are very similar in each of 3 groups of FAST stages of AD in all layers of the EC, ECII, and CA1 studied herein. These data indicate that neurons that developed neurofibrillary degeneration during the 9 years of FAST stages 3–4 died in this stage of AD. A similar number of neurons with NFTs and number of lost neurons during the 4 years of FAST stages 5–6 and during the 7 years of FAST stage 7 indicate that in these 3 structures, neurofibrillary degeneration is the main mechanism leading to neuron death. This pattern is consistent with our regression-based estimates of 3.4-year survival of NFT-positive neurons in CA1 and 5.4-year survival in the subiculum of individuals with AD (56) as well as 4.7-year survival in the amygdala of DS/AD subjects (41).
Regional Differences Between Magnitude of Neurofibrillary Degeneration-Dependent and -Independent Neuronal Loss
In contrast to the similar number of lost neurons and neurons with NFTs in the EC, ECII, and CA1, the subiculum neuronal loss (2.2 million) exceeds the number of NFTs (1.38 million) by 59%. The prevalence of neuronal loss caused by mechanisms other than neurofibrillary degeneration is a dominant feature in a majority of examined brain regions, and the ratio between NFTs and neuronal loss is stage-dependent. The number of lost neurons exceeds 3.8 times the number of neurons with NFTs in the amygdala, 51 times in the thalamus in FAST stages 3–4, and 15 times in the caudate nucleus in FAST stage 7. Severe loss of Purkinje cells (2.89 million) known to be free of neurofibrillary degeneration is observed in FAST stages 5–6. The detected patterns indicate that neuronal loss is driven by neurofibrillary degeneration in structures affected in the earliest stage of AD, whereas in other structures, including the amygdala, thalamus, and caudate nucleus, non-NFT pathology appears to be the major, or in the case of Purkinje cells, the only, contributor to neuronal death. More severe neuronal loss in comparison to the number of neurons with NFTs detected in this and other studies (11, 16, 17) indicates that in AD, more than 1 mechanism is involved in neuronal loss. Our estimates of neuronal loss in aging without dementia (18) revealed a comparable rate of neuronal loss per million neurons/day in structures with neurofibrillary degeneration, including the EC (−14), EC islands (−26), CA1 (−12), and amygdala (−21), and in structures free of neurofibrillary degeneration including the thalamus (−18), caudate nucleus (−15), Purkinje cells (−23), and dentate nucleus (−18). Similarities in topography of neuronal loss without neurofibrillary degeneration in aging and in the AD cohort suggest that mechanisms of nonneurofibrillary degeneration/death are prominent in all adults/elderly with and without AD.
Potential Contribution of TDP-43 and Lewy Body Pathology to Structural and Functional Changes in AD
TAR DNA-binding protein of 43 kDa (TDP-43) acting as a transcriptional repressor is binding both RNA and DNA and modulates gene splicing (57). TDP-43 pathology with TDP-43 cytoplasmic and nuclear filamentous inclusions containing posttranslationally modified protein (58) is considered the primary factor in frontotemporal lobar degeneration (FTLD-TDP) and amyotrophic lateral sclerosis (primary TDP-43 proteinopathies) and the secondary factor in other neurodegenerative disorders, including AD (59). Screening of TDP-43 pathology was focused on the amygdala, known as the first brain structure affected with this degeneration and one of the most affected in subjects with progressing AD pathology (60, 61). Josephs et al (61) classified cases without TDP-43 immunoreactivity in the amygdala as TDP-43-negative and cases with any amount of TDP-43 immunoreactivity in the amygdala as TDP-43-positive. In their cohort, 57% of cases with AD pathology were TDP-43-positive and TDP pathology was associated with advanced Braak stages, a higher proportion of Lewy bodies and hippocampal sclerosis. Extension of TDP-43 degeneration beyond the amygdala is associated with increasing likelihood of clinical expression of AD dementia (59, 62, 63). In AD, more severe TDP-43 pathology is associated with enhanced regional atrophy including hippocampal atrophy (63). However, a low percentage of neurons with signs of TDP-43 degeneration (0.1% in FAST 3–4, 0.9% in FAST 5–6, and 2.3% in FAST 7), a low percentage of neurons with Lewy bodies (0.06% in FAST 3–4, 2.5% in FAST 5–6, and 1.7% in FAST 7), and no significant correlation between the percentage of neurons with TDP-43 inclusions and Lewy bodies and duration of AD suggest a limited contribution of these proteinopathies to neuronal loss and functional deterioration in the examined AD cohort. The contrast between the low percentage of TDP-43 and Lewy body affected neurons and the increasing percentage of neurons with NFTs in FAST 3–4 (12.7%), FAST 5–6 (28.0%), and FAST 7 (40.2%) reflects a leading role of neurofibrillary degeneration in neuronal loss and functional decline in the examined AD cohort.
Contribution of Senescence to Neuronal Loss in Aging and AD
Mechanisms of neuronal loss in aging subjects in the absence of β-amyloidosis and neurofibrillary degeneration as well as in early stages of AD are not clear, but growing evidence suggests a contribution from senescence. The number of senescent cells increases late in life in normal tissues and especially in tissues affected by age-related changes. Cellular senescence is characterized by a durable cell cycle arrest. Despite a permanent cessation of neuron division, some neurons reenter the cell cycle, and this reentry results in neuron death. Busser et al (64) proposed that dysregulation of various components of the cell cycle is a significant contributor to regionally specific neuronal death in AD.
Although there is no single marker of senescence, the most common features of senescence detected in cortical and hippocampal neurons and Purkinje cells include (1) activation of lysosomal enzyme senescence-associated β-galactosidase (SA-β-gal); (2) persistent DNA damage response detected by γH2AX foci; (3) expression of tumor suppressors, such as p21 CIP1/WAF1 (encoded by Cdkn1α) and p16 INK4A (encoded by Cdkn2α), and p53-dependent release of alarmin HMGB1 as a mediator of senescent phenotypes (65); and (4) lipofuscin accumulation and nuclear envelope protein degradation (66–68).
Several studies have revealed a link between the appearance of cell cycle-related proteins, neuronal loss, and cognitive impairment in AD subjects (64, 69–71). Yang et al (72) reported higher density of cell cycle-positive neurons in MCI than in the next stages of AD and suggested that cell cycle-induced neuronal death and the rate of neuronal loss are high in all stages of AD. This study reveals regional patterns of increase in neurofibrillary degeneration independent of neuronal loss in MCI in the subiculum, amygdala, and thalamus and a very strong increase in the caudate nucleus in FAST stage 7 and Purkinje cells in FAST stages 5–6. The link between neuron reentry in the cell cycle and neurofibrillary degeneration suggests region- and stage-specific interactions between mechanisms of neurofibrillary degeneration and mechanisms of senescence-related neuronal death (72, 73). This assumption is supported by results of experimental clearance of senescent glial cells with senolytics, which ameliorate secretome-related pathology and prevent tau-dependent pathology and cognitive decline (74). Another supportive argument is application of Dasatinib and Quercetin to eliminate senescent cells in a mouse model of AD, which reduces brain injury and inflammation and slows the pace of memory loss (75).
β-Amyloidosis
Some pathological aspects of AD pathology, especially β-amyloidosis, are not detectable as linear correlations of amyloid volume and stage/duration of AD, but both β-amyloidosis and neurofibrillary degeneration contribute to structural brain deterioration and cognitive impairment (2). The increase of Aβ load early after the onset of clinical symptoms and the absence of a substantial increase during clinical progression of AD were reported by Serrano-Pozo et al (76, 77). In this study, average amyloid load in the EC, hippocampus, subcortical nuclei, and cerebellum was estimated as 2.1% in MCI/mild AD (FAST stages 3–4). The amyloidosis reached a plateau with a 3.8% amyloid load in subjects diagnosed with moderate/moderately severe AD (FAST stages 5–6) and 3.6% in severe AD. However, estimates of amyloidosis do not predict cognitive status in AD (78, 79). This study also demonstrates an absence of correlations between amyloid load and the majority of examined parameters, including age and duration of AD, number of all neurons and of neurons with NFTs, and volume of examined structures.
Amyloid load is the function of a dynamic cascade of events leading to Aβ fibrillization in cored plaques by microglial cells and amyloid core degradation by astrocytes leading to plaque disintegration (80–82). Experimental depletion of microglial cells with CSF1R inhibitor confirms a key role of microglia in fibrillar plaque formation in transgenic mice models of AD and reveals mechanisms preventing/restricting amyloid plaque formation (83, 84). Neuronal loss and a corresponding focal reduction of the Aβ pool, and fluctuation in the number of uniquely activated microglial cells and astrocytes in plaques are associated with permanent formation/degradation of individual plaques, but changes in the volume of Aβ and amyloid load are relatively inert and a poor correlate of neurodegeneration and functional deterioration. However, these methodological limitations do not diminish the substantial role of massive amyloidosis in AD.
This study of neuronal loss and degeneration including β-amyloidosis highlights the relationship between neuronal loss and decline of amyloid load. Numerous reports postulated that amyloid deposition in plaques is associated with neurons (85–88). Ubiquitous, chemically and structurally diversified fibrillar and nonfibrillar Aβ-deposits are formed at the terminal fields of axonal projections, including the perforant pathway, with projections of stellate neurons of ECII to the molecular layer of the hippocampal dentate gyrus. The primary role of neuronal projections in fibrillar and nonfibrillar amyloid deposition is documented by fibrillar plaque formation in the molecular layer of the dentate gyrus in the human hippocampus and diffuse Aβ deposition in the same area in several species of animals (88). One of the final effects of neuronal loss is a decline of Aβ in projection areas limiting the extracellular pool of Aβ and diffuse or fibrillar amyloid deposit formation. Neuronal loss is a direct cause of the decrease in amyloid load in structures innervated by a degenerating/dying neuronal population. The high brain region-specific percentage of lost neurons in MCI/mild AD (FAST stages 3–4) and the ceiling level of the rate of neuronal loss in moderate/moderately severe AD (FAST stages 5–6) detected in this study are measures of the reduction in the number of neurons and are predictors of a proportional reduction of the pool of extracellular neuronal origin Aβ available for oligomerization, fibrillization, and deposition in plaques. It suggests that neuronal loss might be a critical factor for decline/arrest of new amyloid plaque formation in FAST stages 5–6 and 7 in AD as well as in aging subjects with DS/AD (88). Reduced volume of amyloid in the majority of examined structures in FAST 7 is not a sign of recovery but instead reflects a reduction in the pool of Aβ of neuronal origin due to a ceiling in the rate of neuronal loss in FAST stages 5–6 and the decline in the number of neurons to a floor level in severe AD.
Study Limitations
A very narrow racial and ethnic profile of the examined cohort represented mainly by white individuals eliminates the possibility of a study of the contribution of racial and ethnic factors to the reported patterns of structural changes and functional decline.
This and other morphometric studies of AD-associated neuronal loss concentrate on the contribution to neuronal loss of immunocytochemically detectable and measurable neurofibrillary degeneration, TDP-43 inclusions and Lewy bodies. This research design may underestimate the contribution of soluble toxic oligomeric proteins to structural alterations including neuronal loss. Significant loss of Purkinje cells detected in aging subjects in absence of neurofibrillary degeneration and amyloid deposits and in subjects diagnosed with AD in absence of neurofibrillary degeneration but in presence of significant load of diffuse amyloid deposits suggests that in AD, other mechanisms contribute to neuronal loss in parallel to those reported in this study (18).
Recent studies reveal a significant role of coincident pathologies commonly observed within AD populations. They recommend expansion of standard postmortem neuropathological evaluations with estimates of not only TDP-43 proteinopathy and Lewy bodies but also amyloid angiopathy, common in AD and considered an additional potent cause of cognitive impairment within AD population (89). Lack of estimates of an association between the almost ubiquitous but topographically variable amyloid angiopathy and multiregional but structure-specific rate of neuronal loss limits insight of this report in a potentially significant component of AD pathology.
Closing Remarks
The report relies on a rather limited sample of cases representing FAST stages 3–4, 5–6, and 7. The attempt of modeling of clinicopathological staging of AD pathology is still sort of a panoramic overview of the process in 25 subjects diagnosed with AD selected using a set of inclusion and exclusion criteria to exclude comorbid conditions and non-AD pathology and to characterize “pure” AD. However, the examined cohort still reflects a low prevalence of TDP-43 and Lewy body pathology, affecting a strikingly marginal percentage of neurons in comparison to common and severe neurofibrillary degeneration defining the mainstream of AD neuronal loss and functional decline.
A similar ratio between the number of neurons with NFTs and neuronal loss in the ECII and CA1 suggests that in structures affected in the earliest stages of AD, neuronal loss is driven by neurofibrillary degeneration, whereas in the amygdala, thalamus, caudate nucleus, and in the case of Purkinje cells, non-NFT pathology—most likely, senescence—appears to be a significant contributor to neuronal death. The highest percentage of neuronal loss in MCI/mild AD, the highest rate of neuronal loss in the moderate/moderately severe stages of AD, and the plateau/decrease in amyloid load in late stages of AD suggest that reduction in neuronal number and the pool of neuronal origin Aβ contribute to reduction in the volume (mm3) and load (%) of amyloid deposits. Confirmation that microglial cells are a critical factor in amyloid deposition in plaques and demonstration that CSF1R inhibitors reduce the number of microglia and prevent/reduce amyloid plaque formation reveal mechanisms for therapeutic control of brain amyloidosis.
Development of treatments reducing neurofibrillary degeneration- and senescence-related neuronal loss, prevention of brain amyloidosis with microglial cell CSF1R inhibitors, and application of these treatments in MCI/mild AD may prevent/slow down senescence and AD-related mechanisms of neuronal loss, brain amyloidosis, and corresponding functional decline.
Supplementary Material
ACKNOWLEDGMENTS
Brain tissue of AD subjects and some brains of control subjects were provided by the Aging and Dementia Research Center at the New York University Langone Medical Center. Most control brains were obtained from the Brain and Tissue Bank for Developmental Disabilities and Aging at the NYS Institute for Basic Research in Developmental Disabilities, and the Brain and Tissue Bank for Developmental Disorders of the National Institute of Child Health and Human Development at the University of Maryland. The authors thank Dr. Humi Imaki for her contribution to morphometric studies, Mrs. Maureen Marlow for manuscript editing, and Mrs. Jadwiga Wegiel and Mrs. En Wu Zheng for histology and immunocytochemistry.
The study was supported in part by funds from the New York State Office for People with Developmental Disabilities and grants from the NIH National Institute of Child Health and Human Development (R01 HD43960 to JW) and National Institute on Aging (P30AG066512 and AG060882 to TW, ML, BR, JW).
The authors have no conflicts of interest to declare.
Supplementary Data can be found at academic.oup.com/jnen.
REFERENCES
- 1. Gaugler J, James B, Johnson T, et al. Alzheimer’s disease facts and figures 2013. Alzheimer’s Disease Association; 2013. Available at: http://www.alz.org/downloads/factsfigures_2013.pdf
- 2. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol 2012;71:362–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wisniewski T, Goni F.. Immunotherapeutic approaches for Alzheimer disease. Biochem Pharmacol 2014;88:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hyman BT, Van Hoesen GW, Damasio AR, et al. Alzheimer’s disease: Cell specific pathology isolates the hippocampal formation. Science 1984;225:1168–70 [DOI] [PubMed] [Google Scholar]
- 5. Hyman BT, Van Hoesen GW, Kromer LJ, et al. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann Neurol 1986;20:472–81 [DOI] [PubMed] [Google Scholar]
- 6. Braak H, Braak E.. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59 [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E.. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:351–7 [DOI] [PubMed] [Google Scholar]
- 8. Rössler M, Zarski R, Bohl J, et al. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer’s disease. Acta Neuropathol 2002;103:363–9 [DOI] [PubMed] [Google Scholar]
- 9. Gomez-Isla T, Price JL, McKeel DW Jr, et al. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996;16:4491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bobinski M, Wegiel J, Tarnawski M, et al. Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer’s disease. J Neuropathol Exp Neurol 1997;56:414–20 [DOI] [PubMed] [Google Scholar]
- 11. Kril JJ, Patel S, Harding AJ, et al. Neuron loss from the hippocampus of Alzheimer’s disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol 2002;103:370–6 [DOI] [PubMed] [Google Scholar]
- 12. von Gunten A, Kövari E, Rivara CB, et al. Stereologic analysis of hippocampal Alzheimer’s disease pathology in the oldest-old: Evidence for sparing of the entorhinal cortex and CA1 field. Exp Neurol 2005;193:198–206 [DOI] [PubMed] [Google Scholar]
- 13. West MJ, Coleman PD, Flood DG, et al. Differences in the pattern of hippocampal neuronal loss in normal aging and Alzheimer's disease. Lancet 1994;344:769–72 [DOI] [PubMed] [Google Scholar]
- 14. Reisberg B, Ferris SH, de Leon MJ, et al. The Functional Assessment Staging (FAST). Psychopharmacol Bull 1988;24:653–9 [PubMed] [Google Scholar]
- 15. Reisberg B, Franssen E, Bobinski M, et al. Overview of methodologic issues for pharmacologic trials in mild, moderate, and severe Alzheimer’s disease. Int Psychogeriatr 1996;8:159–93 [DOI] [PubMed] [Google Scholar]
- 16. Gomez-Isla T, Hollister R, West M, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer disease. Ann Neurol 1997;41:17–24 [DOI] [PubMed] [Google Scholar]
- 17. von Gunten A, Kovari E, Bussiere T, et al. Cognitive impact of neuronal pathology in the entorhinal cortex and CA1 field in Alzheimer’s disease. Neurobiol Aging 2006;27:270–7 [DOI] [PubMed] [Google Scholar]
- 18. Wegiel J, Flory M, Kuchna I, et al. Multiregional age-associated reduction of brain neuronal reserve without association with neurofibrillary degeneration or β-amyloidosis. J Neuropathol Exp Neurol 2017;76:439–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Folstein MF, Folstein SE, McHugh PR.. The Mini-Mental State; a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–98 [DOI] [PubMed] [Google Scholar]
- 20. Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–72 [DOI] [PubMed] [Google Scholar]
- 21. Reisberg B, Franssen E, Shah MA, et al. Clinical diagnosis of dementia: A review In: Maj M, Sartorus N, eds. Dementia. West Sussex, UK: John Wiley and Sons Ltd; 2000:69–115 [Google Scholar]
- 22. Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 1986;83:4913–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated τ precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res 1989;477:90–9 [DOI] [PubMed] [Google Scholar]
- 24. Kim KS, Miller DL, Sapienza VJ, et al. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun 1988;2:121–30 [Google Scholar]
- 25. Amaral DG, Insausti R.. Hippocampal formation In: Paxinos G, ed. The Human Nervous System. San Diego, CA: Academic Press Inc; 1990:711–55 [Google Scholar]
- 26. Lorente de No R. Studies on the structure of the cerebral cortex. II. Continuation of the study of the Ammonic system. J Psychol Neurol 1934;46:113–77 [Google Scholar]
- 27. Rosene DL, van Hoesen GW.. The hippocampal formation of the primate brain: A review of some comparative aspects of cytoarchitecture and connections In: Jones EG, Peters A, eds. Cerebral Cortex, Vol. 6 New York, NY: Plenum; 1987:345–456 [Google Scholar]
- 28. Duvernoy HM. The Human Hippocampus. An Atlas of Applied Anatomy. Munich, Germany: J.F. Bergmann-Verlag München; 1988 [Google Scholar]
- 29. Schumann CM, Amaral DG.. Stereological analysis of amygdala neuron number in autism. J Neurosci 2006;26:7674–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fearnley JM, Lees AJ.. Aging and Parkinson's disease: Substantia nigra regional selectivity. Brain 1991;114:2283–301 [DOI] [PubMed] [Google Scholar]
- 31. Fearnley JM, Lees AJ.. Pathology of Parkinson's disease In: Calne DB, ed. Neurodegenerative Diseases. Philadelphia, PA: Saunders Company; 1993:545–54 [Google Scholar]
- 32. Vogels OJ, Broere CA, ter Laak HJ, et al. Cell loss and shrinkage in the nucleus basalis Meynerti complex in Alzheimer's disease. Neurobiol Aging 1990;11:3–13 [DOI] [PubMed] [Google Scholar]
- 33. West MJ, Gundersen HJ.. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 1990;296:1–22 [DOI] [PubMed] [Google Scholar]
- 34. Gould W. Stata Statistical Software. Release 16. College Station, TX: Stata Corp LLC; 2019. [Google Scholar]
- 35. Brookmeyer R, Corrada MM, Curriero RC, et al. Survival following a diagnosis of Alzheimer’s disease. Arch Neurol 2002;59:1764–7 [DOI] [PubMed] [Google Scholar]
- 36. Mann DM, Esiri MM.. The pattern of acquisition of plaques and tangles in the brain of patients under 50 years of age with Dow’s syndrome. J Neurol Sci 1989;89:169–79 [DOI] [PubMed] [Google Scholar]
- 37. Wisniewski KE, Wisniewski HM, Wen GY.. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 1985;17:278–82 [DOI] [PubMed] [Google Scholar]
- 38. McCarron M, McCallion P, Reilly E, et al. A prospective 14-years longitudinal follow-up of dementia in persons with Down syndrome. J Intellect Disabil Res 2014;58:61–70 [DOI] [PubMed] [Google Scholar]
- 39. Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement 2015;11:700–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zigman WB. Atypical aging in Down syndrome. Dev Disabil Res Rev 2013;18:51–67 [DOI] [PubMed] [Google Scholar]
- 41. Wegiel J, Wisniewski HM, Morys J, et al. Neuronal loss and β-amyloid removal in the amygdala of people with Down syndrome. Neurobiol Aging 1999;20:259–69 [DOI] [PubMed] [Google Scholar]
- 42. Amieva H, Letenneur L, Dartigues JF, et al. Annual rate and predictors of conversion to dementia in subjects presenting mild cognitive impairment criteria defined according to a population-based study. Dement Geriatr Cogn Disord 2004;18:87–93 [DOI] [PubMed] [Google Scholar]
- 43. Yaffe K, Petersen RC, Lindquist K, et al. Subtype of mild cognitive impairment and progression to dementia and death. Dement Geriatr Cogn Disord 2006;22:312–9 [DOI] [PubMed] [Google Scholar]
- 44. Petersen RC, Knopman DS.. MCI is a clinically useful concept. Int Psychogeriatr 2006;18:394–402 [DOI] [PubMed] [Google Scholar]
- 45. Jicha GA, Parisi JE, Dickson DW, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol 2006;63:674–81 [DOI] [PubMed] [Google Scholar]
- 46. Markesbery WR, Schmitt FA, Kryscio RJ, et al. Neuropathologic substrate of mild cognitive impairment. Arch Neurol 2006;63:38–46 [DOI] [PubMed] [Google Scholar]
- 47. Kluger A, Ferris SH, Golomb J, et al. Neuropsychological prediction of decline to dementia in nondemented elderly. J Geriatr Psychiatry Neurol 1999;12:168–79 [DOI] [PubMed] [Google Scholar]
- 48. Reisberg B, Ferris SH, Anand R, et al. Clinical assessment of cognitive decline in normal aging and primary degenerative dementia: Concordant ordinal measures In Pichot P, Berner P, Wolf R, Thau K, eds. Psychiatry, Vol. 5 New York, NY: Plenum Press; 1985:333–8 [Google Scholar]
- 49. Reisberg B, Franssen E, Sclan SG, et al. Stage specific incidence of potentially remediable behavioral symptoms in aging and Alzheimer’s disease: A study of 120 patients using the BEHAVE-AD. Bull Clin Neurosci 1989;54:95–112 [Google Scholar]
- 50. Franssen EH, Reisberg B, Kluger A, et al. Cognition independent neurologic symptoms in normal aging and probable Alzheimer’s disease. Arch Neurol 1991;48:148–54 [DOI] [PubMed] [Google Scholar]
- 51. Franssen EH, Somen LEM, Torossian CL, et al. Equilibrium and limb coordination in mild cognitive impairment and mild Alzheimer’s disease. J Am Geriatr Soc 1999;47:463–99 [DOI] [PubMed] [Google Scholar]
- 52. Reisberg B, Finkel S, Overall S, et al. The Alzheimer’s disease Activities of Daily Living International Scale (ADL-IS). Int Psychogeriatr 2001;13:163–81 [DOI] [PubMed] [Google Scholar]
- 53. de Leon MJ, Golomb J, George AE, et al. The radiologic prediction of Alzheimer’s disease: The atrophic hippocampal formation. AJNR Am J Neuroradiol 1993;14:897–906. [PMC free article] [PubMed] [Google Scholar]
- 54. Grundman M, Petersen RC, Ferris SH, et al. Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch Neurol 2004;61:59–66 [DOI] [PubMed] [Google Scholar]
- 55. Morsch R, Simon W, Coleman PD.. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 1999;58:188–97 [DOI] [PubMed] [Google Scholar]
- 56. Bobinski M, Wegiel J, Tarnawski M, et al. Duration of neurofibrillary changes in the hippocampal pyramidal neurons. Brain Res 1998;799:156–8 [DOI] [PubMed] [Google Scholar]
- 57. Buratti E, Baralle FE.. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of vCFTR exon 9. J Biol Chem 2001;276:36337–43 [DOI] [PubMed] [Google Scholar]
- 58. Igaz LM, Kwong LK, Xu Y, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 2008;173:182–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wilson AC, Dugger BN, Dickson DW, Wang D-S.. TDP-43 in aging and Alzheimer’s disease – a review. Int J Clin Exp Pathol 2011;4:147–55 [PMC free article] [PubMed] [Google Scholar]
- 60. Hu WT, Josephs KA, Knopman DS, et al. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 2008;116:215–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Josephs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 2014;127:811–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. James BD, Wilson RS, Boyle PA, et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s type dementia. Brain 2016;139:2983–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Josephs KA, Whitwell JL, Knopman DS, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008;70:1850–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Busser J, Geldmacher DS, Herrup K.. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci 1998;18:2801–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Davalos AR, Kawahara M, Malhotra GK, et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescence phenotypes. J Cell Biol 2013;201:613–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Geng YQ, Guan JT, Xu XH, Fu YC.. Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem Biophys Res Commun 2010;396:866–9 [DOI] [PubMed] [Google Scholar]
- 67. Jurk D, Wang C, Miwa S, et al. Postmitotoc neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012;11:996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moreno-Blas D, Gorostieta Salas E, Pommer-Alba A, et al. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019;11:6175–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Golde TE, Miller VM.. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res Ther 2009;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McShea A, Harris PL, Webster KR, et al. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol 1997;150:1933–9 [PMC free article] [PubMed] [Google Scholar]
- 71. Vincent J, Jicha G, Rosado M, et al. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons in Alzheimer’s disease brain. J Neurosci 1997;17:3588–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang Y, Mufson EJ, Herrup K.. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci 2003;23:2557–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018;17:e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bussian TJ, Aziz A, Meyer CF, et al. Clearance of senescent glial cells prevents taudependent pathology and cognitive decline. Nature 2018;562:578–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 2019;22:719–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Serrano-Pozo A, Mielke ML, Gomez-Isla T, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol 2011;179:1373–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Serrano-Pozo A, Mielke ML, Muzitansky A, et al. Stable size distribution of amyloid plaques over the course of Alzheimer disease. J Neuropathol Exp Neurol 2012;71:694–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Arriagada PV, Marzloff K, Hyman BT.. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992;42:1681–8 [DOI] [PubMed] [Google Scholar]
- 79. Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003;60:1495–500 [DOI] [PubMed] [Google Scholar]
- 80. Wegiel J, Wang K-C, Tarnawski M, et al. Microglial cells are the driving force in fibrillar plaque formation whereas astrocytes are a leading factor in plaque degradation. Acta Neuropathol 2000;100:356–64 [DOI] [PubMed] [Google Scholar]
- 81. Wegiel J, Wang K-C, Imaki H, et al. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APPsw mice. Neurobiol Aging 2001;22:49–61 [DOI] [PubMed] [Google Scholar]
- 82. Wegiel J, Imaki H, Wang K-C, et al. Origin and turnover of microglial cells in fibrillar plaques of APPsw transgenic mice. Acta Neuropathol 2003;105:393–402 [DOI] [PubMed] [Google Scholar]
- 83. Sosna J, Philipp S, Albay IIR, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener 2018;13:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Spangenberg E, Severson PL, Hohsfield LA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nature Commun 2019;10:3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cork LC, Masters C, Beyreuther K, et al. Development of senile plaques. Relationships of neuronal abnormalities and amyloid deposits. Am J Pathol 1990;137:1383–92 [PMC free article] [PubMed] [Google Scholar]
- 86. Pappolla MA, Omar RA, Vinters HV.. Image analysis microspectroscopy shows that neurons participate in the genesis of a subset of early primitive (diffuse) senile plaques. Am J Pathol 1991;39:599–607 [PMC free article] [PubMed] [Google Scholar]
- 87. Probst A, Langui D, Ipsen S, et al. Deposition of beta/A4 protein along plasma membrane in diffuse senile plaques. Acta Neuropathol 1991;83:21–9 [DOI] [PubMed] [Google Scholar]
- 88. Wegiel J, Wisniewski HM.. Projections of neurons in neuritic plaque formation. NeuroSci News 1999;2:34–9 [Google Scholar]
- 89. Thomas DX, Bajaj S, McRAE-McKee K, et al. Association of TDP-43 proteinopathy, cerebral amyloid angiopathy, and Lewy bodies with cognitive impairment in individuals with or without Alzheimer’s disease neuropathology. Sci Rep 2020;10:14579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.