

Abstract
Mutations of epigenetic regulators are pervasive in human tumors. ASXL1 is frequently mutated in myeloid malignancies. We previously found that ASXL1 forms together with BAP1 a complex that can deubiquitinylate mono-ubiquitinylated lysine 119 on histone H2A (H2AK119ub1), a Polycomb repressive mark. However, a complete mechanistic understanding of ASXL1 in transcriptional regulation and tumor suppression remains to be defined. Here, we find that depletion of Asxl1 confers murine 32D cells to IL3-independent growth at least partly due to sustained activation of PI3K/AKT signaling. Consistently, Asxl1 is critical for the transcriptional activation of Pten, a key negative regulator of AKT activity. Then we confirm that Asxl1 is specifically enriched and required for H2AK119 deubiquitylation at the Pten promoter. Interestingly, ASXL1 and PTEN expression levels are positively correlated in human blood cells and ASXL1 mutations are associated with lower expression levels of PTEN in human myeloid malignancies. Furthermore, malignant cells with ASXL1 downregulation or mutations exhibit higher sensitivity to the AKT inhibitor MK2206. Collectively, this study has linked the PTEN/AKT signaling axis to deregulated epigenetic changes in myeloid malignancies. It also provides a rationale for mechanism-based therapy for patients with ASXL1 mutations.
Keywords: tumor suppressor, Polycomb, H2A ubiquitylation, PTEN, AKT
Introduction
Polycomb group (PcG) proteins are highly conserved epigenetic regulators, which are essential for the maintenance of gene silencing (Simon and Kingston, 2009; Di Croce and Helin, 2013). These proteins form at least two well-characterized complexes, namely Polycomb repressive complex 2 (PRC2) and PRC1. Biochemically, PRC1 catalyzes mono-ubiquitinylation of H2AK119 (H2AK119ub1), while PRC2 catalyzes tri-methylation of histone H3 Lys 27 (H3K27me3) (Muller and Verrijzer, 2009; Simon and Kingston, 2009). Additional sex combs (Asx) was initially identified in Drosophila as a member of PcG genes as its mutants showed a posterior transformation phenotype (Simon et al., 1992), typical of compromised PcG functions. However, Asx mutants also exhibited anterior homoeotic transformation similar to the loss of Trithorax group (TrxG) functions. These phenotypes led to the hypothesis that Asx plays dual roles in both silencing and activation of Hox genes by balancing PcG and TrxG proteins (Sinclair et al., 1998; Fisher et al., 2010). Through biochemical complex purification, Scheuermann et al. (2010) found that Asx interacts with Calypso, a ubiquitin carboxy-terminal hydrolase, and they specifically remove mono-ubiquitinylation from H2A. Surprisingly, the deregulated H2A deubiquitylation in the Drosophila mutants lacking Asx or Calypso was correlated with derepression of PcG-targeted Hox genes. Therefore, this complex was named Polycomb repressive deubiquitinase complex (PR-DUB) (Scheuermann et al., 2010). Interestingly, we have previously identified a similar complex composed of ASX-like (ASXL) and BAP1 (human homologue of Calypso) with specific H2A deubiquitylation activity in humans (Wu et al., 2015).
Additional sex combs-like 1 (ASXL1) is one of the three mammalian homologs of the Drosophila Asx. Frequent somatic ASXL1 alterations have been reported in patients with myeloid malignancies, including 11%–21% of myelodysplastic syndrome (MDS), 43%–58% of chronic myelomonocytic leukemia (CMML), and 5%–25% of acute myeloid leukemia (AML) (Gelsi-Boyer et al., 2009; Boultwood et al., 2010; Inoue et al., 2013). Moreover, ASXL1 mutations have been repeatedly identified to be associated with adverse prognosis of MDS and AML patients (Gelsi-Boyer et al., 2012; Itzykson et al., 2013). Recently, two independent groups have reported that the constitutive or conditional deletion of Asxl1 in the hematopoietic system in mice leads to the development of MDS-like defects, including dysplastic neutrophils and cytopenias, which may transform into myeloid leukemia with age (Abdel-Wahab et al., 2013; Wang et al., 2014). Using genetic models, Asxl1 loss collaborates with oncogenic NRasG12D mutation (Abdel-Wahab et al., 2012; Wu et al., 2015) or haploinsufficiency of Nf1 (Zhang et al., 2018b) in bone marrow cells to induce myeloid malignancies. These studies suggest that ASXL1 mutations may function as an early/initiating event in the development of myeloid malignancies, and additional genetic events may cooperate with Asxl1 loss to induce leukemia. However, the exact molecular mechanisms for ASXL1 mutations in myeloid transformation remain to be elucidated.
MDS, a ‘preleukemia’ stage, generally represents a failure of cellular differentiation but progresses to AML when additional genetic and epigenetic events provide growth advantages (Itzykson and Fenaux, 2014). To address how Asxl1 loss promotes myeloid transformation, we took advantage of 32D cells, an immortalized myeloblast-like cell line originally derived from long-term cultures of murine bone marrow and strictly dependent on cytokine IL3 for the undifferentiated state and cell cycle progression (Greenberger et al., 1983). We found that Asxl1 downregulation in 32D cells confers IL3 independent growth, which may be at least partly due to the sustained activation of PI3K/AKT signaling. Mechanistically, we revealed that Asxl1 binds and deubiquitylates H2AK119 at the Pten promoter and therefore is required for the transcriptional activation of Pten. Finally, we showed that ASXL1-downregulated or mutated malignant myeloid cells are more sensitive to AKT inhibitor. This study provides a novel mechanistic insight into how ASXL1 loss promotes myeloid transformation and suggests a potential therapeutic strategy for myeloid malignancies with ASXL1 mutations.
Results
Asxl1 loss confers IL3-independent growth of 32D cells
To gain insight into the roles of ASXL1 loss in myeloid transformation, we tried to investigate how Asxl1 downregulation affects the proliferation of myeloid progenitor 32D cells. We generated two Asxl1-knockdown (KD) 32D cell lines (Asxl1 sh_A and Asxl1 sh_B) by stably expressing two short hairpin RNAs (shRNAs). The knockdown efficiency for Asxl1 was confirmed by quantitative real-time RT-PCR (RT-qPCR) analysis and western blotting (WB) assays (Supplementary Figure S1A; Figure 1A). The proliferation of the control (Scr sh) and Asxl1-KD cells cultured with or without IL3 was assessed by the MTS assay. As expected, the control cells completely stop growing after 24 h withdrawal of IL3. However, we found that Asxl1-KD cells are able to grow in the absence of IL3 at a comparable rate to the control cells cultured with IL3 (Figure 1B). To further confirm this effect, we tried to knock out Asxl1 in 32D cells through CRISPR/Cas9 techniques. Among the generated mutant lines, we chose a line with homozygous frameshift mutation leading to a premature stop code just adjoining to the ASXN domain coding region (Asxl1−/−) (Figure 1C). WB assay confirmed that the protein expression of Asxl1 is completely abrogated in Asxl1−/− cells compared with the control (wild-type, WT) (Figure 1D). As shown by the cell growth curve, Asxl1−/− 32D cells can be maintained without IL3 though they grow a little bit slower than WT cells cultured with IL3 (Figure 1E). We also examined the effects of Asxl1 depletion on cell apoptosis and cell cycle progression using flow cytometry. As shown in Supplementary Figure S2A and B, IL3 withdrawal for 24 h causes a mass of cell apoptosis and G1 arrest in WT cells. However, these effects are attenuated in Asxl1−/− cells, though the Asxl1−/− cells showed partial G2 arrest even in the presence of IL3. These data suggest that the adequate depletion of Asxl1 can bypass IL3 withdrawal-induced cell death, indicative of loss-of-function mutations of ASXL1 in myeloid malignancies.
Figure 1.
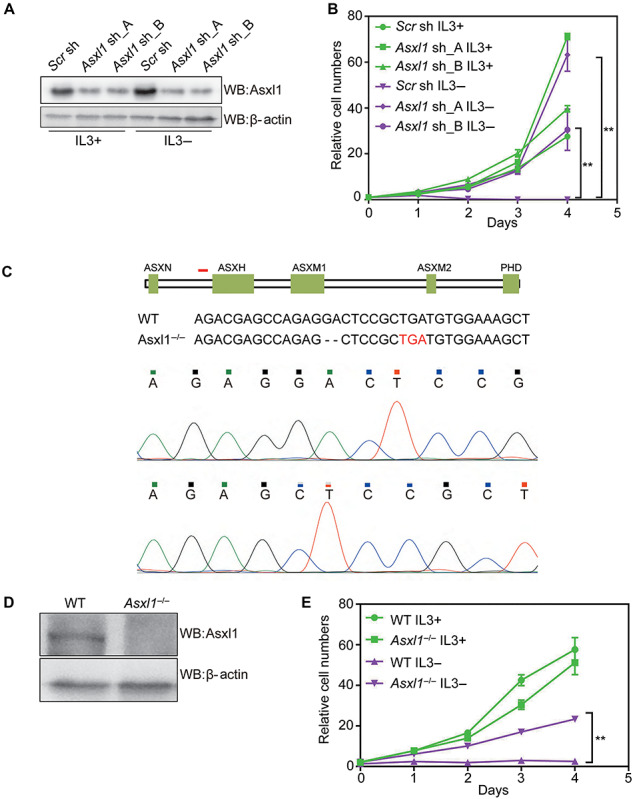
Depletion of Asxl1 confers IL3-independent growth of 32D cells. (A) WB analysis of Asxl1 in control (Scr sh) and Asxl1-KD 32D cells (Asxl1 sh_A and Asxl1 sh_B) in the presence or absence of IL3. (B) Cell growth curves of 32D cells expressing the indicated shRNAs under culture conditions with or without IL3. MTS assay was employed at the specified time points to follow the rate of proliferation. (C) The schematic showing nucleotide deletion of Asxl1 by the CRISPR/Cas9 method. The short red line marks the location of designed sgRNA. The premature stop codon is highlighted in red in the below sequences. Sequencing peaks around the sgRNA docking site is shown at the bottom. (D) WB analysis of total protein lysates of WT and Asxl1-KO 32D cells. β-actin served as a loading control. (E) Cell growth curves of WT and Asxl1-KO 32D cells under culture conditions with or without IL3. Significance level was determined using Student’s two-sided t-tests. The error bars denote SD, n = 3; **P ≤ 0.01.
Asxl1 depletion results in sustained activation of PI3K/AKT signaling pathway in the absence of IL3
Three oncogenic signaling pathways, PI3K/AKT, JAK/STAT3, and MAPK/ERK, have been reported to promote the cellular growth and clonogenicity downstream of cytokine IL3 (Ekman et al., 2000; Reddy et al., 2000; Kandasamy et al., 2010; Yuzugullu et al., 2016). Hence, we wondered whether Asxl1 loss confers activation of these signaling pathways in the absence of IL3. Taking advantage of the above Asxl1-KD (Asxl1 sh_A) or KO (Asxl1−/−) 32D cells, we compared the phosphorylation levels of AKT (p-AKT 473), STAT3 (p-STAT3 705) and ERK1/2 (p-ERK1/2) with control cells cultured in the presence or absence of IL3. As shown in Figure 2A and B, there was a dramatic decrease of p-AKT 473 and p-STAT3 705 and a slight decrease of p-ERK1/2 levels when IL3 was withdrawn in control or WT cells, but significant levels of p-AKT 473 but not p-STAT3 705 and p-ERK1/2 were sustained in Asxl1-KD or KO cells even without IL3. Consistently, the phosphorylation levels of S6k1, a well-known effector of AKT, is also upregulated upon depletion of Asxl1. These data indicate that the activation of the PI3K/AKT signaling pathway induced by the downregulation of Asxl1 is at least partly responsible for the cell survival in the deprivation of IL3.
Figure 2.
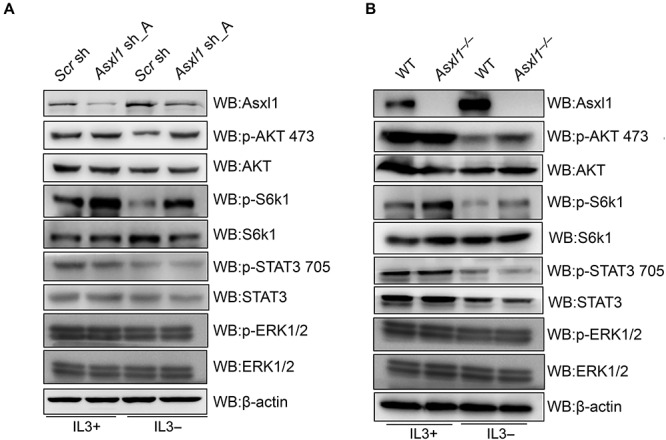
Loss of Asxl1 induces sustained activation of AKT in the absence of IL3. (A and B) WB analysis of Asxl1, p-AKT 473, AKT, p-S6k1, S6k1, p-STAT3 705, STAT3, p-ERK1/2, and ERK1/2 levels in control and Asxl1-KD (Asxl1 sh_A) cells (A) or in WT and Asxl1-KO 32D cells (B) under culture conditions with or without IL3 for 48 h. β-actin served as a loading control.
Asxl1 is essential for the transcriptional activation of Pten
We next sought to delineate mechanisms for the sustained AKT activation in Asxl1-downregulated cells. The tumor suppressor PTEN is the main negative regulator of the PI3K/AKT signaling pathway. Loss of PTEN leads to a constitutive AKT activity (p-AKT), which is frequently found in human cancer including hematological malignancies (Gutierrez et al., 2009). Similarly, PTEN depletion was found to allow Ba/F3 cells, a murine IL3-dependent pro-B cell line, to modestly grow in culture medium lacking IL3 (Yuzugullu et al., 2016). Hence, it is likely that Pten inactivation accounts for the hyperactive AKT and IL3-independent growth of 32D cells induced by Asxl1 loss. To determine whether Pten is a primary responsive gene to the absence of IL3, we detected the mRNA levels of Pten after 24 and 48 h of IL3 withdrawal by RT-qPCR analysis. As shown in Figure 3A, a gradual increase of Pten mRNA levels was observed after prolonged removal of IL3. Then we investigated the effect of Asxl1 deletion on Pten expression. Though Pten mRNA or protein levels are not affected by Asxl1 deletion in the presence of IL3, the Pten induction by IL3 withdrawal for 24 h is significantly inhibited (Figure 3B and C). Similar results were also observed in Asxl1-KD cells (Supplementary Figure S3). Therefore, Pten inactivation upon loss of Asxl1 may be responsible for the IL3-independent transformation of 32D cells.
Figure 3.
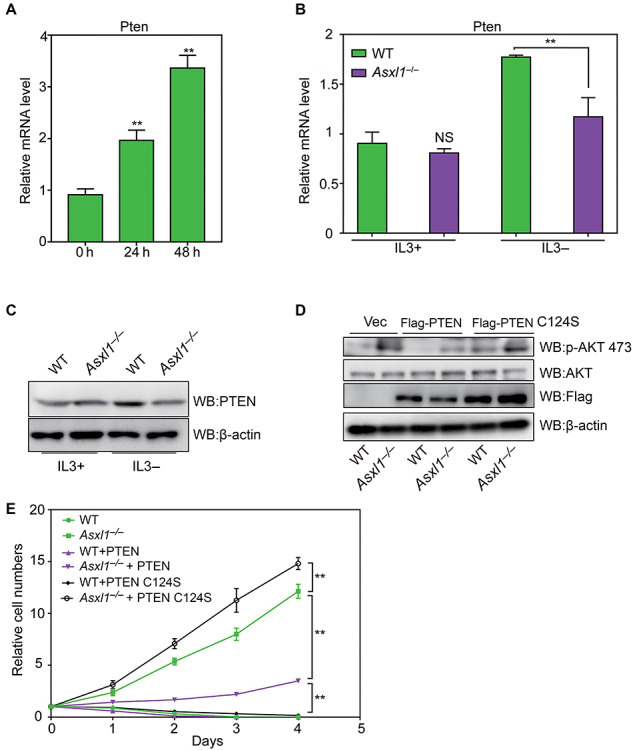
Pten inactivation due to Asxl1 loss contributes to IL3-independed growth of 32D cells. (A) RT-qPCR analysis of relative mRNA levels of Pten after IL3 withdrawal at the specified time points. (B and C) Expression levels of Pten in the presence or absence of IL3 for 24 h were determined by RT-qPCR (B) and WB (C) analyses. (D) WB analysis of ectopic expression of Flag-tagged PTEN and PTEN C124S as well as p-AKT 473 and total AKT. (E) Cell growth curves of designated groups of 32D cells after withdrawal of IL3 at the specified time points. Significance level was determined using Student’s two-sided t-tests. The error bars denote SD, n = 3; **P ≤ 0.01, NS indicates not significant.
To further confirm the causal role of Pten inactivation in transformation, we ectopically expressed Flag-tagged PTEN-WT or PTEN C124S, a dominant negative mutant (Papa et al., 2014) in Asxl1−/− 32D cells to assess their effects on cell survival under IL3-deficient condition and performed WB assay to confirm their expression levels and the effects on AKT activation (Figure 3D). We found that PTEN-WT overexpression strongly inhibits but not abolishes the IL3-independent growth induced by Asxl1 loss. In contrast, PTEN C124S overexpressing cells show a significantly elevated cell growth rate compared to Asxl1−/− cells (Figure 3E). These data suggest that Pten lies downstream of Asxl1 to antagonize cellular transformation.
Asxl1 is required for H2AK119 deubiquitylation at the Pten locus
Then we proceeded to investigate how Asxl1 loss contributes to Pten inactivation. As shown in Figure 4A, IL3 withdrawal leads to a significant and rapid increase in Asxl1 expression levels, but not Bap1 expression levels. Accordingly, IL3 absence for 24 h results in a dramatic decrease of global H2AK119ub1 levels, in parallel with a modest decrease of H3K27me3 levels. Moreover, H2AK119ub1 loss is largely rescued by Asxl1 deletion (Figure 4B), indicative of a crucial role of Asxl1 in H2A deubiquitylation and transcriptional regulation. To further prove that the transcriptional activation of Pten after IL3 withdrawal depends on ASXL1-mediated H2AK119 deubiquitylation, we overexpressed Asxl1-WT or Asxl1 d400 (deletion of 1–400 amino acids at the N-terminus, loss of Bap1 binding and thereby loss of deubiquitylation activity) in the absence of IL3. As shown in Supplementary Figure S4A, overexpressed Asxl1-WT but not Asxl1 d400 mutant significantly restores the Pten expression in Asxl KO cells. These data suggest that Asxl1-induced Pten expression relies on the interaction with Bap1 and H2AK119 deubiquitylation activity. Hence, we would like to find out whether Pten induction by IL3 withdrawal requires locus-specific H2AK119 deubiquitylation by Asxl1.
Figure 4.
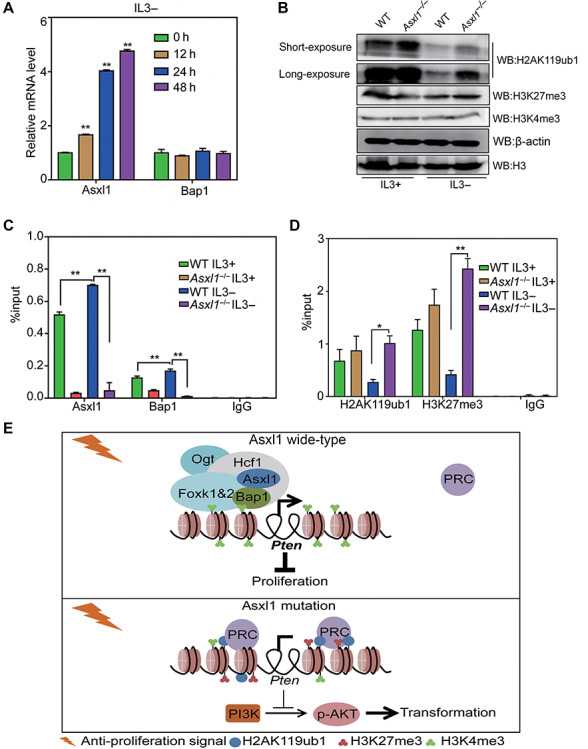
Asxl1 is required for H2AK119 deubiquitylation at the Pten promoter. (A) RT-qPCR analysis of relative mRNA levels of Asxl1 and Bap1 after IL3 withdrawal at the indicated time points. (B) WB analysis of H2AK119ub1, H3K27me3, and H3K4me3 in WT and Asxl1-KO 32D cells under culture conditions with or without IL3. H3 and β-actin were used as loading controls. (C and D) ChIP-qPCR analysis of Asxl1 and Bap1 (C) as well as H2AK119ub1 and H3K27me3 (D) in WT and Asxl1−/− 32D cells under culture conditions with or without IL3. IgG was used as a negative control. Significance level was determined using Student’s two-sided t-tests. The error bars denote SD, n = 3; *P ≤ 0.05, **P ≤ 0.01. (E) The model for the role of Asxl1 in regulation of the Pten promoter. Anti-proliferative signals like IL3 deprivation lead to enrichment of Asxl1 and Bap1 at the Pten promoter, accompanied with H2AK119 deubiquitylation and decreased H3K27me3 levels for Pten activation, thereby inhibiting cell proliferation. Asxl1 loss leads to insufficiency of H2AK119 deubiquitylation and thereby maintains Pten silencing, which allows sustained activation of AKT signaling for cellular transformation.
First we queried previously published chromatin immunoprecipitation-sequencing (ChIP-seq) data for Asxl1 (Zhang et al., 2018a). As shown by the track in Supplementary Figure S4B, Asxl1 binds to the Pten promoter in mouse bone marrow stromal cells, accompanied with the enrichment of RNA polymerase II (PolII). Then we performed ChIP-qPCR analyses in 32D cells to validate whether Asxl1 and Bap1 are bound to the Pten promoter and examined the accompanying changes of histone modifications in different conditions. As shown in Figure 4C and D, Asxl1 and Bap1 are specifically bound to the Pten promoter, and their enrichment is further induced by the absence of IL3 in WT cells. Accordingly decreased levels of H2AK119ub1 and H3K27me3 at the locus are observed upon IL3 withdrawal. In contrast, the deubiquitylase complex is dissociated and the repressive histone marks remain unaffected even at the absence of IL3 in Asxl1−/− cells. Therefore, Asxl1 and Bap1 bind to and deubiquitylate H2AK119 at the Pten promoter in response to the cytokine deprivation.
Based on the above findings, we raise a model of deregulated Asxl1 function in myeloid transformation (Figure 4E). In normal conditions, the cell growth of myeloid progenitors is under strict control. Upon anti-proliferation signals, Asxl1 cooperates with Bap1 and specifically deubiquitylates H2AK119 at the promoters of tumor suppressor genes for their activation, thereby leading to cell growth retardation. However, the loss of Asxl1 results in retention of H2AK119ub1 and PRC on tumor suppressor genes for the maintenance of their silencing state and therefore cell survival under an unfavorable extracellular environment. In this context, the inactive Pten after loss of Asxl1 allows the constitutive activation of AKT and ultimately cell proliferation independent of growth factors.
Conserved ASXL1–PTEN/AKT axis in humans
Next, we tried to investigate if these mechanisms are conserved in human hematopoietic cells. We first analyzed public database to investigate the correlation between ASXL1 and PTEN expression in human hematopoietic cells. Notably, we find a remarkable positive correlation between ASXL1 expression and PTEN expression by analyzing the published expression profiling data (GSE37642) from bone marrow mononuclear cells of AML patients (Figure 5A) and GTEx dataset for normal whole blood cells (Figure 5B).
Figure 5.
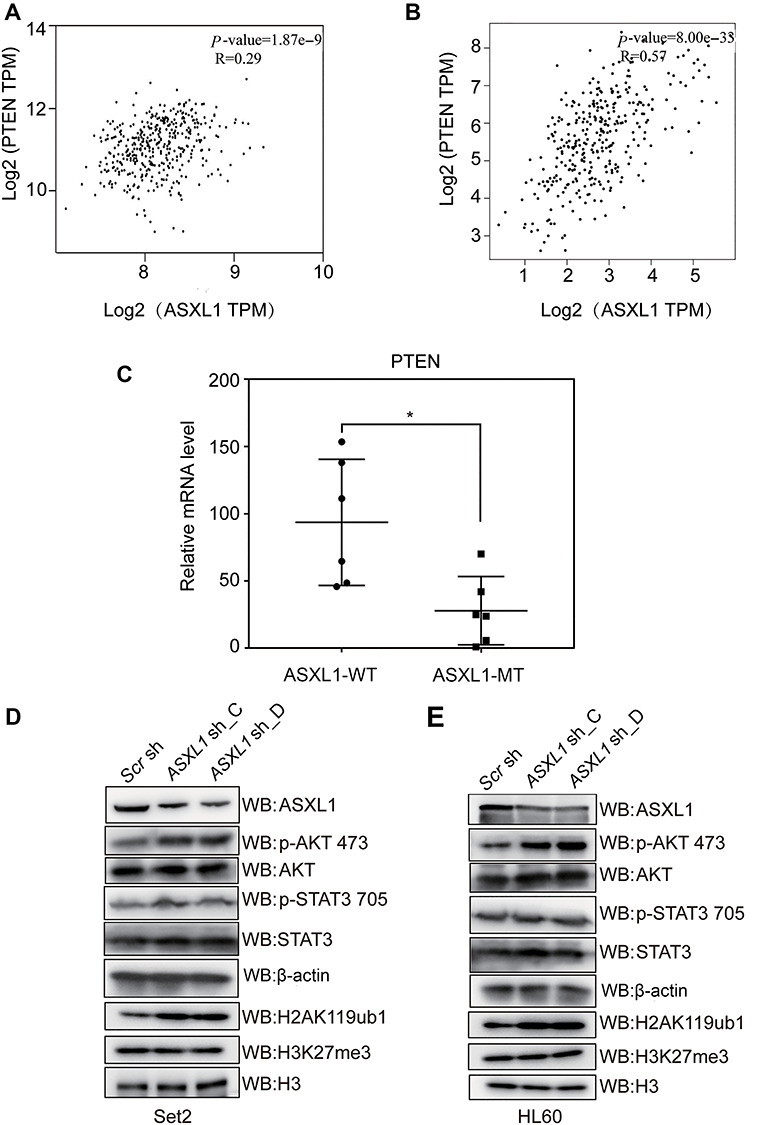
Conserved ASXL1–PTEN/AKT axis in human leukemia. (A and B) Correlation analysis of ASXL1 and PTEN expression levels in bone marrow mononuclear cells of AML patients from GEO dataset (GSE37642) (A) and human whole blood cells from GTEx expression dataset (B). Significance level was determined using Pearson’s correlation test. (C) RT-qPCR analysis of PTEN expression levels in primary bone marrow mononuclear cells from MDS patients with (ASXL1-MT) or without (ASXL1-WT) ASXL1 mutations. The bar in the middle of each group represents the median. Significance level was determined using the two-tailed Student’s t-tests. *P ≤ 0.05. (D and E) WB analysis of ASXL1, p-AKT 473, AKT, p-STAT3 705, STAT3, H2AK119ub1, and H3K27me3 levels in Set2 cells (D) and HL60 cells (E) transduced with two different shRNA constructs targeting ASXL1 (ASXL1 sh_C and ASXL1 sh_D) or scramble shRNA (Scr sh). β-actin and H3 served as loading controls.
To find out whether deregulated PTEN/AKT signaling is correlated with ASXL1 mutations or depletion in human malignancy, we first harvested the bone marrow mononuclear cells from clinically diagnosed MDS/CMML patients. The cells with or without ASXL1 mutations (ASXL1-MT or ASXL1-WT) were confirmed by targeted genomic DNA sequencing analysis (Supplementary Table S3; Wang et al., 2013). Through RT-qPCR analysis, we found that the PTEN expression levels are significantly lower in ASXL1-MT compared with the ASXL1-WT group (Figure 5C). Furthermore we efficiently knocked down ASXL1 (ASXL1 sh_C and ASXL1 sh_D) in Set2 and HL60 cells (Figure 5A and B; Supplementary Figure S5), two human myeloid leukemia cell lines which harbor no ASXL1 mutations. Similar to the observation in 32D cells, ASXL1 depletion leads to the activation of AKT (Figure 5D and E), though PTEN protein levels are barely detectable in these two leukemia cell lines. We also observed a slightly increased p-STAT3 level after knockdown in Set2 cells, which may be a synergistic effect at the specific background. These data indicate that the ASXL1–AKT axis is likely to be a conserved tumor suppressive mechanism, even though there may exist other regulatory hubs in addition to PTEN.
Downregulated expression of ASXL1 confers sensitivity to AKT inhibitor in myeloid leukemia cells
Given the link of ASXL1 downregulation to AKT activation, we were wondering whether ASXL1 downregulation in human myeloid malignancy is associated with higher sensitivity to AKT inhibitors. To address this question, we treated the control and ASXL1-KD Set2 and HL60 cells with AKT inhibitor MK2206 and STAT3 inhibitor Stattics. The inhibition ratio was measured after 48 h treatment of a concentration gradient of the two inhibitors to assess their anti-proliferation ability. And the IC50 value was calculated accordingly. As shown in Figure 6A and B, we observed significantly lower IC50 of AKT inhibitor MK2206 in ASXL1-KD cells compared with control cells but that not for STAT3 inhibitor Sttatics. The inhibition ratio of each concentration in these two cell lines is also presented as sigmoid curves (Supplementary Figure S4C and D). These data suggest that ASXL1-mutated malignant myeloid cells are probably more sensitive to AKT inhibition.
Figure 6.
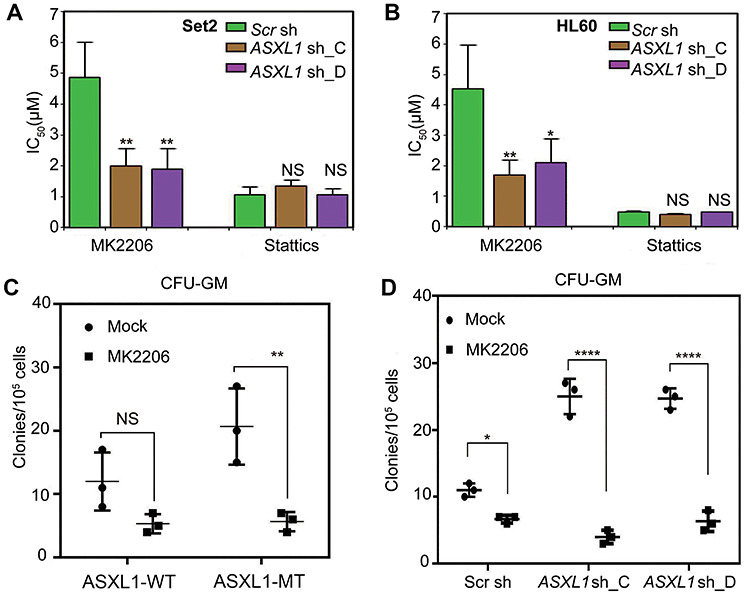
Decreased expression levels of ASXL1 in human leukemia cells are associated with AKT inhibitor sensitivity. (A and B) The inhibition ratios were measured by MTS assay after 48 h treatment of a concentration gradient of the AKT inhibitor MK2206 and the STAT3 inhibitor Stattics in Set2 cells (A) and HL60 cells (B). The IC50 values were calculated accordingly. The error bars denote SD, n = 3. (C) Colony formation assay in methylcellulose was performed using primary bone marrow mononuclear cells isolated from MDS/CMML patients with (ASXL1-MT) or without (ASXL1-WT) ASXL1 mutation under treatment of the AKT inhibitor MK2206. CFU-GM was classified according to standard morphologic criteria. Data represent the counts of colonies from three independent experiments. (D) ASXL1-WT cells were transduced with two different shRNA constructs targeting ASXL1 (ASXL1 sh_C and ASXL1 sh_D) or scramble shRNA (Scr sh). CFU-GM assay was performed to assess the effect of the AKT inhibitor MK2206. Data represent the counts of colonies from three independent experiments. Statistical significance was determined by the two-tailed Student’s t-test. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, NS indicates not significant.
Clinical relevance of the ASXL1−PTEN/AKT axis
We next tested the effect of MK2206 on the growth of primary bone marrow mononuclear cells with or without ASXL1 mutations by colony-forming unit-granulocyte-macrophage (CFU-GM) assay. Consistent with our previous report (Wu et al., 2015), ASXL1-MT cells from MDS patients formed significantly more colonies than ASXL1-WT cells. Notably, MK2206 significantly impaired the colony formation of ASXL1-MT cells, but showed marginal effects on ASXL1-WT cells (Figure 6C). To exclude the effects of cellular heterogeneity in MSD patient samples, we knocked down the expression of ASXL1 in primary bone marrow mononuclear cells and compared the sensitivity to AKT inhibitors with the control group. CFU-GM assay showed that MK2206 mildly inhibits the colony formation of control cells (Scr sh) but much more strikingly inhibits the colony formation of the ASXL1-downregulated cells (ASXL1 sh_C and ASXL1 sh_D) (Figure 6D). Hence, the loss of ASXL1 confers AKT inhibitor sensitivity in human hematopoietic malignant cells.
Discussion
The loss-of-function mutations of ASXL1 have been frequently found in myeloid malignancies and have been correlated with poor prognosis (Boultwood et al., 2010; Gelsi-Boyer et al., 2010; Wang et al., 2014). Nevertheless, there were also evidence showing that gain-of-function of ASXL1 contributes to myeloid malignancies by artificial or transgenic overexpression of ASXL1 C-terminal truncated mutants (Balasubramani et al., 2015; Yang et al., 2018). However, the protein expression of either full-length or N-terminal ASXL1 could not be detected in the clinical samples with ASXL1 mutations (Abdel-Wahab et al., 2012). In this study, we generated cell models with homozygous deletion of Asxl1 and found that the complete deletion of Asxl1 confers cell survival in the deprivation of growth factor (Figure 1; Supplementary Figure S1). It indicates that ASXL1 plays tumor suppressive roles in hematopoietic cells.
In addition to the functional evidence, our mechanistic studies also support that ASXL1 acts as a tumor suppressor. It is well-recognized that ASXL1 and BAP1 specifically form the H2AK119 deubiquitylase complex, though it still remains controversial whether the complex promotes (Scheuermann et al., 2010; Abdel-Wahab et al., 2012) or opposes (Campagne et al., 2019) Polycomb repressive functions. More insightful context-dependent regulatory mechanisms await further investigation. Here we identified PTEN, one of the best known tumor suppressor genes, as one of the key target genes of ASXL1 and BAP1 (Figure 4). And we demonstrate that ASXL1 is required for H2AK119 deubiquitylation at the PTEN promoter and the transcriptional activation of PTEN in response to IL3 deprivation. PTEN is a phosphatase that de-phosphorylates the phosphatidylinositol-3,4,5-trisphosphate (PIP3), and therefore counteracts the activation of PI3K/AKT pathway which plays critical role in the development of myeloid malignancies. PTEN deletion in murine hematopoietic compartment has been reported to promote the development of myeloid malignancies such as myeloproliferative disease (MPD) (Tesio et al., 2013) and CML (Peng et al., 2010). However, PTEN is rarely found to be genetically altered in human myeloid malignancies, unlike the widespread PTEN mutations in solid tumors. Actually PTEN has been frequently found to be inactivated through different pathways in human myeloid leukemia (Worby and Dixon, 2014; Morotti et al., 2015) as well as in our MDS/CMML samples. Hence the epigenetic regulation of PTEN expression by ASXL1 that we have demonstrated may at least partly account for its tumor suppressive mechanisms in hematopoietic systems though PTEN-independent regulatory mechanisms may also exist.
In short, our study has demonstrated that PTEN could be a hub through which ASXL1 restricts the activation of PI3K/AKT pathway to safeguard normal cellular identity. Consistently, ASXL1-mutated or depleted malignant cells are more sensitive to AKT inhibitor (Figure 6). Therefore, it provides a rationale for mechanism-based therapy in patients with genetic or even epigenetic lesions of ASXL1. Given that mutations of ASXL1 and RAS (Abdel-Wahab et al., 2012) or NF1 (Zhang et al., 2018b) often co-occur in myeloid malignancies, it is tempting to generate more relevant models for the understanding of pathophysiological mechanisms and to test potential combination therapies.
Materials and methods
Cloning and plasmid preparation
Specific guide RNA for mouse Asxl1 was designed and cloned into the vector PX459 (Ran et al., 2013). Specific oligonucleotides against human ASXL1 and mouse Asxl1 were designed and cloned into pLKO.1 vector according to the recommended Addgene protocol. Human PTEN cDNA derived from HEK293 cells was cloned and ligated into pCDH-CMV-MCS-EF1-puro vector. The PTEN C124S mutant was cloned by PCR site-directed mutagenesis. The sequences for oligos and cloning can be found, respectively, in Supplementary Tables S1 and S2.
Cell culture
32D cells (gift from Gerald de Haan lab, University Medical Center Groningen) were cultured in RPMI-1640 media supplemented with 10 ng/μl IL3, 10% (v/v) fetal bovine serum (FBS), and 1% penicillin–streptomycin (Gibco). For the IL3 withdrawal experiments, IL3 was removed from the medium. HL60 cells (ATCC) were grown in RPMI-1640 supplemented with 10% FBS. Set2 cells (ATCC) were maintained in RPMI-1640 supplemented with 20% FBS. All cells were maintained at 37°C with 5% CO2. Stable cell lines were generated by lentivirus stable transfection as described (Wu et al., 2015). Inhibitors used in this study include MK2206 (Selleck, S1078) and Stattics (Selleck, S7024).
Primary bone marrow mononuclear cells and genomic DNA sequencing analysis
Approval was obtained from the Ethics Committees of the Institute of Hematology, Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC), and informed consent was provided according to the guidelines of the declaration of Helsinki. The genomic DNA sequence analysis for ASXL1 was performed as described (Wang et al., 2013).
MTS proliferation assay, cell apoptosis assay, and cell cycle assay
For MTS assay, 32D cells were seeded in 96-well plates at a density of 1 × 103 cells per well in 100 μl of growth medium. For the measurement, cells at exponential phase were incubated in dark with 100 μl of MTS solution (medium: MTS:PMS = 100:20:1) at 37°C for 1.5 h. The absorbance was measured at 490 nm. Assays were performed in replicates of 6 wells/condition. Cell apoptosis was analyzed by annexin V-FITC/propidium iodide (PI) double staining using an Annexin V-FITC Apoptosis Detection Kit (Keygen Biotec). For cell cycle assay, cells were stained by Propidium iodide as previously reported (Azizi et al., 2019).
RT-qPCR and immunoblotting
Total RNA was extracted with Trizol reagent (Ambion, catalog no. 15596018) and subjected to reverse transcription with Superscript Reverse Transcriptase (Invitrogen, catalog no. EP0441). RT-qPCR reactions were performed using SYBR Green Master Mix (Roche catalog no. 4913914001) on 7500 Fast real-time PCR system (Applied Biosystems). The PCR conditions were performed according to the manufacturer’s instructions. The Ct values were normalized to RPLPO gene. The ΔΔCt method was used for quantifying the relative expression of target genes. Primer sequences are available in Supplementary Table S2. To obtain whole-cell protein extracts or histones, cells were lysed or extracted as previously described (Wu et al., 2013). Membranes were blotted with the corresponding primary antibodies: anti-PTEN (CST 9188), anti-AKT (CST 4691), anti-p-AKT 473 (CST 4060), anti-Stat3 (Abcam, 68153), anti-p-Stat3 (Abcam 76315), anti-β-actin (CST 4970), anti-H2AK119ub1 (CST 8240S), anti-H3K27me3 (CST 9733), and home-made anti-Bap1 and anti-Asxl1 antibodies as previously described (Wu et al., 2015). After careful washing, bound antibodies were detected with HRP-linked anti-mouse or anti-rabbit IgG (CST), followed by electrochemiluminescence (ECL, PerkinElmer) determination.
Quantitative ChIP assay
The regular chromatin preparation was performed as described (Zhao et al., 2018). The resulting fragmented chromatin extract was precleared with Protein A/G beads (ThermoFisher) and then incubated overnight with primary antibodies and normal anti-rabbit IgG (Cell Signaling Technology) as controls. After stringent washes, elution, and reverse cross-linking, DNA was purified using PCR purification kits (QIAGEN). Primer sequences for amplification of the Pten promoter are available in Supplementary Table S2.
Colony-forming unit granulocyte macrophage assay
A total of 105 primary bone marrow mononuclear cells isolated from different MDS/CMML patients with wild-type ASXL1 or mutated-type of ASXL1 were cultured in semisolid Metho-Cult H4434 Classic methylcellulose (StemCell Technologies) according to the manufacturer’s instructions. The colony numbers were assessed after 14 days in culture.
Statistics
Unless otherwise noted, all grouped data are presented as mean ± SD. Unpaired two-tailed student’s t-test was used to compare two groups. Bivariate correlation analysis (Pearson’s r test) was used to examine the correlation of two variables in human specimens. Significant differences for all quantitative data were considered when *P ≤ 0.05, **P ≤ 0.01, and ****P ≤ 0.0001.
Supplementary Material
Acknowledgements
We thank Gerald de Haan (University Medical Center Groningen) for 32D cells and Zhijian Xiao (Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College) for the human leukemia cell lines.
Funding
This work was supported by the National Natural Science Foundation of China (31570774, 31701126, and 31900464), National Key Research and Development Program (2017YFA0504102), Natural Science Foundation of Tianjin Municipal Science and Technology Commission (17JCZDJC35200 and 18JCQNJC82300), Open Grant from the Chinese Academy of Medical Sciences (157-Zk19-02), and Talent Excellence Program from Tianjin Medical University and Research Project of Tianjin Education Commission (2018KJ075).
Conflict of interest: none declared.
Author contributions: X.W. supervised the project; X.W. and L.C. designed the project and wrote the manuscript; L.C., Y.K., and X.X. performed most of the experiments; B.Y., R.L., Y.W., M.C., Z.D., and H.Z. repeated some results; Q.L. analyzed the data. F.J. and J.C. prepared the ASXL1 and BAP1 antibodies. All authors read and approved the final version of the manuscript.
References
- Abdel-Wahab, O., Adli, M., LaFave, L.M., et al. (2012). ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab, O., Gao, J., Adli, M., et al. (2013). Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 210, 2641–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi, R., Salemi, Z., Fallahian, F., et al. (2019). Inhibition of didscoidin domain receptor 1 reduces epithelial–mesenchymal transition and induce cell-cycle arrest and apoptosis in prostate cancer cell lines. J. Cell. Physiol. 234, 19539–19552. [DOI] [PubMed] [Google Scholar]
- Balasubramani, A., Larjo, A., Bassein, J.A., et al. (2015). Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1–BAP1 complex. Nat. Commun. 6, 7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boultwood, J., Perry, J., Pellagatti, A., et al. (2010). Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 24, 1062–1065. [DOI] [PubMed] [Google Scholar]
- Campagne, A., Lee, M.K., Zielinski, D., et al. (2019). BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat. Commun. 10, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce, L., and Helin, K. (2013). Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155. [DOI] [PubMed] [Google Scholar]
- Ekman, N., Arighi, E., Rajantie, I., et al. (2000). The Bmx tyrosine kinase is activated by IL-3 and G-CSF in a PI-3K dependent manner. Oncogene 19, 4151–4158. [DOI] [PubMed] [Google Scholar]
- Fisher, C.L., Lee, I., Bloyer, S., et al. (2010). Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev. Biol. 337, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer, V., Brecqueville, M., Devillier, R., et al. (2012). Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J. Hematol. Oncol. 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer, V., Trouplin, V., Adelaide, J., et al. (2009). Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 145, 788–800. [DOI] [PubMed] [Google Scholar]
- Gelsi-Boyer, V., Trouplin, V., Roquain, J., et al. (2010). ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br. J. Haematol. 151, 365–375. [DOI] [PubMed] [Google Scholar]
- Greenberger, J.S., Eckner, R.J., Sakakeeny, M., et al. (1983). Interleukin 3-dependent hematopoietic progenitor cell lines. Fed. Proc. 42, 2762–2771. [PubMed] [Google Scholar]
- Gutierrez, A., Sanda, T., Grebliunaite, R., et al. (2009). High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 114, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, D., Kitaura, J., Togami, K., et al. (2013). Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J. Clin. Invest. 123, 4627–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzykson, R., and Fenaux, P. (2014). Epigenetics of myelodysplastic syndromes. Leukemia 28, 497–506. [DOI] [PubMed] [Google Scholar]
- Itzykson, R., Kosmider, O., Renneville, A., et al. (2013). Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 31, 2428–2436. [DOI] [PubMed] [Google Scholar]
- Kandasamy, K., Mohan, S.S., Raju, R., et al. (2010). NetPath: a public resource of curated signal transduction pathways. Genome Biol. 11, R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotti, A., Panuzzo, C., Crivellaro, S., et al. (2015). The role of PTEN in myeloid malignancies. Hematol. Rep. 7, 5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, J., and Verrijzer, P. (2009). Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr. Opin. Genet. Dev. 19, 150–158. [DOI] [PubMed] [Google Scholar]
- Papa, A., Wan, L., Bonora, M., et al. (2014). Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, C., Chen, Y., Yang, Z., et al. (2010). PTEN is a tumor suppressor in CML stem cells and BCR–ABL-induced leukemias in mice. Blood 115, 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, F.A., Hsu, P.D., Wright, J., et al. (2013). Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, E.P., Korapati, A., Chaturvedi, P., et al. (2000). IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene 19, 2532–2547. [DOI] [PubMed] [Google Scholar]
- Scheuermann, J.C., deAyala Alonso, A.G., Oktaba, K., et al. (2010). Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, J., Chiang, A., and Bender, W. (1992). Ten different Polycomb group genes are required for spatial control of the abdA and AbdB homeotic products. Development 114, 493–505. [DOI] [PubMed] [Google Scholar]
- Simon, J.A., and Kingston, R.E. (2009). Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 10, 697–708. [DOI] [PubMed] [Google Scholar]
- Sinclair, D.A., Milne, T.A., Hodgson, J.W., et al. (1998). The Additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development 125, 1207–1216. [DOI] [PubMed] [Google Scholar]
- Tesio, M., Oser, G.M., Baccelli, I., et al. (2013). Pten loss in the bone marrow leads to G-CSF-mediated HSC mobilization. J. Exp. Med. 210, 2337–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Ai, X., Gale, R.P., et al. (2013). TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk. Res. 37, 305–311. [DOI] [PubMed] [Google Scholar]
- Wang, J., Li, Z., He, Y., et al. (2014). Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby, C.A., and Dixon, J.E. (2014). PTEN. Ann. Rev. Biochem. 83, 641–669. [DOI] [PubMed] [Google Scholar]
- Wu, X., Bekker-Jensen, I.H., Christensen, J., et al. (2015). Tumor suppressor ASXL1 is essential for the activation of INK4B expression in response to oncogene activity and anti-proliferative signals. Cell Res. 25, 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., Johansen, J.V., and Helin, K. (2013). Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 49, 1134–1146. [DOI] [PubMed] [Google Scholar]
- Yang, H., Kurtenbach, S., Guo, Y., et al. (2018). Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 131, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzugullu, H., Von, T., Thorpe, L.M., et al. (2016). NTRK2 activation cooperates with PTEN deficiency in T-ALL through activation of both the PI3K–AKT and JAK–STAT3 pathways. Cell Discov. 2, 16030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P., Chen, Z., Li, R., et al. (2018a). Loss of ASXL1 in the bone marrow niche dysregulates hematopoietic stem and progenitor cell fates. Cell Discov. 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P., He, F., Bai, J., et al. (2018b). Chromatin regulator Asxl1 loss and Nf1 haploinsufficiency cooperate to accelerate myeloid malignancy. J. Clin. Invest. 128, 5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X., Wang, X., Li, Q., et al. (2018). FBXL10 contributes to the development of diffuse large B-cell lymphoma by epigenetically enhancing ERK1/2 signaling pathway. Cell Death Dis. 9, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.