

Abstract
Cannabis is the most commonly used illicit drug among pregnant women. Moreover, over half of pregnant women who are consuming cannabis are also consuming alcohol; however, the consequences of combined prenatal alcohol and cannabis exposure on fetal development are not well understood. The current study examined behavioral development following exposure to ethanol (EtOH) and/or CP-55,940 (CP), a cannabinoid receptor agonist. From postnatal days (PD) 4–9, a period of brain development equivalent to the third trimester, Sprague–Dawley rats received EtOH (5.25 g/kg/day) or sham intubation, as well as CP (0.4 mg/kg/day) or vehicle. All subjects were tested on open field activity (PD 18–21), elevated plus maze (PD 25), and spatial learning (PD 40–46) tasks. Both EtOH and CP increased locomotor activity in the open field, and the combination produced more severe overactivity than either exposure alone. Similarly, increases in thigmotaxis in the Morris water maze were caused by either EtOH or CP alone, and were more severe with combined exposure, although only EtOH impaired spatial learning. Finally, developmental CP significantly increased time spent in the open arms on the elevated plus maze. Overall, these data indicate that EtOH and CP produce some independent effects on behavior, and that the combination produces more severe overactivity in the open field. Importantly, these data suggest that prenatal cannabis disrupts development and combined prenatal exposure to alcohol and cannabis may be particularly damaging to the developing fetus, which has implications for the lives of affected individuals and families and also for establishing public health policy.
Keywords: behavior, cannabinoid, development, ethanol, teratology
1 |. INTRODUCTION
The adverse consequences of prenatal alcohol exposure have been extensively studied over the past 40 years (Riley, Infante, & Warren, 2011). Individuals exposed to alcohol prenatally may suffer from a range of physical, neurological, and behavioral consequences referred to as fetal alcohol spectrum disorders (FASD). FASD include growth deficits, facial dysmorphology, as well as altered cognitive, emotional, and behavioral functions (Riley et al., 2011). Approximately 1 in 10 women in the United States report some alcohol consumption during pregnancy (Centers for Disease Control and Prevention, 2015; Substance Use and Mental Health Services Administration, 2014). Thus, FASD continue to pose a serious public health problem in the United States, as well as around the globe (Roozen et al., 2016).
Prenatal alcohol exposure alters development of numerous brain regions, leading to structural and functional alterations in such areas as the basal ganglia, cerebellum, cortex and hippocampus (Moore, Migliorini, Infante, & Riley, 2014) as well as white matter tracts (Taylor et al., 2015; Uban et al., 2017). Alcohol-induced neuropathology can lead to behavioral alterations and impairments in learning, attention, executive functioning, and emotional regulation, all of which can cause serious problems in school and daily life (Khoury, Milligan, & Girard, 2015; Mattson, Crocker, & Nguyen, 2011; Norman et al., 2013). Similarly, animal studies have also shown that developmental alcohol exposure alters brain development, producing a variety of behavioral alterations, including hyperactivity, increased anxiety-related behaviors, and impaired learning and memory (Schneider, Moore, & Adkins, 2011).
However, alcohol is not the only drug of abuse consumed by pregnant women. Recent reports show that 5.4% of pregnant women reported using illicit drugs (Substance Use and Mental Health Services Administration, 2014), with higher rates of use among younger pregnant women (15–17 years; 14.6%; Substance Use and Mental Health Services Administration, 2014). The most commonly used illicit drug among women of reproductive age is cannabis (Substance Use and Mental Health Services Administration, 2014), with prevalence rates of 3–4% among pregnant women in Western countries (Ebrahim & Gfroerer, 2003; El Marroun et al., 2011). Similar to alcohol, the primary psychoactive constituent in cannabis (Δ9-tetrahydrocannabinol [THC]) and its metabolites can freely cross the placenta and directly affect the fetus (Gómez et al., 2003). Of particular concern are the steadily increasing amounts of THC in cannabis products. In 2016, the average potency of THC in cannabis-related products was 11% (Botticelli, 2017); this number has continually risen from 3.4% in 1993 and is even higher among synthetic variations (Botticelli, 2016; Mehmedic et al., 2010). Importantly, many women perceive cannabis as safe to use during pregnancy (Saint Louis, 2017).
Although the dangers of alcohol consumption during pregnancy are well established, much less is known of the consequences of prenatal cannabis exposure, particularly at the high levels consumed today. There are few prospective clinical studies (such as Generation R; Jaddoe et al., 2012) examining the effects of prenatal cannabis exposure (Huizink, 2014), and results from these and retrospective studies are mixed, likely due to differences in cannabis exposure levels, prospective versus retrospective approaches, confounding of other drug use like tobacco, age, and nature of outcome measures, as well as a host of other methodological, maternal, and environmental factors. In general, evidence suggests that prenatal cannabis exposure may alter emotional, behavioral, and cognitive development in clinical populations, particularly in executive functioning domains (Huizink, 2014). Importantly, the consequences of prenatal exposure to the increased potencies of THC and synthetic cannabinoids available today will not be known for years to come. Animal studies are similarly inconsistent; while prenatal THC exposure may not cause neuronal cell death in areas such as the hippocampus, cortex, and thalamus (Hansen et al., 2008), animal model studies do illustrate that prenatal cannabinoid exposure can produce hyperactivity (Huizink & Mulder, 2006), increase anxiety-related behaviors (Goldschmidt, Richardson, Cornelius, & Day, 2004), and impair working (Smith, Fried, Hogan, & Cameron, 2006) and long-term memory (Mereu et al., 2003), although behavioral results are mixed and there are a lot of inconsistencies reported across the literature (Schneider et al., 2011). In addition, perinatal cannabinoid exposure can impair reproductive functioning in male mice (D’Alterio & Bartke, 1979) and disrupts neurodevelopmental CB1 signaling later in adulthood (de Salas-Quiroga et al., 2015). Similar to clinical studies, mixed results likely vary based on differences in timing, dose and form of cannabinoid, outcome measures, and nature of control groups (Abel, Rockwood, & Riley, 1986; Huizink, 2014; Schneider, 2009; Trezza et al., 2012).
Moreover, the effects of concurrent exposure to alcohol and cannabis are not known, despite the high rates of co-use reported among women of child-bearing age. A recent analysis found that 8.7% of females use both alcohol and cannabis, with 5.5% reporting simultaneous use, suggesting that individuals who use both drugs tend to consume them at the same time (Subbaraman & Kerr, 2015). Higher rates of simultaneous alcohol and cannabis use (15.3%) were observed in the 18–29 years age group (Subbaraman & Kerr, 2015), which is consistent with other recent data showing that 19.5% of individuals under age 21 reported smoking cannabis within 2 hr of consuming alcohol (Substance Use and Mental Health Services Administration, 2014).
Importantly, cannabis is also the illicit drug most commonly used simultaneously with alcohol among women who binge drink during pregnancy (Bhuvaneswar, Chang, Epstein, & Stern, 2007); ~50% of pregnant women who have reported consuming cannabis were also drinking alcohol (Substance Abuse and Mental Health Services Administration, 2015). These numbers may be even higher than reports suggest; accurate data reflecting concurrent use and exposure levels among pregnant women are difficult to obtain, not only because women who consume either alcohol or cannabis frequently under-report their usage (Lange, Shield, Koren, Rehm, & Popova, 2014; Lendoiro et al., 2013; Midanik, 1988), but also because of the rapidly changing legalization, accessibility, and potency of cannabis. In addition, ~50% of pregnancies are unplanned. Thus, fetuses may be exposed to these drugs before pregnancy is recognized, which is alarming given the high rates of concurrent use.
The possibility that these drugs may interact with one another is strengthened by the role of endogenous cannabinoids in alcohol-induced neurodegenerative pathways (Subbanna, Shivakumar, Psychoyos, Xie, & Basavarajappa, 2013), depression of synaptic activity (Nagre, Subbanna, Shivakumar, Psychoyos, & Basavarajappa, 2015), and impaired DNA methylation (Basavarajappa, Ninan, & Arancio, 2008). Furthermore, alcohol can alter the development of endocannabinoid systems, including neuronal communication and circuit formation, which may lead to deficits in neuronal plasticity (Basavarajappa, 2015). Thus, concurrent exposure during early development may be even more detrimental to the developing fetus than either drug alone.
However, there is limited research on co-exposure to prenatal alcohol and cannabis on brain and behavioral development. Most clinical (Fried & O’Connell, 1987; Fried, O’Connell, & Watkinson, 1992; Fried & Watkinson, 1990; Richardson, Ryan, Willford, Day, & Goldschmidt, 2002) and animal studies (Hansen et al., 2008; Subbanna et al., 2013) have focused on the effects of each drug separately (Fried et al., 1992; Fried & O’Connell, 1987; Fried & Watkinson, 1990; Richardson et al., 2002), rather than the combination of effects. Research that has examined the combined effects suggest that concurrent cannabinoid and alcohol exposure can significantly increase fetal toxicity (Abel, Tan, & Subramanian, 1987) and synergistically increase neurotoxicity in the hippocampus (Hansen et al., 2008) in rodents. In addition, exposure to cannabinoids on gestational day (GD) eight produces cranio-logical, ocular, and brain abnormalities in a dose-dependent manner (Gilbert et al., 2016), whereas concurrent alcohol and cannabinoid exposure yielded synergistic effects, showing greater physical abnormalities than exposure to either alcohol or cannabinoids alone (Fish et al., 2017; Fish, Gilbert, Sulik, & Parnell, 2016). Nonetheless, research on behavioral development following co-exposure is lacking. Limited previous clinical (Goldschmidt et al., 2004) and animal studies (Abel & Subramanian, 1990) have failed to find any interactions of prenatal alcohol and THC on behavioral development, but these studies investigated low doses of THC or alcohol, notably lower than the THC levels consumed by individuals today.
To examine the possible consequences of combined developmental exposure to alcohol and cannabinoids on behavioral development, we used a rat model of drug exposure during the 3rd trimester brain growth spurt equivalent (PD 4–9). Importantly, this is a period of development particularly sensitive to ethanol (Goodlett, Thomas, & West, 1991; Olney et al., 2002) and a period in which the endogenous cannabinoid system plays an important role in neuronal development (Fernández-Ruiz, Berrendero, Hernández, & Ramos, 2000). The brain growth spurt is characterized by neuronal maturation, including axonal growth, dendritic arborization, as well as high rates of synaptogenesis, gliogenesis, and myelination (Dobbing & Sands, 1979; Gauda, 2006). Additionally, CB1 receptor levels rapidly increase in numerous brain regions during this time (Belue, Howlett, Westlake, & Hutchings, 1995; Berrendero, Sepe, Ramos, Di Marzo, & Fernández-Ruiz, 1999; Rodriguez de Fonseca, Ramos, Bonnin, & Fernández-Ruiz, 1993).
The current study used the synthetic CB1 and CB2 receptor agonist CP-55,940 (CP) to mimic the effects of THC. CP is as an ingredient in synthetic marijuana preparations (Berkovitz, Arieli, & Marom, 2011) and is commonly used in cannabinoid research (Tai & Fantegrossi, 2014) as it has a similar action, peak effect, and neurobehavioral effects as THC (McGregor, Issakidis, & Prior, 1996). The dose of CP-55,940 (CP; 0.4 mg/kg/day) was chosen based on previous studies (Gilbert et al., 2016; LaFleur, Wilson, Morgan, & Henderson-Redmond, 2018; Maguire & France, 2016; Minervini, Dahal, & France, 2017) and was administered to model a moderate–high cannabinoid dose in humans (Desrosiers et al., 2014; Farquhar et al., 2019; Javadi-Paydar et al., 2018; Nguyen et al., 2016; Taffe, Creehan, & Vandewater, 2015; Wakley, Wiley, & Craft, 2014).
All subjects were tested on a battery of behavioral tests to investigate effects of combined exposure to alcohol and cannabinoids on behavioral development. The behavioral battery included an open-field activity chamber (activity levels), an elevated plus maze (anxiety-related behaviors), and a Morris water maze task (visuospatial learning). Following the behavioral battery, gross brain weights (g) were measured. The objective of the current study is to lay the foundation for future studies examining multiple dose combinations of alcohol and cannabinoids.
2 |. MATERIALS AND METHODS
The goal of the current study was to examine the effects of combined neonatal alcohol and cannabinoid exposure on behavioral development (see Figure 1). All procedures and behavioral paradigms included in this study were approved by the San Diego State University (SDSU) Institutional Animal Care and Use Committee (IACUC) and are in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals.
FIGURE 1.
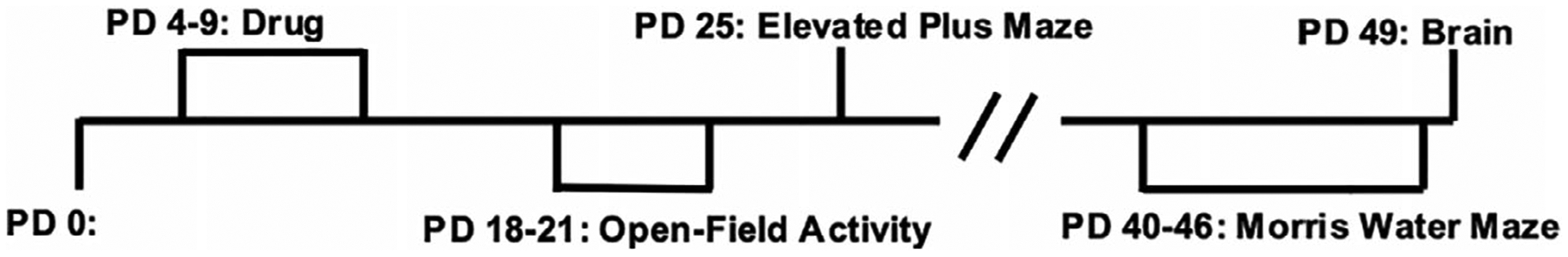
Timeline of drug exposure, behavioral testing, and tissue collection
2.1 |. Subjects
For breeding, one male and one female Sprague–Dawley rat were housed together overnight at the SDSU Animal Care facilities at the Center for Behavioral Teratology. Once a seminal plug was present, dams were individually housed in standard plastic cages (gestational [GD] 0) and left undisturbed until the day of delivery (typically GD 22), except for routine husbandry. On postnatal day (PD) 1, the day after birth, litters were pseudo-randomly culled to eight pups with equal numbers of male and female subjects (whenever possible). Subjects were then randomly assigned to each exposure group; no more than one sex pair per litter was used within each exposure condition to control for potential litter effects. Subjects were generated from a total of 18 litters; additional outcome measures are reported in a separate publication (Breit, Zamudio, & Thomas, 2019).
2.2 |. Developmental alcohol and cannabinoid exposure
From PD 4–9, a time period equivalent to the human third trimester “brain growth spurt,” half of the subjects were intragastrically intubated with ethanol (EtOH, 5.25 g/kg/day) dissolved in an artificial milk diet (11.9% v/v, 0.0275 mL/g, twice per day, 2 hr apart), followed by two additional feedings of milk diet only (Goodlett & Johnson, 1997). Briefly, the intubation procedure involved lubricating thin, silastic tubing (PE-50) with corn oil, inserting the tubing into the pup’s mouth to be swallowed into the stomach, and injecting the milk diet through the tubing (Goodlett & Johnson, 1997). The other half of subjects received sham intubations, with no EtOH or milk exposure. In addition, all subjects were injected (i.p. 10 mL/kg) with either CP (0.4 mg/kg/day) or the vehicle (VEH; 10% dimethylsulfoxide [DMSO] and sterilized saline). Injections utilized a 30-gauge needle with a guide to allow only the tip of the needle to enter the i.p. area. During the drug administrations, all pups were removed from the dam simultaneously and maintained on a heating pad with littermates and nesting material throughout the exposure period. Each intubation and injection took approximately 1 min per pup to complete; litters were returned to the dam within 10 min of separation.
Offspring were individually identified with a nontoxic marker until PD 7, when they were tattooed with nontoxic veterinary tattoo ink, allowing the experimenter to remain blind during later behavioral testing. Subjects were weaned on PD 21 and group-housed by sex on PD 28. All subjects were housed at a constant humidity and temperature (21 ± 1°C) in plastic cages with woodchips, and exposed to a 12-hr light/dark cycle, receiving food and water ad libitum.
2.2.1 |. Drug preparation
Ethanol (11.9% v/v) was added to an artificial milk diet (West, Hamre, & Pierce, 1984). CP (Enzo Life Sciences, NY) was dissolved into a stock solution (5 mg of CP dissolved into 2 mL of 100% DMSO [Sigma-Aldrich, MO]) and kept at −20 °C until daily injection volumes were made. Daily injection volumes were prepared by combining the CP stock solution with the vehicle to the appropriate final dose (0.4 mg/kg/day).
2.2.2 |. Behavioral testing
Activity levels
From PD 18–21, activity levels in an open field were measured in all subjects during the dark cycle (starting at 18:00). Subjects remained with the dam and littermates while habituated to the dark testing room for 30 min and then each subject was placed in an individual open-field activity chamber (16 × 16 × 14 in; Hamilton-Kinder), equipped with fans for ventilation and white noise, for 60 min. Activity levels were recorded in 5-min bins via interruptions of infrared beams, indicating overall activity levels, location and time spent in different areas of the chamber, and behaviors such as rearing.
Anxiety-related behaviors
On PD 25, anxiety-related behaviors were measured in all subjects using an elevated plus maze test. The plus sign-shaped maze was elevated 50 cm above the floor, with an open center, two exposed arms, and two enclosed arms (50 × 10 cm); the maze was made of clear Plexiglas except for the black sides of the enclosed arms (40 cm). Subjects were placed in the center of the maze and allowed to roam freely for 5 min. A video camera recorded subjects’ locations on the maze (open vs. closed arms), time spent in each area, as well as grooming and rearing behaviors, which were later coded using OdLog software. Time spent in the center of the maze was coded as an open area.
Visuospatial learning and memory
From PD 40–46, all subjects were tested for visuospatial memory performance using a Morris water maze. Subjects were required to use spatial cues in the room to remember the location of a clear Plexiglas platform (4-in diameter) hidden 1 in below the water surface of a black tank (48-in diameter) filled with water. All data were recorded via a video tracking system interfaced with Water2020 software (HVS Image).
For acquisition, subjects were tested for 4 trials per day over 6 consecutive days with an inter-trial interval of 3–5 min. For each trial, the platform location remained consistent, while the starting position changed. Subjects were given 60 s to find and swim to the platform; if subjects failed to find the platform, they were guided. Once on the platform, subjects were given 10 additional seconds to observe spatial location. Distance and latency traveled to find the platform, as well as heading angle, and thigmotaxis (time spent within the outer 4.8 in of the pool) were measured. Twenty-four hours after the last acquisition trial, subjects were given a 60-s probe trial, in which the platform was removed and memory of the platform location was measured by examining time spent and passes through the platform area (within three times the diameter of the platform).
2.2.3 |. Brain collection
Brains were collected from all subjects via intracardial perfusion between PD 47–51. Subjects were given a lethal dose of pentobarbital (10% pentobarbital in sterile saline, 3 mL/kg, i.p.) and perfused with a 4% paraformaldehyde solution. Once perfused, the forebrain and cerebellum were collected and weighed.
2.3 |. Statistical analyses
All data were analyzed using the Statistical Package for the Social Sciences (SPSS, version 24). All analyses used a 2 (EtOH exposure: EtOH, Sham) × 2 (CP exposure: CP, VEH) × 2 (sex: female, male) ANOVA. Repeated measures ANOVAs for Day, 5-min Time Bin (Bin), and/or Trial were used when applicable. Post hoc analyses were conducted using Student–Newman–Keuls. Means (M) and standard errors of the mean (SEM) are reported when applicable. All significance levels were set at p < 0.05. In addition, to determine if CP had effects by itself or altered ethanol’s effects, planned comparisons were made between CP and controls and between CP + EtOH and EtOH groups.
3 |. RESULTS
At least 10 subjects per exposure group and sex completed all behavioral tasks (EtOH + CP: 23 [13 F, 10 M], EtOH + VEH: 30 [16 F, 14 M], Sham + CP: 30 [15 F, 15 M], Sham + VEH: 28 [13 F, 15 M]).
3.1 |. Body weights
On the first day of the open-field activity testing (PD 18), subjects exposed to EtOH during development weighed significantly less than sham-intubated subjects (F[1,103] = 38.19, p < 0.001; Figure 2). There was also a main effect of CP (F[1,103] = 7.40, p < 0.01); although there was no significant interaction of EtOH*CP on body weights during this period, the main effect of CP was driven by significant reductions in body weight among subjects exposed to EtOH + CP (F[1,49] = 7.6, p < 0.01). In addition, although there were no effects of sex on body weight during open-field activity, males weighed more than females during the elevated plus maze (F[1,103] = 16.33, p < 0.001; males = 88.8 ± 1.6 g, females = 79.7 ± 1.6 g) and Morris water maze (F[1,103] = 112.65, p < 0.001; males = 216.0 ± 3.3 g, females = 168.0 ± 3.1 g) tasks (data not shown). However, sex did not interact with either developmental EtOH or CP exposure at any time.
FIGURE 2.
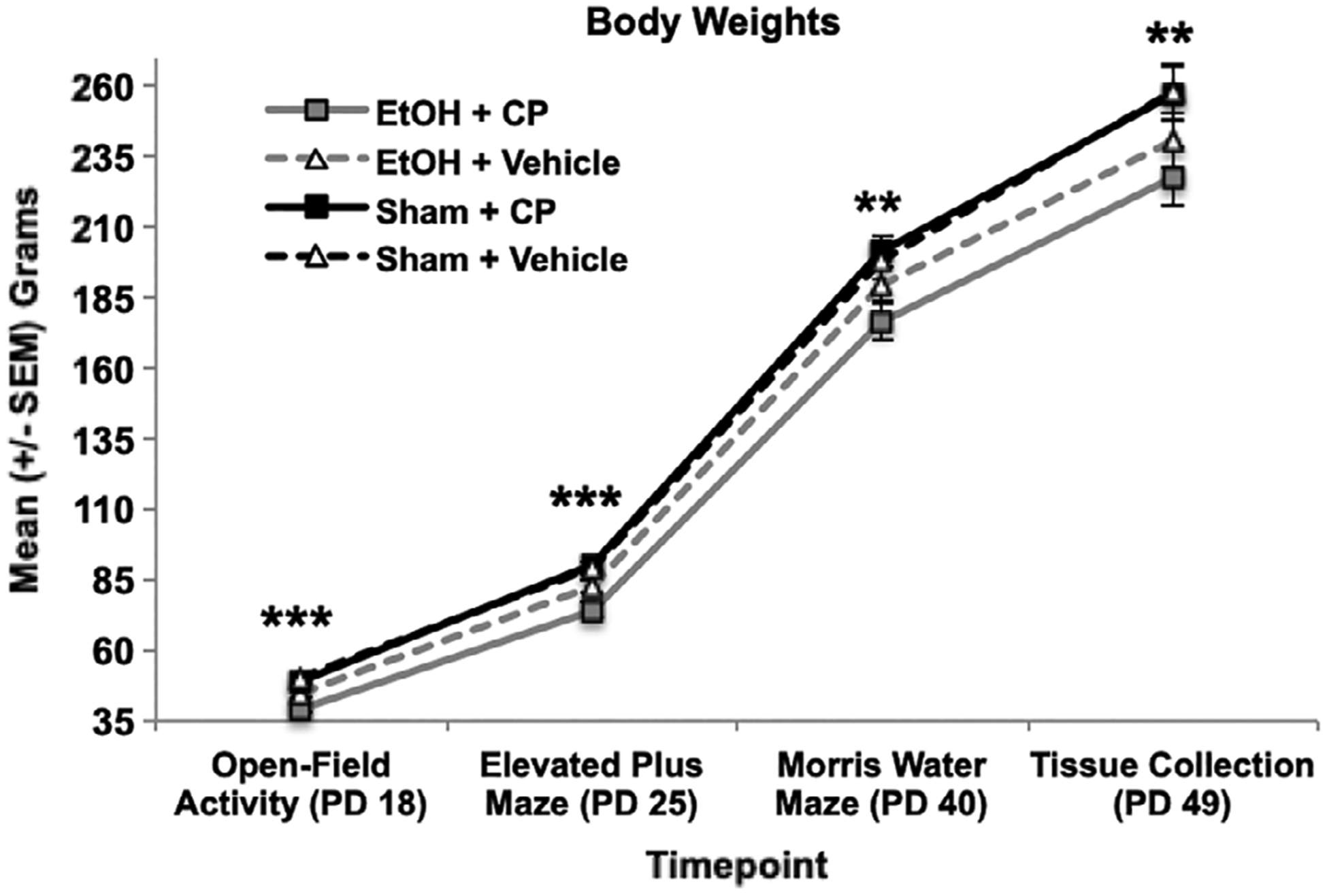
Developmental EtOH reduced body weight, an effect that persisted throughout early adulthood. CP exposure exacerbated the EtOH growth reductions up to PD 25.
*** = EtOH + CP < EtOH + Vehicle and EtOH + < + Sham;
** = EtOH < Sham
On PD 25, during the elevated plus maze task, there was an interaction of EtOH*CP exposure on body weights (F[1,103] = 6.26, p < 0.05). Subjects exposed to combined EtOH+CP during development weighed less than those exposed to EtOH alone (F[1,51] = 7.32, p < 0.01), whereas subjects exposed to only CP did not weigh significantly different from controls (F[1,56] = 0.15, p = 0.70). However, by the first day of the Morris water maze test (PD 40), only a main effect of developmental EtOH exposure persisted (F[1,103] = 8.45, p < 0.01), as EtOH-exposed subjects weighed less than their sham-intubated counterparts, regardless of CP exposure. EtOH-related reductions in body weight persisted until sacrifice (PD 49) (F[1,103] = 11.41, p < 0.01). In contrast, developmental CP exposure had no significant long-lasting effects on body weights.
3.2 |. Activity levels
All activity measures were initially analyzed using a 2 (EtOH) × 2 (CP) × 2 (Sex) ANOVA with 4 (Day) × 12 (Bin) as repeated measures. Overall, all subjects habituated within and across testing days on all activity measures, producing main effects of Day (p’s < 0.001), Bin (p’s < 0.001), and interactions of Day*Bin (p’s < 0.001) regardless of developmental EtOH and/or CP exposure. However, habituation differences among exposure groups are noted, if applicable. No differences between male or female subjects were observed in any activity measure.
3.2.1 |. Overall activity
Subjects exposed to EtOH during development were more active than sham-intubated subjects, as evident by increased total distance traveled (F[1,103] = 28.9, p < 0.001; Figure 3a). In addition, EtOH-exposed subjects habituated slower within sessions, producing a Bin*EtOH interaction (F[11,1133] = 4.2, p < 0.001; Figure 3b shown averaged across Days); EtOH-exposed subjects significantly habituated across Days similar to sham-intubated subjects. Although the interaction of EtOH and CP failed to reach statistical significance, CP by itself did significantly increase total distance traveled in sham, but not EtOH-exposed, subjects (F[1,56] = 5.1, p < 0.05; Figure 3a shown summed across Days). In addition, locomotor activity of subjects exposed to CP did not habituate to the same level as controls, producing a Day*Bin*CP interaction (F[33,3399] = 1.5, p < 0.05). Similar effects were observed in the total number of beam breaks (data not shown).
FIGURE 3.
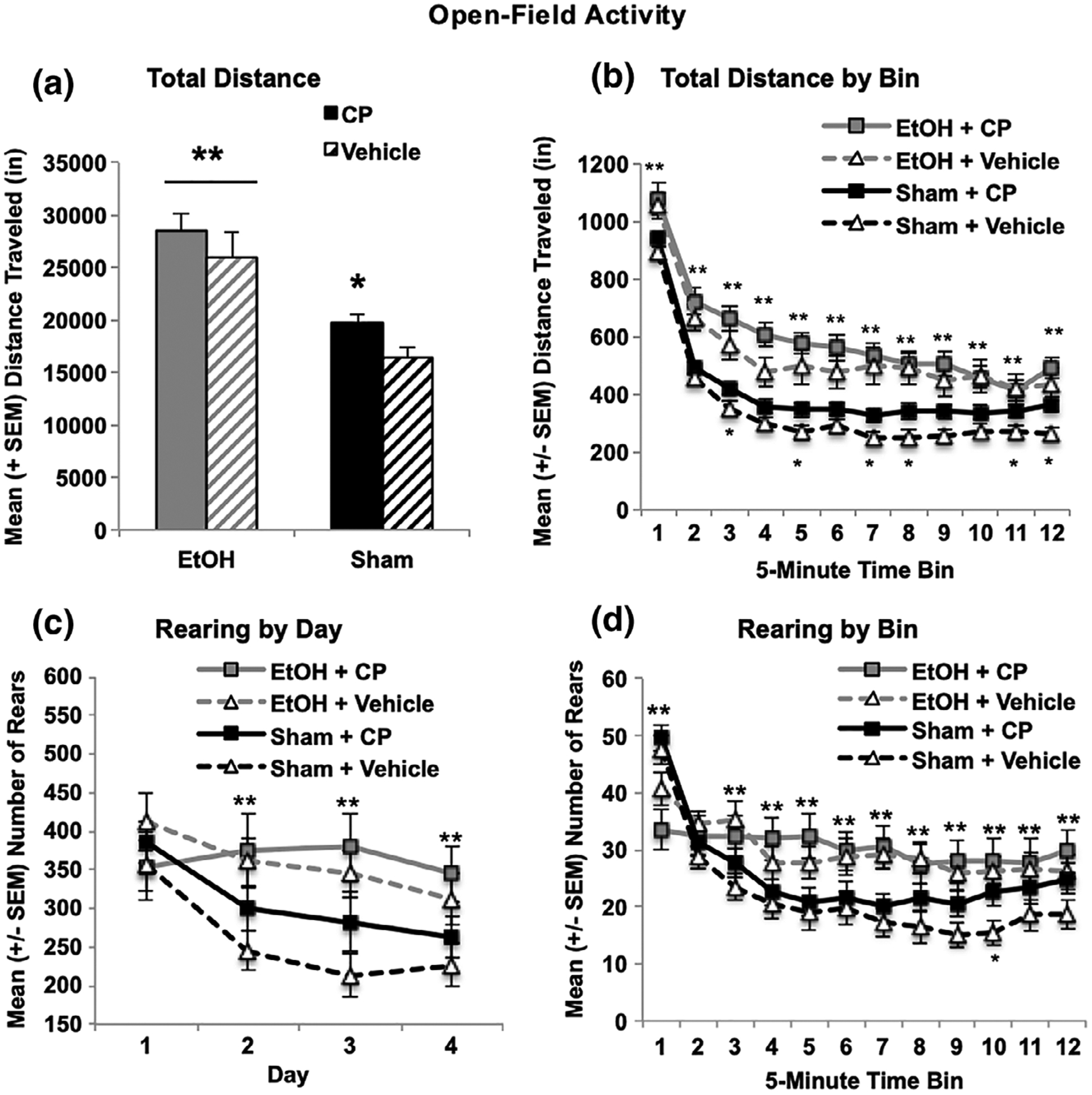
EtOH exposure during the third trimester equivalent increased overall locomotor activity in the open-field chamber (a, total distance traveled summed across days) and slowed habituation within sessions (b, total distance per bin averaged across days). Similarly, subjects exposed to EtOH showed less habituation of rearing across days compared to their Sham counterparts (c). CP exposure also increased locomotor activity and reduced habituation of locomotor activity (a,b) and rearing across testing (d). * = CP + Sham > Vehicle + Sham; ** = EtOH sig differs from Sham
3.2.2 |. Center-related activity
In contrast, when locomotor activity in the center of the chamber was examined (inner 8 × 8 in space), the combination of developmental EtOH + CP exposure produced more severe overactivity than either exposure by itself. Separately, both EtOH and CP increased locomotor activity in the center of the chamber, producing main effects of EtOH (F[1,103] = 53.1, p < 0.001) and CP (F[1,103] = 8.1, p < 0.01; Figure 4a shown summed across Days). CP significantly increased locomotor activity in both sham (F[1,54] = 5.4, p < 0.05) and EtOH-exposed subjects (F[1,49] = 3.8, p < 0.05). Moreover, subjects exposed to combined EtOH + CP took significantly longer to habituate compared to all other groups, and subjects exposed to CP by itself did not habituate to the level of controls (Figure 4c), producing a 3-way interaction of Bin*EtOH*CP (F[11,1133] = 1.9, p < 0.05). Similar reductions in habituation among subjects exposed to the combination of EtOH + CP were evident across days, although there were no statistically significant interactions of Day with either drug exposure (Figure 4b). Importantly, although the combination of EtOH and CP increased locomotor activity in the center of the chamber, only developmental EtOH exposure significantly increased the time spent in the center of the chamber (F[1,103] = 16.4, p < 0.001; Figure 4d,e). CP exposure had no significant effect on time spent in the center, nor did EtOH and CP interact on this measure. Similar effects were seen with entries into the center of the chamber (data not shown).
FIGURE 4.
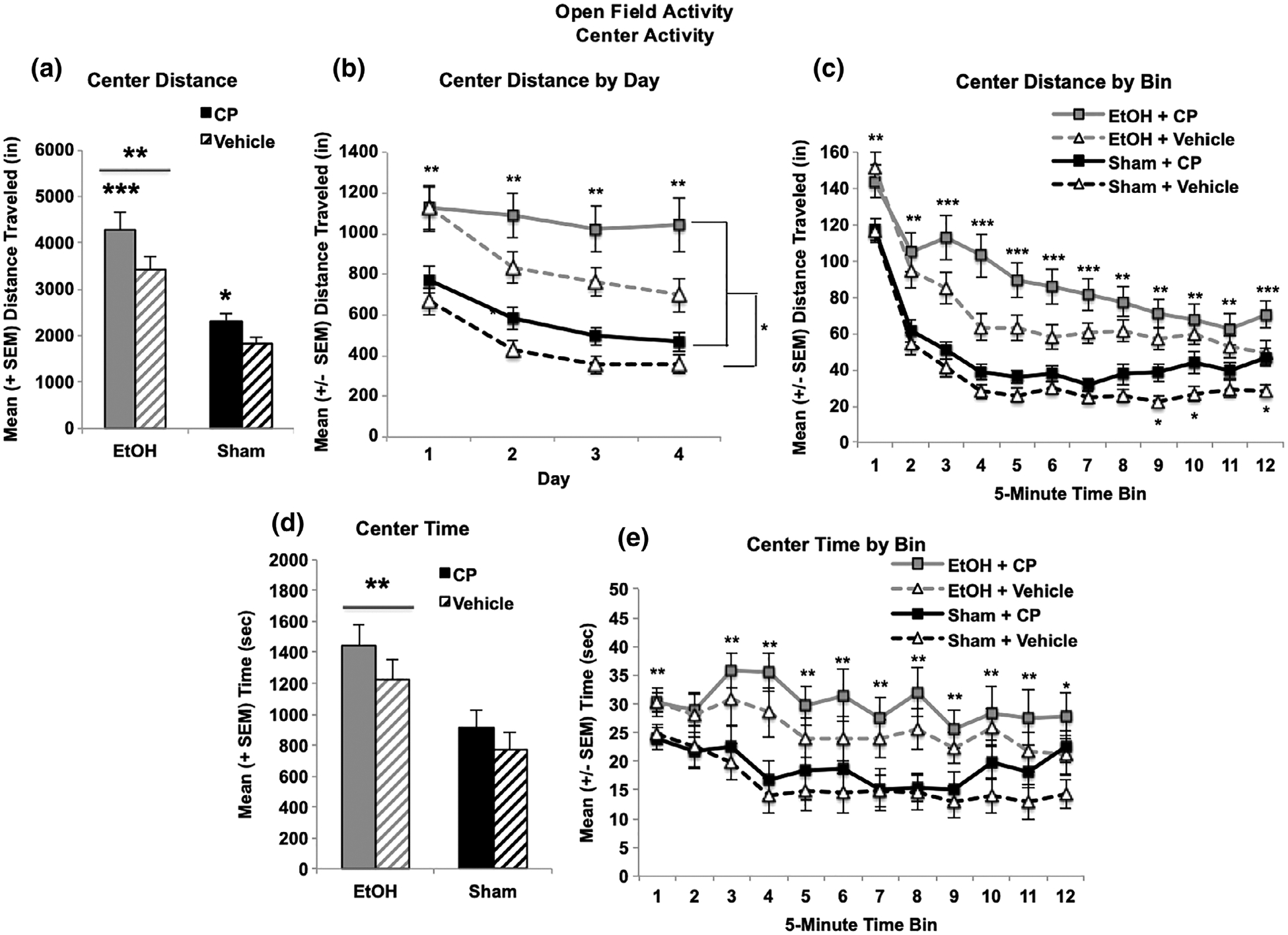
Both developmental EtOH and CP exposure separately increased locomotor activity in the center of the chamber (a). The combination of CP and EtOH significantly impaired habituation during sessions (b) and both EtOH and CP reduced habituation across days (c). However, only developmental EtOH exposure significantly increased the time spent in the center of the chamber (d,e). *** = EtOH + CP > all other groups, EtOH > Sham; ** = EtOH > Sham; * = CP > Vehicle
3.2.3 |. Exploratory activity
Overall, EtOH exposure increased rearing in the chamber (F[1,103] = 7.1, p < 0.01), regardless of CP exposure. A Day*EtOH interaction was present (F[3,309] = 5.0, p < 0.01), as EtOH-exposed subjects exhibited less habituation over days (Figure 3c shown summed across Day), rearing significantly more during the last 3 days of open field activity testing (PD 19–21; p’s < 0.01). In addition, a three-way interaction between Day*Bin*CP was evident (F[33,3399] = 1.7, p < 0.01), as subjects exposed to CP exhibited less habituation in rearing across and within sessions (Figure 3d shown averaged across Days).
3.3 |. Anxiety-related behaviors
The elevated plus maze utilizes rats’ natural aversion to open spaces and other behaviors to infer levels of anxiety. All measures on this task were analyzed using a 2 (EtOH) × 2 (CP) × 2 (Sex) ANOVA. Neither EtOH nor CP exposure during development significantly altered the frequency of open, closed, or total arm entries (Figure 5b). However, developmental CP exposure increased the time spent on the open arms of the elevated plus maze (F[1,103] = 5.91, p < 0.05; Figure 5a). Although an interaction of EtOH*CP was not significant, it should be noted that this effect was driven by a significant increase in open arm time among subjects exposed only to CP compared to controls (F[1,56] = 9.39, p < 0.01); CP had no significant effect among EtOH-exposed subjects (F[1,51] = 0.43, p = 0.52). Importantly, since CP exposure did not alter the frequency (Figure 5b) or the ratio of different arm entries, CP-exposed subjects specifically spent more time in the open areas of the maze without an increase in activity level. Finally, the main effect of EtOH on open arm time failed to reach statistical significance (F[1,103] = 2.46, p = 0.12); however, when comparing only the EtOH-exposed group to controls, developmental EtOH exposure also increased open arm time (F[1,54] = 5.31, p < 0.05).
FIGURE 5.
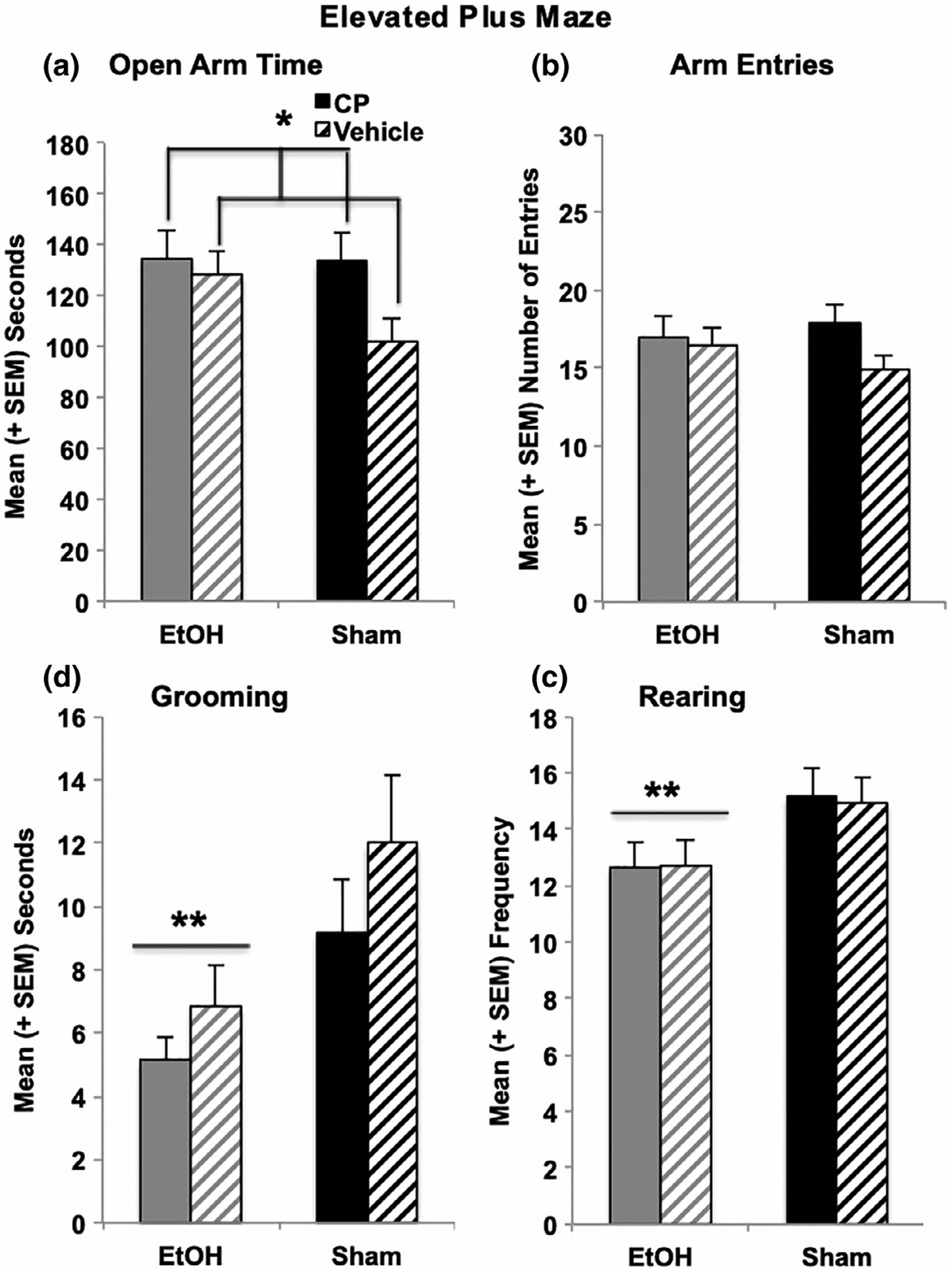
CP exposure during the 3rd trimester equivalent increased the total time spent on the open arms of the elevated plus maze, driven by a significant CP-related increase among sham-intubated subjects (a). In contrast, EtOH exposure decreased the time spent grooming (c) and the number of rearing (d) behaviors on the maze. No groups differed in overall activity on the plus maze (b). * = CP > Vehicle; ** = EtOH < Sham
In addition, EtOH exposure decreased the time spent grooming (F[1,103 = 9.39, p < 0.01; Figure 5c), grooming frequency (F[1,103] = 4.55, p < 0.05), and average grooming bout duration (F[1,103] = 4.98, p < 0.05). Developmental EtOH exposure also reduced the frequency of rearing, an exploratory behavior (F[1,103] = 10.19. p < 0.01; Figure 5d), but did not affect the total time spent rearing or average rearing bout. Neither grooming nor rearing behaviors were significantly altered by developmental CP exposure. Moreover, there were no interactions between EtOH*CP or effects of Sex on any behaviors on the elevated plus maze.
3.4 |. Spatial learning
3.4.1 |. Acquisition
All acquisition data were analyzed using a 2 (EtOH) × 2 (CP) × 2 (Sex) ANOVA with 6 (Day) as a repeated measure. Although performance in all subjects improved over acquisition days (effects of Day, p’s < 0.001), subjects exposed to EtOH during development exhibited impairments in spatial learning. EtOH-exposed subjects learned more slowly than controls and required longer path lengths to find the platform compared to controls, producing an interaction of Day*EtOH (F[5,515] = 2.19, p = 0.05), as well as a main effect of EtOH (F[1,103] = 39.90, p < 0.001; Figure 6a). Similar effects were seen with latency to find the platform, even though EtOH-exposed subjects did swim significantly faster. In addition, EtOH-exposed subjects were less precise in aiming toward the platform, exhibiting larger heading angles (F[1,103] = 15.73, p < 0.001; Figure 6b). Finally, males performed better than females on all acquisition measures, exhibiting shorter path lengths (F[1,103] = 7.22, p < 0.001; males = 6.3 ± 0.4 m, females = 7.8 ± 0.4 m), as well as smaller heading angles (F[1,103] = 8.71, p < 0.001; males = 42.0 ± 2.0°, females = 50.1 ± 1.9°).
FIGURE 6.
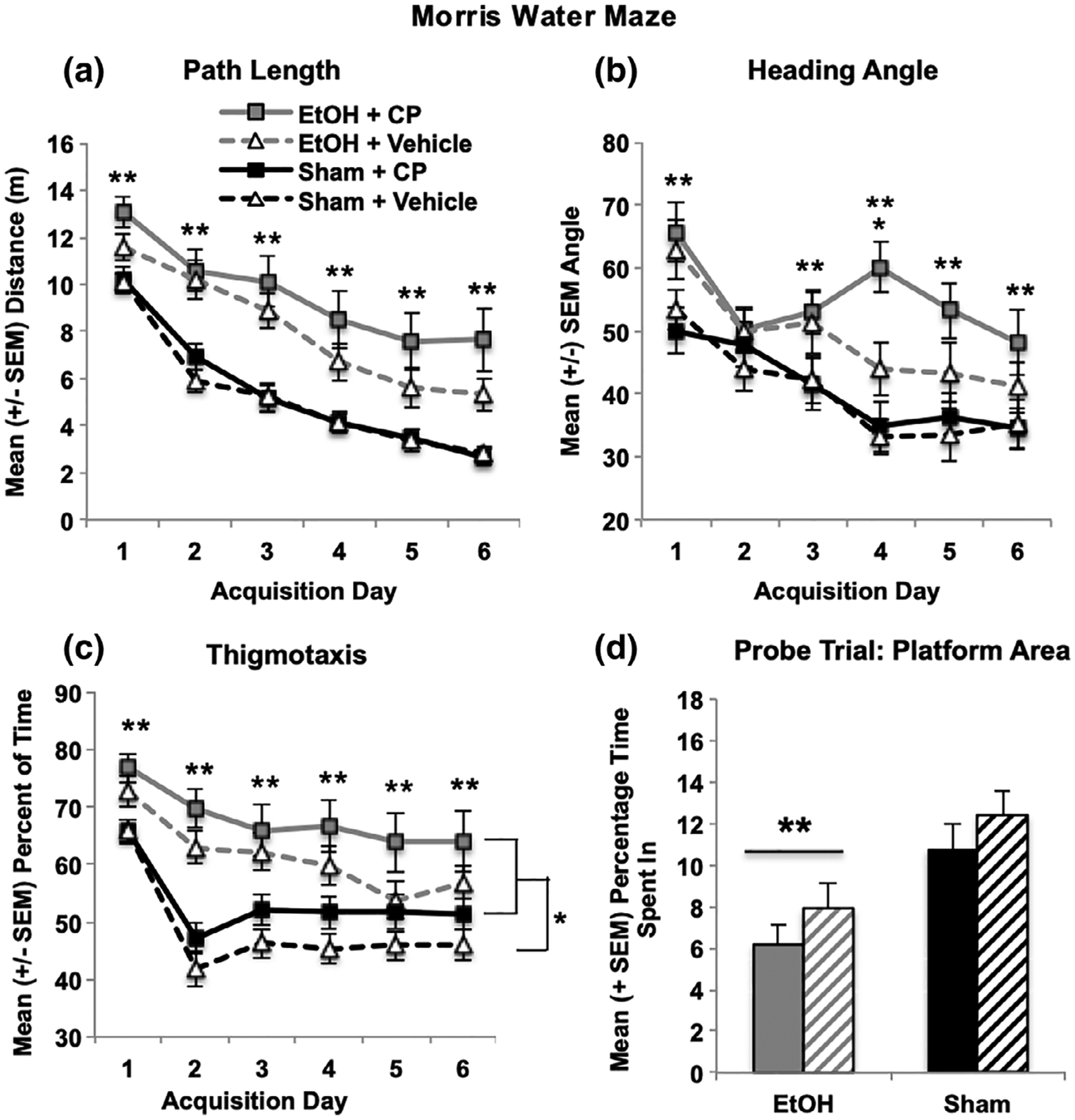
Developmental EtOH exposure impaired performance on the Morris water maze spatial learning task, leading to increased path lengths to find the platform (a), as well as less precise direction of swimming toward the target (increased heading angle; b), whereas EtOH and CP separately increased thigmotaxis during acquisition (c). Only developmental EtOH exposure significantly impaired spatial memory during the probe test, reducing time spent in the target platform area (d). ** EtOH differs sig from sham; *CP > Vehicle
In contrast, both developmental EtOH and CP exposure separately increased thigmotaxis during acquisition (Figure 6c), but did not interact. Subjects exposed to EtOH spent more time (F[1,103] = 26.23, p < 0.001) and did not decline in thigmotaxis as quickly as sham-intubated subjects, producing an interaction of Day*EtOH (F[5,515] = 5.92, p < 0.001). Similarly, subjects exposed to CP spent more time in thigmotaxis across days (F[1,103] = 4.20, p < 0.05). Regardless of developmental EtOH or CP exposure, females spent more time in thigmotaxis than male subjects (F[1,103] = 8.74, p < 0.01; males = 53.8 ± 1.8% time, females = 61.3 ± 1.8% time). The same effects were seen in thigmotaxic path lengths (data not shown).
3.4.2 |. Test (probe)
Performance on the probe trial was analyzed with a 2 (EtOH) × 2 (CP) × 2 (Sex) ANOVA. Only developmental EtOH exposure significantly impaired performance during the probe trial. EtOH-exposed subjects spent less time in the platform area (F[1,103] = 14.41, p < 0.001; Figure 6d) and the platform quadrant (F[1,103] = 4.82, p < 0.05), and made fewer passes through the platform area and quadrant (data not shown). No main effects or interactions with developmental CP exposure were observed in any measure. Overall, females performed worse than males regardless of drug exposure, spending less time in the platform area (F[1,103] = 4.88, p < 0.05; males = 11.7 ± 0.9% time, females = 7.3 ± 0.7% time) and platform quadrant (F[1,103] = 9.24, p < 0.01; males = 51.7 ± 2.1% time, females = 41.2 ± 2.3% time), and made fewer passes through each as well (p’s < 0.01; data not shown).
3.5 |. Brain weights
Developmental EtOH exposure reduced total gross brain weights (F[1,103] = 11.88, p < 0.01; Figure 7a), but did not alter the total brain–body weight ratios. EtOH exposure also decreased the forebrain (F[1,103] = 79.17, p < 0.001; Figure 7b) and cerebellum (F[1,103] = 59.72, p < 0.001; Figure 7c) weights, as well as the forebrain- (F[1,103] = 17.86, p < 0.001) and cerebellum-body weight ratios (F[1,103] = 37.03, p < 0.001; data not shown). Developmental CP exposure did not significantly alter gross weights of any brain region. Finally, regardless of EtOH or CP exposure, male subjects had larger total brain (F[1,103] = 10.67, p < 0.01; males = 1.6 ± 0.04 g, females = 1.4 ± 0.04 g), forebrain (F[1,103] = 25.72, p < 0.01; males = 1.3 ± 0.01 g, females = 1.1 ± 0.01 g), and cerebellum (F[1,103] = 10.91, p < 0.01; males = 0.23 ± 0.006 g, females = 0.20 ± 0.005 g) weights compared to females, as well as greater body weight ratios for each (p’s < 0.001; data not shown).
FIGURE 7.
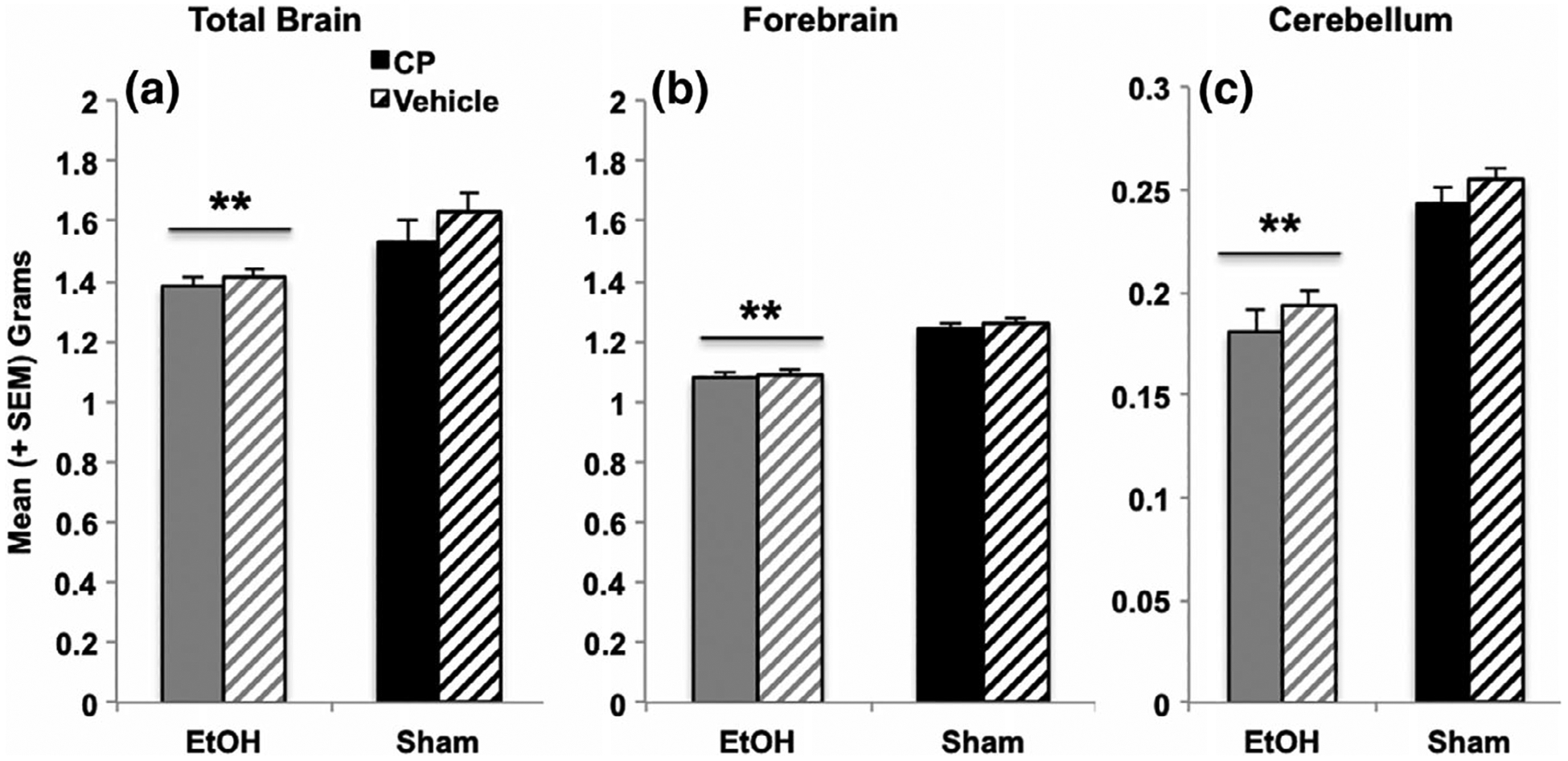
Developmental EtOH exposure reduced total brain weights (a) as well as forebrain (b) and cerebellum weights (c), whereas CP exposure had no long-lasting effects on brain weights. ** = EtOH < Sham
4 |. DISCUSSION
The current study suggests that exposure to combined alcohol and cannabinoids during early postnatal development may alter behavioral development in a task-specific manner. Importantly, not only may early cannabinoid exposure, by itself, alter later behavioral performance, but also concurrent exposure to alcohol and cannabinoids during development may disrupt behavioral development more than prenatal exposure to either drug alone, an effect particularly evident in open field activity.
First, exposure to developmental alcohol increased overall activity levels, with elevations in locomotor activity, rearing, as well as entries and time spent in the center of the chamber. Consistent with the results of this study, past preclinical research has consistently shown that rats exposed to alcohol during early development display increased activity levels (Thomas, Abou, & Dominguez, 2009; Thomas, Biane, O’bryan, O’neill, & Dominguez, 2007; Tran, Cronise, Marino, Jenkins, & Kelly, 2000), which corresponds to clinical reports that children with FASD are commonly hyperactive (Mattson et al., 2011), although some clinical data fail to find difference in activity levels (Glass et al., 2014). Similarly, developmental CP exposure alone also increased locomotor activity, rearing, and center travel and entries. Preclinical data examining activity levels following prenatal cannabis exposure are more limited and inconsistent. One study has shown that limited prenatal THC exposure (GD 10–12) increased activity levels from PD 9–21, yet did not increase time spent within the center of the chamber (Borgen, Davis, & Pace, 1973), which is consistent with the findings of the current study. However, other preclinical studies have shown no difference or even decreased activity levels in early adulthood (Fride & Mechoulam, 1996). Limited clinical studies report that children (El Marroun et al., 2009; Hofman et al., 2004; Jaddoe et al., 2010; Jaddoe et al., 2012) and adolescents (Fried & Watkinson, 1988) prenatally exposed to marijuana may exhibit hyperactivity, further indicating that exposure to cannabinoids may have long-lasting effects on activity levels.
The effects of combined developmental alcohol and THC were largely additive, with subjects exposed to both drugs exhibiting the most severe increases in activity levels. However, there were some synergistic effects, as subjects exposed to combined developmental alcohol and cannabinoids habituated more slowly compared to all other groups. This alteration may represent unique deficits in simple learning, as habituation represents learning of the environment. While these measures have not been well examined in preclinical studies, clinical reports do suggest that children (Goldschmidt, Day, & Richardson, 2000) and adolescents (Fried & Watkinson, 1988) prenatally exposed to marijuana exhibit impairments in attention and impulsivity in addition to hyperactivity. It would be important to investigate the effects of combined alcohol and cannabinoids on habituation of other behaviors to better understand the nature of this behavioral change.
The effects of both drugs on emotional development are more complex to interpret. Increases in locomotion and time spent in the center of the open field chamber may indicate that the EtOH-exposed subjects are less anxious or are more inclined to take risks (Prut & Belzung, 2003), suggesting that developmental alcohol exposure may affect emotional development as well as activity. In fact, subjects exposed to developmental alcohol exposure did increase the time spent in the open arms of the elevated plus maze. Ethanol exposure itself also reduced grooming behaviors, an ambiguous behavior as it has been interpreted as a representation of increased or decreased anxiety levels (Rodgers & Dalvi, 1997). In contrast, subjects exposed to CP did increase the time spent in the open arms in the elevated plus maze, suggesting that developmental cannabinoid exposure leads to reductions in anxiety. Yet CP exposure did not significantly affect time spent in the center of the open field. Importantly, the contexts of the open field and elevated plus maze vary considerably and subjects were tested at different ages. Nevertheless, based on elevated plus maze performance, one could conclude that CP exposure during the third trimester equivalent reduces anxiety, although given behaviors in the open field, one must be cautious of making firm conclusions of the effects of both drugs on stress and emotional development.
Moreover, both alcohol and CP, by themselves, increased thigmotaxis in the spatial learning task. The combination produced additive effects, as subjects exposed to both drugs were more thigmotaxic than subjects exposed to only one drug. Thigmotaxis has classically been described as increased fear or anxiety (Barnett, 1963), but more recent research suggests it may also represent a cognitive impairment of selection-response (Devan, McDonald, & White, 1999). Given the results of the open-field activity and elevated plus maze tasks, where drug-exposed subjects spend more time in open areas, the increased thigmotaxis among subjects could suggest a specific difference in cognitive strategy during the spatial learning task.
Interestingly, exposure to alcohol, and not CP, during the third trimester equivalent impaired spatial learning and memory, producing longer path lengths to find the platform and larger heading angles during acquisition, as well as impaired performance on the probe trial. This spatial learning deficit is consistent with past literature from our lab and others (Berman & Hannigan, 2000; Girard, Xing, Ward, & Wainwright, 2000; Thomas et al., 2007). Subjects exposed to cannabinoids during development did not show these same impairments; this is not consistent with some past literature suggesting that prenatal THC can impair spatial learning and memory (O’shea & Mallet, 2005), although a different task was used in that study. It is also somewhat surprising, given the high density of cannabinoid receptors in the hippocampus. One possibility is that the spatial learning task was tested at the oldest age and the effects of CP may not be long-lasting. Nevertheless, the current study found no evidence that early postnatal exposure to cannabinoids influences spatial memory, nor that cannabinoids exacerbate impairments related to developmental alcohol exposure.
Developmental alcohol exposure decreased body weights throughout behavioral testing. Although cannabinoid exposure, by itself, did not significantly affect body growth, it did exacerbate ethanol-related body weight reductions up through subjects’ early adolescent periods (PD 25), but did not have lasting effects. This is consistent with past clinical (Riley et al., 2011) and preclinical data (Ryan, Williams, & Thomas, 2008; Thomas et al., 2009; Thomas, Garrison, & O’Neill, 2004) showing that prenatal alcohol exposure can have long-lasting effects on body weight, whereas the effects of prenatal cannabis exposure on physical development have been described as subtle and/or brief in past clinical reviews (Day et al., 1991; Day & Richardson, 1991; Fergusson, Horwood, & Northstone, 2002; Fried & O’Connell, 1987; Huizink, 2014; Hurd et al., 2005). Similarly, developmental alcohol exposure led to long-lasting reductions in brain weight, whereas developmental cannabinoid exposure had no significant effects. Brain weights are a gross measure of neuropathology; however, they indicate how devastating early alcohol may be on brain development. Relatively little is known of the effects of prenatal cannabis exposure on more precise measures of neuropathology, but is desperately needed. Future research would benefit from detailed neuropathological analyses following combined prenatal alcohol and cannabinoid exposure, as past research has shown that the combination can be neurotoxic when either drug alone is not (Hansen et al., 2008).
The current study found limited overall sex differences in behavioral development, and these differences were independent of developmental alcohol or cannabinoid exposure. Previous research has shown sex-dependent effects of developmental alcohol exposure can be found in spatial learning (Goodlett & Peterson, 1995; Kelly, Goodlett, Hulsether, & West, 1988; Zimmerberg, Sukel, & Stekler, 1991), stress responsivity (Weinberg, 1992; Weinberg, Sliwowska, Lan, & Hellemans, 2008), and neuropathology (Barron, Tiernan, & Riley, 1988; Zimmerberg & Mickus, 1990; Zimmerberg & Scalzi, 1989), although the patterns have been inconsistent and often sex interactions are not seen (Thomas et al., 2004; Thomas et al., 2009; Thomas, La Fiette, Quinn, & Riley, 2000). Similarly, sex-dependent effects of prenatal cannabis exposure have also been reported on emotional behaviors, motor performance, and physiology (Biscaia et al., 2003; Navarro, Rubio, & Rodríguez de Fonseca, 1994; Pérez-Rosado, Manzanares, Fernández-Ruiz, & Ramos, 2000; Rubino et al., 2008). In general, these studies suggest that females may be more sensitive to the effects of prenatal drug exposure. Although females performed worse on the Morris water maze, regardless of exposure group, no other sex differences were observed on behavior.
Importantly, there are some limitations to the present study. First, the present study exposed rats to CP-55,940, a synthetic cannabinoid. Although currently available for consumption in clinical populations, results may not necessarily generalize to all other cannabinoids. On the one hand, CP has many similarities to THC. Although CP is more potent than THC, the two substances have similar peak effects, durations of action, and neurobehavioral effects (McGregor et al., 1996). Moreover, cross-tolerance can develop between THC and CP (Fan, Compton, Ward, Melvin, & Martin, 1994), indicating that these substances share common characteristics. However, although CP-55,940 is a cannabinoid receptor agonist similar to THC, it is not structurally chemically identical and there may be differences partially due to CP’s higher affinity for CB1 and CB2 receptors (Fantegrossi, Moran, Radominska-Pandya, & Prather, 2014) and shorter half-life (Fouda, Lukaszewicz, & Luther, 1987; Grotenhermen, 2003). Moreover, natural cannabis products contain numerous additional constituents, including cannabidiol and cannabivarin (ElSohly & Slade, 2005), which could have additional effects on development. Thus, the consequences of early cannabinoid exposure may vary depending on the particular drug and constituent combination consumed.
A second limitation is the examination of single doses of both CP and ethanol. The present study was aimed to determine how exposure to moderate/high levels of each drug affected behavioral outcome. The dose of CP was chosen to reflect the amount of cannabinoids consumed by women of childbearing age in the general population (Botticelli, 2017) and is based on doses used in other studies, including developmental (Gilbert et al., 2016) and adult (Gilbert et al., 2016; LaFleur et al., 2018; Maguire & France, 2016; Minervini et al., 2017) CP exposure. The dose of ethanol used in the current study is a well-established model of moderate/high binge-like third trimester exposure (Idrus, McGough, Spinetta, Thomas, & Riley, 2011; Klintsova et al., 2002; Ryan et al., 2008; Thomas et al., 2000; Thomas et al., 2007). Given the robust effects of ethanol, it is possible that more subtle synergistic effects of ethanol and cannabinoid exposure may be masked, effects that could be evident with a lower dose of ethanol. For example, combined exposure to cannabinoids and alcohol during early postnatal development has been shown to be synergistically neurotoxic, when each drug is administered at a sub-threshold level (Hansen et al., 2008). Future studies should determine how the combinations of various doses of ethanol and/or cannabinoids affect behavioral development, including sub-threshold doses of each drug. Nevertheless, the present data illustrate that cannabinoid exposure, by itself, can alter behavioral development and that the combination may produce more severe effects on some outcome measures.
Indeed, we have found that the addition of CP with ethanol can exacerbate other outcome measures (Breit et al., 2019). For example, CP, by itself, had no significant effect on mortality rates or body weights during drug exposure (PD 4–9). Nevertheless, when combined with ethanol, it did exacerbate ethanol’s effects, even though maternal behavior was not affected (dams and pups were videotaped) nor were the presence of milkbands reduced in the pups. This is consistent with the exacerbation of ethanol-related body weight reductions seen up to PD 25 in the present study, although there was catch-up by PD 40. CP also increased peak blood alcohol concentrations (BACs), but only in females, one of the few sex differences we have found (BACs: EtOH males = 274 ± 22 mg/dL; EtOH + CP males = 285 ± 26 mg/dL; EtOH females = 279 ± 15 mg/dL; EtOH + CP females = 348 ± 17 mg/dL). Consistent with increased peak BACs, long-lasting motor coordination deficits (tested at PD 30–32) associated with third trimester equivalent ethanol were more severe in females co-exposed to ethanol and CP. However, we have not seen any other interactions between sex and drug exposures. It is somewhat surprising that we did not see more synergistic effects of CP and ethanol on the present behaviors, particularly among females.
It is also important to acknowledge that the present study utilized a within-subjects design, where all subjects performed each behavioral task, and each test occurred at different ages. Age of testing was based on the ontogeny of the behavior and the developmental ages at which these tasks are sensitive to early alcohol exposure (Idrus et al., 2011; Osborn, Kim, Steiger, & Weinberg, 1998; Ryan et al., 2008) and the testing order was designed to minimize carryover effects. However, it is possible that variation of outcomes across behavioral tasks may be affected by age of testing and future studies will need to determine if the task-specific effects of CP alone and in combination with ethanol depend on domain specificity and/or permanence of effects. Nevertheless, the present data do indicate that cannabinoid exposure may disrupt behavioral development by itself and may exacerbate some behavioral alterations induced by developmental alcohol exposure.
5 |. CONCLUSIONS
In conclusion, the current study suggests that exposure to alcohol and cannabinoids during a period of development equivalent to late gestation may have individual, additive, and synergistic effects on behavioral development, depending on behavioral outcome. Only developmental alcohol exposure impaired spatial learning and produced gross brain pathology. In contrast, developmental cannabinoid exposure increased time spent in the open arms of the plus maze. However, both drugs increased open field activity levels and increased thigmotaxis in the water maze, with largely additive consequences. Importantly, combined exposure to alcohol and cannabinoids may specifically impair simple learning processes, such as habituation, in a synergistic manner. These results show that cannabinoids and the combination of ethanol and cannabinoids can produce severe behavioral alterations, findings that have important implications for individuals exposed to both alcohol and marijuana during gestation, including an increased risk of behavioral problems later in life.
ACKNOWLEDGMENTS
Supported by National Institute on Alcohol Abuse and Alcoholism grant AA025425 to Dr. Thomas and an NIH Loan Repayment Program award to Dr. Breit. Special thanks to Dr. Nirelia Idrus, Cristina Rodriguez, and the Center for Behavioral Teratology at San Diego State University for assisting in data collection and interpretation.
REFERENCES
- Abel E, & Subramanian M (1990). Effects of low doses of alcohol on delta-9-tetrahydrocannabinol’s effects in pregnant rats. Life Sciences, 47(18), 1677–1682. [DOI] [PubMed] [Google Scholar]
- Abel E, Tan S, & Subramanian M (1987). Effects of Δ9-tetrahydrocannabinol, phenobarbital, and their combination on pregnancy and offspring in rats. Teratology, 36(2), 193–198. [DOI] [PubMed] [Google Scholar]
- Abel EL, Rockwood GA, & Riley EP (1986). The effects of early marijuana exposure In Handbook of Behavioral Teratology (pp. 267–288). Boston, MA: Springer. [Google Scholar]
- Barnett SA (1963). The rat: A study in behavior. Chicago, IL: Routledge. [Google Scholar]
- Barron S, Tiernan SB, & Riley EP (1988). Effects of prenatal alcohol exposure on the sexually dimorphic nucleus of the preoptic area of the hypothalamus in male and female rats. Alcoholism: Clinical and Experimental Research, 12(1), 59–64. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS (2015). Fetal alcohol spectrum disorder: Potential role of endocannabinoids signaling. Brain Sciences, 5(4), 456–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Ninan I, & Arancio O (2008). Acute ethanol suppresses glutamatergic neurotransmission through endocannabinoids in hippocampal neurons. Journal of Neurochemistry, 107(4), 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belue RC, Howlett AC, Westlake TM, & Hutchings DE (1995). The ontogeny of cannabinoid receptors in the brain of postnatal and aging rats. Neurotoxicology and Teratology, 17(1), 25–30. [DOI] [PubMed] [Google Scholar]
- Berkovitz R, Arieli M, & Marom E (2011). Synthetic cannabinoids–The new legal high drugs. Harefuah, 150(12), 884–887, 937. [PubMed] [Google Scholar]
- Berman RF, & Hannigan JH (2000). Effects of prenatal alcohol exposure on the hippocampus: Spatial behavior, electrophysiology, and neuroanatomy. Hippocampus, 10(1), 94–110. [DOI] [PubMed] [Google Scholar]
- Berrendero F, Sepe N, Ramos JA, Di Marzo V, & Fernández-Ruiz JJ (1999). Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse, 33(3), 181–191. [DOI] [PubMed] [Google Scholar]
- Bhuvaneswar CG, Chang G, Epstein LA, & Stern TA (2007). Alcohol use during pregnancy: Prevalence and impact. Primary Care Companion to the Journal of Clinical Psychiatry, 9(6), 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscaia M, Marín S, Fernández B, Marco EM, Rubio M, Guaza C, … Viveros MP (2003). Chronic treatment with CP 55,940 during the peri-adolescent period differentially affects the behavioural responses of male and female rats in adulthood. Psychopharmacology, 170(3), 301–308. 10.1007/s00213-003-1550-7 [DOI] [PubMed] [Google Scholar]
- Borgen LA, Davis WM, & Pace HB (1973). Effects of prenatal Δ9-tetrahydrocannabinol on the development of rat offspring. Pharmacology Biochemistry and Behavior, 1(2), 203–206. [Google Scholar]
- Botticelli M (2016). National drug control strategy: Data supplement 2015. [Google Scholar]
- Botticelli M (2017). National drug control strategy: Data supplement 2016. [Google Scholar]
- Breit KR, Zamudio B, & Thomas JD (2019). Altered motor development following late gestational alcohol and cannabinoid exposure in rats. 10.1101/513713. [DOI] [PMC free article] [PubMed]
- Centers for Disease Control and Prevention. (2015). Fetal alcohol spectrum disorders (FASD). [Google Scholar]
- D’Alterio S, & Bartke A (1979). Perinatal exposure to cannabinoids alters male reproductive function in mice. Science, 205(4413), 1420–1422. [DOI] [PubMed] [Google Scholar]
- Day N, Sambamoorthi U, Taylor P, Richardson G, Robles N, Jhon Y, … Jasperse D (1991). Prenatal marijuana use and neonatal outcome. Neurotoxicology and Teratology, 13(3), 329–334. [DOI] [PubMed] [Google Scholar]
- Day NL, & Richardson GA (1991). Prenatal marijuana use: Epidemiology, methodologic issues, and infant outcome. Clinics in Perinatology, 18(1), 77–91. [PubMed] [Google Scholar]
- de Salas-Quiroga A, Díaz-Alonso J, García-Rincón D, Remmers F, Vega D, Gómez-Cañas M, … Galve-Roperh I (2015). Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proceedings of the National Academy of Sciences, 112(44), 13693–13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers NA, Himes SK, Scheidweiler KB, Concheiro-Guisan M, Gorelick DA, & Huestis MA (2014). Phase I and II cannabinoid disposition in blood and plasma of occasional and frequent smokers following controlled smoked cannabis. Clinical Chemistry, 60(4), 1–13. [DOI] [PubMed] [Google Scholar]
- Devan B, McDonald R, & White N (1999). Effects of medial and lateral caudate-putamen lesions on place-and cue-guided behaviors in the water maze: Relation to thigmotaxis. Behavioural Brain Research, 100(1–2), 5–14. [DOI] [PubMed] [Google Scholar]
- Dobbing J, & Sands J (1979). Comparative aspects of the brain growth spurt. Early Human Development, 3(1), 79–83. [DOI] [PubMed] [Google Scholar]
- Ebrahim SH, & Gfroerer J (2003). Pregnancy-related substance use in the United States during 1996–1998. Obstetrics & Gynecology, 101(2), 374–379. [DOI] [PubMed] [Google Scholar]
- El Marroun H, Hudziak JJ, Tiemeier H, Creemers H, Steegers EA, Jaddoe VW, … Huizink AC (2011). Intrauterine cannabis exposure leads to more aggressive behavior and attention problems in 18-month-old girls. Drug and Alcohol Dependence, 118(2–3), 470–474. [DOI] [PubMed] [Google Scholar]
- El Marroun H, Tiemeier H, Steegers EA, Jaddoe VW, Hofman A, Verhulst FC, … Huizink AC (2009). Intrauterine cannabis exposure affects fetal growth trajectories: The generation r study. Journal of the American Academy of Child & Adolescent Psychiatry, 48(12), 1173–1181. [DOI] [PubMed] [Google Scholar]
- ElSohly MA, & Slade D (2005). Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sciences, 78(5), 539–548. [DOI] [PubMed] [Google Scholar]
- Fan F, Compton DR, Ward S, Melvin L, & Martin BR (1994). Development of cross-tolerance between delta 9-tetrahydrocannabinol, CP 55,940 and WIN 55,212. Journal of Pharmacology and Experimental Therapeutics, 271(3), 1383–1390. [PubMed] [Google Scholar]
- Fantegrossi WE, Moran JH, Radominska-Pandya A, & Prather PL (2014). Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to Δ9-THC: Mechanism underlying greater toxicity? Life Sciences, 97(1), 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar CE, Breivogel CS, Gamage TF, Gay EA, Thomas BF, Craft RM, & Wiley JL (2019). Sex, THC, and hormones: Effects on density and sensitivity of CB1 cannabinoid receptors in rats. Drug and Alcohol Dependence, 194, 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergusson DM, Horwood LJ, & Northstone K (2002). Maternal use of cannabis and pregnancy outcome. BJOG: An International Journal of Obstetrics & Gynaecology, 109(1), 21–27. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Berrendero F, Hernández ML, & Ramos JA (2000). The endogenous cannabinoid system and brain development. Trends in Neurosciences, 23(1), 14–20. [DOI] [PubMed] [Google Scholar]
- Fish EW, Boschen KE, Murdaugh LB, Mendoze-Romero HN, Williams K, & Parnell SE (2017). Alcohol exacerbates the teratogenic effects of prenatal cannabinoid exposure in a C57BL/6J mouse model. Birth Defects Research, 109(9), 688–688. [Google Scholar]
- Fish EW, Gilbert MT, Sulik KK, & Parnell SE (2016). Ethanol and the synthetic cannabinoid, CP-55,940, are synergistically teratogenic in a mouse model. Alcoholism: Clinical and Experimental Research, 40(S1), 287A–287A. [Google Scholar]
- Fouda HG, Lukaszewicz J, & Luther EW (1987). Selected ion monitoring analysis of CP-55,940, a cannabinoid derived analgetic agent. Biomedical & Environmental Mass Spectrometry, 14(11), 599–602. [DOI] [PubMed] [Google Scholar]
- Fride E, & Mechoulam R (1996). Ontogenetic development of the response to anandamide and Δ9-tetrahydrocannabinol in mice. Developmental Brain Research, 95(1), 131–134. [DOI] [PubMed] [Google Scholar]
- Fried P, & O’Connell C (1987). A comparison of the effects of prenatal exposure to tobacco, alcohol, cannabis and caffeine on birth size and subsequent growth. Neurotoxicology and Teratology, 9(2), 79–85. [DOI] [PubMed] [Google Scholar]
- Fried P, & Watkinson B (1988). 12-and 24-month neurobehavioural follow-up of children prenatally exposed to marihuana, cigarettes and alcohol. Neurotoxicology and Teratology, 10(4), 305–313. [DOI] [PubMed] [Google Scholar]
- Fried PA, O’Connell CM, & Watkinson B (1992). 60-and 72-month follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol: Cognitive and language assessment. Journal of Developmental & Behavioral Pediatrics, 13(6), 383–391. [PubMed] [Google Scholar]
- Fried PA, & Watkinson B (1990). 36-and 48-month neurobehavioral follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol. Journal of Developmental & Behavioral Pediatrics, 11(2), 49–58. [PubMed] [Google Scholar]
- Gauda EB (2006). Knowledge gained from animal studies of the fetus and newborn: Application to the human premature infant. ILAR Journal, 47 (1), 1–4. [Google Scholar]
- Gilbert MT, Sulik KK, Fish EW, Baker LK, Dehart DB, & Parnell SE (2016). Dose-dependent teratogenicity of the synthetic cannabinoid CP-55,940 in mice. Neurotoxicology and Teratology, 58, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard T, Xing HC, Ward G, & Wainwright P (2000). Early postnatal ethanol exposure has long-term effects on the performance of male rats in a delayed matching-to-place task in the Morris water maze. Alcoholism: Clinical and Experimental Research, 24(3), 300–306. [PubMed] [Google Scholar]
- Glass L, Graham DM, Deweese BN, Jones KL, Riley EP, & Mattson SN (2014). Correspondence of parent report and laboratory measures of inattention and hyperactivity in children with heavy prenatal alcohol exposure. Neurotoxicology and Teratology, 42, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L, Day NL, & Richardson GA (2000). Effects of prenatal marijuana exposure on child behavior problems at age 10. Neurotoxicology and Teratology, 22(3), 325–336. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Richardson GA, Cornelius MD, & Day NL (2004). Prenatal marijuana and alcohol exposure and academic achievement at age 10. Neurotoxicology and Teratology, 26(4), 521–532. [DOI] [PubMed] [Google Scholar]
- Gómez M, Hernández M, Johansson B, De Miguel R, Ramos JA, & Fernández-Ruiz J (2003). Prenatal cannabinoid exposure and gene expression for neural adhesion molecule L1 in the fetal rat brain. Developmental Brain Research, 147(1), 201–207. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, & Johnson TB (1997). Neonatal binge ethanol exposure using intubation: Timing and dose effects on place learning. Neurotoxicology and Teratology, 19(6), 435–446. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, & Peterson SD (1995). Sex differences in vulnerability to developmental spatial learning deficits induced by limited binge alcohol exposure in neonatal rats. Neurobiology of Learning and Memory, 64(3), 265–275. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Thomas JD, & West JR (1991). Long-term deficits in cerebellar growth and rotarod performance of rats following “binge-like” alcohol exposure during the neonatal brain growth spurt. Neurotoxicology and Teratology, 13(1), 69–74. [DOI] [PubMed] [Google Scholar]
- Grotenhermen F (2003). Pharmacokinetics and pharmacodynamics of cannabinoids. Clinical Pharmacokinetics, 42(4), 327–360. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Krutz B, Sifringer M, Stefovska V, Bittigau P, Pragst F, … Ikonomidou C (2008). Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Annals of Neurology, 64(1), 42–52. [DOI] [PubMed] [Google Scholar]
- Hofman A, Jaddoe VW, Mackenbach JP, Moll HA, Snijders RF, Steegers EA, … Büller HA (2004). Growth, development and health from early fetal life until young adulthood: The generation r study. Paediatric and Perinatal Epidemiology, 18(1), 61–72. [DOI] [PubMed] [Google Scholar]
- Huizink A (2014). Prenatal cannabis exposure and infant outcomes: Overview of studies. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 52, 45–52. [DOI] [PubMed] [Google Scholar]
- Huizink AC, & Mulder EJ (2006). Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neuroscience & Biobehavioral Reviews, 30(1), 24–41. [DOI] [PubMed] [Google Scholar]
- Hurd Y, Wang X, Anderson V, Beck O, Minkoff H, & Dow-Edwards D (2005). Marijuana impairs growth in mid-gestation fetuses. Neurotoxicology and Teratology, 27(2), 221–229. [DOI] [PubMed] [Google Scholar]
- Idrus NM, McGough NNH, Spinetta MJ, Thomas JD, & Riley EP (2011). The effects of a single memantine treatment on behavioral alterations associated with binge alcohol exposure in neonatal rats. Neurotoxicology and Teratology, 33(4), 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaddoe VW, Van Duijn CM, Franco OH, Van Der Heijden AJ, Van IIzendoorn MH, De Jongste JC, … Raat H (2012). The generation R study: Design and cohort update 2012. European Journal of Epidemiology, 27(9), 739–756. [DOI] [PubMed] [Google Scholar]
- Jaddoe VW, van Duijn CM, van der Heijden AJ, Mackenbach JP, Moll HA, Steegers EA, … Hofman A (2010). The generation R study: Design and cohort update 2010. European Journal of Epidemiology, 25(11), 823–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadi-Paydar M, Nguyen JD, Kerr TM, Grant Y, Vandewater SA, Cole M, & Taffe MA (2018). Effects of Δ9-THC and cannabidiol vapor inhalation in male and female rats. Psychopharmacology, 235(9), 2541–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SJ, Goodlett CR, Hulsether SA, & West JR (1988). Impaired spatial navigation in adult female but not adult male rats exposed to alcohol during the brain growth spurt. Behavioural Brain Research, 27(3), 247–257. [DOI] [PubMed] [Google Scholar]
- Khoury JE, Milligan K, & Girard TA (2015). Executive functioning in children and adolescents prenatally exposed to alcohol: A meta-analytic review. Neuropsychology Review, 25(2), 149–170. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Scamra C, Hoffman M, Napper RM, Goodlett CR, & Greenough WT (2002). Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats: II. A quantitative stereological study of synaptic plasticity in female rat cerebellum. Brain Research, 937(1–2), 83–93. [DOI] [PubMed] [Google Scholar]
- LaFleur RA, Wilson RP, Morgan DJ, & Henderson-Redmond AN (2018). Sex differences in antinociceptive response to Δ−9-tetrahydrocannabinol and CP 55,940 in the mouse formalin test. Neuroreport, 29(6), 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S, Shield K, Koren G, Rehm J, & Popova S (2014). A comparison of the prevalence of prenatal alcohol exposure obtained via maternal self-reports versus meconium testing: A systematic literature review and meta-analysis. BMC Pregnancy and Childbirth, 14(1), 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendoiro E, González-Colmenero E, Concheiro-Guisán A, de Castro A, Cruz A, López-Rivadulla M, & Concheiro M (2013). Maternal hair analysis for the detection of illicit drugs, medicines, and alcohol exposure during pregnancy. Therapeutic Drug Monitoring, 35(3), 296–304. [DOI] [PubMed] [Google Scholar]
- Maguire DR, & France CP (2016). Additive antinociceptive effects of mixtures of the kappa opioid receptor agonist spiradoline and the cannabinoid receptor agonist CP55940 in rats. Behavioural Pharmacology, 27(1), 69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson SN, Crocker N, & Nguyen TT (2011). Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychology Review, 21(2), 81–101. 10.1007/s11065-011-9167-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor IS, Issakidis CN, & Prior G (1996). Aversive effects of the synthetic cannabinoid CP-55,940 in rats. Pharmacology Biochemistry and Behavior, 53(3), 657–664. [DOI] [PubMed] [Google Scholar]
- Mehmedic Z, Chandra S, Slade D, Denham H, Foster S, Patel AS, … ElSohly MA (2010). Potency trends of Δ9THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. Journal of Forensic Sciences, 55(5), 1209–1217. [DOI] [PubMed] [Google Scholar]
- Mereu G, Fà M, Ferraro L, Cagiano R, Antonelli T, Tattoli M, … Cuomo V (2003). Prenatal exposure to a cannabinoid agonist produces memory deficits linked to dysfunction in hippocampal long-term potentiation and glutamate release. Proceedings of the National Academy of Sciences, 100(8), 4915–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midanik LT (1988). Validity of self-reported alcohol use: A literature review and assessment. British Journal of Addiction, 83(9), 1019–1029. [DOI] [PubMed] [Google Scholar]
- Minervini V, Dahal S, & France CP (2017). Behavioral characterization of κ opioid receptor agonist spiradoline and cannabinoid receptor agonist CP55940 mixtures in rats. Journal of Pharmacology and Experimental Therapeutics, 360(2), 280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EM, Migliorini R, Infante MA, & Riley EP (2014). Fetal alcohol spectrum disorders: Recent neuroimaging findings. Current Developmental Disorders Reports, 1(3), 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagre NN, Subbanna S, Shivakumar M, Psychoyos D, & Basavarajappa BS (2015). CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT 1, DNMT 3A, and DNA methylation. Journal of Neurochemistry, 132(4), 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro M, Rubio P, & Rodríguez de Fonseca F (1994). Sex-dimorphic psychomotor activation after perinatal exposure to (−)-Δ9-tetrahydrocannabinol. An ontogenic study in wistar rats. Psychopharmacology, 116(4), 414–422. 10.1007/bf02247471 [DOI] [PubMed] [Google Scholar]
- Nguyen JD, Aarde SM, Vandewater SA, Grant Y, Stouffer DG, Parsons LH, … Taffe MA (2016). Inhaled delivery of Δ9-tetrahydrocannabinol (THC) to rats by e-cigarette vapor technology. Neuropharmacology, 109, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AL, O’brien JW, Spadoni AD, Tapert SF, Jones KL, Riley EP, & Mattson SN (2013). A functional magnetic resonance imaging study of spatial working memory in children with prenatal alcohol exposure: Contribution of familial history of alcohol use disorders. Alcoholism: Clinical and Experimental Research, 37(1), 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’shea M, & Mallet P (2005). Impaired learning in adulthood following neonatal Δ9-THC exposure. Behavioural Pharmacology, 16(5–6), 455–461. [DOI] [PubMed] [Google Scholar]
- Olney JW, Wozniak DF, Jevtovic-Todorovic V, Farber NB, Bittigau P, & Ikonomidou C (2002). Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathology, 12(4), 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn J, Kim C, Steiger J, & Weinberg J (1998). Prenatal ethanol exposure differentially alters behavior in males and females on the elevated plus maze. Alcoholism: Clinical and Experimental Research, 22(3), 685–696. [PubMed] [Google Scholar]
- Pérez-Rosado A, Manzanares J, Fernández-Ruiz J, & Ramos JA (2000). Prenatal Δ9-tetrahydrocannabinol exposure modifies proenkephalin gene expression in the fetal rat brain: Sex-dependent differences. Developmental Brain Research, 120(1), 77–81. [DOI] [PubMed] [Google Scholar]
- Prut L, & Belzung C (2003). The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. European Journal of Pharmacology, 463(1–3), 3–33. [DOI] [PubMed] [Google Scholar]
- Richardson GA, Ryan C, Willford J, Day NL, & Goldschmidt L (2002). Prenatal alcohol and marijuana exposure: Effects on neuropsychological outcomes at 10 years. Neurotoxicology and Teratology, 24(3), 309–320. [DOI] [PubMed] [Google Scholar]
- Riley EP, Infante MA, & Warren KR (2011). Fetal alcohol spectrum disorders: An overview. Neuropsychology Review, 21(2), 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers R, & Dalvi A (1997). Anxiety, defence and the elevated plus-maze. Neuroscience & Biobehavioral Reviews, 21(6), 801–810. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Fonseca F, Ramos JA, Bonnin A, & Fernández-Ruiz JJ (1993). Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport, 4(2), 135–138. [DOI] [PubMed] [Google Scholar]
- Roozen S, Peters GJY, Kok G, Townend D, Nijhuis J, & Curfs L (2016). Worldwide prevalence of fetal alcohol spectrum disorders: A systematic literature review including meta-analysis. Alcoholism: Clinical and Experimental Research, 40(1), 18–32. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Guidali C, Braida D, Capurro V, Castiglioni C, … Sala M (2008). Chronic Δ 9-tetrahydrocannabinol during adolescence provokes sex-dependent changes in the emotional profile in adult rats: Behavioral and biochemical correlates. Neuropsychopharmacology, 33(11), 2760–2771. [DOI] [PubMed] [Google Scholar]
- Ryan SH, Williams JK, & Thomas JD (2008). Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: Effects of varying the timing of choline administration. Brain Research, 1237, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint Louis C (2017). Pregnant women turn to marijuana, perhaps harming infants. The New York Times. Retrieved from https://www.nytimes.com/2017/02/02/health/marijuana-and-pregnancy.html?emc=eta1&_r=0 [Google Scholar]
- Schneider M (2009). Cannabis use in pregnancy and early life and its consequences: Animal models. European Archives of Psychiatry and Clinical Neuroscience, 259(7), 383–393. [DOI] [PubMed] [Google Scholar]
- Schneider ML, Moore CF, & Adkins MM (2011). The effects of prenatal alcohol exposure on behavior: Rodent and primate studies. Neuropsychology Review, 21(2), 186–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Fried PA, Hogan MJ, & Cameron I (2006). Effects of prenatal marijuana on visuospatial working memory: An fMRI study in young adults. Neurotoxicology and Teratology, 28(2), 286–295. [DOI] [PubMed] [Google Scholar]
- Subbanna S, Shivakumar M, Psychoyos D, Xie S, & Basavarajappa BS (2013). Anandamide–CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic, and memory deficits. Journal of Neuroscience, 33(15), 6350–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbaraman MS, & Kerr WC (2015). Simultaneous versus concurrent use of alcohol and cannabis in the national alcohol survey. Alcoholism: Clinical and Experimental Research, 39(5), 872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. (2015). Behavioral health trends in the United States: Results from the 2014 national survey on drug use and health. [Google Scholar]
- Substance Use and Mental Health Services Administration. (2014). Results from the 2013 national survey on drug use and health: Summary of national findings. [Google Scholar]
- Taffe MA, Creehan KM, & Vandewater SA (2015). Cannabidiol fails to reverse hypothermia or locomotor suppression induced by Δ9-tetrahydrocannabinol in sprague-dawley rats. British Journal of Pharmacology, 172(7), 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai S, & Fantegrossi WE (2014). Synthetic cannabinoids: Pharmacology, behavioral effects, and abuse potential. Current Addiction Reports, 1(2), 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PA, Jacobson SW, van der Kouwe A, Molteno CD, Chen G, Wintermark P, … Meintjes EM (2015). A DTI-based tractography study of effects on brain structure associated with prenatal alcohol exposure in newborns. Human Brain Mapping, 36(1), 170–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Abou EJ, & Dominguez HD (2009). Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicology and Teratology, 31(5), 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Biane JS, O’bryan KA, O’neill TM, & Dominguez HD (2007). Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behavioral Neuroscience, 121(1), 120–130. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garrison M, & O’Neill TM (2004). Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicology and Teratology, 26(1), 35–45. [DOI] [PubMed] [Google Scholar]
- Thomas JD, La Fiette MH, Quinn VR, & Riley EP (2000). Neonatal choline supplementation ameliorates the effects of prenatal alcohol exposure on a discrimination learning task in rats. Neurotoxicology and Teratology, 22 (5), 703–711. [DOI] [PubMed] [Google Scholar]
- Tran TD, Cronise K, Marino MD, Jenkins WJ, & Kelly SJ (2000). Critical periods for the effects of alcohol exposure on brain weight, body weight, activity and investigation. Behavioural Brain Research, 116(1), 99–110. [DOI] [PubMed] [Google Scholar]
- Trezza V, Campolongo P, Manduca A, Morena M, Palmery M, Vanderschuren L, & Cuomo V (2012). Altering endocannabinoid neurotransmission at critical developmental ages: Impact on rodent emotionality and cognitive performance. Frontiers in Behavioral Neuroscience, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uban K, Herting M, Wozniak J, Sowell E, & CIFASD. (2017). Sex differences in associations between white matter microstructure and gonadal hormones in children and adolescents with prenatal alcohol exposure. Psychoneuroendocrinology, 83, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakley AA, Wiley JL, & Craft RM (2014). Sex differences in antinociceptive tolerance to delta-9-tetrahydrocannabinol in the rat. Drug and Alcohol Dependence, 143, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg J (1992). Prenatal ethanol effects: Sex differences in offspring stress responsiveness. Alcohol, 9(3), 219–223. [DOI] [PubMed] [Google Scholar]
- Weinberg J, Sliwowska JH, Lan N, & Hellemans K (2008). Prenatal alcohol exposure: Foetal programming, the hypothalamic-pituitary-adrenal axis and sex differences in outcome. Journal of Neuroendocrinology, 20(4), 470–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JR, Hamre KM, & Pierce DR (1984). Delay in brain growth induced by alcohol in artificially reared rat pups. Alcohol, 1(3), 213–222. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B, & Mickus LA (1990). Sex differences in corpus callosum: Influence of prenatal alcohol exposure and maternal undernutrition. Brain Research, 537(1), 115–122. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B, & Scalzi LV (1989). Commissural size in neonatal rats: Effects of sex and prenatal alcohol exposure. International Journal of Developmental Neuroscience, 7(1), 81–86. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B, Sukel HL, & Stekler JD (1991). Spatial learning of adult rats with fetal alcohol exposure: Deficits are sex-dependent. Behavioural Brain Research, 42(1), 49–56. [DOI] [PubMed] [Google Scholar]