

Abstract
Background:
Inflammasome-mediated neuroinflammation plays an important role in the pathogenesis of early brain injury (EBI) following subarachnoid hemorrhage (SAH). The activation of the TGR5 receptor has been shown to be neuroprotective in a variety of neurological diseases. This study aimed to investigate the effects of the specific synthetic TGR5 agonist, INT-777, in attenuating NLRP3-ASC inflammasome activation and reducing neuroinflammation after SAH.
Methods:
One hundred and eighty-four male Sprague Dawley rats were used. SAH was induced by the endovascular perforation. INT-777 was administered intranasally at 1 h after SAH induction. To elucidate the signaling pathway involved in the effect of INT-777 on inflammasome activation during EBI, TGR5 knockout CRISPR and PKA inhibitor H89 were administered intracerebroventricularly and intraperitoneally at 48 h and 1 h before SAH. The SAH grade, short- and long-term neurobehavioral assessments, brain water content, western blot, immunofluorescence staining, and Nissl staining were performed.
Results:
The expressions of endogenous TGR5, p-PKA, and NLRP3-ASC inflammasome were increased after SAH. INT-777 administration significantly decreased NLRP3-ASC inflammasome activation in microglia, reduced brain edema and neuroinflammation, leading to improved short-term neurobehavioral functions at 24 h after SAH. The administration of TGR5 CRISPR or PKA inhibitor (H89) abolished the anti-inflammation effects of INT-777, on NLRP3-ASC inflammasome, pro-inflammatory cytokines (IL-6, IL-1β, and TNF-a), and neutrophil infiltration at 24 h after SAH. Moreover, early administration of INT-777 attenuated neuronal degeneration in hippocampus on 28 d after SAH.
Conclusions:
INT-777 attenuated NLRP3-ASC inflammasome-dependent neuroinflammation in the EBI after SAH, partially via TGR5/cAMP/PKA signaling pathway. Early administration of INT-777 may serve as a potential therapeutic strategy for EBI management in the setting of SAH.
Keywords: Subarachnoid hemorrhage, Early brain injury, TGR5, INT-777, NLRP3-ASC inflammasome, Neuroinflammation
1. Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a life-threatening hemorrhagic stroke, accounting for 5–10% of all stroke types, with a mortality and disability rate of 25–50% (Lawton and Vates 2017; Kamp, Steiger, and van Lieshout 2020). Early brain injury (EBI) begins immediately after an aneurysm ruptures, and lasts up to 72 h; EBI has been proven to be the primary cause of poor outcomes after SAH (Cahill, Calvert, and Zhang 2006; Peng, Pang, et al. 2019).
Over the past few decades, researchers have been elucidated the pathological mechanisms underlying EBI including sharply elevated intracranial pressure, decreased cerebral perfusion pressure and cerebral blood flow as well as cerebral vasospasm, all of which consecutively initiate various of cascades of pathophysiological events such as oxidative stress, neuroinflammation, blood-brain barrier (BBB) dysfunction, and apoptosis (Conzen et al. 2019; Koseki et al. 2020). SAH-induced neuroinflammation has been considered one of the key devastating pathophysiological processes in EBI induced by SAH (Pang et al. 2018; Suzuki 2019). Thus, therapeutically targeting neuroinflammation would attenuate EBI and benefit neurological outcomes in the setting of SAH. Previous studies have demonstrated that the brain’s residual microglial/macrophage activation and neutrophil infiltration releases massive pro-inflammatory cytokines which would magnify the inflammatory responses and further aggravate the neurobehavioral deficits and delayed cognitive dysfunctions (Zuo, Huang, et al. 2019; Xu et al. 2020). While microgliosis following neuronal damage is a normal physiologic response to injury, however, persistent and self-propagating microgliosis can result in sustained neurological dysfunctions (Alawieh et al. 2018; Figueiredo et al. 2019; Alawieh et al. 2020). The selective polarization of microglia toward anti-inflammatory phenotype attenuated neuroinflammatory response and white matter injury in EBI after SAH (Peng, Pang, et al. 2019; Zheng et al. 2020).
Inflammasomes are a part of the innate immune system, and plays a central role in microglia activation, which involves multimeric protein complexes sensing various environmental and cellular stress signals (Freeman et al. 2017; Luo, Reis, and Chen 2019). The NLRP3 (NOD-, LRR- and pyrin domain-containing 3) inflammasome activation leads to oligomerization, which can trigger the helical fibrillar assembly of a downstream adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD). ASC results in caspase1 activation and promotes pro-interleukin-1 beta (pro-IL-1β) processing and release of the mature cytokine IL-1β (Latz, Xiao, and Stutz 2013; Broz and Dixit 2016). NLRP3–ASC inflammasome activation plays a central role in microglia activation that induces an increase in cytokine and chemokine concentrations (Heneka, McManus, and Latz 2018; Stancu et al. 2019). Microglial NLRP3-ASC inflammasome activation exacerbates Tau-associated neuroinflammatory changes in Alzheimer’s disease (AD) (Stancu et al. 2019). The inhibition of NLRP3–ASC inflammasome reduces the innate immune response ameliorates neuroinflammation, attenuates neuronal pyroptosis, and delays neuronal degeneration in animal models of diabetic cardiomyopathy (Ye et al. 2017), AD (Venegas et al. 2017; Stancu et al. 2019), cerebellar ataxias (Kojic et al. 2018), neurodegenerative disorders (Gordon et al. 2018), intracerebral hemorrhage (Yang et al. 2018), and ischemic stroke (She et al. 2019). Furthermore, the activation of NLRP3 inflammasome has been demonstrated to take part in neuronal apoptosis following SAH (Dong et al. 2016). The signaling pathway that suppressed NLRP3 inflammasome activation could promote neurogenesis after SAH (Zuo et al. 2018). However, there is currently no research exerting the role of TGR5 targeting the NLRP3-ASC inflammasome-mediated neuroinflammation in EBI after SAH.
Trans-membrane G protein-coupled receptor-5 (TGR5), is a novel membrane-bound bile acid receptor found in the gastrointestinal tract and in immune cells; has pleiotropic actions in metabolic diseases and immunomodulation (Guo et al. 2016; Shapiro et al. 2018). The expression of TGR5 was also reported in neurons, astrocytes, and microglia cells (Keitel et al. 2010). Recent studies have demonstrated that the TGR5 activation reduces BBB breakdown after ischemic stroke (Liang et al. 2020a), alleviates microglial activation in hepatic encephalopathy (McMillin et al. 2015) and acute brain injury (Yanguas-Casas et al. 2017), as well as provides a beneficial effect against neuronal apoptosis and neuroinflammation in an animal model of AD (Wu et al. 2018). On the contrary, the reduction of endogenous TRG5 expressions exacerbated neuroinflammation associated with microglial activation and reduced postsynaptic plasticity (Jena et al. 2018). INT-777, as a novel specific semisynthetic TGR5 agonist, has been shown to alleviate cardiomyocyte injury by inhibiting inflammatory responses and oxidative stress (Deng et al. 2019), attenuated NLRP3 inflammasome-mediated inflammatory bowel diseases (Chen, Le, et al. 2019), mitigated impaired urinary concentration in lithium-induced nephrogenic diabetes insipidus (Wang et al. 2016), and improved cognitive impairment and synaptic dysfunction in mice model of AD (Wu et al. 2018). In our previous study, we have shown that activation of the TGR5 receptor with INT-777 attenuated oxidative stress and neuronal apoptosis in a rat model of endovascular perforation model of SAH (Zuo, Zhang, et al. 2019). Specifically, INT-777 administration significantly increased intracellular cAMP levels and the phosphorylation of PKCε (Zuo, Zhang, et al. 2019). An elevation of intracellular cAMP levels can result in the phosphorylation of PKA and further induced the phosphorylation and the ubiquitination of NLRP3, which blocks NLRP3 inflammasome-dependent inflammation and NLRP3 inflammasome-related metabolic disorders (Guo et al. 2016). The inhibition of cAMP or PKA increases bone marrow-derived macrophages-mediated inflammation (Song et al. 2018).
In this present study, we hypothesized that INT-777 would attenuate the neurological deficits and suppress NLRP3-ASC inflammasome dependent neuroinflammation via TGR5/cAMP/PKA signaling pathway in EBI after SAH. Moreover, these beneficial effects of INT-777-attenuated EBI improved delayed neurological impairments after experimental SAH (shown in fig. s1).
2. Materials and methods
2.1. Animals
Adult male Sprague-Dawley rats (n=184; weighting=280–320g; Harlan, Indianapolis, USA) were used in this study. All animals were housed in a room with controlled humidity (60 ±5%) and temperature (25 ±1°C), under a 12 h light and dark cycle with libitum access to food and water. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Loma Linda University, which comply with the National Institutes of Health Guidelines for the Care and Use of laboratory Animals in Neuroscience Research and ARRIVE guidelines.
2.2. Experimental design
Animals were randomly assigned to four separated experiments as shown in Fig. s2.
2.2.1. Experiment 1
We characterized the time course of endogenous expressions of TGR5 receptors, PKA, and NLRP3-ASC inflammasome in the sham group and SAH groups. The rats were randomly divided into six groups (n=6/group): Sham, SAH-3 h, SAH-6 h, SAH-12 h, SAH-24 h, and SAH-72 h. Western blot analysis was performed to assess the protein levels of TGR5, PKA, NLRP3-ASC inflammasome in the ipsilateral (left) hemisphere cerebral cortex. Additionally, the cellular localization of TGR5 with neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP) or calcium-binding adaptor molecule 1 (Iba-1) was evaluated by double immunofluorescence staining in the Sham and SAH-24 h group (n=2/group).
2.2.2. Experiment 2
To evaluate the neuroprotective effects of INT-777 on short-term neurological outcome after SAH, three different dosages of INT-777 were used based on our previous study. Rats were randomly assigned to five groups (n=6/group): Sham, SAH+vehicle (10% dimethyl sulfide), SAH+INT-777 (10 μg/kg), SAH+INT-777 (30 μg/kg), and SAH+INT-777 (90 μg/kg). INT-777 was administrated intranasally (i.n.) at 1 h after SAH. The SAH grading score, neurobehavioral performance (including modified Garcia test and beam balance test) and brain water content were assessed at 24 h after SAH in all groups. Based on the short-term neurological outcomes and brain water content results, the best dose of INT-777 (30 μg/kg) was chosen and used for the following long-term outcome and mechanism experiments.
To explore the effects of INT-777 on microglia/macrophage activation and NLRP3-ASC inflammasome activation at 24 h after SAH, rats were randomly assigned to three groups (n=10/group): Sham, SAH+vehicle (10% dimethyl sulfide), SAH+INT-777 (30 μg/kg). Immunofluorescence staining (n=4/group) of Iba-1 with CD68, NLRP3, and ASC was performed, and CD68, NLRP3, and ASC-positive Iba-1 cells in the peri-hemorrhage area were counted at 24 h after SAH. Western blot (n=6/group) was performed to evaluate NLRP3-ASC inflammasome activation and neuroinflammation in the ipsilateral (left) hemisphere at 24 h after SAH. Brain samples of these three groups were shared with experiment 4.
2.2.3. Experiment 3
To evaluate the effects of INT-777 on long-term neuronal degeneration after SAH, rats were randomly assigned to three groups (n=3/group): Sham, SAH+vehicle (10% dimethyl sulfide), SAH+INT-777 (30 μg/kg). Nissl staining was performed to assess the neuronal degeneration on 28 day after SAH.
2.2.4. Experiment 4
To explore the underlying mechanism of TGR5/cAMP/PKA/NLRP3-ASC signaling pathway underlying the anti-inflammatory effects of INT-777 after SAH, the TGR5 knockout CRISPR was administration by intracerebroventricular injection (i.c.v.) at 48 h before SAH and the selective PKA inhibitor H89 was administration by intraperitoneally (i.p.) at 1 h before SAH, respectively. Rats were randomly assigned to seven groups (n=10/group): Sham, SAH+vehicle (10% dimethyl sulfide, i.n.), SAH+INT-777, SAH+INT-777+TGR5 CRISPR, SAH+INT-777+scrambled CRISPR, SAH+INT-777+H89, and SAH+INT-777+vehicle (10% dimethyl sulfide, i.p.). The ipsilateral (left) hemisphere of each group was collected for western blot analysis (n=6/group) and immunofluorescence staining (n=4/group) after neurological performance and SAH grade evaluation at 24 h after SAH.
2.3. SAH model
Experimental SAH model was performed in rats using the endovascular perforation method as previously described (Peng, Zuo, et al. 2019). Rats were anesthetized with isoflurane (4% induction, 2.5% maintenance). After intubation, the rats were connected to the rodent ventilator (Harvard Apparatus, Holliston, USA) in the supine position with 70% medical air and 30% oxygen gas during surgery (the heart rate, respiration, skin color, and pedal reflex were assessed intraoperatively every 5 minutes to confirm the anesthetic status and prevent distress). After exposing the carotid artery and its bifurcation, a sharp 4–0 monofilament nylon suture was inserted into the left internal carotid artery from the external carotid artery and perforated the bifurcation of the anterior and middle cerebral arteries. Afterward, the nylon suture withdrawn immediately and isoflurane was reduced to 1.5%. After the operation, the animals were pulled out the endotracheal tube and transferred into a heated chamber for recovery, maintained at a temperature of 37.5 °C. Rats in the sham group underwent the same procedures, except for artery vessel wall puncture.
2.4. SAH Grading
The severity of SAH was evaluated according to the SAH grading scale system after euthanasia as previously described (Sugawara et al. 2008). The basal cistern was divided into six segments, each with a score from 0 to 3 according to the subarachnoid blood clot. The total score was calculated by adding the scores of all six segments (maximum SAH grade=18). Rats with a score of 8 or less were excluded from this study.
2.5. Drug administration
INT-777 or vehicle was given by intranasal administration at 1 h after SAH as previously described (Sun et al. 2019). Briefly, animals were anesthetized under 2% isoflurane and placed in a supine position. A total volume of 30 μL vehicle (10% dimethyl sulfide) or INT-777 (MedChem Express, NJ, USA) at three different doses (10 μg/kg, 30 μg/kg, and 90 μg/kg) were administered into the right and left nares, alternating 10 μL in one naris per 5 minutes. H89 was diluted in 10% dimethyl sulfide (DMSO) and was administrated by intraperitoneally (i.p.) at 1 h before SAH.
2.6. Intracerebroventricular drug administration
TGR5 knockout CRISPR (Santa Cruz Biotechnology, Dallas, TX, USA) or scrambled CRISPR was given i.c.v at 48 h before SAH according to our previous study (Zuo, Zhang, et al. 2019). Briefly, animals were placed in a stereotaxic frame under anesthesia with isoflurane (4% induction, 2.5% maintenance) in mixed gases (65% medical air/35% oxygen gas). The needle of a 10 μL Hamilton syringe (Microliter 701, Hamilton Company, USA) was inserted through a burr hole perforated through the skull into the left lateral ventricle at the following coordinates relative to bregma: 0.9 mm posterior, 1.5 mm lateral, and 3.2 mm beneath the horizontal plane of the skull. The rate of i.c.v injection was controlled at a rate of 1 μL/min with an infusion pump (Stoelting, Harvard Apparatus, Holliston, USA). After completing the injection, the needle was kept in situ for 5 minutes and then retracted slowly for 5 minutes. The burr hole was sealed with bone wax immediately after removal of the needle and the rats were awakened from anesthesia after sutures.
2.7. Assessment short-term neurological performance
We calculated the mortality and assessed neurobehavioral outcomes at 24 h after SAH. The short-term neurobehavioral outcomes were assessed blindly using the 18 points modified Garcia scoring system and 4 points beam balance test as previously described (Zuo, Huang, et al. 2019). Higher scores indicated better neurological function.
2.8. Brain water content
Brain edema was assessed by measuring brain water content using wet-dry method as previously described (Zhu et al. 2018). The rats were anesthetized and euthanized at 24 h after SAH, and the brains were quickly removed and separated into four parts (right hemisphere, left hemisphere, cerebellum, and brain stem). Afterwards, each part of the brain was weighed immediately to obtain the wet weight, and then put into an oven at 100 ℃ for 72 h. The dried brain was weighed again. The percentage of brain water content was calculated as follows: (wet weight − dry weight) /wet weight × 100%.
2.9. Immunofluorescence staining
The rats were deeply anesthetized (5% isoflurane), then underwent a trans-cardiac perfusion with 100–150 mL of cold PBS and 100 mL of 10% formalin. Whole brains were rapidly collected and fixed in 10% formalin (4 °C for 24 h), followed by dehydration with 30% sucrose (4 °C for 72 h). Brain samples were embedded into OCT (Scigen Scientific Gardena, CA, USA) and then frozen at − 80 °C. The brains were cut into 8–10 μm thick coronal brain sections using a cryostat (CM3050S, Leica Microsystems, Bannockburn, Germany) and mounted on normal Poly-L-Lysine coated slides. Slices were washed with 0.01 M of PBS three times for 5–10 min then incubated in 0.3% Triton X-100 in 0.01 M of PBS for 10 min at room temperature. After being blocked with 5% donkey serum in 0.01 M of PBS for 2 h at room temperature, the sections were incubated at 4 °C overnight with the following primary antibody: anti-NeuN (1:200, Abcam), anti-GFAP (1:200, Abcam), anti-Iba-1 (1:200, Abcam), anti-TGR5 (1:200, Abcam), anti-CD68 (1:200, Santa Cruz Biotechnology), anti-NLRP3 (1:200, Thermo Fisher Scientific), anti-ASC (1:200, Santa Cruz Biotechnology), rabbit anti-IL-1β (1:200, Abcam), and mouse anti-MPO (1:200, Santa Cruz Biotechnology). Then, the slices were incubated with fluorescence-conjugated secondary antibodies (1:500, Jackson ImmunoResearch) for 1h at room temperature. The slides were visualized and photographed with a fluorescence microscope (DMi8, Leica Microsystems). The numbers of Iba-1-positive cells were identified and counted in three different fields of the left basal cortex from five random coronal sections per rat, and the positive cells were quantified under a microscopic field of 200x magnification; data were expressed as cells/field. To assess the neuroinflammation level, six randomly selected field of views were examined to count IL-1β- and MPO-positive cells under microscopic field of 400x and 200x magnification. The fluorescence intensity was quantified by ImageJ software (ImageJ 1.5, NIH, USA).
2.10. Nissl staining
Nissl staining was performed to evaluate neuronal damage in the hippocampus as previously shown (Ai et al. 2019). The coronal brain slices (15–18 μm thick) were dehydrated in 95% and 70% ethanol for 1 min and then stained with 0.5% cresyl violet (Sigma-Aldrich, MO, USA) for 2 min, followed by dehydration with 100% ethanol and xylene for 1 min. The slices were mounted with DPX (Sigma-Aldrich, USA) and viewed for degenerating neurons in hippocampal regions (CA1-CA3, DG) using a microscope (Olympus, BX51) under microscopic field of 200x magnification. The degenerative neurons were characterized by a shrunken cytoplasm and condensed staining. The mean number of surviving neurons was counted and the neuronal density loss was estimated.
2.11. Western blot analysis
Rats were deeply anesthetized (5% isoflurane) and transcardially perfused with chilled PBS followed by decapitation at 24 h after SAH. The brain sections were separated into ipsilateral and contralateral hemispheres. The ipsilateral hemisphere brain tissues were snap frozen in liquid nitrogen and stored in −80 °C freezer for storage until use. Brain samples were homogenized in RIPA lysis buffer (Santa Cruz Biotechnology) with protease inhibitor for 15 min and then centrifuged at 14,000 g (4 °C, 30 min). The supernatant was collected and protein concentration was measured by detergent compatible assay (DC Protein Assay, Bio-Rad Laboratories). Equal amounts of protein were loaded onto 10% SDS-PAGE gel for electrophoresis and then transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat blocking grade milk (Bio-Rad) for 2 h at 37 °C and incubated overnight at 4 °C with the following primary antibodies: anti-TGR5 (1:1000, Abcam), anti-cAMP (1:1000, Abcam), anti-p-PKA (1:1000, Cell Signaling Technology), anti-PKA (1:1000, Cell Signaling Technology), anti-NLRP3 (1:1000, Thermo Fisher Scientific), anti-ASC (1:1000, Santa Cruz Biotechnology), anti-IL-1β (1:1000, Abcam), anti-IL-6 (1:1000, Abcam), anti-TNF-a (1:1000, Abcam), and anti-β-actin (1:5000, Santa Cruz Biotechnology). The membranes were incubated with the appropriate peroxidase conjugated secondary antibodies (1:5000, Santa Cruz Biotechnology) for 1 h at 37 °C. Bands were then visualized with ECL Plus chemiluminescence reagent kit (Amersham Bioscience, Pittsburgh, PA) and quantified with the ImageJ software (ImageJ 1.5, NIH, USA).
2.12. Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 (Graph Pad Software, San Diego, CA, USA). All data were presented as mean ±SD. One-way ANOVA followed by Tukey’s post hoc test was used for comparison among multiple groups. Two-way ANOVA was used to analyze the long-term neurobehavioral results. P < 0.05 was considered statistically significant.
3. Results
3.1. Expression levels of endogenous TGR5, p-PKA, NLRP3, ASC and cellular locations of TGR5 after SAH
The protein levels of endogenous TGR5, p-PKA, NLRP3, and ASC in ipsilateral (left) hemisphere among groups of sham, 3 h, 6 h, 12 h, 24 h, and 72 h after SAH were measured by western blot. The results showed that the expression of endogenous TGR5, p-PKA, NLRP3, and ASC increased in a time-dependent manner, and peaked at 24 h after SAH (P < 0.05 compared with sham group, Fig. 1A–E).
Figure 1. Time course of TGR5, p-PKA, and NLRP3-ASC inflammasome expressions as well as cellular localization of TGR5 receptor after SAH.
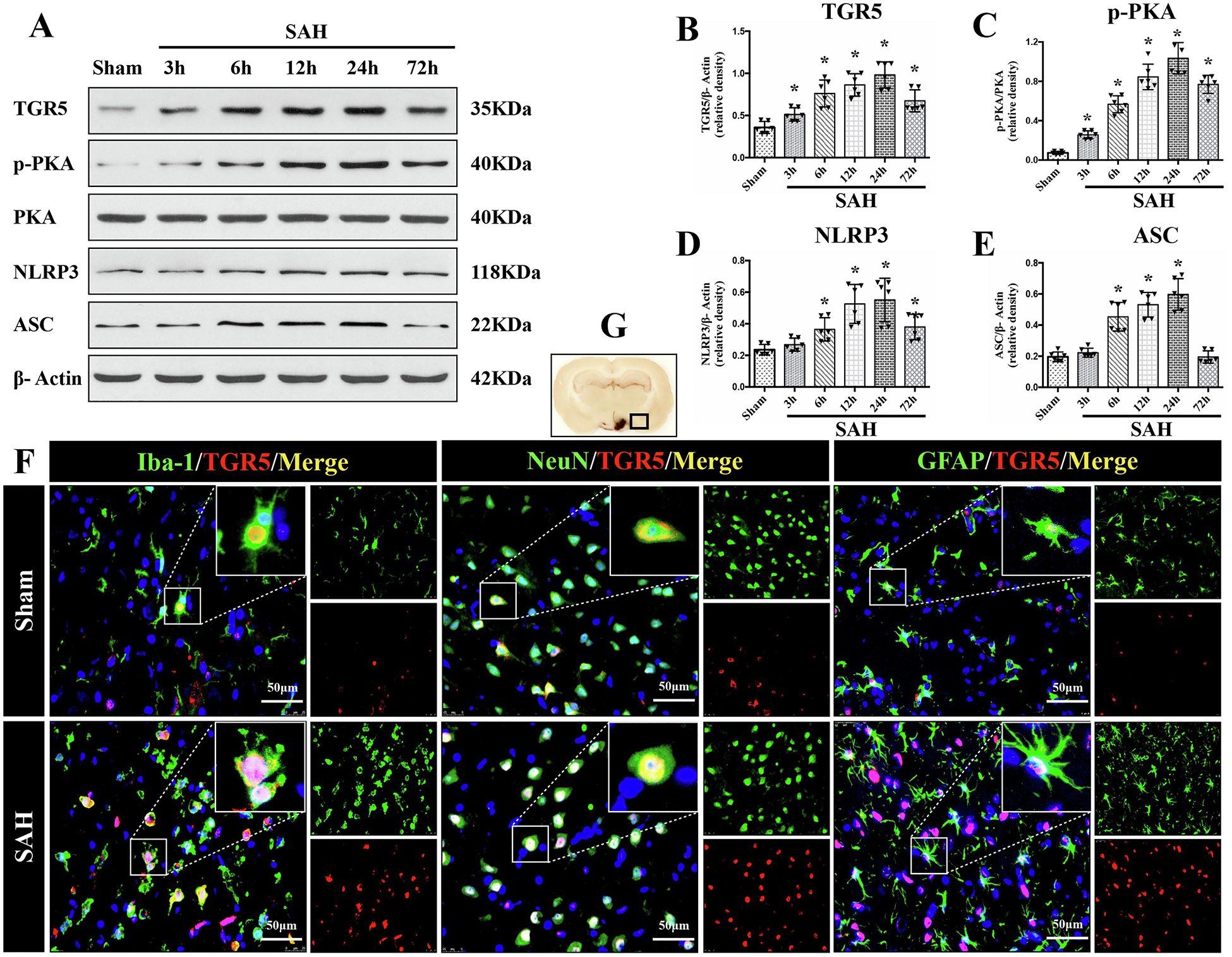
Representative western blot (WB) bands of time course (A) and densitometric quantification of TGR5 (B-E) in the ipsilateral hemisphere after SAH. n=6 per group. *P<0.05 vs. Sham group; One-way ANOVA, Tukey’s post hoc test. (C) Double immunofluorescence staining (IF) of TGR5 (red) with microglia (Iba-1, green), neurons (NeuN, green), and astrocytes (GFAP, green) in the ipsilateral basal cortex at 24 h after SAH. Cell nuclei were counterstained with DAPI (blue). A small black square within coronal section of brain indicated the location of where the immunofluorescence staining images were taken. n=2 per group. Scale bar=50 μm. Data was represented as mean ± SD. *P<0.05 vs. Sham group.
Double immunofluorescence staining of TGR5 receptors with microglia (Iba-1), neurons (NeuN), and astrocytes (GFAP) was performed in the sham group and SAH-24 h group. The cellular colocalization of TGR5 with Iba-1, NeuN, and GFAP were detected in left hemisphere peri-hemorrhage cortex and the number of TGR5-positive microglia, neurons and astrocytes were increased at 24 h after SAH (Fig. 1F).
3.2. SAH grading score
Subarachnoid blood clots were distributed around the circle of Willis and ventral brain stem after SAH induction with a significant difference in sham group (Fig. 2A). The average SAH grading scores among all SAH groups showed no significant differences (Fig. 2B).
Figure 2. Intranasal administration of INT-777 improved short-term neurobehavioral outcome and attenuated brain edema at 24 h after SAH.
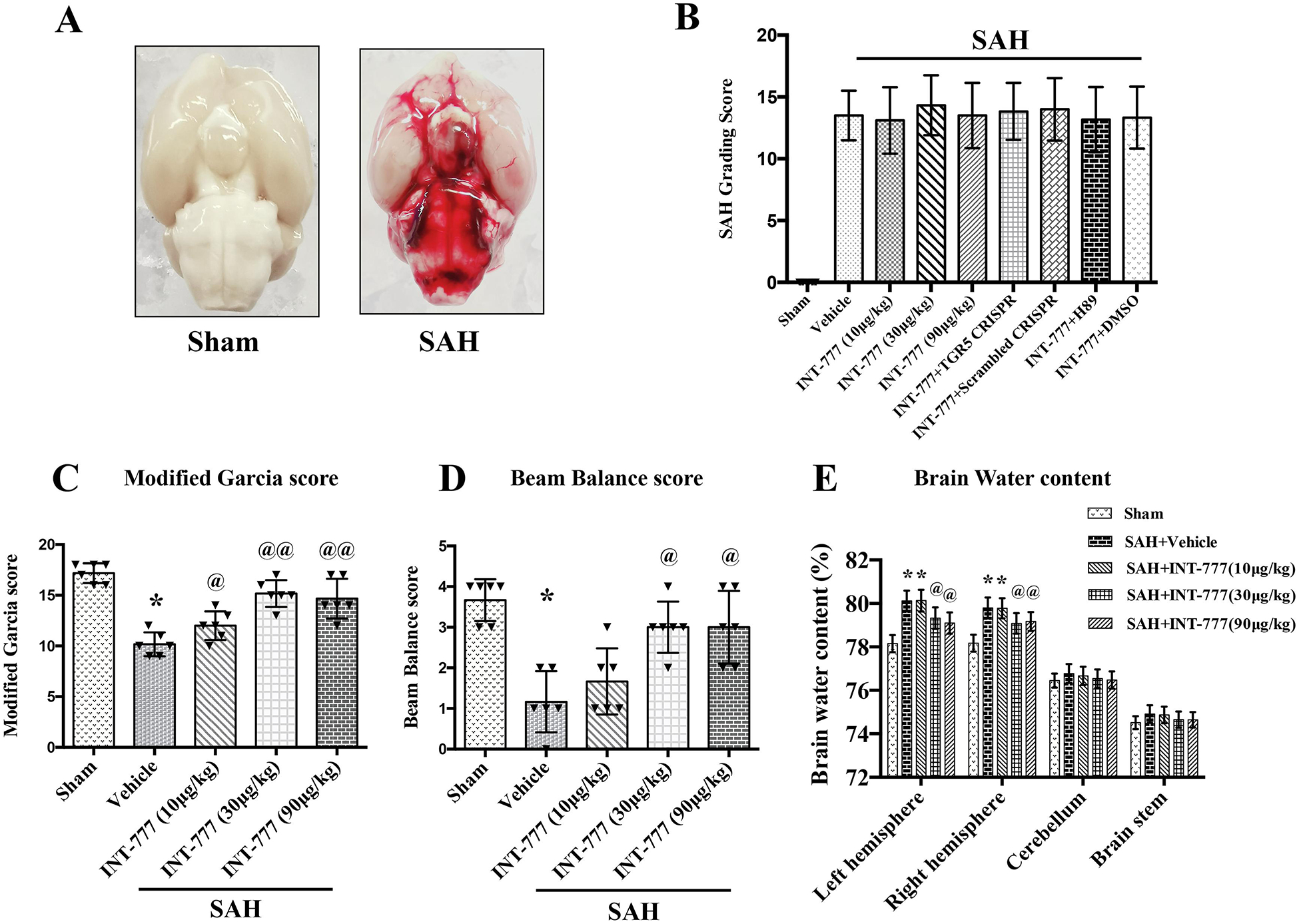
(A) Representative brain pictures of Sham and SAH rats (subarachnoid blood clots were mainly presented around the circle of Willis). (B) SAH grading scores of all groups. (C-D) INT-777 administration improved neurobehavioral performance on the modified Garcia and beam balance test at 24 h after SAH. (E) Quantitative analysis of brain water content in the left/right hemisphere, cerebellum, and brain stem at 24 h after SAH. n=6 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. n=6 per group. *P<0.05, **P<0.01 vs. Sham group; @P<0.05, @@P<0.01 vs. SAH+Vehicle group.
3.3. Intranasal administration of INT-777 improved short-term neurological deficits and attenuated brain edema at 24 h after SAH
The Modified Garcia test, beam balance test, and brain water content were evaluated at 24 h after SAH. The results showed that rats in the SAH+vehicle group performed significant neurological impairments compared with the Sham group (Fig. 2C–D). The medium (30μg/kg) and high (90μg/kg) doses of INT-777 administration significantly improved the neurological deficits on modified Garcia test and beam balance test at 24 h after SAH compared with the vehicle group (Fig. 2C–D). Consistently, the brain water contents in the ipsilateral (left) and contralateral (right) hemisphere were significantly increased in the SAH groups compared to the sham group (Fig. 2E), which was significantly decreased by the medium and high doses of INT-777 treatment (Fig. 2E). The brain water contents in the cerebellum and brain stem showed no significant differences between the sham and SAH groups. Based on these results, 30μg/kg of INT-777 was chosen as the optimal dose in the subsequent studies.
3.4. Intranasal administration of INT-777 attenuated microglia activation at 24 h after SAH
To characterize the response of resident microglia after SAH insult, Iba-1 and CD68 staining were performed to evaluate microglia activation in the ipsilateral basal cortex at 24 h after SAH. Iba-1 is constitutively expressed in both resting and active microglia, while CD68 is only labeled the activated microglia. The ramified morphology of resting microglia were identified by Iba-1 staining in the sham group (Fig. 3A). In contrast, rats in the SAH+vehicle group showed increases in Iba-1 positive cells with activated microglia morphology of rod shape or larger body with short/thick processes. Rats treated with INT-777 had fewer Iba-1 positive microglia and less morphology transformation to the activated microglia status (Fig. 3A). Co-immunofluorescence staining of Iba-1 with CD68 further confirmed a significant increase in microglial cell activation in the peri-hemorrhage area at 24 h after SAH (Fig. 3A). Quantitative analysis showed that INT-777 administration significantly reduced the number of Iba-1-positive microglia and CD68-positive activated microglia (Fig. 3H–I).
Figure 3. Intranasal administration of INT-777 reduced microglial NLRP3-ASC inflammasome activation-mediated neuroinflammation within ipsilateral hemisphere at 24 h after SAH.
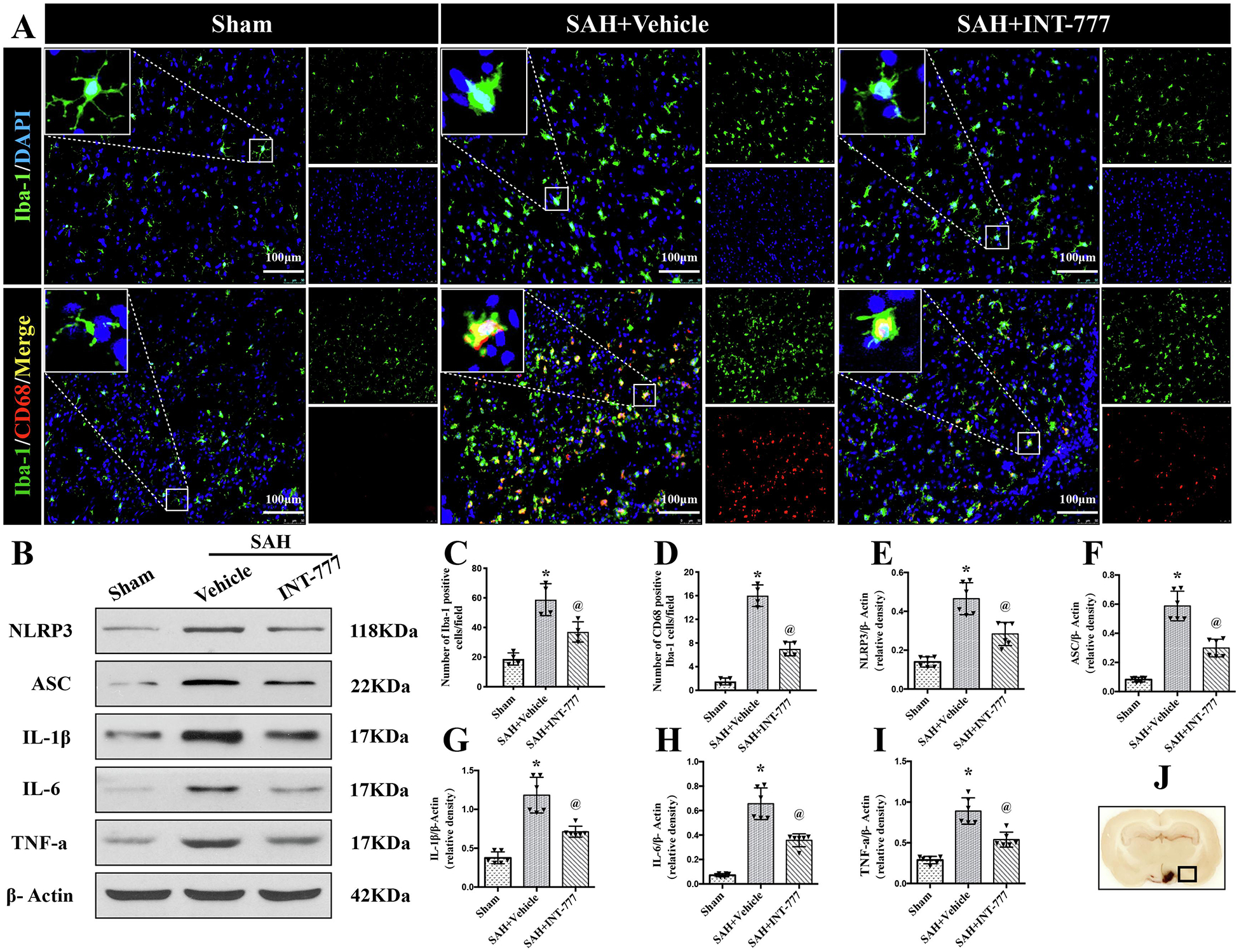
(A) Representative immunofluorescence microphotographs of microglia (Iba-1, green) and CD68 (red)-positive activated microglia in the ipsilateral basal cortex for sham, SAH+Vehicle, and SAH+INT-777 groups. Nuclei were stained with DAPI (blue). Scale bar=100 μm (J) A small black squares in the coronal section of brain indicated the area used for microphotographs. (C-D) Quantitative analysis of Iba-1-positive and CD68-positive microglia. n=4 per group. (B, E-I) Representative western blot bands and densitometric quantification of NLRP3, ASC, IL-1β, IL-6, and TNF-a in the ipsilateral hemisphere at 24 h after SAH, n=6 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group.
3.5. Intranasal administration of INT-777 reduced SAH-induced ipsilateral hemisphere NLRP3-ASC inflammasome activation and release of pro-inflammatory cytokine at 24 h after SAH
To better understand the effect of INT-777 on innate immune response involving inflammasome after SAH, the double immunofluorescence staining of NLRP3, ASC with Iba-1 was performed. In sham group, Iba-1-positive resting cells were prominent and there were no NLRP3 and ASC expression at 24 h after SAH (Fig. 4A). After SAH, NLRP3-ASC inflammasomes were double stained in most of the Iba-1 positive activated microglia around the ipsilateral basal cortex area (Fig. 4A). Compared with sham group, almost all Iba-1-positive cells showed large cell bodies at 24 h after SAH, while almost no NLRP3 and ASC expression were observed in Iba-1-positive cells in sham group (Fig. 4A). However, INT-777 significantly downregulated NLRP3-ASC inflammasomes expression compared to the SAH+vehicle group (Fig. 4A). Quantitative analysis showed that INT-777 treatment reduced the number of NLRP3-positive and ASC-positive microglia (Fig. 4C–D).
Figure 4. Intranasal administration of INT-777 reduced ipsilateral hemisphere NLRP3-ASC inflammasome activation in microglia at 24 h after SAH.
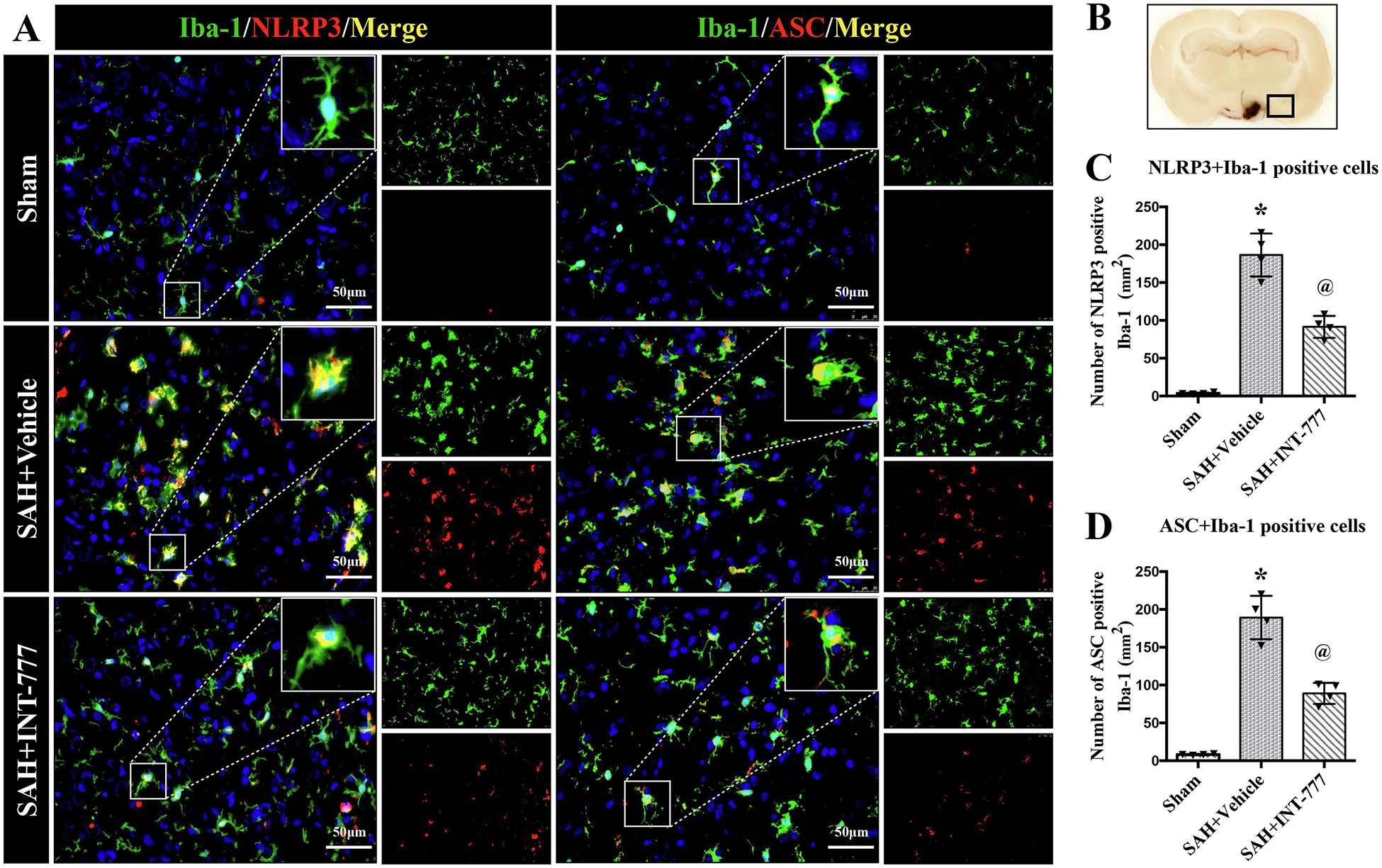
(A) Representative immunofluorescence micrographs of NLRP3 (red) or ASC (red) with Iba-1 (green) in the ipsilateral basal cortex for sham, SAH+Vehicle, and SAH+INT-777 groups. Nuclei were stained with DAPI (blue). Scale bar=50 μm. (B) A small black squares in the coronal section of brain indicated the area used for microphotographs. (C-D) Quantitative analysis of NLRP3-positive and ASC-positive microglia. n=4 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group.
To evaluate the downstream signaling after the inflammasomes activation, the western blot assay of NLRP3-ASC inflammasomes and pro-inflammatory cytokine (including IL-1β, IL-6, and TNF-a) protein levels within the ipsilateral hemisphere was performed at 24 h after SAH. Compared to their levels in the sham group, NLRP3, ASC, IL-1β, IL-6, and TNF-a were remarkably increased at 24 h after SAH (Fig. 3B–G). However, INT-777 treatment inhibited these effects compared to the SAH+vehicle group (Fig. 3B–G).
3.6. Intranasal administration of INT-777 decreased neuronal degeneration in hippocampus region on 28 d after SAH
Nissl staining of the hippocampus was performed at 28 d after SAH. Within the CA1, CA2, CA3, and DG, regions of ipsilateral hippocampus, there was more neuron loss and shrinkage morphology of neurons in the SAH+vehicle group compared with the sham group (Fig. 5A–D). However, these neurons degeneration were improved in SAH+INT-777 group (Fig. 5A–D).
Figure 5. INT-777 administration decreased neuronal degeneration in hippocampus region on 28 d after SAH.
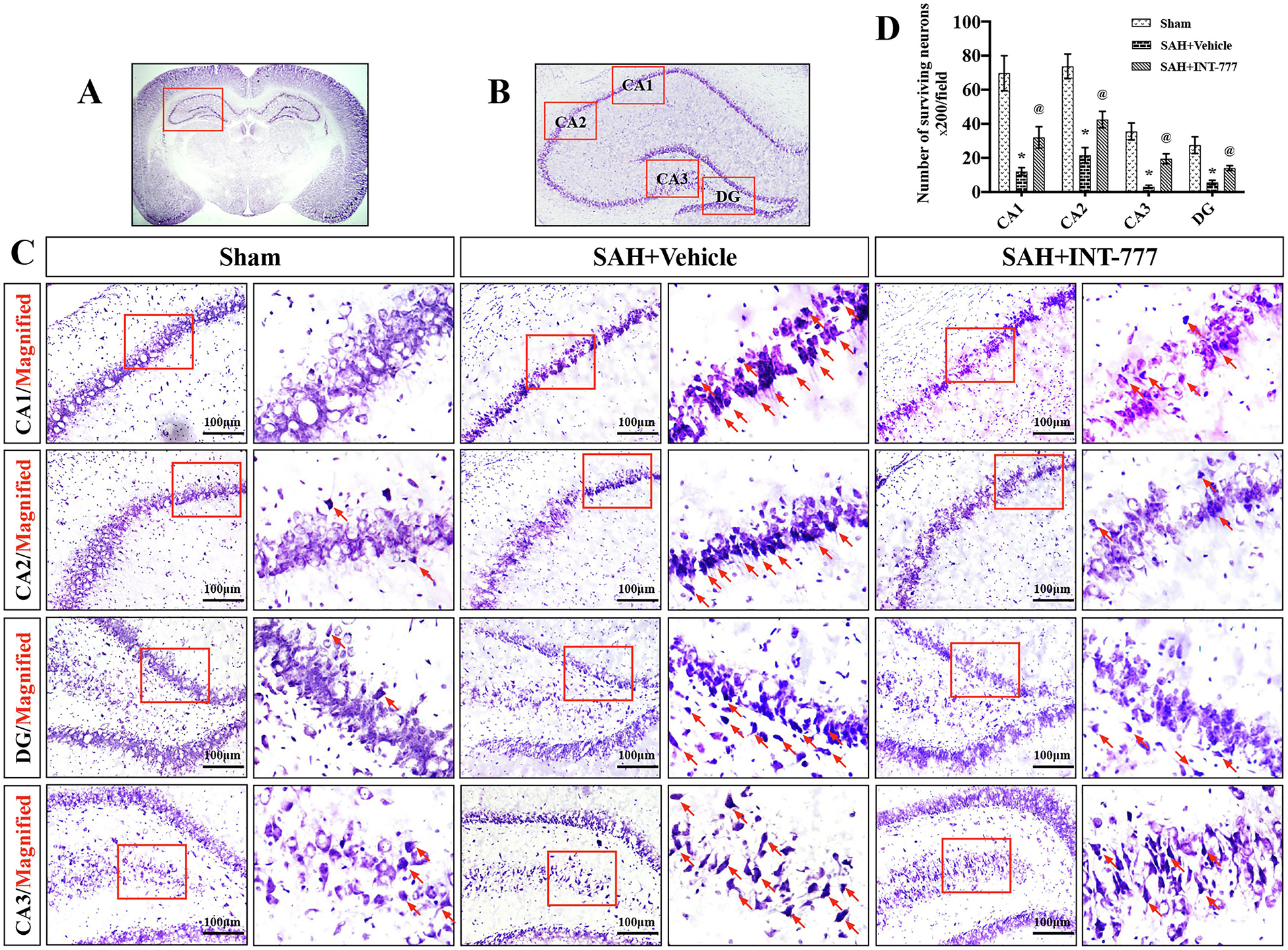
(A-C) INT-777 decreased the number of Nissl staining-identified degenerative neurons in different regions of hippocampus (CA1, CA2, CA3, and DG). Scale bar=100 μm (D) Quantifications of Nissl staining-identified neuron survival in CA1, CA2, CA3, and DG regions. The small red squares in the coronal section of brain indicated the area observed. Red arrows indicated degenerative neurons with condensed staining and shrunken cytoplasm. n=3 per group. Vehicle: 10% dimethyl sulfide. DG, dentate gyrus. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group.
3.7. TGR5 knockout CRISPR and PKA inhibitor H89 reversed the neuroprotective effects of INT-777 on neurobehavioral outcomes after SAH
The pretreatment of TGR5 knockout CRISPR or PKA inhibitor H89 significantly reversed the neurobehavioral benefits of INT-777 on the modified Garcia score and the beam balance score (Fig. s3).
3.8. Intranasal administration of INT-777 attenuated NLRP3-ASC inflammasome-mediated neuroinflammation via TGR5/cAMP/PKA signaling pathway at 24 h after SAH
Western blots results showed that the expressions of TGR5, cAMP, p-PKA, NLRP3, ASC, and pro-inflammatory cytokines including IL-1β, IL-6, and TNF-a were significantly increased at 24 h after SAH compared with sham group (Fig. 6A–I). INT-777 treatment further increased the expression levels of TGR5, cAMP, and p-PKA, but decreased the expressions of NLRP3, ASC, and pro-inflammatory cytokines IL-1β, IL-6, and TNF-a compared with the SAH+vehicle group (Fig. 6A–I). The expressions of TGR5 was significantly decreased by TGR5 knockout CRISPR compared to the scrambled CRISPR (Fig. 6A–B). Additionally, TGR5 knockout reversed the effects of INT-777 on the downstream protein levels. In SAH+INT-777+TGR5 knockout CRISPR group, the protein levels of cAMP and p-PKA were lower, but pro-inflammatory cytokine IL-1β, IL-6, and TNF-a were higher than that in the SAH+INT-777+scrambled CRISPR group (Fig. 6A, B–I).
Figure 6. TGR5 knockout CRISPR abolished the anti-neuroinflammation effects of INT-777 at 24 h after SAH.
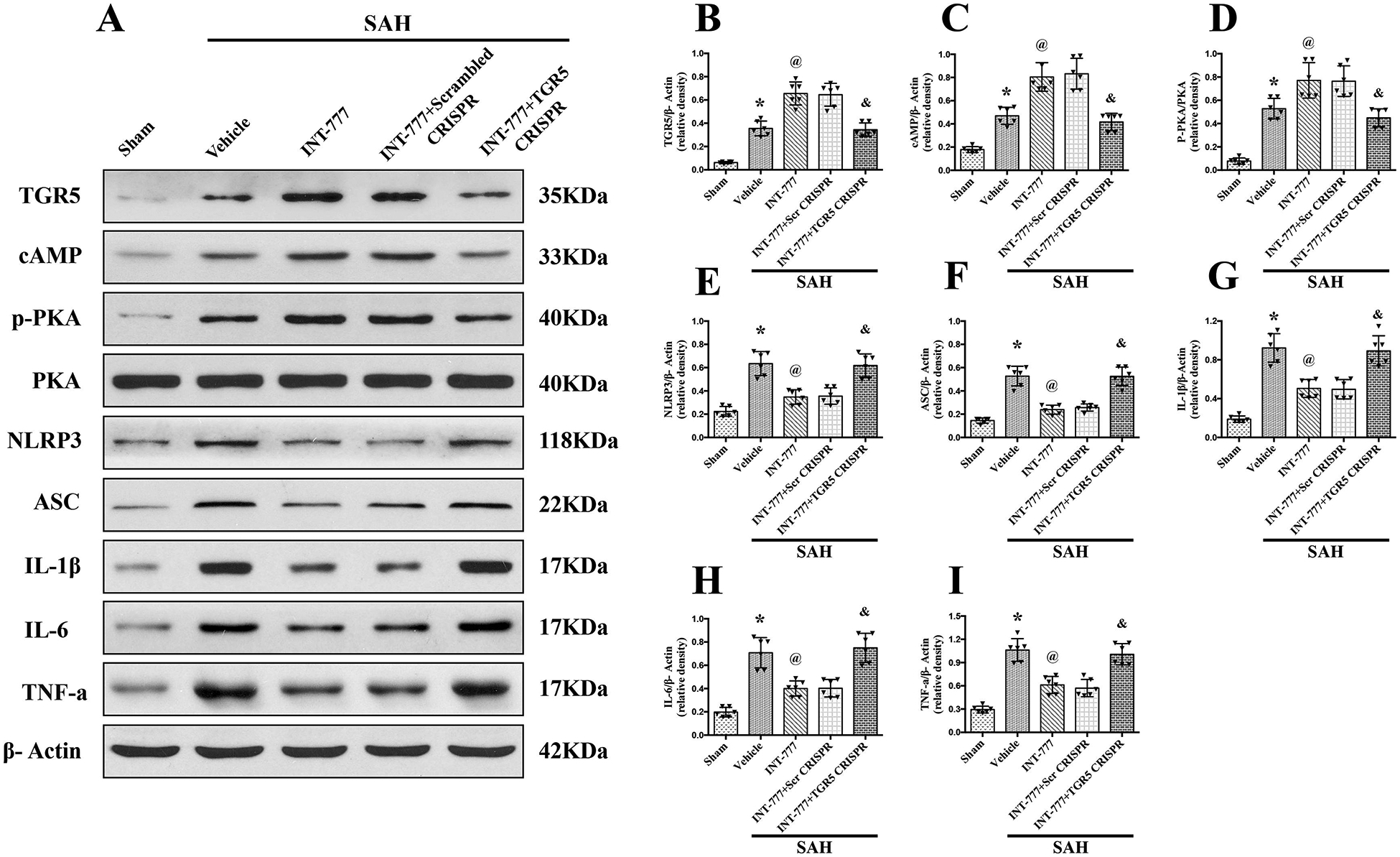
(A) Representative western blot bands and (B-I) quantification of TGR5, cAMP, p-PKA/PKA, NLRP3, ASC, IL-1β, IL-6, and TNF-a. Vehicle: 10% dimethyl sulfide. n=6 per group. Data was represented as mean ± SD. *P<0.05 vs. Sham group, @P<0.05 vs. SAH+Vehicle group, &P<0.05 vs. SAH+Scrambled CRISPR group.
To further clarify the role of downstream molecules, the selective PKA inhibitor H89 was administrated i.p. at 1 h before SAH. Without affecting the expression levels of TGR5 and cAMP, H89 significantly suppressed the expression of p-PKA but increased protein levels of NLRP3, ASC, and pro-inflammatory cytokines IL-1β, IL-6, and TNF-a at 24 h after SAH in the SAH+ INT-777+H89 group compared with the SAH+INT-777+DMSO group (Fig. 7A–I).
Figure 7. PKA selective inhibitor H89 abolished the anti-neuroinflammation effects of INT-777 at 24 h after SAH.

(A) Representative western blot bands and (B-I) quantification of TGR5, cAMP, p-PKA/PKA, NLRP3, ASC, IL-1β, IL-6, and TNF-a. Vehicle: 10% dimethyl sulfide. n=6 per group. Data was represented as mean ± SD. *P<0.05 vs. Sham group, @P<0.05 vs. SAH+Vehicle group, &P<0.05 vs. SAH+INT-777+DMSO group.
3.9. Intranasal administration of INT-777 attenuated the microglia activation associated IL-1β secretion and neutrophil infiltration at 24 h after SAH, which were reversed by TGR5 CRISPR or PKA inhibitor
Compared with the sham group, microglia were notably activated, in terms of large, round amoeba morphology, and thick, short projections (Fig. 8B). The double immunofluorescence staining of Iba-1 and IL-1β showed that the activated Iba-1 positive microglia were one of the sources of pro-inflammatory IL-1β at 24 h after SAH. Moreover, the number of activated microglia and MPO-positive cells were significantly increased in SAH+vehicle group (Fig. 8,9A–C). INT-777 treatment significantly attenuated IL-1β secretion and reduced the number of MPO-positive cells, which were reversed by TGR5 CRISPR or PKA inhibitor (Fig. 8,9A–C).
Figure 8. INT-777 administration attenuated microglia activation associated IL-1β secretion, which were reversed by TGR5 knockout CRISPR or PKA inhibitor at 24 h after SAH.
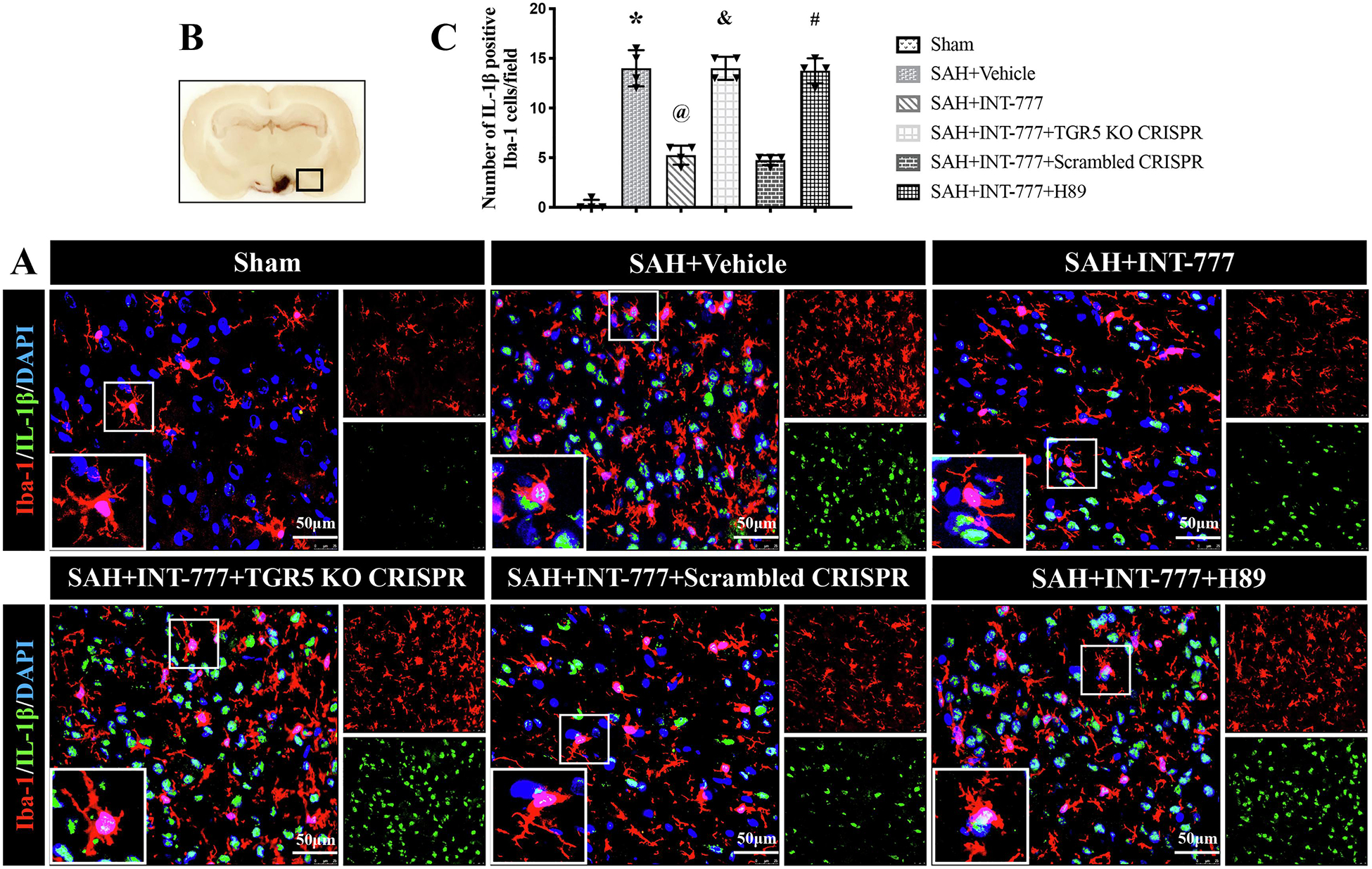
(A) Representative immunofluorescence microphotographs of IL-1β (green) with Iba-1 (red) in the ipsilateral basal cortex for different groups. Nuclei were stained with DAPI (blue). Scale bar=50 μm. (B) A small black squares in the coronal section of brain indicated the area used for microphotographs. (C) Quantitative analysis of IL-1β-positive microglia. n=4 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group; &P<0.05 vs. SAH+Scrambled CRISPR group; #P<0.05 vs. SAH+INT-777 group.
Figure 9. INT-777 administration attenuated neutrophil infiltration, which were reversed by TGR5 knockout CRISPR or PKA inhibitor at 24 h after SAH.
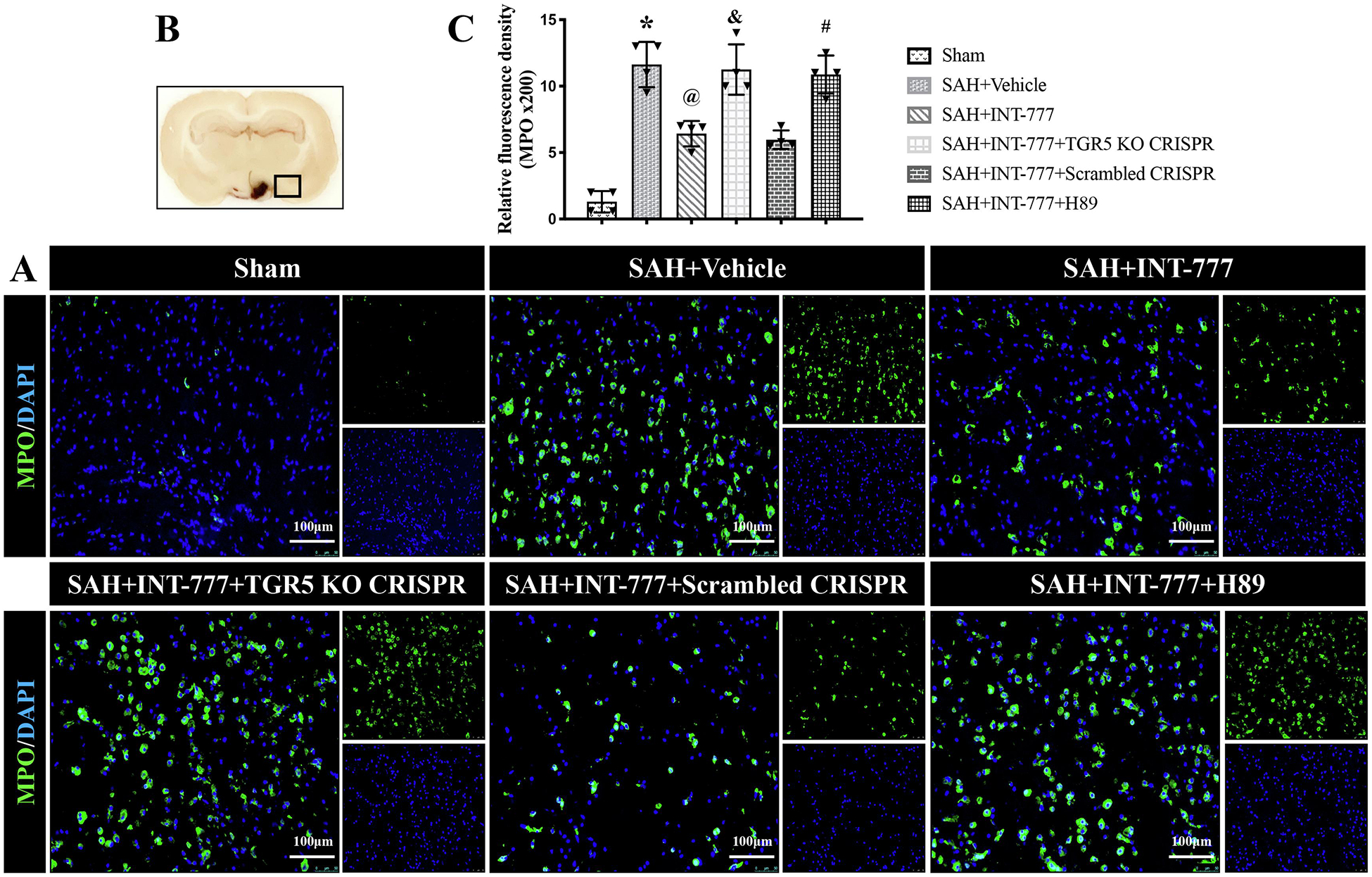
(A) Representative microphotographs of MPO immunofluorescence staining in the ipsilateral basal cortex for different groups. Nuclei were stained with DAPI (blue). Scale bar=100 μm. (B) A small black squares in the coronal section of brain indicated the area used for microphotographs. (C) Quantitative analysis of MPO-positive cells. n=4 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group; &P<0.05 vs. SAH+Scrambled CRISPR group; #P<0.05 vs. SAH+INT-777 group.
4. Discussion
EBI describes the immediate injury to the brain after SAH before onset of delayed vasospasm. During the EBI period, a ruptured aneurysm brings on many physiological derangements such as elevated intracranial pressure (ICP), decreased cerebral blood flow (CBF), and global cerebral ischemia. These events initiate secondary injuries such as blood-brain barrier disruption, neuroinflammation, and oxidative cascades that all ultimately lead to cell death (Fujii et al., 2013). High intracranial hypertension is associated with a strong activation of the inflammatory cascade after SAH (Graetz et al., 2010). NLRP3-ASC inflammasome-mediated neuroinflammation is critical pathologic process contributing to poor outcomes after stroke including SAH. Thus, the therapeutic approach targeting the NLRP3-ASC inflammasome could potentially improve neurological outcomes after SAH. In the present study, we found that the endogenous protein levels of the TGR5 receptor, p-PKA, NLRP3, and ASC were increased after SAH in rats, and peaked at 24 h after SAH in rats. The TGR5 receptors were expressed in microglia, neurons, and astrocytes. Moreover, the activation of TGR5 with INT-777 significantly improved the short-term neurobehavioral deficits, accompanied by a reduction in the activation of the NLRP3-ASC inflammasome-mediated pro-inflammatory responses at 24 h after SAH. Mechanistically, INT-777 treatment upregulated the protein levels of TGR5, cAMP, and p-PKA but down-regulated the protein levels of NLRP3, ASC, IL-1β, IL-6, and TNF-a within ipsilateral hemisphere at 24 h after SAH. TGR5 knockout CRISPR or selective PKA inhibitor H89 abolished the beneficial effects of INT-777 on neurological deficits, NLRP3-ASC inflammasome-mediated neuroinflammation, and neutrophil infiltration. Furthermore, we found that INT-777 alleviates delayed neuronal degeneration in the hippocampus after SAH. Taken together, our findings suggest that INT-777 could inhibit the activation of NLRP3-ASC inflammasome in activated microglia at 24 h after SAH, partially by up-regulating the TGR5/cAMP/PKA signaling pathway. The administration of INT-777 may serve as an effective therapeutic strategy against EBI and delayed brain injury after SAH.
The inflammatory responses involving microglial/macrophage activation and neutrophil infiltration in EBI after SAH exacerbated brain injury and neuronal degradation within the cerebral cortex and hippocampus, leading to neurological dysfunctions (Xu et al. 2020). Activated microglia released various pro-inflammatory cytokines, ultimately leading to axonal injury and neuronal cell loss, thus aggravating secondary brain injury after SAH (Schneider et al. 2015). Modulation of microglial polarization toward anti-inflammatory phenotype attenuated inflammation-induced brain injury after SAH (Xie et al. 2017; Li, Liu, et al. 2018; Peng, Pang, et al. 2019; Zheng et al. 2020). Previous studies showed that microglial NLRP3–ASC inflammasome activation induces the release of pro-inflammatory cytokines and chemokine (Heneka, McManus, and Latz 2018). Inhibition of microglial NLRP3-ASC inflammasome activation is capable of inhibiting exogenously seeded and non-exogenously seeded Tau pathology in AD (Stancu et al. 2019). Microglial NLRP3-ASC inflammasome activation stimulates neurotoxicity by increasing inflammation (Sarkar et al. 2019), leading to the formation of amyloid-β oligomers, contributing to pathology in AD (Venegas et al. 2017), drives progressive dopaminergic neuropathology in Parkinson’s disease (Gordon et al. 2018), and finally exacerbates the brain damage and the pro-inflammatory response after recurrent strokes (He et al. 2020). The inhibition of the NLRP3–ASC inflammasome suppressed infarct volume growth and neurological damage in a brain ischemia/reperfusion model in mice (Ito et al. 2015). Furthermore, the activation of the NLRP3-ASC inflammasome contributed to neuronal apoptosis, oxidative stress related endothelial cells injury, micro-thrombosis, and inflammatory response following SAH (Dong et al. 2016; Shao et al. 2016; Zhang et al. 2017; Zhuang et al. 2019). In our present study, we showed a strong inflammatory response, exemplified by increased M1 microglia/macrophage polarization (CD68 positive microglia), which was consistent with previous investigations (Qin et al. 2017; Xie et al. 2017; Li, Liu, et al. 2018). We also observed up-regulation of endogenous expression of NLRP3-ASC inflammasomes in a time-dependent manner in the left/ipsilateral hemisphere and activated microglia are most likely the sources of NLRP3-ASC in the acute stage after SAH in rats, accompanied by the impairment of neurological functions at 24 h after SAH.
The TGR5 receptor is a membrane-bound bile acid receptor and is also expressed in microglia, neurons, and astrocytes (Keitel et al. 2010; Eggink et al. 2018). There is increasing evidence demonstrating that TGR5 activation exerts strong neuroprotective effects in the neurological diseases including acute neuroinflammation (Yanguas-Casas et al. 2017), ischemic stroke (Liang et al. 2020a, 2020b), hepatic encephalopathy (McMillin et al. 2015), and AD (Wu et al. 2018). After SAH, the expression of endogenous TGR5 receptor in our study was significantly increased and was colocalized with activated microglia. The up-regulation of endogenous TGR5 receptor after SAH may suggest its role as an endogenous neuroprotective response to deleterious stimuli in the acute stage after SAH in our previous study (Zuo, Zhang, et al. 2019). INT-777, a selective TGR5 agonist, through binding to the side chain of TGR5 receptor, leads to methylation or sulphation of TGR5 receptor and enhances its activity (Pellicciari et al. 2009; Schaap, Trauner, and Jansen 2014). INT-777 exerts its anti-inflammatory, anti-apoptotic, and anti-oxidative stress effects in AD (Wu et al. 2018; Wu et al. 2019), ischemic stroke (Liang et al. 2020b, 2020a), endometriosis (Lyu et al. 2019), kidney disease in obesity and diabetes (Wang et al. 2016), diabetic cardiomyopathy (Deng et al. 2019), diabetic nephropathy (Xiao et al. 2020), and intestinal epithelial renewal (Sorrentino et al. 2020). We previously demonstrated the anti-apoptosis and anti-oxidative stress effects of INT-777 in a rat mode of SAH. However, the effect of TGR5 receptor activation by INT-777 on neuroinflammation after SAH has not been elucidated. In the present study, we demonstrated that INT-777 administration significantly improved neurobehavioral deficits, and decreased expression levels of NLRP3-ASC inflammasome and pro-inflammatory cytokine IL-1β, IL-6, and TNF-a after SAH. Importantly, immunofluorescence staining results showed that INT-777 reduced the numbers of NLRP3-ASC inflammasome in activated microglia and neutrophil infiltration at 24 h after SAH, which suggested anti-inflammation effects were through suppression of NLRP3-ASC activation.
Recently, more emphasis has been placed on the investigation of delayed brain injuries after SAH (Peng, Pang, et al. 2019; Sun et al. 2019; Zhang et al. 2019; Zuo, Zhang, et al. 2019; Zuo, Huang, et al. 2019; Xu et al. 2020). We previously found that INT-777 administration significantly improved the long-term performance on the rotarod test and water maze test in SAH rats (Zuo, Zhang, et al. 2019). Specifically in the present study, we further assessed hippocampal neurons in brain slices and found that SAH resulted in severe neuronal damage and degeneration in CA1, CA2, CA3, and dentate gyrus sectors, yet early INT-777 administration reversed such delayed injuries. Severe EBI may directly lead to poor prognosis or be associated with delayed cerebral ischemia after SAH (Macdonald 2014; Ahn et al. 2018; Suzuki 2019). Thus, the early INT-777 treatment attenuated EBI which could alleviate long-term neurobehavioral dysfunctions after SAH.
We further investigated the possible signaling pathway underlying the anti-neuroinflammation effect of TGR5 activation with INT-777 after SAH. Previous studies showed that the activated TGR5-cAMP-PKA axis was identified as a unique regulatory mechanism promoting stimulus-secretion coupling of pancreatic β-cells (Maczewsky et al. 2019), increasing glucose induced insulin secretion [71], and ameliorating insulin resistance in skeletal muscles (Vettorazzi et al. 2016), and ameliorating insulin resistance in skeletal muscles (Huang et al. 2019). Specifically, in vivo and in vitro models of acute neuroinflammation, the TGR5 receptor-mediated the activation of cAMP-PKA signaling pathway in microglia induced anti-inflammatory markers, while inhibiting pro-inflammatory responses (Yanguas-Casas et al. 2017). Furthermore, the PKA-induced phosphorylation and ubiquitination of NLRP3 served as a critical brake on NLRP3-ASC inflammasome activation-mediated metabolic disorder (Guo et al. 2016). Even though the TGR5 receptor and NLRP3-ASC inflammasome have been reported to participate in cAMP-PKA signaling pathway, the interaction between TGR5 receptor activation with INT-777 and NLRP3-ASC inflammasome ubiquitination after SAH needs to be further elucidated.
INT-777 increasing TGR5 protein levels has been reported in a mouse model of AD and liver ischemia/reperfusion injury (Yang et al. 2017; Wu et al. 2018). In the present study, we found that INT-777 administration significantly increased the protein levels of TGR5, cAMP and the phosphorylation of PKA, while inhibiting NLRP3-ASC inflammasome activation in the ipsilateral/left hemisphere at 24 h after SAH. In addition, INT-777 administration markedly attenuated inflammatory responses, reduced neutrophil infiltration, as evidenced by the reduction of pro-inflammatory molecules (IL-1β, IL-6, and TNF-a), as well as IL-1β-positive activated microglia cells, and MPO-positive cells. However, TGR5 knockout CRISPR or PKA specific inhibitor H89 significantly reversed such neuroprotective effects of INT-777. Collectively, INT-777 attenuates NLRP3-ASC inflammasome-mediated neuroinflammation, at least in part, via up-regulation of TGR5/cAMP/PKA signaling pathway after SAH in rats.
This study has several limitations. First, previous studies have shown that the activation of the TGR5 receptor with INT-777 exerts protective effects, including anti-oxidative stress, anti-apoptosis, anti-necrosis, promoting dendritic spine generation, and alleviating BBB permeability (Li, Yang, et al. 2018; Wu et al. 2018; Deng et al. 2019; Wu et al. 2019; Zuo, Zhang, et al. 2019; Liang et al. 2020b). Thus, we could not exclude those neuroprotective mechanisms underlying the neurological benefits of INT-777. Second, we focus on NLRP3 inflammasome which mainly expressed in microglia, which can produce pro-inflammatory cytokines to induce inflammatory response (Gordon et al. 2018; Heneka, McManus, and Latz 2018; Ising et al. 2019). However, NLRP1 inflammasome is mainly expressed in neurons, which is closely related to pyroptosis (Tan et al. 2014; Tan et al. 2015; Chen, Zuo, et al. 2019; Huang et al. 2020). Future study is needed to elucidate the effects of INT777 on neuronal pyroptosis through NLRP1 inflammasome signaling after SAH. Third, a more specific time window and potential mechanisms for long-term outcomes are worth investigating in future studies. Last but not least, in this study, rats with slight SAH were excluded from this study. The correlation between the degrees of neuroinflammation in brain tissues with the SAH grade was not evaluated. Given that neuroinflammatory response is secondary to the pathological factors such as extent of bleeding and brain tissue damage, it is certain that the degree of the neuroinflammation is associated with the degree of SAH severity (Lucke-Wold, Logsdon et al. 2016; Zheng and Wong 2017). Nevertheless, future study is needed to elucidate the specific contribution of TGR5 signaling and NLRP3-ASC inflammasome to mild SAH.
5. Conclusions
Our results showed that INT-777 administration suppressed NLRP3-ASC inflammasome-mediated neuroinflammation and improved neurological deficits partially through activating the TGR5/cAMP/PKA signaling pathway in a rat model of SAH. Early administration of INT-777 may be a therapeutic and preventive strategy against delayed hippocampal injury after SAH due to its attenuation of EBI.
Supplementary Material
Figure s1. Schematic summary. INT-777 attenuates NLRP3-ASC inflammasome-dependent neuroinflammation via TGR5/cAMP/PKA signaling pathway and improves long-term neurological deficits after subarachnoid hemorrhage in rats.
Figure s2. Experimental design. SAH, subarachnoid hemorrhage; WB, western blot; BWC, brain water content; IF, immunofluorescence; i.n, intranasal; i.c.v, intracerebroventricular; i.p, intraperitoneal; DMSO, dimethyl sulfoxide.
Figure s3. Effects of TGR5 knockout CRISPR and PKA inhibitor on neurobehavioral outcome at 24 h after SAH. (A-B) TGR5 knockout CRISPR and PKA inhibitor H89 pretreatment reversed the neurobehavioral benefits of INT-777 at 24 h after SAH, n=6 per group.
The expressions of endogenous TGR5, p-PKA, and NLRP3-ASC inflammasome were increased after SAH in rats, and peaked at 24 h after SAH.
INT-777 administration significantly improved the short-term neurobehavioral dysfunctions, and reduced brain edema, accompanied by a reduction in the activation of the NLRP3-ASC inflammasome in activated microglia and inflammatory response after SAH in rats.
INT-777 alleviates delayed neuronal degeneration in the hippocampus after SAH.
INT-777 administration suppressed NLRP3-ASC inflammasome-mediated neuroinflammation, at least in part, through activating the TGR5/cAMP/PKA signaling pathway after SAH.
Funding
This study was supported by grants from the National Institutes of Health NS081740 and NS 082184 to John H. Zhang, grants QNRC2016263, H201654, and GSWS2019080 from Jiangsu provincial health and family planning commission of China to Dr. G. Zuo, and grants [2017]5631, [2018]5764, [2017]5724 from Guizhou Provincial Science and Technology Platform and Talent Team Project of China to Dr. X. Hu.
Abbreviations
- AD
Alzheimer’s disease
- ANOVA
Analysis of variance
- ARRIVE
Animal Research: Reporting of In Vivo Experiments
- ASC
Apoptosis-associated speck-like protein containing a CARD
- BBB
blood-brain barrier
- CNS
Central nervous system
- DMSO
Dimethyl sulfoxide
- EBI
Early brain injury
- GFAP
Glial fibrillary acidic protein
- i.c.v
Intracerebroventricular
- i.n
Intranasally
- i.p
Intraperitoneal
- I/R
Ischemia/reperfusion
- Iba-1
Ionized calcium binding adaptor molecule 1
- IL-1β
Interleukin-1 beta
- MPO
Myeloperoxidase
- NeuN
Neuronal nuclear
- SAH
Subarachnoid hemorrhage
- SD
Standard deviation
- NLRP3
NOD-, LRR- and pyrin domain-containing 3
- TGR5
Trans-membrane G protein-coupled receptor-5
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics approval and consent to participate
All animal experiments performed in this study were accrodance with the National Institutes of Health Guide for the Care and Use of Laboratory Aniamls which was approved by Loma Linda Univesity Institutional Animal Care and Use Committee.
Conflict of Interest
The authors declare no conflict of interest.
Availability of data and materials
The data support the findings of this study and are available from the corresponding author upon reasonable request.
Reference
- Ahn SH, Savarraj JP, Pervez M, Jones W, Park J, Jeon SB, Kwon SU, Chang TR, Lee K, Kim DH, Day AL, and Choi HA. 2018. ‘The Subarachnoid Hemorrhage Early Brain Edema Score Predicts Delayed Cerebral Ischemia and Clinical Outcomes’, Neurosurgery, 83: 137–45. [DOI] [PubMed] [Google Scholar]
- Ai QD, Chen C, Chu S, Zhang Z, Luo Y, Guan F, Lin M, Liu D, Wang S, and Chen N. 2019. ‘IMM-H004 therapy for permanent focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3 inflammasome activation’, Transl Res, 212: 36–53. [DOI] [PubMed] [Google Scholar]
- Alawieh A, Langley EF, Weber S, Adkins D, and Tomlinson S. 2018. ‘Identifying the Role of Complement in Triggering Neuroinflammation after Traumatic Brain Injury’, J Neurosci, 38: 2519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh AM, Langley EF, Feng W, Spiotta AM, and Tomlinson S. 2020. ‘Complement-Dependent Synaptic Uptake and Cognitive Decline after Stroke and Reperfusion Therapy’, J Neurosci, 40: 4042–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, and Dixit VM. 2016. ‘Inflammasomes: mechanism of assembly, regulation and signalling’, Nat Rev Immunol, 16: 407–20. [DOI] [PubMed] [Google Scholar]
- Cahill J, Calvert JW, and Zhang JH. 2006. ‘Mechanisms of early brain injury after subarachnoid hemorrhage’, J Cereb Blood Flow Metab, 26: 1341–53. [DOI] [PubMed] [Google Scholar]
- Chen S, Zuo Y, Huang L, Sherchan P, Zhang J, Yu Z, Peng J, Zhang J, Zhao L, Doycheva D, Liu F, Zhang JH, Xia Y, and Tang J. 2019. ‘The MC4 receptor agonist RO27–3225 inhibits NLRP1-dependent neuronal pyroptosis via the ASK1/JNK/p38 MAPK pathway in a mouse model of intracerebral haemorrhage’, Br J Pharmacol, 176: 1341–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Le TH, Du Q, Zhao Z, Liu Y, Zou J, Hua W, Liu C, and Zhu Y. 2019. ‘Genistein protects against DSS-induced colitis by inhibiting NLRP3 inflammasome via TGR5-cAMP signaling’, Int Immunopharmacol, 71: 144–54. [DOI] [PubMed] [Google Scholar]
- Conzen C, Becker K, Albanna W, Weiss M, Bach A, Lushina N, Steimers A, Pinkernell S, Clusmann H, Lindauer U, and Schubert GA. 2019. ‘The Acute Phase of Experimental Subarachnoid Hemorrhage: Intracranial Pressure Dynamics and Their Effect on Cerebral Blood Flow and Autoregulation’, Transl Stroke Res, 10: 566–82. [DOI] [PubMed] [Google Scholar]
- Deng L, Chen X, Zhong Y, Wen X, Cai Y, Li J, Fan Z, and Feng J. 2019. ‘Activation of TGR5 Partially Alleviates High Glucose-Induced Cardiomyocyte Injury by Inhibition of Inflammatory Responses and Oxidative Stress’, Oxid Med Cell Longev, 2019: 6372786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Fan C, Hu W, Jiang S, Ma Z, Yan X, Deng C, Di S, Xin Z, Wu G, Yang Y, Reiter RJ, and Liang G. 2016. ‘Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling’, J Pineal Res, 60: 253–62. [DOI] [PubMed] [Google Scholar]
- Eggink HM, Tambyrajah LL, van den Berg R, Mol IM, van den Heuvel JK, Koehorst M, Groen AK, Boelen A, Kalsbeek A, Romijn JA, Rensen PCN, Kooijman S, and Soeters MR. 2018. ‘Chronic infusion of taurolithocholate into the brain increases fat oxidation in mice’, J Endocrinol, 236: 85–97. [DOI] [PubMed] [Google Scholar]
- Figueiredo CP, Barros-Aragao FGQ, Neris RLS, Frost PS, Soares C, Souza INO, Zeidler JD, Zamberlan DC, de Sousa VL, Souza AS, Guimaraes ALA, Bellio M, Marcondes de Souza J, Alves-Leon SV, Neves GA, Paula-Neto HA, Castro NG, De Felice FG, Assuncao-Miranda I, Clarke JR, Da Poian AT, and Ferreira ST. 2019. ‘Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice’, Nat Commun, 10: 3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman L, Guo H, David CN, Brickey WJ, Jha S, and Ting JP. 2017. ‘NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes’, J Exp Med, 214: 1351–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, and Zhang JH. 2013. ‘Early Brain Injury, an Evolving Frontier in Subarachnoid Hemorrhage Research’, Transl Stroke Res, 4: 432–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS, Rowe DB, O’Neill LA, Kanthasamy AG, Schroder K, Cooper MA, and Woodruff TM. 2018. ‘Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice’, Sci Transl Med, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graetz D, Nagel A, Schlenk F, Sakowitz O, Vajkoczy P, and Sarrafzadeh A. 2010. ‘High Icp as Trigger of Proinflammatory Il-6 Cytokine Activation in Aneurysmal Subarachnoid Hemorrhage’, Neurol Res, 7: 728–35. [DOI] [PubMed] [Google Scholar]
- Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, Lu L, and Wang D. 2016. ‘Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome’, Immunity, 45: 802–16. [DOI] [PubMed] [Google Scholar]
- He XF, Zeng YX, Li G, Feng YK, Wu C, Liang FY, Zhang Y, Lan Y, Xu GQ, and Pei Z. 2020. ‘Extracellular ASC exacerbated the recurrent ischemic stroke in an NLRP3-dependent manner’, J Cereb Blood Flow Metab, 40: 1048–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, and Latz E. 2018. ‘Inflammasome signalling in brain function and neurodegenerative disease’, Nat Rev Neurosci, 19: 610–21. [DOI] [PubMed] [Google Scholar]
- Huang J, Lu W, Doycheva DM, Gamdzyk M, Hu X, Liu R, Zhang JH, and Tang J. 2020. ‘IRE1alpha inhibition attenuates neuronal pyroptosis via miR-125/NLRP1 pathway in a neonatal hypoxic-ischemic encephalopathy rat model’, J Neuroinflammation, 17: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Ma S, Ning M, Yang W, Ye Y, Zhang L, Shen J, and Leng Y. 2019. ‘TGR5 agonist ameliorates insulin resistance in the skeletal muscles and improves glucose homeostasis in diabetic mice’, Metabolism, 99: 45–56. [DOI] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buee L, and Heneka MT. 2019. ‘NLRP3 inflammasome activation drives tau pathology’, Nature, 575: 669–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, and Morita R. 2015. ‘Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury’, Nat Commun, 6: 7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena PK, Sheng L, Di Lucente J, Jin LW, Maezawa I, and Wan YY. 2018. ‘Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity’, FASEB J, 32: 2866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp MA, Steiger HJ, and van Lieshout JH. 2020. ‘Experimental Aneurysmal Subarachnoid Hemorrhage: Tiding Over’, Transl Stroke Res, 11: 1–3. [DOI] [PubMed] [Google Scholar]
- Keitel V, Gorg B, Bidmon HJ, Zemtsova I, Spomer L, Zilles K, and Haussinger D. 2010. ‘The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain’, Glia, 58: 1794–805. [DOI] [PubMed] [Google Scholar]
- Kojic M, Gaik M, Kiska B, Salerno-Kochan A, Hunt S, Tedoldi A, Mureev S, Jones A, Whittle B, Genovesi LA, Adolphe C, Brown DL, Stow JL, Alexandrov K, Sah P, Glatt S, and Wainwright BJ. 2018. ‘Elongator mutation in mice induces neurodegeneration and ataxia-like behavior’, Nat Commun, 9: 3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseki H, Miyata H, Shimo S, Ohno N, Mifune K, Shimano K, Yamamoto K, Nozaki K, Kasuya H, Narumiya S, and Aoki T. 2020. ‘Two Diverse Hemodynamic Forces, a Mechanical Stretch and a High Wall Shear Stress, Determine Intracranial Aneurysm Formation’, Transl Stroke Res, 11: 80–92. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, and Stutz A. 2013. ‘Activation and regulation of the inflammasomes’, Nat Rev Immunol, 13: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton MT, and Vates GE. 2017. ‘Subarachnoid Hemorrhage’, N Engl J Med, 377: 257–66. [DOI] [PubMed] [Google Scholar]
- Li B, Yang N, Li C, Li C, Gao K, Xie X, Dong X, Yang J, Yang Q, Tong Z, Lu G, and Li W. 2018. ‘INT-777, a bile acid receptor agonist, extenuates pancreatic acinar cells necrosis in a mouse model of acute pancreatitis’, Biochem Biophys Res Commun, 503: 38–44. [DOI] [PubMed] [Google Scholar]
- Li R, Liu W, Yin J, Chen Y, Guo S, Fan H, Li X, Zhang X, He X, and Duan C. 2018. ‘TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway’, J Neuroinflammation, 15: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Matei N, McBride DW, Xu Y, Tang J, Luo B, and Zhang JH. 2020a. ‘Activation of TGR5 protects blood brain barrier via the BRCA1/Sirt1 pathway after middle cerebral artery occlusion in rats’, J Biomed Sci, 27: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2020b. ‘Correction to: Activation of TGR5 protects blood brain barrier via the BRCA1/Sirt1 pathway after middle cerebral artery occlusion in rats’, J Biomed Sci, 27: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, Huber JD, Rosen CL, and Simard JM. 2016. ‘Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review’, Int J Mol Sci, 4: 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Reis C, and Chen S. 2019. ‘NLRP3 Inflammasome in the Pathophysiology of Hemorrhagic Stroke: A Review’, Curr Neuropharmacol, 17: 582–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu D, Tang N, Wang J, Zhang Y, Chang J, Liu Z, and Liu H. 2019. ‘TGR5 agonist INT-777 mitigates inflammatory response in human endometriotic stromal cells: A therapeutic implication for endometriosis’, Int Immunopharmacol, 71: 93–99. [DOI] [PubMed] [Google Scholar]
- Macdonald RL 2014. ‘Delayed neurological deterioration after subarachnoid haemorrhage’, Nat Rev Neurol, 10: 44–58. [DOI] [PubMed] [Google Scholar]
- Maczewsky J, Kaiser J, Gresch A, Gerst F, Dufer M, Krippeit-Drews P, and Drews G. 2019. ‘TGR5 Activation Promotes Stimulus-Secretion Coupling of Pancreatic beta-Cells via a PKA-Dependent Pathway’, Diabetes, 68: 324–36. [DOI] [PubMed] [Google Scholar]
- McMillin M, Frampton G, Tobin R, Dusio G, Smith J, Shin H, Newell-Rogers K, Grant S, and DeMorrow S. 2015. ‘TGR5 signaling reduces neuroinflammation during hepatic encephalopathy’, J Neurochem, 135: 565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J, Peng J, Matei N, Yang P, Kuai L, Wu Y, Chen L, Vitek MP, Li F, Sun X, Zhang JH, and Jiang Y. 2018. ‘Apolipoprotein E Exerts a Whole-Brain Protective Property by Promoting M1? Microglia Quiescence After Experimental Subarachnoid Hemorrhage in Mice’, Transl Stroke Res, 9: 654–68. [DOI] [PubMed] [Google Scholar]
- Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, Schoonjans K, and Auwerx J. 2009. ‘Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity’, J Med Chem, 52: 7958–61. [DOI] [PubMed] [Google Scholar]
- Peng J, Pang J, Huang L, Enkhjargal B, Zhang T, Mo J, Wu P, Xu W, Zuo Y, Peng J, Zuo G, Chen L, Tang J, Zhang JH, and Jiang Y. 2019. ‘LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats’, Redox Biol, 21: 101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Zuo Y, Huang L, Okada T, Liu S, Zuo G, Zhang G, Tang J, Xia Y, and Zhang JH. 2019. ‘Activation of GPR30 with G1 attenuates neuronal apoptosis via src/EGFR/stat3 signaling pathway after subarachnoid hemorrhage in male rats’, Exp Neurol, 320: 113008. [DOI] [PubMed] [Google Scholar]
- Qin C, Fan WH, Liu Q, Shang K, Murugan M, Wu LJ, Wang W, and Tian DS. 2017. ‘Fingolimod Protects Against Ischemic White Matter Damage by Modulating Microglia Toward M2 Polarization via STAT3 Pathway’, Stroke, 48: 3336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Rokad D, Malovic E, Luo J, Harischandra DS, Jin H, Anantharam V, Huang X, Lewis M, Kanthasamy A, and Kanthasamy AG. 2019. ‘Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells’, Sci Signal, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap FG, Trauner M, and Jansen PL. 2014. ‘Bile acid receptors as targets for drug development’, Nat Rev Gastroenterol Hepatol, 11: 55–67. [DOI] [PubMed] [Google Scholar]
- Schneider UC, Davids AM, Brandenburg S, Muller A, Elke A, Magrini S, Atangana E, Turkowski K, Finger T, Gutenberg A, Gehlhaar C, Bruck W, Heppner FL, and Vajkoczy P. 2015. ‘Microglia inflict delayed brain injury after subarachnoid hemorrhage’, Acta Neuropathol, 130: 215–31. [DOI] [PubMed] [Google Scholar]
- Shao A, Wu H, Hong Y, Tu S, Sun X, Wu Q, Zhao Q, Zhang J, and Sheng J. 2016. ‘Hydrogen-Rich Saline Attenuated Subarachnoid Hemorrhage-Induced Early Brain Injury in Rats by Suppressing Inflammatory Response: Possible Involvement of NF-kappaB Pathway and NLRP3 Inflammasome’, Mol Neurobiol, 53: 3462–76. [DOI] [PubMed] [Google Scholar]
- Shapiro H, Kolodziejczyk AA, Halstuch D, and Elinav E. 2018. ‘Bile acids in glucose metabolism in health and disease’, J Exp Med, 215: 383–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She Y, Shao L, Zhang Y, Hao Y, Cai Y, Cheng Z, Deng C, and Liu X. 2019. ‘Neuroprotective effect of glycosides in Buyang Huanwu Decoction on pyroptosis following cerebral ischemia-reperfusion injury in rats’, J Ethnopharmacol, 242: 112051. [DOI] [PubMed] [Google Scholar]
- Song N, Fang Y, Sun X, Jiang Q, Song C, Chen M, Ding J, Lu M, and Hu G. 2018. ‘Salmeterol, agonist of beta2-aderenergic receptor, prevents systemic inflammation via inhibiting NLRP3 inflammasome’, Biochem Pharmacol, 150: 245–55. [DOI] [PubMed] [Google Scholar]
- Sorrentino G, Perino A, Yildiz E, El Alam G, Sleiman MB, Gioiello A, Pellicciari R, and Schoonjans K. 2020. ‘Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration’, Gastroenterology. [DOI] [PubMed] [Google Scholar]
- Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brone B, Huaux F, Octave JN, Terwel D, and Dewachter I. 2019. ‘Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo’, Acta Neuropathol, 137: 599–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Ayer R, Jadhav V, and Zhang JH. 2008. ‘A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model’, J Neurosci Methods, 167: 327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Enkhjargal B, Reis C, Zhang T, Zhu Q, Zhou K, Xie Z, Wu L, Tang J, Jiang X, and Zhang JH. 2019. ‘Osteopontin-Enhanced Autophagy Attenuates Early Brain Injury via FAK-ERK Pathway and Improves Long-Term Outcome after Subarachnoid Hemorrhage in Rats’, Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H 2019. ‘Inflammation: a Good Research Target to Improve Outcomes of Poor-Grade Subarachnoid Hemorrhage’, Transl Stroke Res, 10: 597–600. [DOI] [PubMed] [Google Scholar]
- Tan CC, Zhang JG, Tan MS, Chen H, Meng DW, Jiang T, Meng XF, Li Y, Sun Z, Li MM, Yu JT, and Tan L. 2015. ‘NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model’, J Neuroinflammation, 12: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, and Yu JT. 2014. ‘Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease’, Cell Death Dis, 5: e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, and Heneka MT. 2017. ‘Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease’, Nature, 552: 355–61. [DOI] [PubMed] [Google Scholar]
- Vettorazzi JF, Ribeiro RA, Borck PC, Branco RC, Soriano S, Merino B, Boschero AC, Nadal A, Quesada I, and Carneiro EM. 2016. ‘The bile acid TUDCA increases glucose-induced insulin secretion via the cAMP/PKA pathway in pancreatic beta cells’, Metabolism, 65: 54–63. [DOI] [PubMed] [Google Scholar]
- Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, Lucia S, Adorini L, D’Agati VD, Levi J, Rosenberg A, Kopp JB, Gius DR, Saleem MA, and Levi M. 2016. ‘G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes’, J Am Soc Nephrol, 27: 1362–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Lv YG, Du YF, Chen F, Reed MN, Hu M, Suppiramaniam V, Tang SS, and Hong H. 2018. ‘Neuroprotective effects of INT-777 against Abeta1–42-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice’, Brain Behav Immun, 73: 533–45. [DOI] [PubMed] [Google Scholar]
- Wu X, Lv YG, Du YF, Hu M, Reed MN, Long Y, Suppiramaniam V, Hong H, and Tang SS. 2019. ‘Inhibitory effect of INT-777 on lipopolysaccharide-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice’, Prog Neuropsychopharmacol Biol Psychiatry, 88: 360–74. [DOI] [PubMed] [Google Scholar]
- Xiao H, Sun X, Liu R, Chen Z, Lin Z, Yang Y, Zhang M, Liu P, Quan S, and Huang H. 2020. ‘Gentiopicroside activates the bile acid receptor Gpbar1 (TGR5) to repress NF-kappaB pathway and ameliorate diabetic nephropathy’, Pharmacol Res, 151: 104559. [DOI] [PubMed] [Google Scholar]
- Xie Y, Guo H, Wang L, Xu L, Zhang X, Yu L, Liu Q, Li Y, Zhao N, Zhao N, Ye R, and Liu X. 2017. ‘Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage’, Brain Behav Immun, 60: 346–60. [DOI] [PubMed] [Google Scholar]
- Xu W, Mo J, Ocak U, Travis ZD, Enkhjargal B, Zhang T, Wu P, Peng J, Li T, Zuo Y, Shao A, Tang J, Zhang J, and Zhang JH. 2020. ‘Activation of Melanocortin 1 Receptor Attenuates Early Brain Injury in a Rat Model of Subarachnoid Hemorrhage viathe Suppression of Neuroinflammation through AMPK/TBK1/NF-kappaB Pathway in Rats’, Neurotherapeutics, 17: 294–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhou H, Zhuang L, Auwerx J, Schoonjans K, Wang X, Feng C, and Lu L. 2017. ‘Plasma membrane-bound G protein-coupled bile acid receptor attenuates liver ischemia/reperfusion injury via the inhibition of toll-like receptor 4 signaling in mice’, Liver Transpl, 23: 63–74. [DOI] [PubMed] [Google Scholar]
- Yang X, Sun J, Kim TJ, Kim YJ, Ko SB, Kim CK, Jia X, and Yoon BW. 2018. ‘Pretreatment with low-dose fimasartan ameliorates NLRP3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage’, Exp Neurol, 310: 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanguas-Casas N, Barreda-Manso MA, Nieto-Sampedro M, and Romero-Ramirez L. 2017. ‘TUDCA: An Agonist of the Bile Acid Receptor GPBAR1/TGR5 With Anti-Inflammatory Effects in Microglial Cells’, J Cell Physiol, 232: 2231–45. [DOI] [PubMed] [Google Scholar]
- Ye Y, Bajaj M, Yang HC, Perez-Polo JR, and Birnbaum Y. 2017. ‘SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor’, Cardiovasc Drugs Ther, 31: 119–32. [DOI] [PubMed] [Google Scholar]
- Zhang T, Wu P, Budbazar E, Zhu Q, Sun C, Mo J, Peng J, Gospodarev V, Tang J, Shi H, and Zhang JH. 2019. ‘Mitophagy Reduces Oxidative Stress Via Keap1 (Kelch-Like Epichlorohydrin-Associated Protein 1)/Nrf2 (Nuclear Factor-E2-Related Factor 2)/PHB2 (Prohibitin 2) Pathway After Subarachnoid Hemorrhage in Rats’, Stroke, 50: 978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu Q, Zhang Q, Lu Y, Liu J, Li W, Lv S, Zhou M, Zhang X, and Hang C. 2017. ‘Resveratrol Attenuates Early Brain Injury after Experimental Subarachnoid Hemorrhage via Inhibition of NLRP3 Inflammasome Activation’, Front Neurosci, 11: 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng VZ, and Wong GKC. 2017. ‘Neuroinflammation Responses after Subarachnoid Hemorrhage: A Review’, J Clin Neurosci, 42: 7–11. [DOI] [PubMed] [Google Scholar]
- Zheng ZV, Lyu H, Lam SYE, Lam PK, Poon WS, and Wong GKC. 2020. ‘The Dynamics of Microglial Polarization Reveal the Resident Neuroinflammatory Responses After Subarachnoid Hemorrhage’, Transl Stroke Res, 11: 433–49. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun C, Xie Z, Wu P, Mo J, Tang J, Xie Z, and Zhang JH. 2018. ‘Aggf1 attenuates neuroinflammation and BBB disruption via PI3K/Akt/NF-kappaB pathway after subarachnoid hemorrhage in rats’, J Neuroinflammation, 15: 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang K, Zuo YC, Sherchan P, Wang JK, Yan XX, and Liu F. 2019. ‘Hydrogen Inhalation Attenuates Oxidative Stress Related Endothelial Cells Injury After Subarachnoid Hemorrhage in Rats’, Front Neurosci, 13: 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo G, Zhang T, Huang L, Araujo C, Peng J, Travis Z, Okada T, Ocak U, Zhang G, Tang J, Lu X, and Zhang JH. 2019. ‘Activation of TGR5 with INT-777 attenuates oxidative stress and neuronal apoptosis via cAMP/PKCepsilon/ALDH2 pathway after subarachnoid hemorrhage in rats’, Free Radic Biol Med, 143: 441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Huang L, Enkhjargal B, Xu W, Umut O, Travis ZD, Zhang G, Tang J, Liu F, and Zhang JH. 2019. ‘Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARgamma/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats’, J Neuroinflammation, 16: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Wang J, Liao F, Yan X, Li J, Huang L, and Liu F. 2018. ‘Inhibition of Heat Shock Protein 90 by 17-AAG Reduces Inflammation via P2×7 Receptor/NLRP3 Inflammasome Pathway and Increases Neurogenesis After Subarachnoid Hemorrhage in Mice’, Front Mol Neurosci, 11: 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure s1. Schematic summary. INT-777 attenuates NLRP3-ASC inflammasome-dependent neuroinflammation via TGR5/cAMP/PKA signaling pathway and improves long-term neurological deficits after subarachnoid hemorrhage in rats.
Figure s2. Experimental design. SAH, subarachnoid hemorrhage; WB, western blot; BWC, brain water content; IF, immunofluorescence; i.n, intranasal; i.c.v, intracerebroventricular; i.p, intraperitoneal; DMSO, dimethyl sulfoxide.
Figure s3. Effects of TGR5 knockout CRISPR and PKA inhibitor on neurobehavioral outcome at 24 h after SAH. (A-B) TGR5 knockout CRISPR and PKA inhibitor H89 pretreatment reversed the neurobehavioral benefits of INT-777 at 24 h after SAH, n=6 per group.