

Abstract
Background &Aims
Increased hepatocyte death contributes to the pathology of acute and chronic liver diseases. The role of hepatocyte pyroptosis and extracellular inflammasome release in liver disease is unknown.
Methods
We used primary mouse and human hepatocytes, hepatocyte-specific L351P Nlrp3KICreA mice and GsdmdKO mice to investigate pyroptotic cell death in hepatocytes and its impact on liver inflammation and damage. Extracellular NLRP3 inflammasomes were isolated from mutant NLRP3-YFP HEK cells and internalization was studied in LX2 and primary human hepatic stellate cells. We further utilized a cohort of 154 adult subjects with biopsy-proven NAFLD (Sir Charles Gairdner Hospital, Nedlands, Western Australia).
Results
We demonstrated that primary mouse and human hepatocytes can undergo pyroptosis upon NLRP3 inflammasome activation with subsequent release of NLRP3 inflammasome proteins that amplify and perpetuate inflammasome-driven fibrogenesis. Pyroptosis was inhibited by blocking caspase-1 and Gasdermin D activation. The activated form of caspase-1 was detected in the livers and in serum from patients with NASH and correlated with disease severity. Nlrp3KICreA mice showed spontaneous liver fibrosis under normal chow diet, and increased sensitivity to liver damage and inflammation after treatment with low dose LPS. Mechanistically, hepatic stellate cells engulfed extracellular NLRP3 inflammasome particles leading to increased IL-1ß secretion and α-SMA expression. This effect was abrogated when cells were pre-treated with the endocytosis inhibitor Cytochalasin B.
Conclusion
These results identify hepatocyte pyroptosis and release of inflammasome components as a novel mechanism to propagate liver injury and liver fibrosis development.
Keywords: Pyroptosis, NLRP3, inflammasome, specks, ASC, fibrosis, NASH, liver, hepatocytes
Lay summary
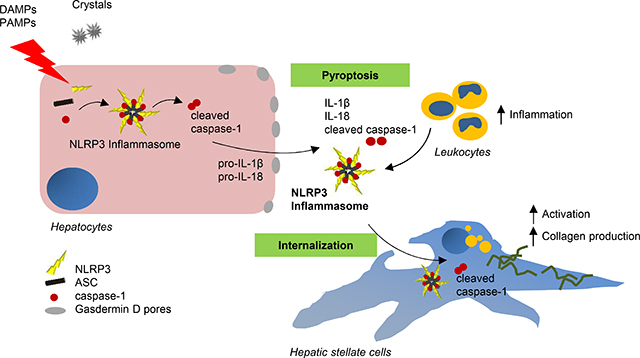
Our findings identify a novel mechanism of inflammation in the liver. Experiments in cell cultures, mice and human samples show that a specific form of cell death, called pyroptosis, leads to the release of complex inflammatory particles, the NLRP3 inflammasome, from inside hepatocytes into the extracellular space. From there they are taken up by other cells and thereby mediate inflammatory and pro-fibrogenic stress signals. The discovery of this mechanism may lead to novel treatments for chronic liver diseases in the future.
Graphical Abstract
Introduction
Hepatocyte damage represents a central mechanism involved in inflammation and disease progression in a variety of acute and chronic liver disorders [1, 2]. Recent evidence suggests a crucial role of pyroptosis in liver diseases [2]. Pyroptosis is dependent on inflammasome mediated caspase-1 activation and results in the formation of plasma membrane pores due to Gasdermin D (GSDMD) insertion, leading to the release of intracellular proteins, ion decompensation, water influx and cell swelling [3]. Inflammasomes are cytoplasmic multiprotein complexes typically consisting of an inflammasome sensor molecule such as NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3), theadaptor protein ASC (apoptosis-associated speck-like protein containing CARD), and the effector molecule pro-caspase-1 [4–6]. Upon stimulation, NLRP3 binds to ASC and builds the inflammasome complex leading to caspase-1 activation, which subsequently proteolytically activates proinflammatory cytokines Interleukin-1ß (IL-1ß) and IL-18, and the cytosolic protein GSDMD [3, 7, 8]. Recent findings in macrophages revealed that during pyroptosis inflammasome oligomers composed of NLRP3 and ASC are released into extracellular space where they can be phagocytosed by neighboring macrophages [9]. Little is known about the role of inflammasome activation and pyroptosis in non-myeloid cells and whether they respond to both sterile and infectious stressors [10–12]. A central question that remains unanswered is whether pyroptotic cell death occurs in hepatocytes and its potential role in-vivo in the development of liver injury and fibrosis.
Previous studies by our group and others have reported a crucial role of NLRP3 inflammasome activation in the development and progression of liver diseases [13–15]. As the liver is a “first pass” organ, continually challenged with diverse microbial particles from the intestine as well as endogenous metabolic stress signals, we hypothesized that hepatocytes are capable of undergoing NLRP3-mediated pyroptotic cell death and release extracellular NLRP3 inflammasome complexes into the extracellular space. We also hypothesize that extracellular inflammasomes can be internalized by hepatic stellate cells leading to their activation and liver fibrogenesis. This intercellular crosstalk between hepatocytes and hepatic stellate cells may reveal a novel mechanism of cell-to-cell communication which can spread inflammation and trigger liver fibrogenesis independent of IL-1ß-secretion.
Material and Methods
Antibodies details are given in Supplemental table 1.
Mouse strains
The Nlrp3 knock-in mouse strain with a leucine 351 to proline (L351P) substitution was described previously [16]. Mice expressing Cre recombinase under control of the hepatocyte specific albumin promoter (Alb1-Cre mice) were obtained from Jax (#016832). Gasdermin D knock-out (GsdmdKO) mice were kindly provided by Dr. V. Dixit at Genentech and Dr. G. Mbalaviele at Washington University (St. Louis, USA) [17, 18].
Generation of hepatocytes specific NLRP3 mutant mice (L351P Nlrp3KICreA)
Floxed L351P Nlrp3 KI mice were bred with Alb1-Cre mice. Those mice co-express Cre-recombinase to delete a neomycin cassette inserted in reverse orientation that when present causes gene silencing. This allowed us to generate a hepatocyte-specific Nlrp3 heterozygous gain of function mutant mouse strain (L351P Nlrp3KICreA) with constitutively activated NLRP3 [16]. Animal studies were performed following the ARRIVE guidelines. The University of California, San Diego Institutional Animal Care and Use Committee approved the protocol (Animal protocol number S11200). To increase basal protein levels of Nlrp3, mice were injected with lipopolysaccharide (LPS) as previously described [19]. Briefly, mice received a single i.p. injection 500ng/kg LPS (E.coli 0111:B4) for 4h.
Liver histology and immunostaining
Liver tissue was collected and stained for α-SMA, TUNEL positive cells and picrosirius red as previously described [20]. To detect caspase-1 activity in frozen liver tissue slides, we used the FAM-Flica caspase-1 Assay Kit (ImmunoChemistry Technologies, Bloomington, MN, USA) according to manufacturer’s instructions. Flica-positive cells were quantified by ImageJ and normalized to the total number of DAPI-stained cells. F4/80 and Cd11b staining was performed as described previously [21]. Positive cells were counted in 10X magnification images and normalized to DAPI stained cells. Immunofluorescent staining of α-SMA was performed on aceton fixedcells and relative fluorescence intensity was normalized on total cell count.
Cell culture
Human hepatoma cell line (HepG2) and human immortalized hepatic stellate cells (LX2) were obtained and cultured as previously described [22,23]
Primary murine hepatocyte isolation
Primary hepatocytes were isolated using a two-step perfusion method as previously described [24]. Isolated hepatocytes were seeded on collagen-coated culture dishes and used for experiments on the following day.
Primary human hepatocytes and stellate cells
Primary human hepatocytes were isolated from 4 different donors at Leipzig University. Liver tissue samples were obtained from macroscopically healthy tissue that remained from resected human liver of patients with primary or secondary liver tumors (or benign local liver diseases). Written informed consent of the patients for the use of tissue for research purposes was obtained according to the ethical guidelines of Leipzig University Hospital (Az.:17716/lk). PHH were isolated as described previously by Damm et al. [25]. Primer sequences for gene expression are shown in Supplemental table 2. Human primary hepatic stellate cells (HSCs) (n=2) were provided in-house at the UCSD [26].
Cell stimulation
Primary hepatocytes isolated from Nlrp3KICreA mice, primary human hepatocytes, human HepG2 and LX2 and primary stellate cells were cultured as previously described [20, 22, 23]. After 24h seeding, cells were set on serum-free media overnight before treatment. HepG2 cells and murine hepatocytes were stimulated with LPS (1 μg/ml; 6h) alone or followed by Nigericin (Nig) (20 μM; 1h). Palmitate (0.33 mM; 24h) was added to serum-free media containing 0.5% fatty-acid free BSA before stimulation with LPS (1 ug/ml; 6h). Caspase-1 Inhibitor IV (Calbiochem, 50 μM) and Nlrp3 inhibitor (MCC950, 1 μM) were added to the media 1h before stimulation. DMSO was used as a solvent control. Supernatant was collected for cytotoxicity analysis using the LDH Cytotoxicity Assay Kit (Pierce, ThermoFisher Scientific) according to the manufactureŕs instructions. Primary human hepatocytes were stimulated in serum-free media. To activate the NLRP3 inflammasome, cells were first treated with or without the NLRP3 inhibitor MCC950(1 μM)or caspase-1 inhibitor Ac-YVAD-cmk (25 mg/ml) for 1h and then primed with LPS (1 μg/ml) for 18h. Then, cells were stimulated with Nigericin (20 μM, 2h). Pyroptosis was measured by immunostaining of Flica reagent FAM-YVAD-FMK to stain caspase-1 activity and propidium iodide (PI) as previously described [27].
NLRP3 mutant cell line and particle isolation
A stable mutant NLRP3 (p.D303N)-YFP HEK cell line [9] was used to isolate overactive YFP-labeled NLRP3-Inflammasome oligomeric particles as previously described [28]. As adequate control, WT mock transfected HEK cells were used and the same particle isolation protocol was performed. Fluorescent particles were quantified in a fluorescence microscope using a Neubauer hemocytometer and 1:10 dilution. NLRP3-YFP oligomers were then calculated as particles/μl solution and five NLRP3-YFP particles/ seeded cell were used for in-vitro analysis. Isolated mutant NLRP3-YFP oligomers or preparations from WT mock controls were used to stimulate hepatic stellate cells for 24 and 48h. Internalization of extracellular inflammasomes in target cells was analyzed with an Olympus FV1000 Confocal Microscope (Shinjuku) by repeat x-y scanning at 12.5 resolution pixels and unchanged laser power as well as by western blot using anti-GFP (YFP) antibody.
ASC plasmid transfection
DNA constructs encoding YFP-tagged ASC (1 μg DNA) were transiently transfected in HepG2 cells using Lipofectamine™ 2000 according to the manufacturer's instructions (ThermoFisher Scientific). Transfected HepG2 cells were then stimulated as indicated above for 48h to assess the release of ASC-YFP specks. Nuclei were stained with the Hoechst 33342 (Sigma-Aldrich), and then both cells and supernatant were analyzed with a fluorescence microscope (Olympus IX71). Images from 10 fields were captured at 20X magnification and later ASC-YFP specks were counted. Further, transfected HepG2 cells were treated with LPS and Nigericin or palmitate to activate inflammasome signaling and pyroptosis. Then, supernatant was transferred to LX2 cells and uptake of fluorescent-labeled ASC specks and gene expression was analyzed.
siRNA knock-down
Knockdown (KD) of GSDMD was performed by transfection of siRNA oligonucleotides targeting human GSDMD (Dharmacon, Catalogue No. L-016207–00-0005). Scrambled (control) or GSDMD-specific siRNA was transfected into HepG2 cells as per manufacture’s protocol. Forty-eight hours after transfection, cells were incubated with LPS alone or followed with Nig or PA in OPTI-MEM medium.
Immunoblot analysis
Livers or cells were homogenized in RIPA buffer (Cell Signaling, Danvers, MA, USA) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO, USA) and immunoblot analysis was performed as described previously [20]. Densitometric analysis was performed with ImageJ after background subtraction and normalized to loading control (GAPDH) or No-stain™ protein labeling reagent (ThermoFisher Scientific). List of antibodies and dilutions is provided in Supplemental table 1. To detect extracellular inflammasome components, cell-free supernatant was concentrated using Amicon Ultracel −3K (Millipore) and centrifuged at 3800 × g at 4°C for 1h.
Real-time PCR
Total RNA was isolated from liver tissue and cells and analyzed as previously described [27]. The sequences of the primers used for quantitative PCR are provided in Supplemental table 2.
Human Study Cohort
To examine the relationship between serum markers of NLRP3 activation and severity of liver injury based on histology, we utilized a cohort of 154 adult subjects with biopsy-proven NAFLD (Supplemental table 3). Subjects had been seen at a hepatology or bariatric surgery clinic at a tertiary center (Sir Charles Gairdner Hospital) and undergone liver biopsy as part of their clinical care. Patients underwent a standardized assessment including anthropometric and blood pressure measures, a standardized questionnaire including quantification of alcohol consumption, and collection of fasting serum samples on the day of liver biopsy. Each participant had an average daily alcohol consumption <20 g in females and <30 g in males. Alternative causes of liver disease including viral hepatitis, hemochromatosis, alpha-1 antitrypsin deficiency, auto-immune hepatitis, Wilson’s disease and drug induced liver injury were excluded using standard clinical, laboratory and histological criteria. Liver biopsies were assessed by an expert liver histopathologist and scored according to the NASH–CRN histological system [29]. Patients provided informed written consent and the study was approved by the Sir Charles Gairdner Hospital Human Research Ethics Committee.
Serum analysis
Serum stored at −80°C from the biopsy proven NAFLD cohort (n=154) was analysed for caspase-1 activity by fluorometric assay (Abcam, Cambridge MA). Briefly, for the functional assay of serum caspase-1 activity, 10 μl of serum was aliquoted onto a 96 well plate and incubated on ice for 10 min with 40 μl of cell lysis buffer; 50 μl of 2x reaction buffer containing 10 nM DTT and 5 μl of 50 μM YVAD-AFC substrate was added to each sample. Following incubation for 90 min at 37°C, activity was read on a fluorometer (Ex400/Em505). Samples read as fold increase minus untreated control at 30 min intervals.
Statistics
Statistical analyses were performed with Graph Pad Prism (version 7; Graph Pad Software Inc., La Jolla, CA, USA) and IBM SPSS Statistics version 24.0. (IBM Corporation, Armonk, New York, USA). The significance level was set at p< 0.05 for all comparisons. Gaussian distributed data were analyzed using One-way analysis of variance and Bonferroni post-hoc test. Nonparametric data were compared using Kruskal-Wallis test followed by the Dunn’s post-hoc test (*p< 0.05). Two groups were analyzed by Studentś T-test. Data are expressed as mean ± standard error of mean (SEM).
Results
Hepatocytes undergo caspase-1 dependent pyroptotic cell death and release inflammasome oligomers when exposed to canonical NLRP3 inflammasome activators
To test the hypothesis that hepatocytes are capable of undergoing pyroptotic cell death, we initially performed in vitro studies by stimulating isolated primary mouse hepatocytes, human hepatocytes and HepG2 cells with LPS plus Palmitate (PA) or nigericin (Nig) as an additional second signal. Both treatments resulted in a significant increase in the number of active caspase-1 and PI positive cells in primary mouse hepatocytes (Fig. 1A, B) and human hepatocytes (Fig. 2A) as well as in human HepG2 cells (Supplementary Fig.1). The combination of LPS plus Nig and LPS plus PA increased LDH release in primary murine hepatocytes (Fig. 1C) and in HepG2 cells (Supplementary Fig.1). Blocking caspase-1 activity completely abolished LDH release (Fig. 1C, Supplementary Fig. 1) and pyroptotic cell death in human hepatocytes (Fig. 2A, B). We further showed that increased caspase-1 activation was dependent on NLRP3 inflammasome assembly and activation as the NLRP3-inhibitor MCC950 abrogated caspase-1 activation in HepG2 cells (Supplementary Fig.2). Knock-down (KD) of Gasdermin D in HepG2 cells or studies on primary hepatocytes isolated from GsdmdKO mice showed resistance against inflammasome-induced pyroptotic cell death (Fig. 1 G, H) indicating that hepatocyte cell death was dependent on Gasdermin D activation.
Figure 1. Hepatocytes undergo pyroptotic cell death following NLRP3 inflammasome activation.

Representative immunofluorescence images (A) and quantification (B) showing pyroptosis induction by LPS + Nig and LPS + PA in primary mouse hepatocytes (*p< 0.05 compared to control, #p< 0.05 compared to LPS) (scale bar: 100 μm). (C) LDH release in presence of caspase-1 inhibitor (*p< 0.05 compared to control, #p< 0.05 compared to LPS + Nig and LPS + PA, respectively). (D) Representative images showing release of ASC specks from HepG2 cells transfected with a plasmid expressing ASC-YFP and treated with LPS + Nig and LPS + PA (scale bar: 10 0μm). Intra- an dextracellula rspeck swer equantified and compared to control. Western blot (E) and densitometric analysis (F) of cell-free supernatant of HepG2 cells after treatment with LPS and LPS + Nig for NLRP3 inflammasome components (NLRP3, ASC, pro-caspase1, pro-ILf) an dit scleave dmatur eform s(cleaved caspase-1 p20, mature IL-1ß 17kDa). Level of extracellular proteins were calculated as % of their intracellular protein amount (F) (*p< 0.05 vs. control). (G) LDH release of Gasdermin D (KD) silenced or scramble siRNA transfected HepG2 cells treated with LPS + Nig or PA. (H) LDH release frol primar ymurin ehepatocyte so fGsdmdK Ocompare dt oW Tmice treated with LPS + Nig or PA. Data was normalized to control serum-free media (*p< 0.05 vs. control, #p<0.05 vs. WT hepatocytes). Gaussian distributed data were analyzed using One-way analysis of variance and Bonferroni post-hoc test.
Figure 2. Primary human hepatocytes (PHH) undergo pyroptotic cell death and release activated inflammasome proteins and IL-1ß.

PHH (n=3–4) were treated in starvation medium for NLRP3 inflammasome activation with LPS and Nig and in presence of inhibitors for NLRP3 and caspase-1. (A) Flow cytometric analysis of compensated Flica-caspase-1and PI double-positive cells (*p< 0.05). (B) mRMA expression of primary human hepatocytes (n=3) treated with LPS + Nig with or without NLRP3 and caspase-1 inhibitors normalized on housekeeping genes and referred to the control which was set as 1. Statistical significance was tested using One-way ANOVA and Bonferroni post-hoc test (*p< 0.05). Western blot analysis (C) and densitometric analysis (D) of concentrated supernatant and lysate of human hepatocytes (n=3) for NLRP3, cleaved caspase-1 p20, pro-caspase-1, mature IL-1ß 17kDa and pro-IL1ß normalized on whole-lane sample load. Data were referred to starvation media which was set at 1 (D).
We next examined whether the pyroptotic process in hepatocytes was associated with the release of NLRP3 oligomers and other inflammasome components. Cells treated with LPS plus Nig or LPS plus PA lead to the release of ASC-YFP specks by 43.1% and 32.4%, respectively (Fig. 1D). Immunoblot analysis also revealed that hepatocytes release NLRP3 inflammasome components, such as NLRP3, ASC, pro-caspase-1, pro-IL-1ß but also mature IL-1ß and cleaved caspase-1 (p20) into the extracellular space (Fig. 1E–F, 2C–D). Additionally, increased pyroptotic cell death in primary human hepatocytes were associated with increased mRNA expression of NLRP3 and IL1b (Fig. 2B).
Caspase-1 activation occurs in the liver of patients with NASH and can be monitored in circulation
Weexamined for the presence of caspase-1 activation in frozen liver sections from patients with NASH, steatosis and normal controls and found increased caspase-1 activation in NASH livers compared to normal controls (Fig. 3A). Next, serum caspase-1 activity was assessed in a large cohort comprised of 154 subjects with biopsy proven NAFLD (Supplementary table 3). Approximately one third (n=63, 29.7%) had NASH. Serum caspase-1 activity was significantly increased in the presence of hepatocellular ballooning, inflammation and fibrosis, but not steatosis, demonstrating specificity for liver injury (Fig. 3B–E). Additionally, western blot analysis in asub-setof 20 subjects demonstrated the active caspase-1 was significantly higher in patients with NASH than those with steatosis or controls (Supplementary Fig. 4).
Figure 3. Caspase-1 activity increases in liver and serum of NASH patients and correlates with liver injury severity.

(A) Active caspase-1 levels were analyzed in frozen liver tissue sections (n 3 per group). Nuclei were stained with DAPI. Fluorescence intensity was analyzed using ImageJ and normalized on DAPI positive cells. Statistical significance was tested using One-way ANOVA and Bonferroni post-hoc test. (*p< 0.05 vs. control, scale bar: 100 μm). Level of serum caspase-1 activity in patients with present or absent (B) Steatosis, (C) Inflammation, (D) Ballooning and (E) Fibrosis stage. Two groups were analyzed by Studentś T-test (*p< 0.05 vs. control).
Hepatocyte specific NLRP3 over-activation induce pyroptotic cell death ex vivo and in vivo and triggers stellate cell activation and fibrosis in hepatocyte-specific Nlrp3KI CerA mice
To mechanistically investigate the relevance of hepatocyte pyroptosis in liver injury in-vivo, we generated a hepatocyte-specific Nlrp3KI CreA mice. We initially examined the occurrence of pyroptosis ex-vivo in hepatocytes isolated from these mice. Hepatocytes isolated from Nlrp3KICreA mice showed 5-times higher caspase-1/PI- double positive cells than hepatocytes from WT mice indicating pyroptotic cell death (Fig. 4A, B). We next assessed the potential effects of hepatocyte pyroptosis in vivo. Nlrp3KICreA mice were maintained on a regular chow diet for 6 and 9 months. We found an increase in TUNEL positive cells in the livers of Nlrp3KICreA mice at 9 months (1.2- fold vs. WT, p< 0.05) (Fig. 4C) but not at 6 months of age (Supplementary Figure 4) demonstrating hepatocyte death in Nlrp3KICreA mice. Hepatic gene expression analysis revealed that marker genes of fibrogenesis (Timp1: 3.4- fold vs. WT, Fibronectin 1: 1.8- fold vs. WT, p<0.05) and stellate cell activation were up-regulated in Nlrp3KICreA mice at 9 month of age (Fig. 4F). This was in line with increased collagen deposition in livers of Nlrp3KICreA mice measured by Sirius red staining (1.8- fold vs. WT, p< 0.05) (Fig. 4D) as well as increased a-SMA protein expression (1.5- fold vs. WT, p< 0.05) (Fig. 4E). These findings uncover a novel link between hepatocyte NLRP3 activation, pyroptosis and liver fibrogenesis.
Figure 4. Hepatocyte-specific Nlrp3 mutant mice (L351P Nlrp3KICreA) showed increased hepatocyte caspase-1 activation and increased fibrogenesis.

(A) Flica-caspase-1 activity (green) and PI (red) stained Nlrp3KICreA and WT primary hepatocytes (scale bar: 100 μm). (B) Quantification of caspase-1 positive cells (%) normalized on WT hepatocytes. (*p< 0.05 vs. WT hepatocytes). Immunohistological stainings and quantification of livers from WT and Nlrp3KICreA mice for (C) TUNEL positive cells (scale bar: 250 μm), (D) Sirius red collagen disposition (scale bar: 250 μm), and (E) α-SMA protein (scale bar: 100 μm). (F) mRNA expression of Timp1 and Fibronectin 1 (Fn1) in Nlrp3KICreA livers. Two groups were analyzed by Studentś T-test (*p< 0.05).
Hepatocyte-specific Nlrp3 mutant mice are more prone to LPS-induced liver inflammation and hepatocytes cell death
As the endogenous expression level of NLRP3 in hepatocytes are very low [30], we injected Nlrp3KICreA mice with LPS (4hours)toincrease the expression of NLRP3 and exacerbate the effects of our constitutively active hepatocyte-specific transgenic mice. Immunohistology of the livers showed increased leukocyte infiltration (Fig. 5A), TUNEL positive cells (Fig. 5B) and F4/80- and Cd11b- positive cells (Fig. 5C,D) in Nlrp3KICreA mice compared to WT mice treated with LPS. We also measured increased Gasdermin D activation in livers of LPS-injected Nlrp3KICreA mice compared to WT mice (Fig. 5E). These data showed that upon priming with LPS, Nlrp3 hepatocyte-specific Nlrp3KICreA mice were more susceptible to liver cell death and inflammation.
Figure 5. Hepatocyte-specific Nlrp3 mutant mice are more prone to LPS- induced liver inflammation and hepatocytes cell death.

(A) H&E staining, (B) TUNEL positive cells (% of total area), (C) F4/80 positive cells and (D) Cd11b positive cells (% of total cells) in livers from WT and Nlrp3KICreA mice injected with LPS (scale bars: 200 μm). (E) Western blot and densitometric analysis of GasderminD N-terminal (31 kDa) protein in liver lysate from WT (n=3) and Nlrp3KICreA mice (n=3) injected with LPS. Data were normalized on β-actin. Two groups were analyzed by Studentś T-tes (*p< 0.05 vs. WT control).
3. Murine and human hepatic stellate cells internalize extracellular NLRP inflammasome oligomers
The findings of increased HSC activation, fibrogenesis and early fibrotic changes occurring spontaneously in the liver of Nlrp3KICreA mice in conjunction with the strong correlation between circulating caspase-1 activity with liver fibrosis in NASH patients lead us to study the potential mechanisms involved in this process. We designed experiments to test the hypothesis that NLRP3 inflammaosme particles released by hepatocytes during pyroptosis can be internalized by HSC and modulate its phenotype. YFP fluorescent-labeled NLRP3 was isolated from a Nlrp3 mutant HEK cell line carrying the same mutation in the NLRP3 gene (L351P) as our hepatocyte-specific Nlrp3KICreA mice that leads to spontaneous oligomerization and activation of NLRP3 in these cells [9, 28]. The internalization of extracellular NLRP3-YFP inflammasomes were confirmed by confocal microscopy (Fig. 6A) and by immunoblot analysis of LX2 lysate after incubation with NLRP3-YFP inflammasome oligomers (Fig. 6B). Further, the uptake of extracellular NLRP3-YFP inflammasome oligomers induced IL-1ß secretion from HSC indicating that internalized NLRP3 inflammasomes are able to promote IL-1ß secretion in target cells (Fig. 6C). Calcein-AM and F-Actin staining shows uptake of NLRP3-YFP oligomers in LX2 cells (Fig. 6D) and primary human hepatic stellate cells (Fig. 6E) and indicates changes in cell morphology towards myofibroblast-like cell trans-differentiation. Immunofluorescent staining of α-smoothmuscle actin (α-SMA) showed increased mRNA and protein expression (6-fold, p< 0.01) compared to serum-free media Journal treatedLX2 cells (Fig. 6G). Stellate cell activation was dependent on NLRP3-YFP oligomeric uptake as co-stimulation studies with Cytochalasin B, an inhibitor of endocytosis, blocked α-SMA expression and protein level in LX2 cells (Fig. 6F, H). Inhibition of caspase-1 was also able to reduce ACTA2 expression in LX2 cells after treatment with extracellular NLRP3-YFP oligomers (Fig. 6F). To test whether supernatant from pyroptotic hepatocytes also led to stellate cell activation, ASC-YFP transfected HepG2 cells were stimulated with LPS plus Nig and supernatant was transferred to hepatic stellate cells. Gene expression analysis in LX2 cells after incubation with supernatant from pyroptotic hepatocytes revealed increased marker genes for stellate cell activation (ACTA2, COL1A1, TIMP1) as well as IL1b expression (Fig. 6J). These results demonstrated that internalized extracellular NLRP3 inflammasome oligomers in hepatic stellate leads to activation of cells.
Figure 6. Hepatic stellate cells internalize extracellular NLRP3/ASC inflammasome specks that leads to their activation.

(A) Immunocytochemistry of LX2 cells incubated with extracellular NLRP3-YFP inflammasome particles and stained with F-Actin (red) and nuclei (DAPI, blue) (scale bar: 25 μm). (B) Western blot analysis and (C) IL-1ß ELISA of LX2 cells (n=3) treated with vehicle (n=3), WT control particles (WT con) (n=3) and NLRP3-YFP oligomeric particles (n=5). Primary antibody against YFP- tag and GAPDH was used. Data were compared using One-way analysis of variance and Bonferroni post-hoc test (*p< 0.05 compared to untreated control). Representative Calcein-stained and F-actin stained LX2 cells (n=4) (D) and primary human hepatic stellate cells (n=2) (E) stimulated with extracellular NLRP3-YFP oligomeric particles (green) for 24 h. Nuclei were stained with DAPI (blue) (scale bars: 100 μm). (F) mRNA expression of ACTA2 in NLRP3-YFP particles treated LX2 cells with or without caspase-1 inhibitor and endocytosis inhibitor Cytochalasin B. (G) Immunofluorescent α-smooth muscle actin (α-SMA) staining and quantification of LX2 cells stimulated with NLRP3-YFP oligomers or WT control particles for 24 h (scale bar: 100 μm). (H) Quantitated α-SMA immunofluorescent staining of LX2 cells treated with NLRP3-YFP oligomeric particles and Cytochalasin B for 24h. Relative fluorescence intensity (RFU) was normalized on total cells (DAPI). (J) mRNA expression of ACTA2, COL1a1, TIMP1 and IL1b in LX2 cells treated with supernatant of pyroptotic ASC-YFP transfected HepG2 cells compared to supernatant from untreated HepG2 cells. Two groups were analyzed by unpaired Studentś T-test (*p< 0.05).
Discussion
The principal findings of this study describe the presence and significance of hepatocyte pyroptosis and extracellular inflammasomes. Our results identify pyroptotic cell death and release of inflammasome proteins in primary human hepatocytes during NLRP3 inflammasome activation. We revealed that extracellular inflammasome particles can be internalized by hepatic stellate cells leading to their cell activation. Furthermore, liver and serum caspase-1 activity is significantly increased in patients with NASH but not steatosis.
Growing evidence suggests that NLRP3 inflammasome activation is an important driver of various acute and chronic liverdiseases[31,32]. Previous studies from our group demonstrated that global but not myeloid-specific Nlrp3 KI mice showed hepatic pyroptosis in the liver while both developed liver inflammation and fibrosis in different extent [27]. Global Nlrp3KO mice were protected from a diet-induced NASH development[13]. Additionally, Xu and colleagues demonstrated that global GsdmdKO mice were protected from steatosis, inflammation, and stellate cell activation induced by the methionine and choline deficient diet, while in human NASH livers increased GasderminD expression was associated with lobular inflammation and ballooning [33]. However, the mechanisms and the role of hepatocyte pyroptotic cell death were not assessed. These findings lead us in the current study to investigate whether specific NLRP3 activation in hepatocytes results in pyroptotic cell death, the downstream events, and the potential contribution of this process to liver injury. We found that isolated hepatocytes from Nlrp3KICreA mice as well as primary mouse and human hepatocytes, and HepG2 cells stimulated with NLRP3 inflammasome canonical activators LPS and nigericin or palmitate were able to undergo pyroptotic cell death in a caspase-1-dependent manner. Hepatocyte pyroptosis was associated with the presence of all components of the NLRP3 inflammasome in the extracellular space and the release of ASC-YFP inflammasome particles. We could confirm the clinical significance for extracellular activated caspase-1 in NASH patients where serum caspase-1 activity was significantly increased in the presence of hepatocellular ballooning, inflammation and fibrosis. In-vivo, hepatocyte NLRP3 over-activation in Nlrp3KICreA mice developed liver fibrosis accompanied by increased cell death, expression of pro-fibrogenic genes and activation of hepatic stellate cells at 9 months of age, a phenotype that was not observed in mice sacrificed at 6 months of age. The changes observed in the liver were significantly less severe than those that we previously found on the global and myeloid-specific Nlrp3KI mice [13, 27] and similar but to a lesser extent to what we found on the hepatic stellate cell-specific KI mice [34]. These results highlight the cell-specific contribution of NLRP3 activation to the liver injury and fibrosis and have important potential translational implications when designing approaches to treat NLRP3-driven liver disease. We further demonstrated that treatment of the KI mice with low dose LPS worsened the liver phenotype and increased liver inflammation suggesting that a priming stimulus is required for KI hepatocytes to achieve the threshold required for full NLRP3 activation and subsequent pyroptosis. The unexpected findings of fibrotic changes in the hepatocyte-specific KI mice lead us to examine the potential mechanism in vitro. Indeed, we found that hepatic stellate cells internalized extracellular recombinant NLRP3-YFP inflammasome particles leading to increased IL-1ß secretion, protein expression of α-SMA, and morphological changes consistent with a trans-differentiation into collagen producing activated HSC. Effects on α-SMA expression were abrogated when LX2 cells were pre-treated with the endocytosis inhibitor Cytochalasin B. Collectively, these results identified extracellular NLRP3 inflammasome particles as a novel mechanism for intercellular communication during liver injury associated with NLRP3 activation. The findings further demonstrated that these particles have a pro-inflammatory and pro-fibrogenic effect once internalized by HSC (graphical abstract). Future studies to further examine the role of extracellular NLRP3 particles as contributors in spreading and perpetuating NLRP3 inflammasome responses during liver injury as well as their potential role as biomarkers are warranted.
In summary, our data demonstrated that hepatocyte NLRP3 over-activation leads to hepatocyte pyroptosis and secretion of inflammasome complexes into the extracellular space. Caspase-1 activation is present in the livers of NASH patients and can be monitored in serum of these patients correlating with severity of liver injury and particularly with stage of fibrosis. These newly described pro-fibrogenic effect of extracellular NLRP3 inflammasome complexes identify a plethora of pyroptotic stress signals released by hepatocytes and supports the potential for novel therapeutic strategies aiming at modulation of extracellular NLRP3 to treat chronic liver diseases.
Supplementary Material
Highlights.
Human and murine hepatocytes undergo pyroptosis and release NLRP3 inflammasome proteins
Pyroptotic cell death in hepatocytes is dependent on caspase-1 and GasderminD activation
Caspase-1 activity is increased in livers and serum from NASH patients
Nlrp3KICreA mice develop fibrosis and show increased sensitivity to liver damage
Human hepatic stellate cells internalize extracellular NLRP3-YFP oligomeric particles
Extracellular NLRP3 oligomeric particles perpetuate inflammation and fibrogenesis
Acknowledgments
This work was funded by NIH grants R01 DK113592 (AF and HH) and R01AA024206 (AF), the German Research Foundation (DFG) SCHU 3146/1–2 to SG, Leipzig University, and the Spanish Ministry of Economy SAF2017–88276-R, the Fundación Séneca 20859/PI/18, and the European Research Council CoG 614578 to PP; PFIS (FI12/00198) and Mobility of Research Staff (MV16/00004), both from Instituto de Salud Carlos III (Spanish Ministry of Economy) (FA).
Footnotes
Conflict of interest statement: None. The funding sources had no involvement in study design; data collection, analysis and interpretation; in the writing of the manuscript; and in the decision to submit the article for publication.
Data availability statement: All methods and protocols used were peer-reviewed and cited in the reference section. Reagents, antibodies and resources are listed in the CTAT table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147(4):765–783.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wree A, Mehal WZ, Feldstein AE. Targeting Cell Death and Sterile Inflammation Loop for the Treatment of Nonalcoholic Steatohepatitis. Seminars in liver disease 2016;36(1):27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nature reviews. Immunology 2020;20(3):143–57. [DOI] [PubMed] [Google Scholar]
- [4].Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature 2012;481(7381):278–86. [DOI] [PubMed] [Google Scholar]
- [5].Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nature reviews. Immunology 2016;16(7):407–20. [DOI] [PubMed] [Google Scholar]
- [6].Dick MS, Sborgi L, Rühl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nature communications 2016;7(1):11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi J, Gao W, Journal Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends in biochemical sciences 2017;42(4):245–54. [DOI] [PubMed] [Google Scholar]
- [8].He W-t, Wan H, Hu L, Chen P, Wang X, Huang Z et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1ß secretion. Cell research 2015;25(12):1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nature immunology 2014;15(8):738–48. [DOI] [PubMed] [Google Scholar]
- [10].Li C, Wang X, Kuang M, Li L, Wang Y, Yang F et al. UFL1 modulates NLRP3 inflammasome activation and protects against pyroptosis in LPS-stimulated bovine mammary epithelial cells. Molecular immunology 2019;112:1–9. [DOI] [PubMed] [Google Scholar]
- [11].Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J et al. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Experimental cell research 2016;342(2):184–92. [DOI] [PubMed] [Google Scholar]
- [12].Ershaid N, Sharon Y, Doron H, Raz Y, Shani O, Cohen N et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nature communications 2019;10(1):4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wree A, McGeough MD, Pena CA, Schlattjan M, Li H, Inzaugarat ME et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. Journal of molecular medicine (Berlin, Germany) 2014;92(10):1069–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF. Hepatology (Baltimore, Md.) 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, van Rooyen DM et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. Journal of hepatology 2017;66(5):1037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 2009;30(6):875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526(7575):666–71. [DOI] [PubMed] [Google Scholar]
- [18].Xiao J, Wang C, Yao J-C, Alippe Y, Xu C, Kress D et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLOS Biology 2018;16(11):e3000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Martin I, Cabán-Hernández K, Figueroa-Santiago O, Espino AM. Fasciola hepatica fatty acid binding protein inhibits TLR4 activation and suppresses the inflammatory cytokines induced by lipopolysaccharide in vitro and in vivo. Journal of immunology (Baltimore, Md. 1950) 2015;194(8):3924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schuster-Gaul S., Geisler LJ, McGeough MD, Johnson CD, Zagorska A, Li L.i et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI insight 2020;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hachim D, Wang N, Lopresti ST, Stahl EC, Umeda YU, Rege RD et al. Effects of aging upon the host response to implants. Journal of biomedical materials research. Part A 2017;105(5):1281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Povero D, Panera N, Eguchi A, Johnson CD, Papouchado BG, de Araujo Horcel L et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cellular and molecular gastroenterology and hepatology 2015;1(6):646–663.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 2005;54(1):142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li W-C, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods in molecular biology (Clifton, N.J.) 2010;633:185–96. [DOI] [PubMed] [Google Scholar]
- [25].Damm G, Schicht G, Zimmermann A, Rennert C, Fischer N, Kießig M et al. Effect of glucose and insulin supplementation on the isolation of primary human hepatocytes. EXCLI journal 2019;18:1071–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shang L, Hosseini M, Liu X, Kisseleva T, Brenner DA. Human hepatic stellate cell isolation and characterization. Journal of gastroenterology 2018;53(1):6–17. [DOI] [PubMed] [Google Scholar]
- [27].Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (Baltimore, Md.) 2014;59(3):898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Martín-Sánchez F, Gómez AI, Pelegrín P. Isolation of Particles of Recombinant ASC and NLRP3. Bio-protocol 2015;5(10):e1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kleiner DE, Brunt EM, van Natta M, Behling C, Contos MJ, Cummings OW et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltimore, Md.) 2005;41(6):1313–21. [DOI] [PubMed] [Google Scholar]
- [30].Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (Baltimore, Md.) 2011;54(1):133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wree A, Holtmann TM, Inzaugarat ME, Feldstein AE. Novel Drivers of the Inflammatory Response in Liver Injury and Fibrosis. Seminars in liver disease 2019;39(3):275–82. [DOI] [PubMed] [Google Scholar]
- [32].Alegre F, Pelegrin P, Feldstein AE. Inflammasomes in Liver Fibrosis. Seminars in liver disease 2017;37(2):119–27. [DOI] [PubMed] [Google Scholar]
- [33].Xu B, Jiang M, Chu Y, Wang W, Chen Di, Li X et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. Journal of Hepatology 2018;68(4):773–82. [DOI] [PubMed] [Google Scholar]
- [34].Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG et al. NLR Family Pyrin Domain-Containing 3 Inflammasome Activation in Hepatic Stellate Cells Induces Liver Fibrosis in Mice. Hepatology (Baltimore, Md.) 2019;69(2):845–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.