

Abstract
Tissue acidosis is an important secondary injury process in the pathophysiology of traumatic spinal cord injury (SCI). To date, no studies have examined the role of proton extrusion as mechanism of pathological acidosis in SCI. In the present study, we hypothesized that the phagocyte-specific proton channel Hv1 mediates hydrogen proton extrusion after SCI, contributing to increased extracellular acidosis and poor long-term outcomes. Using a contusion model of SCI in adult female mice, we demonstrated that tissue pH levels are markedly lower during the first week after SCI. Acidosis was most evident at the injury site, but also extended into proximal regions of the cervical and lumbar cord. Tissue reactive oxygen species (ROS) levels and expression of Hv1 were significantly increased during the week of injury. Hv1 was exclusively expressed in microglia within the CNS, suggesting that microglia contribute to ROS production and proton extrusion during respiratory burst. Depletion of Hv1 significantly attenuated tissue acidosis, NADPH oxidase 2 (NOX2) expression, and ROS production at 3 d post-injury. Nanostring analysis revealed decreased gene expression of neuroinflammatory and cytokine signaling markers in Hv1 knockout (KO) mice. Furthermore, Hv1 deficiency reduced microglia proliferation, leukocyte infiltration, and phagocytic oxidative burst detected by flow cytometry. Importantly, Hv1 KO mice exhibited significantly improved locomotor function and reduced histopathology. Overall, these data suggest an important role for Hv1 in regulating tissue acidosis, NOX2-mediated ROS production, and functional outcome following SCI. Thus, the Hv1 proton channel represents a potential target that may lead to novel therapeutic strategies for SCI.
Keywords: Spinal cord injury, voltage-gated proton channel, Hv1, acidosis, microglia, oxidative stress, motor function
1. Introduction
After the initial impact in traumatic spinal cord injury (SCI), cells within the injury site and surrounding regions undergo biochemical changes (i.e., secondary injury) that ultimately determines outcome trajectory. Tissue acidosis (pH < 7.0) is a prominent feature of central nervous system (CNS) injury, and is usually associated with acute inflammatory damage and energy failure (Clausen et al., 2005; Jalalvand et al., 2016; Marmarou et al., 1993). Extreme or persistent acidosis is toxic to neurons (de Ceglia et al., 2015; Dodge et al., 2013). Much of our understanding of pathological CNS acidosis is derived from research on ischemic stroke and traumatic brain injury (Gupta et al., 2004; Simon, 2006; Zygun et al., 2004), which differ dramatically in cytoarchitecture, white matter composition, and neural connectivity. Indeed, there is a dearth of information regarding the mechanisms underlying the regulation and genesis of tissue acidosis in traumatic SCI. Furthermore, the signaling pathways linking pH regulation to inflammatory processes has yet to be fully resolved.
The normal function of CNS white matter is highly dependent on a continuous supply of oxygen and glucose, the lack of which can cause severe consequences such as disruption of ionic homeostasis, depolarization and conduction failure of central fibers, ultimately leading to demyelination, axonal degeneration and functional deficits (Friese et al., 2007; Ruffin et al., 2014; Stirling and Stys, 2010). Multiple reports have pointed to attenuation of neuronal death and axonal degeneration through disruption of the acid-sensing ion channel 1 (ASIC1), which is primarily expressed in neurons (Friese et al., 2007; Hu et al., 2011; Koehn et al., 2016). These recent discoveries show the importance of extracellular pH for neuronal survival and post-traumatic functional outcomes. The upstream activator of the proton-gated cationic channels belonging to the neuronal ASIC family is not clear, nor is the cellular source of these extrinsic factors. Injury-induced proton extrusion from non-neuronal cells represents an intriguing possibility. Indeed, earlier studies have found that glial cells can directly alter the composition of the extracellular cerebrospinal fluid, including regulation of its pH levels (Chesler, 2003; Ro and Carson, 2004). Yet to date no studies have investigated the role of microglia in spinal cord acidosis. The voltage-gated proton channel Hv1 represents a compelling candidate mechanism because it is exclusively expressed in microglia within the CNS, and functions to facilitate phagocytosis-induced respiratory burst via NADPH (NOX2)-dependent extrusion of hydrogen (H+) protons in order to alleviate the coincident intracellular acidosis (Clark, 2016; El Chemaly et al., 2014; Musset et al., 2008; Ramsey et al., 2009; Seredenina et al., 2015; Wu, 2014). The end result is the release of reactive oxygen species (ROS) and H+ into the extracellular environment. However, there are still many unanswered questions regarding whether acid extrusion by activated microglia leads to tissue acidosis in the spinal cord.
The apparent connection between microglia, NOX2, and H+ led us to explore a possible role for Hv1 in tissue acidosis after SCI. Prior work supports a therapeutic role for Hv1 in ischemic stroke (Li et al., 2019; Tian et al., 2016; Wu et al., 2012). However, its specific role in the pathophysiology of SCI have yet to be fully addressed. In the present study, we used a contusion-induced traumatic SCI mouse model to demonstrate tissue acidification is mediated by Hv1 which, in turn, contributes to functional outcome and neuronal death in the gray matter.
2. Materials and Methods
2.1. Animals and mouse spinal cord contusion model
Adult female and male C57/BL6 mice at 8–10 weeks of age and 20–25g body weight were obtained from Jackson Laboratories. Hv1 knockout (Hv1 KO) mice (Ramsey et al., 2009) were obtained from Dr. Long-Jun Wu’s laboratory at Mayo Clinic, Rochester, MN and maintained in the UMB animal facility. After induction of anesthesia with isoflurane, a laminectomy was performed and the spinal column was stabilized using lateral clamps over the lateral processes at T9 and T11. The subject mice underwent a midline contusion injury of the spinal cord at T10 level using the Infinite Horizon Spinal Cord Impactor (Precision Systems and Instrumentation) with a force of 60 kilodyne, which is considered to be moderate (Matyas et al., 2017; Wu et al., 2016). Manual bladder expression was carried out at least three times per day for 7–14 days after SCI until reflex bladder emptying was reestablished. For mice used as control animals, only laminectomy was performed after anesthesia. The number of mice at various time points in each experiment is indicated in the figure legends. All procedures were performed under protocols approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee (IACUC).
2.2. Basso mouse scale for locomotion
Mice were placed in a flat, enclosed surface with a diameter of 100 cm and observed for 4 min by two trained observers blinded to the genotype using the Basso mouse scale (BMS) for locomotion (Basso et al., 2006). Animals were rated on a scale of 0–9: 0 being complete paralysis, and 9 being normal locomotion based on hind limb joint movement, weight support, plantar stepping, and coordination. Mice were tested for BMS scores on day 1 and day 3 after injury and weekly thereafter for up to 6 weeks.
2.3. Extracellular pH recordings
At 1d, 3d and 7d after SCI, mice were euthanized with Euthasol and intracardially perfused with 40ml of ice-cold normal saline. After a post-mortem laminectomy, the spinal cord was exposed for measurement with the micro pH electrode (Cat# 9863BN, Thermo Fisher Scientific) connected to an Orion Star A211 benchtop pH meter (Cat# STARA2110, Thermo Fisher Scientific). This glass-tip and stainless-steel needle electrode has a measurement accuracy of up to 0.02 and 4 points were taken from each region (Fig. 1A) to minimize confounding factors. Before each experiment, a three-point calibration (pH 4.0, pH 7.0, pH 10.0) was performed to further ensure accuracy of the readings. For sham animals, readings were taken from similar areas of the cervical, thoracic, and lumbar regions. When testing, the electrode was inserted no deeper than 0.5–1 mm into the spinal cord tissue and remained in position until the value was stable. Between each animal, the micro electrode was cleaned with double distilled water and a thorough wash with the pH electrode cleaning solution kit (Cat# 900020, Thermo Fisher Scientific) was conducted according to manufacturer’s instructions after the end of each experiment. The average of 4 readings within a region were taken as the extracellular pH of that particular spinal cord segment.
Figure 1. Spinal cord injury induces extracellular acidosis and increase of Lactate and reactive oxygen species (ROS).
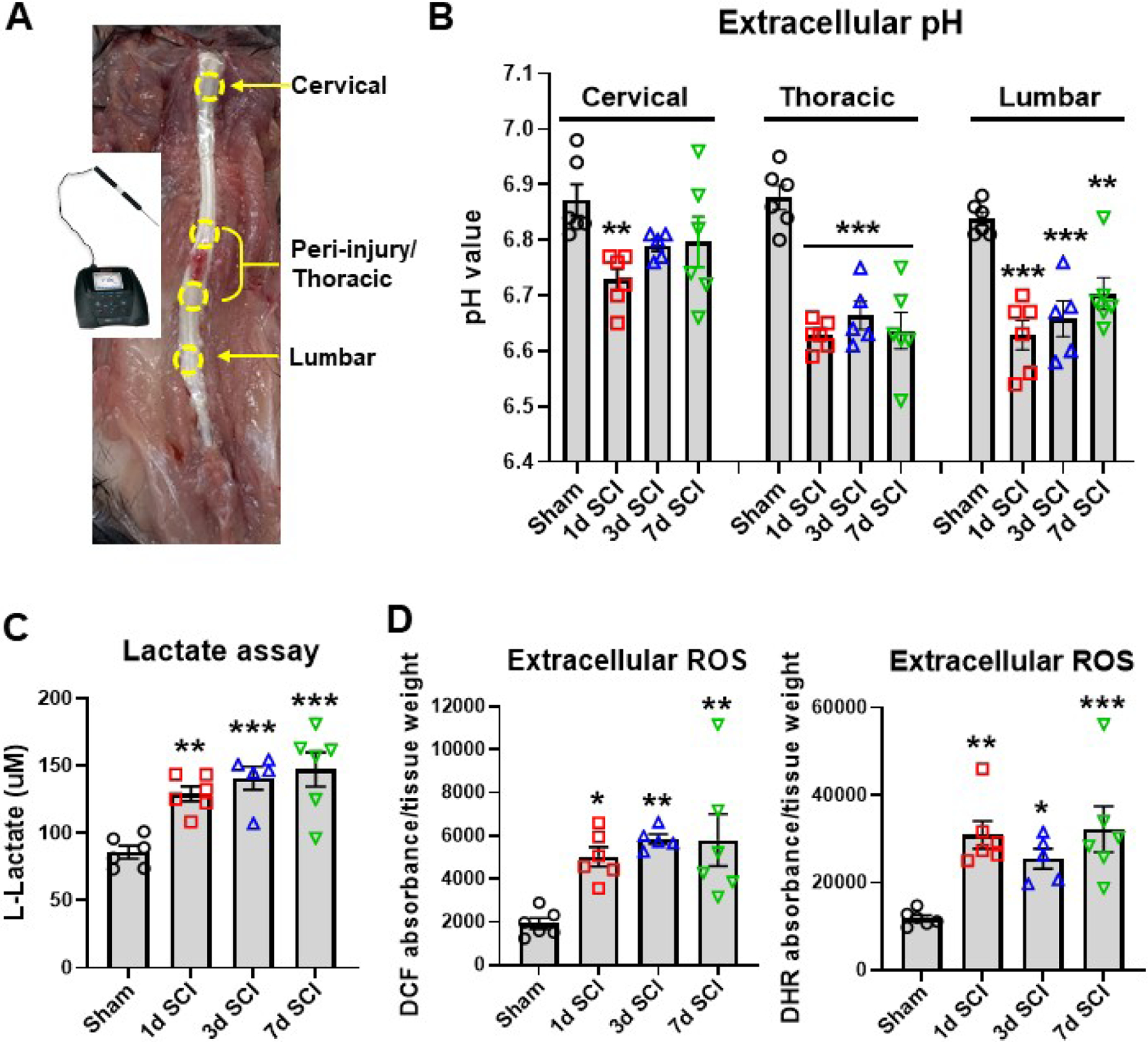
(A) A micro electrode with glass body and stainless-steel needle tip was used in connection with the STARA2110 pH meter to measure localized tissue acidosis within SCI or sham mice. The regions encircled by yellow dotted lines are indicative of the areas examined with the probe. Measurements were taken from three different regions, the cervical, the thoracic peri-injury site, and the lumbar spinal cord regions. (B) Extracellular spinal cord pH at the cervical, thoracic, and lumbar regions was measured in sham mice and day 1, 3, and 7 after SCI. (C) Extracellular lactate concentration in the injury site was determined with the L-lactate Assay kit. (D) Determination of extracellular ROS levels with two different fluorescent dyes (H2DCFDA: DCF; dihydrorhodamine: DHR) showed significant increase in the first week of injury. All data are presented as independent data points. N=6 (sham), 6 (1d SCI), 5 (3d SCI), 6 (7d SCI) mice/group. *p<0.05, **p<0.01, ***p<0.001, vs. Sham group. One-way ANOVA following Dunnett’s multiple comparisons test.
2.4. L-Lactate assay
For assessment of lactate acidosis within the spinal cord injury site, a segment of 5mm length centered on the injury site was taken from the injury site and homogenized in 600 μl of ice-cold molecular grade water (Cat# 351-029-101, Quality Biological) and centrifuged at 150× g for 10 min to harvest extracellular samples. The L-Lactate Assay Kit (Cat# 700510, Cayman Chemical) was used according to manufacturer’s instructions and the final fluorescence intensity were measured with a plate reader at Ex/Em=535/590 (Synergy Hybrid Biotek).
2.5. Measurement of reactive oxygen species in spinal cord tissue
For assessment of ROS within the spinal cord injury site, a segment of 5mm length was taken from the injury site and weighed on by an electronic scale (Cat# ME103TE, Mettler Toledo). The tissue was homogenized in 600 μl of ice-cold molecular grade water (Cat# 351-029-101, Quality Biological) and centrifuged at 1500× g for 10 min to harvest the supernatant. For each sample, 50ul were allocated into a 96-well plate by duplicates. With an additional 150 μl of oxidative stress indicator solution (diluted in molecular grade water) for each sample, the final volume of 200 μl was mixed well by pipette. The fluorogenic dyes, dihydrorhodamine 123 (DHR123, 1:400, Cat# D23806, Invitrogen) and H2DCFDA (DCF, 20.5 μM, Cat# D399, ThermoFisher Scientific) were used to measure hydroxyl, peroxyl and other ROS activity in the sample. The plate was then incubated in the dark for 30 min at 37’C. Final measurements of fluorescent intensity were read with a plate reader (Synergy Hybrid, Biotek) at Ex/Em=490/520 and presented as absorbance units normalized to tissue weight.
2.6. Western blot analysis
At 3 days post-injury, mice spinal cord at a length of 5mm were extracted from the epicenter of the injury groups of either WT or Hv1 KO mice. For sham animals, an equal length of 5mm was extracted at the same time from the approximant area of T10. For sample processing, all tissue samples were immersed in RIPA lysis buffer (Sigma-Aldrich) with the additional supplements of 1x protease inhibitor cocktail (Sigma-Aldrich), phosphatase inhibitor cocktail II and phosphatase inhibitor cocktail III (Sigma-Aldrich) to prevent protein degradation. After SDS-PAGE in gradient 4–20% Tris-glycine gel (Bio-Rad, US) and transferred to 0.2-μm nitrocellulose membrane (Bio-Rad, US). Membranes were blocked with 10% non-fat skim milk in PBST and incubated overnight with primary antibodies diluted in blocking buffer, followed by incubation with the respective secondary HRP-conjugated antibodies for 2h at room temperature. Finally, after visualizing the immunoblots with SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific, US) and imaged with ChemiDoc TM MP system (Bio-Rad, US), the optic density of signal bands were quantified with the Image Lab analysis program (Bio-Rad, US). The primary antibodies used and their respective dilutions are as follows: HVCN1 (1:2000; Cat# AHC-001, Alomone) and GAPDH (1:2000; Cat# AB2302, Millipore).
2.7. Quantitative qPCR analysis
Total RNA was extracted from cultured cells and previously flash frozen spinal cord tissue samples surrounding the epicenter of the lesion site with the miRNeasy Mini Kit (Cat# 74104, Qiagen). Complementary DNA (cDNA) was synthesized by a Verso cDNA RT kit (Cat# AB1453B, Thermo Scientific) per the manufacturer’s protocol. Quantitative PCR for target mRNAs was performed using TaqMan gene expression assays for Hvcn1 (Hv1), Mm01199507_m1 (mouse) and Rn01480592_m1 (rat); Hdc, Mm00456104_m1; Il1β (IL-1β), Mm00434228_m1; Hcar2, Mm01199527_s1; Nlrp3, Mm00840904_m1; Il1rn, Mm00446186_m1; Cd14, Mm01158466_g1; Il1a, Mm00439620_m1; TNFα, Mm00443258_m1; Ptgs2, Mm00478374_m1; Ccl4, Mm00443111_m1; cybb (NOX2), Mm01287743_m1; Nos2, Mm00440502_m1; Ccl5, Mm01302427_m1; Cxcl10, Mm00445235_m1; ITGAM (CD11b), Mm00434455_m1; Cd68, Mm03047343_m1; IL-6, Mm00446190_m1; chil3 (Ym1), Mm00657889_mH; Arginase-1 (Arg1), Mm00475988_m1; IL-10, Mm0–0439614_m1; TGFβ, Mm01178820_m1; SOCS3, Mm01342740_g1; Gfap (GFAP), Mm01253033_m1; GAPDH, Mm99999915_g1 (mouse) and Rn01775763_g1 (rat) (Applied Bioscience) on an QuantStudio 5 Real-Time PCR System (Applied Biosystems). Samples were assayed in duplicate in 1 run (40 cycles), which was composed of 3 stages, 50 ‘C for 2 min, 95 ‘C for 10s for each cycle (denaturation), and finally the transcription step at 60 ‘C for 1 min. Gene expression was normalized by GAPDH and compared to the control sample to determine relative expression levels by the 2−ΔΔCt method.
2.8. Primary cell culture and CD11b+ microglia/macrophages isolation
Primary microglia and astrocytes were cultured from the cerebral cortex of neonatal rats as described (Sabirzhanov et al., 2019b). The cells were grown in Dulbecco’s Modified Eagle’s Medium/F12 (Invitrogen) supplemented with 10 % fetal bovine serum (Invitrogen), 1 % Pen/Strep at 37 °C with 5 % carbon dioxide. When the cells had grown to confluence in 3.5-cm dishes, lipopolysaccharide (LPS) (30 ng/ml) or interferon γ (IFNγ, 60 ng/ml) were applied to the dish for 24 h. The cell lysates were harvested for RNA extraction and Hv1 qPCR analysis.
Primary neuronal cultures were derived from rat embryonic day 18 cortices, as previously described (Sabirzhanov et al., 2019b). Cells were seeded at a density of 1×106 cells/cm2 onto poly-d-lysine-coated 100 mm Petri dishes and maintained in serum-free conditions using the B27 supplement. At 7 days after the culture, neurons were exposed to cell death inducers etoposide (Etop, 25 nM) for 6 h. Total RNA was prepared from whole-cell extracts for Hv1 qPCR.
CD11b+ microglia/macrophages were isolated from sham or d3 injured spinal cord tissue using MACS Separation technology (Miltenyi Biotec, Auburn, CA) as previously described (Sabirzhanov et al., 2019b). Briefly, dissected spinal cord tissue were dissociated by combining an optimized enzymatic treatment (Adult Brain Dissociation Kit; Cat# 130-107-677, Miltenyi Biotec) with gentle mechanical dissociation using the gentleMACS Octo Dissociator with Heaters (130-096-427, Miltenyi Biotec). After removing myelin (Myelin Removal Beads II, Cat# 130-096-733, Miltenyi Biotec), cell suspension was incubated with a magnetic-bead conjugated anti-CD11b antibody (Cat# 130-093-634, Miltenyi Biotec). CD11b+ cells were eluted for RNA extraction and Hv1 qPCR analysis.
2.9. Nanostring analysis
RNA samples were obtained from 5 mm of spinal cord tissue surrounding the lesion area at 3 days post-injury. Total RNA (20 ng/μl) was run on a NanoString nCounter® system for Mouse Neuroinflammation v1.0 panel (NanoString Technologies, Seattle, WA) to profile RNA transcript counts for 757 genes and 13 housekeeping genes. Sample gene transcript counts were normalized prior to downstream analysis and pairwise differential expression analysis was performed with NanoString’s nSolver software Version 4.0. All statistical analysis of NanoString data was performed in the R language using RStudio Version 1.2.5033. Principle component analysis (PCA) was performed with the Euclidean distance measurement method. The four pairwise comparisons were respectively described in this manuscript as follows: (1) Sham/Hv1 KO vs. Sham/WT-Comparison (Comp) 1; (2) SCI/WT vs. Sham/WT-Comp 2; (3) SCI/Hv1 KO vs. Sham/Hv1 KO-Comp 3; and (4) SCI/Hv1 KO vs. SCI/WT-Comp 4. All comparisons “Group 1 vs. Group 2” were interpreted as “Group 1 relative to Group 2” in the text and figures. A p-value of less than 0.05 was used to identify differentially expressed (DE) genes in each comparison. Subsets of DE genes displayed as heatmaps were normalized across samples as z-scores and then averaged to a single value per group before plotting with Graphpad Prism.
2.10. Flow Cytometry
Mice were perfused with 40 ml of cold PBS and 5 mm length of spinal cord tissue surrounding the lesion area was isolated, weighed, and placed in complete Roswell Park Memorial Institute (RPMI) 1640 (Cat# 22400105, Invitrogen) medium and mechanically and enzymatically digested in collagenase/dispase (Cat# 10269638001, 1 mg/ml; Roche Diagnostics), papain (Cat# LS003119, 5 U/ml; Worthington Biochemical), 0.5 M EDTA (Cat# 15575020, 1:1000; Invitrogen), and DNAse I (Cat# 10104159001, 10 mg/ml; Roche Diagnostics) for 1 h at 37 °C on a shaking incubator (200 rpm). The cell suspension was washed twice with RPMI, filtered through a 70-μm filter, and RPMI was added to a final volume of 3 ml and kept on ice. Spinal cord cells were then transferred into FACS tubes and washed with FACS buffer. Cells were then incubated with Fc Block (Cat# 101320, Clone: 93; Biolegend) for 10 min on ice, and stained for the following surface antigens: CD45-eF450 (Cat# 48-0451-82, Clone: 30-F11; eBioscience), CD11b-APC/Fire™750 (Cat# 101262, Clone: M1/70; Biolegend), Ly6C-APC (Cat# 128016, Clone: HK1.4; Biolegend), Ly6G-AF700 (Cat# 128024, Clone: 1A8; Biolegend), and Zombie Aqua fixable viability dye (Cat# 423102, Biolegend). Leukocytes were then washed in FACS buffer, fixed in 2% paraformaldehyde for 8 min, and washed once more prior to adding 500 μl FACS buffer. Intracellular staining for CD68-PE (Cat# 137014, Clone: FA-11; Biolegend) and NOX2/gp91phox-AF647 (Cat# 3889R, 1:500; Bioss Antibodies) was performed as described previously (Ritzel et al., 2020).
ROS production was measured using H2DCFDA (DCF, 5 μM; ThermoFisher Scientific). In brief, each dye indicator was added to RPMI media according to the manufacturer’s instructions, vortexed, and incubated for 30 min in a 37 °C water bath. Data were acquired on a BD LSRFortessa cytometer using FACSDiva 6.0 (BD Biosciences) and analyzed using FlowJo (Treestar Inc.). At least 5 million events were collected for each sample. Countbright™ Absolute Counting Beads (Invitrogen) were used to estimate cell counts per the manufacturer’s instructions. Data were expressed as total counts/mg spinal cord tissue weight. Leukocytes were first gated using a splenocyte reference (SSC-A vs FSC-A). Singlets were gated (FSC-H vs FSC-W), and live cells were gated based on Zombie Aqua exclusion (SSC-A vs Zombie Aqua-Bv510). Resident microglia were identified as the CD45int CD11b+Ly6C− population, whereas peripheral myeloid cells were identified as CD45hiCD11b+ myeloid cells. Within the myeloid subset, monocytes were identified as Ly6ChiLy6G− and neutrophils, Ly6C+Ly6G+. Cell type-matched fluorescence minus one (FMO) controls were used to determine the positivity of each antibody and DCF (Ritzel et al., 2019).
2.11. Tissue processing and histopathology
At 6 weeks after SCI, mice were perfused intracardially with 4% paraformaldehyde and the spinal cord was dissected for further assays with histology. Spinal cord segment containing the lesion area were dissected out and embedded in Tissue-Tek OCT compound (Cat# 4583, Sakura). Serial sections of 20 μm thickness were placed serially on set of 10 slides for 10 sets of slides. One representative slide from each set was then selected for histological staining. For assessment of spared white matter (SWM), myelin was stained with Luxol Fast Blue (LFB) to determine the location of the lesion epicenter (the section with least amount of WM) and calculate residual WM in the rostral and caudal regions (Sabirzhanov et al., 2019b). Images were captured at x2.5 magnification and analyzed using National Institutes of Health ImageJ software (RRID: SCD_003070). The threshold levels of each 8-bit image was set to include LFB-positive tissue exclusively and total LFB-positive area was calculated for each section. For assessment of lesion volume, sections spaced 200 μm apart from 2 mm rostral to 2 mm caudal from the epicenter were stained with GFAP (1:1000; Cat# Z0334, Dako) and DAB (Cat# PK-6100, Vector Labs) as the chromogen for lesion volume assessment (Wu et al., 2015). Quantification was based on the Cavalieri method with a grid spacing of 40 μm on the Stereologer 2000 program (Systems Planning and Analysis). Lesion volume was quantified by outlining the unstained or missing tissue on the injury core.
2.12. Neuronal density
Spinal cord coronal sections at 20 μm thickness was stained with cresyl violet (FD NeuroTechnologies), dehydrated and mounted for analysis. The optical fractionator method of unbiased stereology in the Stereoinvestigator Software (MBF Biosciences) was employed. Neurons in the gray matter were characterized according to previously described methods (Wu et al., 2014). A total of 5 sections were analyzed for each animal and the total number of surviving neurons in each field was divided by volume that region of interest to obtain an end result of counts/mm3, which is reflective of neuronal density in the region.
2.13. Statistical analysis
Quantitative data are all plotted as mean ± standard error of the mean and individual data points are shown for each graph. Animal numbers in each experiment were derived from the power calculation based on effect sizes defined by Cohen (Cohen, 1992) and variability estimated from published data. In most experiments, we included all data. For specific experiments, one mouse in Fig 1 was euthanized due to surgical complications and three mice in Fig 3A were excluded due to tissue damaged. No exclusion criteria were pre-established. All statistical analyses were conducted by using the GraphPad Prism Program, Version 3.02 for Windows (GraphPad Software; RRID:SCR_002798). BMS scores were analyzed using two-way ANOVA with repeated measures followed by Sidak’s multiple comparisons post hoc test. For multiple comparisons, one-way or two-way ANOVA were performed followed by Tukey’s multiple comparisons post hoc test for parametric (normality and equal variance passed) data. Stereological data for lesion volume was analyzed using a Student t test. Statistical analysis in each assay was detailed in figure legends. A p value of < 0.05 was considered statistically significant.
Figure 3. SCI-induced extracellular acidosis, lactate, and ROS production are attenuated by Hv1 deficiency at 3 days post-injury.
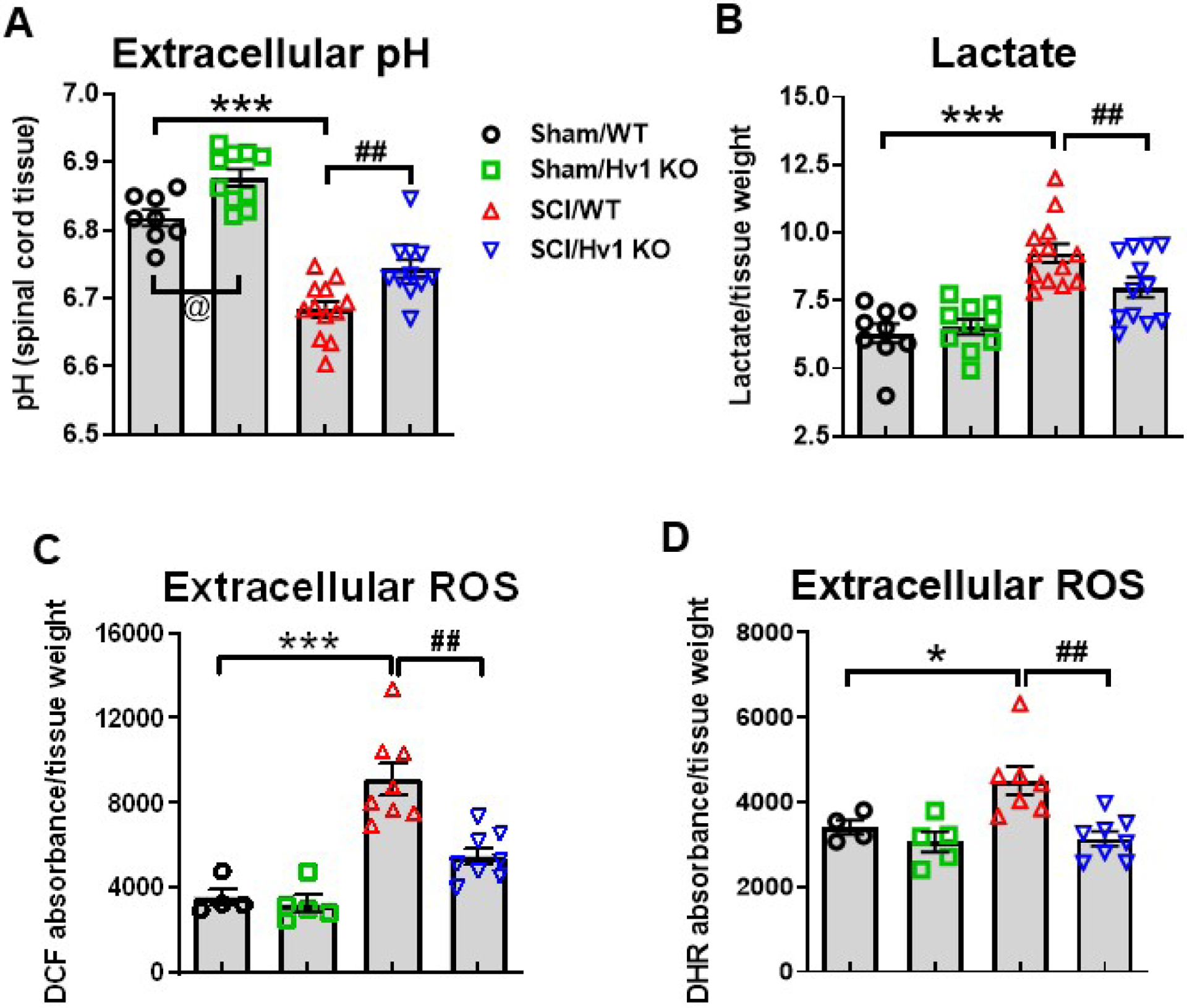
(A) Extracellular peri-lesion spinal cord pH was measured in Sham/WT, Sham/Hv1 KO, SCI/WT, and SCI/Hv1 KO mice (n=8, 10, 12, and 11 mice/group). (B) Extracellular lactate concentration in the injury site was determined with the L-lactate assay kit in Sham/WT, Sham/Hv1 KO, SCI/WT, and SCI/Hv1 KO mice (n=9, 10, 13, and 12 mice/group). (C-D) Extracellular ROS levels as determined by DCF (H2DCFDA) and DHR (dihydrorhodamine) dyes and presented as ratio to tissue weight. Both dyes reflected higher levels of ROS in SCI/WT compared to Sham/WT (n=4), and significant reduction in SCI/Hv1 KO group (n=8) compared to SCI/WT (n=8). All data are presented as independent data points. *p<0.05, ***p<0.001 vs. Sham/WT group; ##p<0.01 vs SCI/WT; @p<0.05 vs Sham/WT. Two-way ANOVA following Tukey’s multiple comparisons test.
3. Results
3.1. Traumatic injury induces tissue acidosis in the spinal cord
As a first step towards exploring and confirming the existence of tissue acidosis after SCI, we measured the extracellular pH levels in the spinal cord with a micro electrode. Taking an average of four readings for each region within the cervical, lumbar and peri-injury/thoracic areas, we were able to determine the extracellular pH of that region for each tested mice (Fig. 1A). In the peri-injury site, we observed a marked decrease in pH starting as early as 1 d and persisting for up to 7d after injury (p<0.001 vs sham, Fig. 1B). At the same time, the ascending cervical and descending lumbar regions also saw significant decreases in pH compared to that taken from sham control mice at 1d after injury. Acidosis was evident in the cervical region at 1d (p<0.01) but returned to baseline levels by d3. Surprisingly, the lumbar region saw a persistent decrease (p<0.001 at 1d, 3d SCI vs sham) in pH similar to that seen in the injury site, continuing for 7d post-injury (p<0.01). Due to the high correlation between acidosis, metabolic failure and neuroinflammation, we then tested the concentration of extracellular lactate and ROS present in a 5 mm segment of spinal cord from the injury site or equivalent area of the thoracic region for sham mice. Starting at 1d SCI (p<0.01), a persistent increase of extracellular L-Lactate concentration could be seen in the injury site, accumulating to its highest levels at 7d post-injury (p<0.001, Fig. 1C). Tissue ROS levels, as measured by either DHR123 or DCF, also showed a significant increase relative to sham control over the first week of injury (DCF: p<0.05 at 1d, p<0.01 at 3d and 7d SCI; DHR: p<0.01 at 1d, p<0.05 at 3d, p<0.001 at 7d SCI), indicating the expected occurrence of metabolic stress and release of ROS into the extracellular microenvironment (Fig. 1D).
3.2. SCI disrupts expression levels of voltage-gated proton channel Hv1
The parallel increases in Lactate, ROS, and tissue acidosis seen during the first week of injury led us to believe that Hv1, a known voltage-dependent proton extruder, may play an important role in this process (DeCoursey, 2015). Using quantitative PCR analysis, we were able to observe a significant increase in Hv1 mRNA expression starting at 3 d post-injury (p<0.05) which persisted up to 28d (p<0.001, Fig. 2A). Taking into account the effects that sex could have on our results, we repeated the experiment in adult male mice and obtained similar results showing increased Hv1 expression levels in the first month of injury (p<0.001, Fig. 2B). As a next step in determining the role of Hv1 in tissue acidosis, we utilized female Hv1 global knockout mice (Hv1 KO) and their wildtype (WT) littermates in a traumatic contusion SCI model. As shown in Fig. 2C, the Hv1 gene was completely ablated in Sham/KO and SCI/KO mice, whereas WT mice at 3 d post-injury showed a similar fold-change increase (p<0.001 vs Sham/WT) as that seen in the prior time course examination. Next, we determined relative protein expression of Hv1 in spinal cord tissue by Western blot analysis. At 3 d post-injury, we found a marked upregulation of Hv1 protein, a nearly four-fold increase between Sham/WT and SCI/WT groups (p<0.001, Fig. 2D–E). As expected, the Hv1 protein expression was absent in KO mice, providing further evidence of the efficacy of our constitutive knockout strain (Fig. 2E).
Figure 2. Hv1 expression levels are significantly upregulated after SCI.
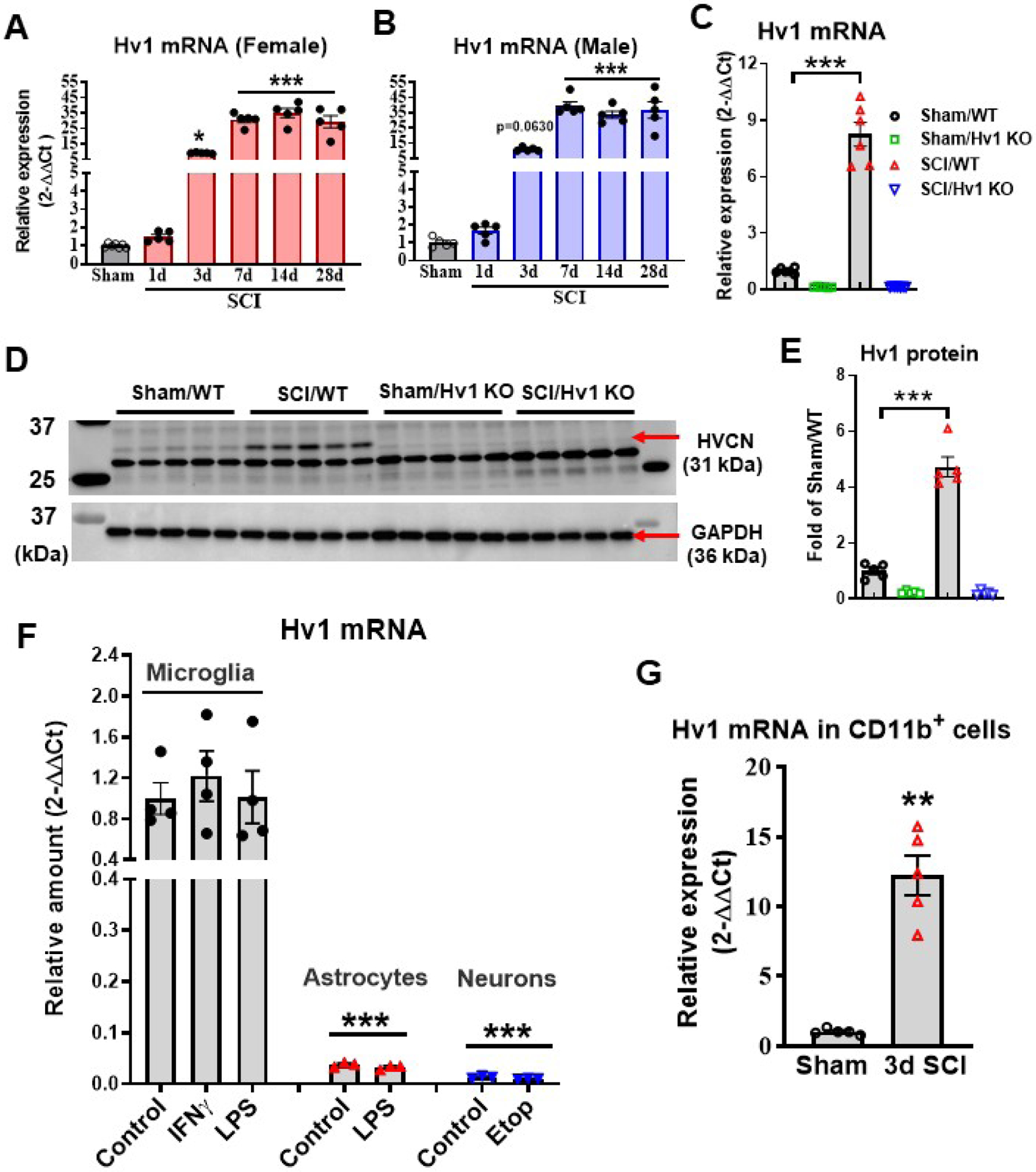
(A-B) qPCR analysis was performed to examine the mRNA expression level of Hv1 in the injured spinal cord in both female and male mice. N=7 (sham) and 6 (1,3,7 d SCI) female mice/group; 5 male mice/group. *p<0.05, ***p<0.001 vs. Sham group. One-way ANOVA following Dunnett’s multiple comparisons test. (C) The mRNA expression level of Hv1 in WT mice was significantly elevated in injured spinal cord tissue at 3 d post-injury. The Hv1 gene in KO mice were completely ablated. N=6 female mice/group. ***p<0.001 vs. Sham/WT group. Two-way ANOVA following Tukey’s multiple comparisons test. (D-E) Hv1 protein expression was determined by Western blot analysis at 3 d post-injury. Representative western blots were indicated in D for Hv1 (HVCN, 31 kDa) and GAPDH (36 kDa). The Hv1 protein expression was absent in KO mice. Protein levels were quantified by densitometry, normalized to GAPDH, and presented as fold change compared with Sham/WT. N=5 female mice/group. ***p<0.001 vs. Sham/WT group. Two-way ANOVA following Tukey’s multiple comparisons test. (F) Hv1 is predominately expressed by microglia in vitro. Hv1 mRNA expression was analyzed by qPCR in primary cultured cortical microglia, astrocytes, and neurons. Microglia were treated with LPS (30 ng/ml) or IFNγ (60 ng/ml) for 24 h, astrocytes were treated with LPS (1 μg/ml) for 24 h, and neurons were exposed to cell death inducer etoposide (Etop, 25 nM) for 6 h. One-way ANOVA was conducted followed by Dunnett’s multiple comparisons test. N=3–4 dishes/group repeated in three independent cultures. ***p<0.001 vs. Control (microglia). (G) Quantification of Hv1 mRNA expression level in CD11b positive cells derived from sham or injured spinal cord tissue at 3 d post-injury. N=5 female mice/group. **p<0.01 vs. Sham group with Mann Whitney test. All data are presented as independent data points.
To compare Hv1 expression levels in CNS different cell types, we determined cell-specific gene expression patterns in cultured microglia, astrocytes, and neurons. As depicted in Fig. 2F, Hv1 mRNA expression was highest in microglia. We found extremely lower basal level of Hv1 in primary cultured astrocytes and neurons than that in cultured microglia (p<0.001 vs Microglia control group). The level of Hv1 mRNA in microglia was approximately 75 times and 26 times lower in neurons and astrocytes, respectively. These data suggest that Hv1 is predominately expressed by microglia. In vitro, we observed that LPS-activated microglia and astrocytes as well as Etop-induced neuron apoptosis failed to stimulate Hv1 expression. After SCI, proliferation and activation of CD11b+ microglia/macrophages peak between 3 and 7 days post-injury. To further confirm the relationship between Hv1, microglia activation, and neuroinflammation, CD11b cells were isolated from injured spinal cord tissues at 3 d post-injury. SCI significantly increased Hv1 mRNA expression in the CD11b+ cells derived from injured mice compare to Sham group (p=0.0079) (Fig. 2G). These data implicate Hv1 in SCI pathogenesis and suggest a possible role for microglia as a modulator of tissue acidosis.
3.3. Hv1 deficiency improves tissue acidosis after SCI
To explore the role that Hv1 plays in H+ extrusion and injury-induced tissue acidosis, we subjected adult female Hv1 KO and WT littermate control mice to traumatic spinal cord contusion. Using a micro electrode probe to measure extracellular pH, we detected a surprising baseline difference of ~0.1 pH unit between sham groups, with Hv1 KO mice exhibiting higher spinal cord tissue pH levels (p<0.05 vs Sham/WT, Fig. 3A). At 3d post-injury, acidosis was observed in the injury site (p<0.001 vs Sham/WT), consistent with our previous results. However, there was a significantly higher pH level in SCI/Hv1 KO mice compared to that of its WT littermates in the SCI/WT group (p<0.01 vs SCI/WT). The attenuation of tissue acidosis in SCI/Hv1 KO mice was accompanied by a significant decrease in lactate concentration compared to WT controls after injury (p<0.01 vs SCI/WT, Fig. 3B). However, it is interesting to note that Lactate concentrations at baseline level did not show the same phenotypic difference as that seen in extracellular pH, suggesting Lactate is not the sole determinant of pH. Similar protective effects were observed in extracellular ROS levels, in which both fluorogenic probes, DCF and DHR123, showed an attenuated increase in oxidative stress of SCI/Hv1 KO compared to SCI/WT (p<0.01, Fig. 3C–D).
3.4. Hv1 enhances transcription of inflammatory-related genes after SCI
To determine the inflammatory consequences of Hv1 depletion in the acute phase of SCI, we evaluated spinal cord tissue at the injury site using Nanostring’s nCounter technology. The Neuroinflammation panel tested a total of 757 genes within three themes: Immunity & Inflammation, Neurobiology & Neuropathology, and Metabolism & Stress. Multidimensional Scaling (MDS) of all normalized gene counts revealed a distinct separation of samples into individual groups across the first two principal coordinates (Fig. 4A). Principal Component Analysis revealed distinct clustering (dashed ellipses) of the four sample groups across the first two principle coordinates, which accounted for 62.2% and 7.5%, respectively, of the total variation across samples. Injury-related effects were captured on Coordinate 1, separating the SCI groups on the right from the left; Phenotypic difference after Hv1 depletion were captured on Coordinate 2. Venn diagram demonstrates the separation of total injury genes (i.e., those genes differentially expressed in SCI/WT vs. Sham/WT (Injury Comparison 1, set 2) and SCI/Hv1 KO vs. Sham/Hv1 KO (Injury Comparison 2, set 3) into those showing phenotypic changes after Hv1 depletion and those that don’t, based on membership in the gene list of Genotype Comparison 2 (SCI/Hv1 KO vs. SCI/WT, set 4) (Fig. 4B). Of the genes that showed differential expression in set 2 and set 3, a total number of 484 genes were regulated after injury in both WT and KO, 30 genes were differentially expressed exclusively in set 2 (WT injury genes) and 31 genes were differentially expressed exclusively in set 3 (KO injury genes). Within the overlapping injury genes of set 2 and set 3, 59 injury genes are modified by Hv1 KO, which may contribute towards the attenuation of injury in SCI/Hv1 KO group samples. Four pairwise comparisons were performed as outlined in Fig. 4C: (1) Sham/Hv1 KO vs. Sham/WT-Genotype Comparison (Comp) 1, set 1; (2) SCI/WT vs. Sham/WT-Injury Comp 1, set 2; (3) SCI/Hv1 KO vs. Sham/Hv1 KO-Injury Comp 2, set 3; (4) SCI/Hv1 KO vs. SCI/WT-Genotype Comp 2, set 4. In the table depicting upregulated and downregulated genes of each set (Fig. 4D), SCI increased transcription counts for the vast majority of genes tested in the Neuroinflammation panel. As many as 160 genes (21.1%) were differentially expressed in the spinal cord of Sham/Hv1 KO relative to Sham/WT mice, indicating that genetic ablation of Hv1 can alter the transcriptional state of the neuroimmune system at baseline levels. Interestingly, the two SCI groups appeared to cluster closer together on the y-axis compared to the Sham groups, which is probably due to the vast majority of inflammatory genes being increased at 3d post-SCI. Volcano plots were used to visualize the Log2 (fold change) and −Log10 (p-values) of each gene in pairwise comparisons, highlighting the genes that meet the cutoff lines for both fold change and p-value (Fig. 4E). Again, the vast majority of genes in Genotype Comp 1 and both Injury Comps’ showed an increase in transcription counts, while a small number of genes showed a significant decrease between SCI/Hv1 KO and SCI/WT. These data demonstrate the importance of Hv1 in the transcriptional regulation of inflammatory mediators.
Figure 4. Hv1 depletion significantly changed transcription levels of neuroinflammatory genes.
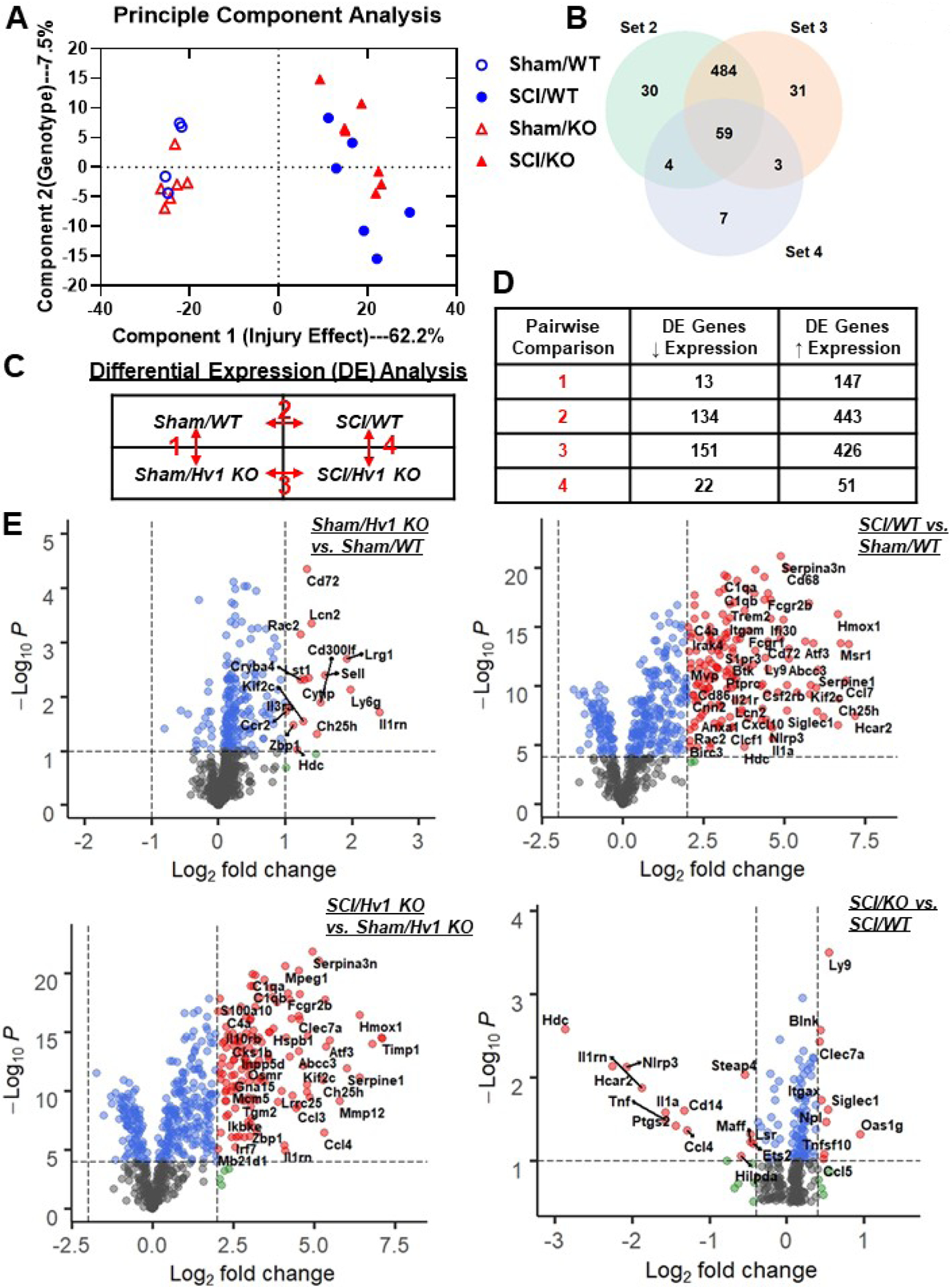
A NanoString nCounter® Neuroinflammation panel was used to assess transcriptional changes at the spinal cord injury site at 3 days post-injury. (A) Principle component analysis (PCA) was performed using all normalized gene counts in the four sample groups: Sham/WT, Sham/Hv1 KO, 3 d SCI/WT, and 3 d SCI/Hv1 KO. The first principle coordinate accounted for the majority of the variation (62.2%) across samples and separated the groups by injury, while the second principal coordinate (7.5%) separated the groups by genotype. (B) Venn diagram of the differential expression genes from Injury Comp 1 (set 2), Injury Comp 2 (set 3) and Genotype Comp 2 (set 4) showed as many as 484 injury genes that overlapped between the two injury comparisons, whereas 59 genes are shown to be differentially expressed in all three comparison groups. (C) Differential expression (DE) analysis was performed on pairwise group comparisons using nSolver (p-value < 0.05). Four pairwise comparisons were performed: (1) Sham/Hv1 KO vs. Sham/Male; (2) SCI/WT vs. Sham/WT; (3) SCI/Hv1 KO vs. Sham/Hv1 KO; and (4) SCI/Hv1 KO vs. SCI/WT. (D) Table depicting the number of differentially expressed genes that showed either up or downregulation within each set of pairwise comparisons. (E) Volcano plots depict the −Log10 (p-values) and log2 (fold change) of each pairwise comparison in differential expression. Dotted lines on the x and y axis show the threshold chosen for each Comp set. Gray dots represent genes that did not pass any threshold criteria. Blue dots represent genes that pass the statistic threshold, but not the fold change threshold set for that comparison. Green dots show genes that pass the fold change threshold, but not the statistic threshold. Red dots with labelled names are genes that pass both the fold change and statistic thresholds.
To answer the question of which cellular processes were affected by global knockout of the Hv1 gene, further analysis of the genes showing differential expression between SCI/Hv1 KO and SCI/WT were made based on their pathway annotations (Fig. 5A–C). We found that many genes related to microglia function, cytokine signaling, and immune response were significantly altered in the injured spinal cord of Hv1 KO mice compared to their WT counterparts. Of note, several genes that have been widely known to be related to cytokine release, neuroinflammation and neuronal death were significantly downregulated in Hv1 KO mice after SCI relative to WT control. Of the genes that showed significant differential expression between SCI/Hv1 KO and SCI/WT, we were able to isolate a subset that was dramatically downregulated (Fig. 5D). These include genes that encode proinflammatory cytokines, including Il1β, Il1rn, Il1α, and Tnf; Nlrp3, which encodes a pyrin-like protein and member of the inflammasome complex; Ccl4, a gene encoder of a mitogen-inducible monokine that has chemokinetic and inflammatory functions; Ptgs, a gene that’s responsible for encoding prostaglandin-endoperoxide synthase, which is involved with inflammation and mitogenesis; Cd14, the encoder of a surface protein that’s preferentially expressed on monocytes/macrophages; Hdc, a gene encoder for histidine decarboxylase, that regulates neurotransmission, gastric acid secretion and inflammation; and finally Hcar2, which encodes hydroxycarboxylic acid receptor, a known mediator of nicotinic acid-induced apoptosis in mature neutrophils.
Figure 5. Depletion of Hv1 modifies injury genes related to neuroinflammation at 3 days post-injury.
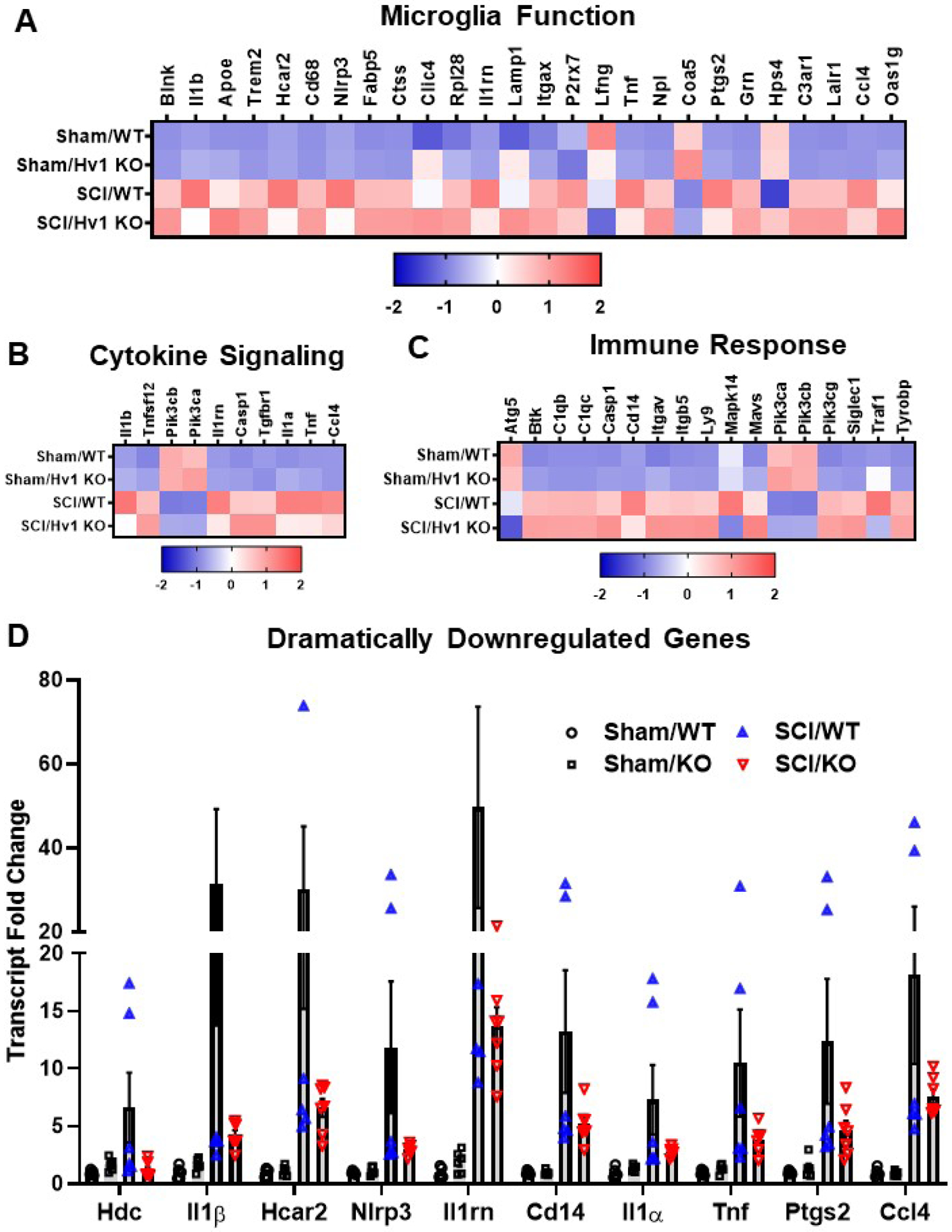
(A) Heatmap of genes related to microglia function that are differentially expressed (DE) by injury in Injury Comp 1 and Genotype Comp 2. Color coding was based on z-score scaling. (B) Heatmap of genes related to cytokine signaling that are DE in Injury Comp 1 and Genotype Comp 2. (C) Heatmap of genes related to both Innate and Adaptive Immune Response that are modified by Hv1 KO in both Injury Comp 1 and Genotype Comp 2. (D) Transcriptional fold change of dramatically downregulated (linear fold change less than or equal to 0.5) genes in Injury Comp 2.
The downregulation of these ten genes were validated with qPCR (Fig. 6). The four genes encoding cytokines were examined at 3d post-SCI, with the mRNA levels of Il1β, Il1rn, and Tnfα (p<0.01, p<0.001, and p<0.001, respectively) showing significant downregulation between SCI/WT and SCI/Hv1 KO. The Il1α gene, however, showed a significant increase between the sham and SCI groups (p<0.001), but was not significantly lower in SCI/Hv1 KO mice relative to SCI/WT control. Expression levels of Cd14, Ccl4, Ptgs, and Nlrp3, a member of the inflammasome complex, were also significantly attenuated in Hv1 KO after SCI compared to injured WT mice (p<0.001). Interestingly, Hdc and Hcar2, two genes that are involved with metabolic processes, also demonstrated an attenuated increase in expression levels in SCI/Hv1 KO mice (p<0.05 vs SCI/WT). These findings highlight a role for Hv1 in the regulation of several key inflammatory signaling pathways at baseline and following SCI.
Figure 6. Validation of dramatically downregulated genes in NanoString.
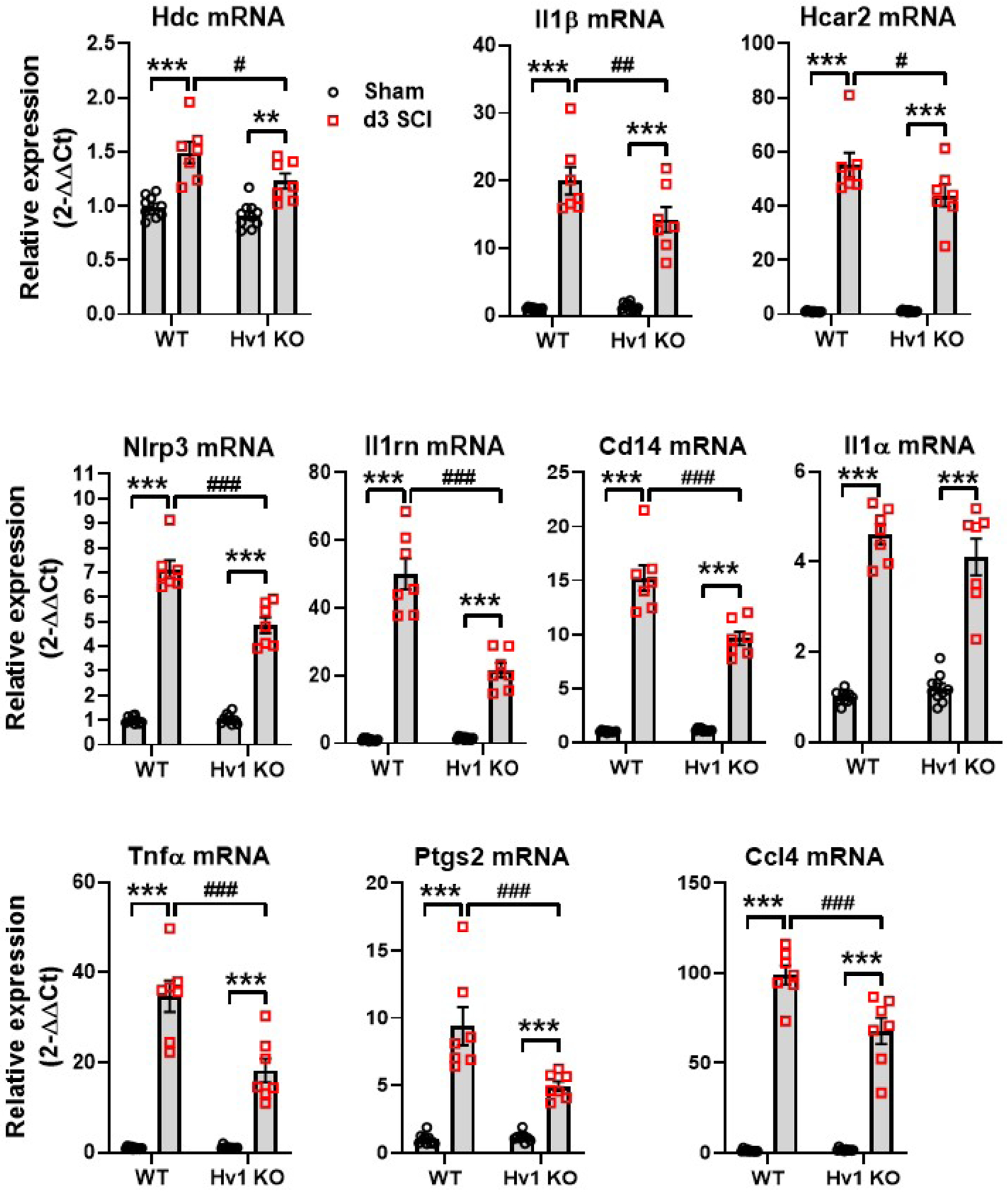
qPCR analysis was performed in injured spinal cord tissue at 3 d post-injury. The expression levels of all ten dramatically downregulated genes was examined, demonstrating a significant decrease for all except the Il1α gene. All data are presented as independent data points. N=10 (Sham/WT), 10 (Sham/Hv1 KO), 7 (SCI/WT), and 7 (SCI/Hv1 KO) mice. **p<0.01, ***p<0.001 vs. Sham groups; #p<0.05, ##p<0.01, ###p<0.001 vs SCI/WT. Two-way ANOVA following Tukey’s multiple comparisons test.
3.5. Hv1 deficiency reduces cellular inflammation and ROS production in phagocytes
Next, we examined the effects of Hv1 deficiency on the neuro-immune response in the injured spinal cord. At 3 d post-injury, CD11b+CD45int microglia proliferation and CD11b+CD45hi leukocyte infiltration (p<0.01 and p<0.05, respectively) were significantly attenuated in SCI/Hv1 KO mice compared to SCI/WT, as assessed by flow cytometry (Fig. 7A–D). Ly6Chi monocyte and Ly6G+ neutrophil infiltration were reduced in equal proportion in the SCI/Hv1 KO group, suggesting that the early attenuation of microgliosis dampens leukocyte entry in a non-specific manner. We then evaluated markers of phagocyte activation and oxidative burst. Protein expression of CD68, an endosomal/lysosomal marker associated with phagocytic activity, was significantly upregulated in microglia after injury (p<0.001), albeit significantly less so in SCI/Hv1 KO mice relative to SCI/WT control (p<0.05, Fig. 7E–F). A similar trend was seen in CD68 expression on the CD11b+CD45hi population of infiltrating myeloid cells, but this change was not statistical (p=0.0927 vs SCI/WT). Protein expression of NOX2 was likewise increased in microglia after injury (p<0.001) but comparably reduced in SCI/Hv1 KO mice relative to the SCI/WT group (p<0.05, Fig. 7G–H). Infiltrating myeloid cells in the injured KO spinal cord had significantly lower NOX2 expression compared to those found in SCI/WT mice (p<0.001). ROS production was significantly attenuated in both microglia and infiltrating myeloid cells of SCI/Hv1 KO mice compared to the injured control group, as determined by DCF fluorescence intensity (p<0.05 vs SCI/WT, Fig. 7I–J). Together, these findings demonstrate that Hv1 activity promotes microglia proliferation, leukocyte infiltration, and phagocytic oxidative burst.
Figure 7. The effects of Hv1 deficiency on the neuro-immune response in the spinal cord at 3 d post-injury.
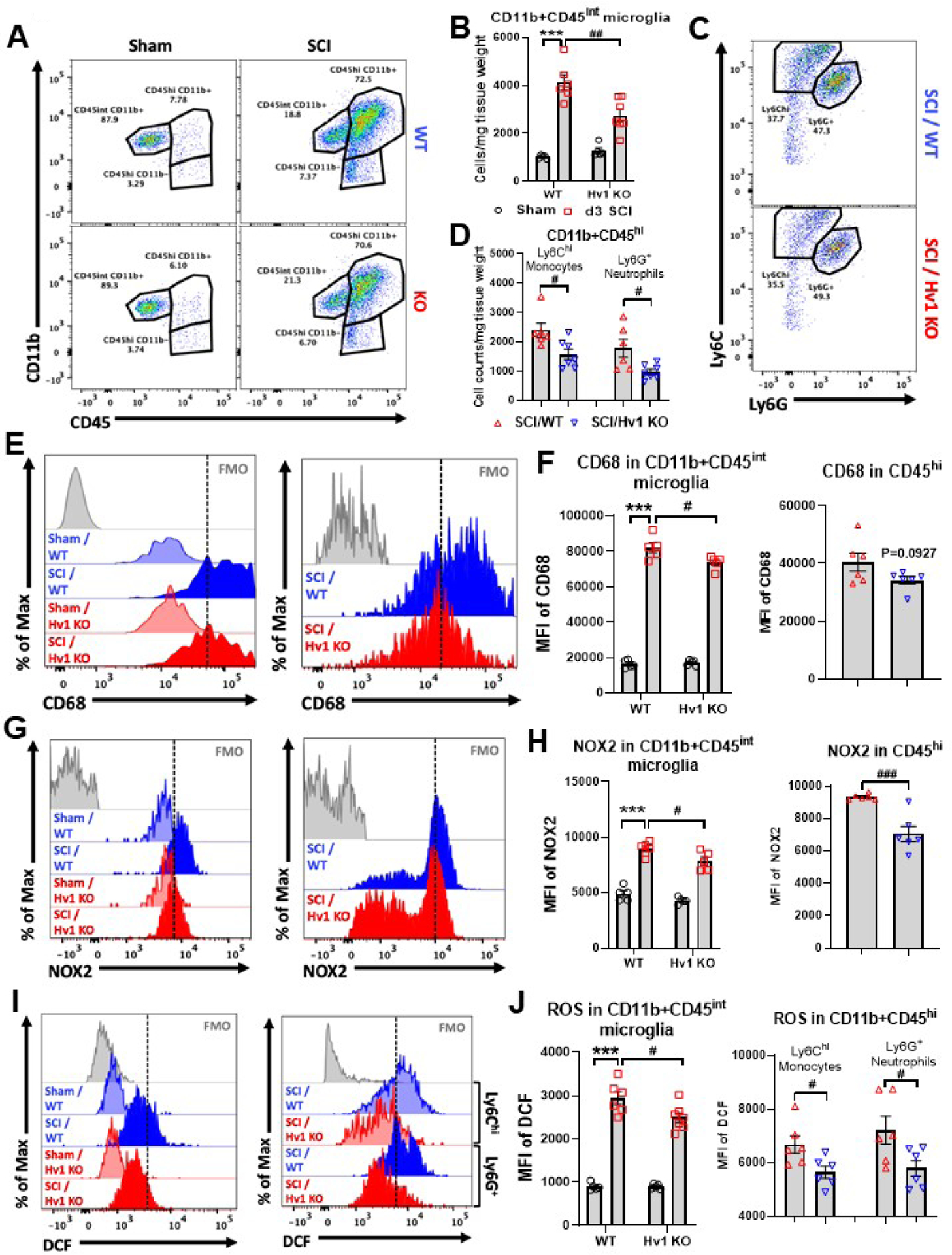
(A-D) Flow cytometry analysis showed that microglia proliferation and leukocyte infiltration were significantly attenuated in SCI/Hv1 KO mice compared to SCI/WT. Representative dot plot of immune cells in the spinal cord of sham and injured mice are shown in A and C. Quantification of CD11b+CD45int microglia counts and CD11b+CD45hi leukocyte are indicated in B and C. N=5 (Sham/WT), 5 (Sham/Hv1 KO), 6 (SCI/WT), and 7 (SCI/Hv1 KO) mice. (E-F) Protein expression of CD68 was significantly upregulated in microglia after injury. Representative histograms (E) show the relative mean fluorescence intensity of CD68+ microglia (left panel) and CD11b+CD45hi leukocyte (right panel). Mean fluorescence intensity of CD68 expression in microglia and leukocyte were quantified in F. N=5/group for E and 6/group for F. (G-H) Protein expression of NOX2 was likewise increased in microglia after injury. Representative histograms (G) show the relative mean fluorescence intensity of NOX2+ microglia (left panel) and CD11b+CD45hi leukocyte (right panel). Mean fluorescence intensity of NOX2 expression in microglia and leukocyte were quantified in H. N=5/group for G and 6/group for H. (I-J) ROS production was significantly attenuated in both microglia and infiltrating myeloid cells of SCI/Hv1 KO mice compared to the injured control group, as determined by DCF (H2DCFDA) fluorescence intensity. Representative histograms (I) show the relative mean fluorescence intensity of DCF in microglia (left panel) and CD11b+CD45hi leukocyte (right panel). Mean fluorescence intensity of DCF expression in microglia and leukocyte were quantified in J. N=5 (Sham/WT), 5 (Sham/Hv1 KO), 6 (SCI/WT), and 7 (SCI/Hv1 KO) mice for I and N=6/group for J. All data are presented as independent data points. Two-way ANOVA following Tukey’s multiple comparisons test for B, and left panels of F, H, J; Unpaired t test for D and right panels of F, H, J.
To further confirm the reduction in neuroinflammation seen in SCI/Hv1 KO mice, we used qPCR to examine the expression levels of Nox2 (Cybb gene) (Henry et al., 2020; Sabirzhanov et al., 2019a; Zhang et al., 2019) and biomarker genes of inflammation. Consistent with our previous results, NOX2 expression was significantly lower in SCI/Hv1 KO group at 3 d post-injury (p<0.001 vs SCI/WT, Fig. 8A). As expected, several upstream pro-inflammatory mediators, including nitric oxide synthase (Nos2), C-C motif chemokine ligand 5 (Ccl5), Cluster of differentiation 11b (Cd11b), Cluster of differentiation 68 (Cd68), and Interleukin 6 (Il6) were upregulated following injury (p<0.001 vs Sham groups, Fig. 8B). Of the six pro-inflammatory genes examined (p<0.001 or p<0.001 vs SCI/WT), only Il6 was not significantly attenuated in SCI/Hv1 KO mice relative to SCI/WT control. Next, we determined the expression level of several anti-inflammatory markers including Chitinase-like protein 3 (Chil3), Arginase 1 (Arg1), Interleukin 10 (Il10), Transforming growth factor β (Tgfβ), and Suppressor of cytokine signaling 3 (Socs3) (Fig. 8C). Consistent with our previous results, SCI caused a significant increase in all anti-inflammatory markers tested in both WT and Hv1 KO mice (p<0.001 vs Sham groups). Notably, two genes, Chil3 and Tgfβ, were significantly more upregulated in SCI/Hv1 KO mice relative to SCI/WT control (p<0.05 vs SCI/WT). Finally, we assessed astrocyte activation by examining Glial fibrillary acidic protein (Gfap) expression (Fig. 8D). Gfap transcription was significantly upregulated after injury (p<0.001 vs Sham groups) but no difference between SCI/WT and SCI/Hv1 KO was seen. These data suggest Hv1 exacerbates NADPH-dependent ROS production by phagocytes which promotes neuroinflammation in the acute stages of SCI.
Figure 8. The effects of Hv1 deficiency on the expression levels of inflammatory biomarker genes in the spinal cord at 3 d post-injury.
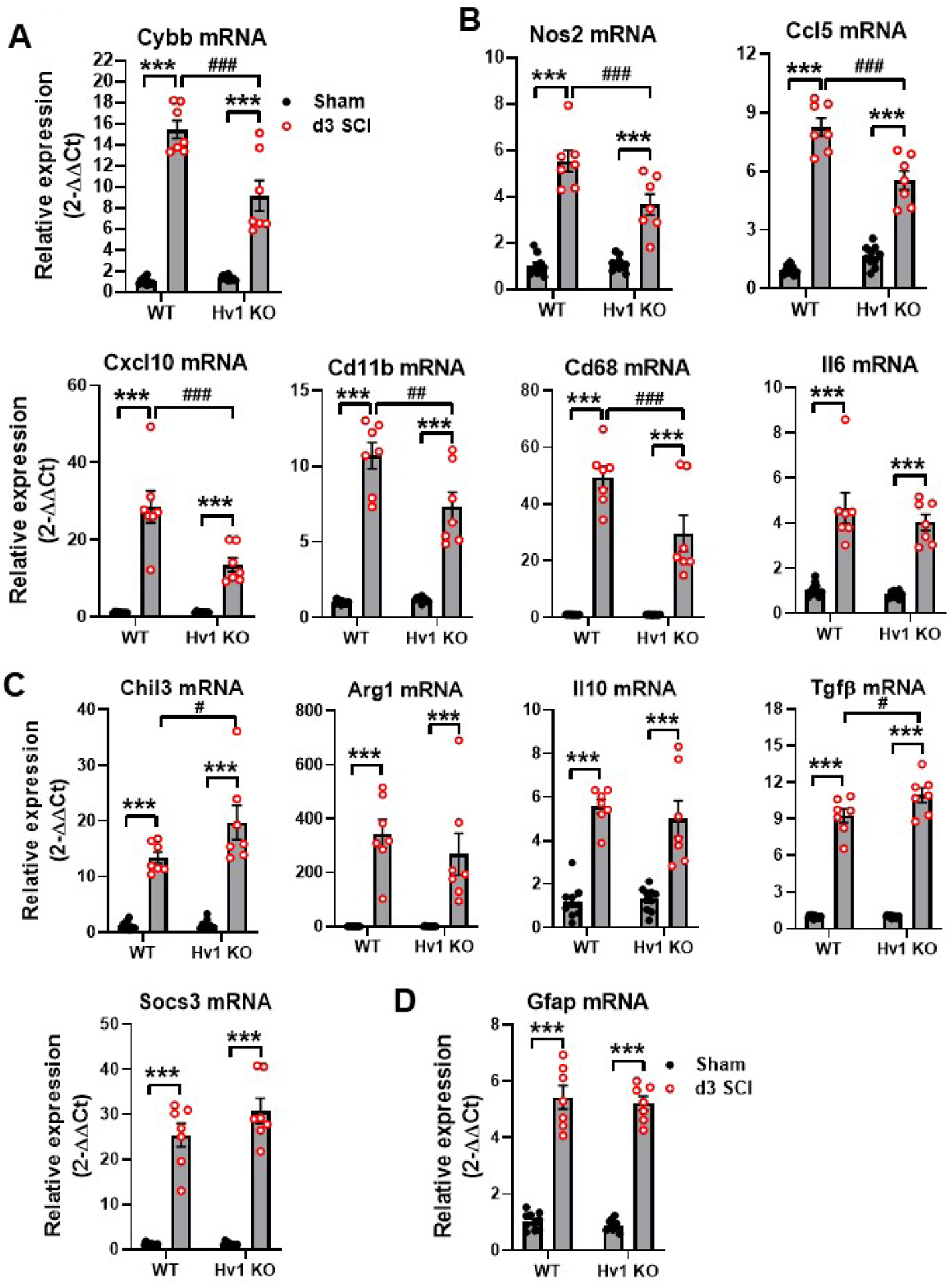
(A) NOX2 (Cybb) expression was significantly lower in SCI/Hv1 KO group compared to SCI/WT group. (B) Pro-inflammatory markers. (C) Anti-inflammatory markers. (D) Astrocyte marker Gfap. All data are presented as independent data points. N=10 (Sham/WT), 10 (Sham/Hv1 KO), 7 (SCI/WT), and 7 (SCI/Hv1 KO) mice. ***p<0.001 vs. Sham groups; #p<0.05, ##p<0.01, ###p<0.001 vs SCI/WT. Two-way ANOVA following Tukey’s multiple comparisons test.
3.6. Depletion of Hv1 improves functional recovery and reduces tissue damage after SCI
Finally, we examined the effects of Hv1 on behavioral outcome and neuropathology after traumatic SCI. Adult female Hv1 KO mice and their WT littermates were subjected to moderate contusion injury on the spinal cord. Hindlimb locomotor function was assessed using the BMS. By day 21, the Hv1 KO mice (n=16) showed significantly higher BMS scores than the WT mice (n=15, Fig. 9A; p<0.01, two-way ANOVA with repeated measurement following by Sidak’s multiple comparisons test), which persisted through 42 d. The factor of Post-injury Days (F(7,203)=100; p<0.0001) in BMS test was found to be significant. The factor of Genotype (F(1,29)=7.839; p=0.0090) as well as the interaction of Postinjury Days × Genotype (F(7,203)=4.751; p<0.0001) were also significant. To quantify improvements in the areas of stepping frequency, coordination, paw position, trunk stability, and tail position, the BMS subscore was evaluated. The Hv1 KO mice demonstrated a recovery in better fine motor control, as shown in the BMS subscore (Fig. 9B), with significant improvement at 21 d, which remained through 42 d after injury. The factors of Post-injury Days (F(7,203)=23.38; p<0.0001), Genotype (F(1,29)=10.39; p=0.0031), and the interaction of Post-injury Days × Genotype (F(7,203)=5.554; p<0.0001) were found to be significant. Therefore, our data indicated that mice lacking Hv1 have better functional recovery after SCI than WT control mice.
Figure 9. Hv1 KO mice show improved functional recovery and alleviated tissue damage.
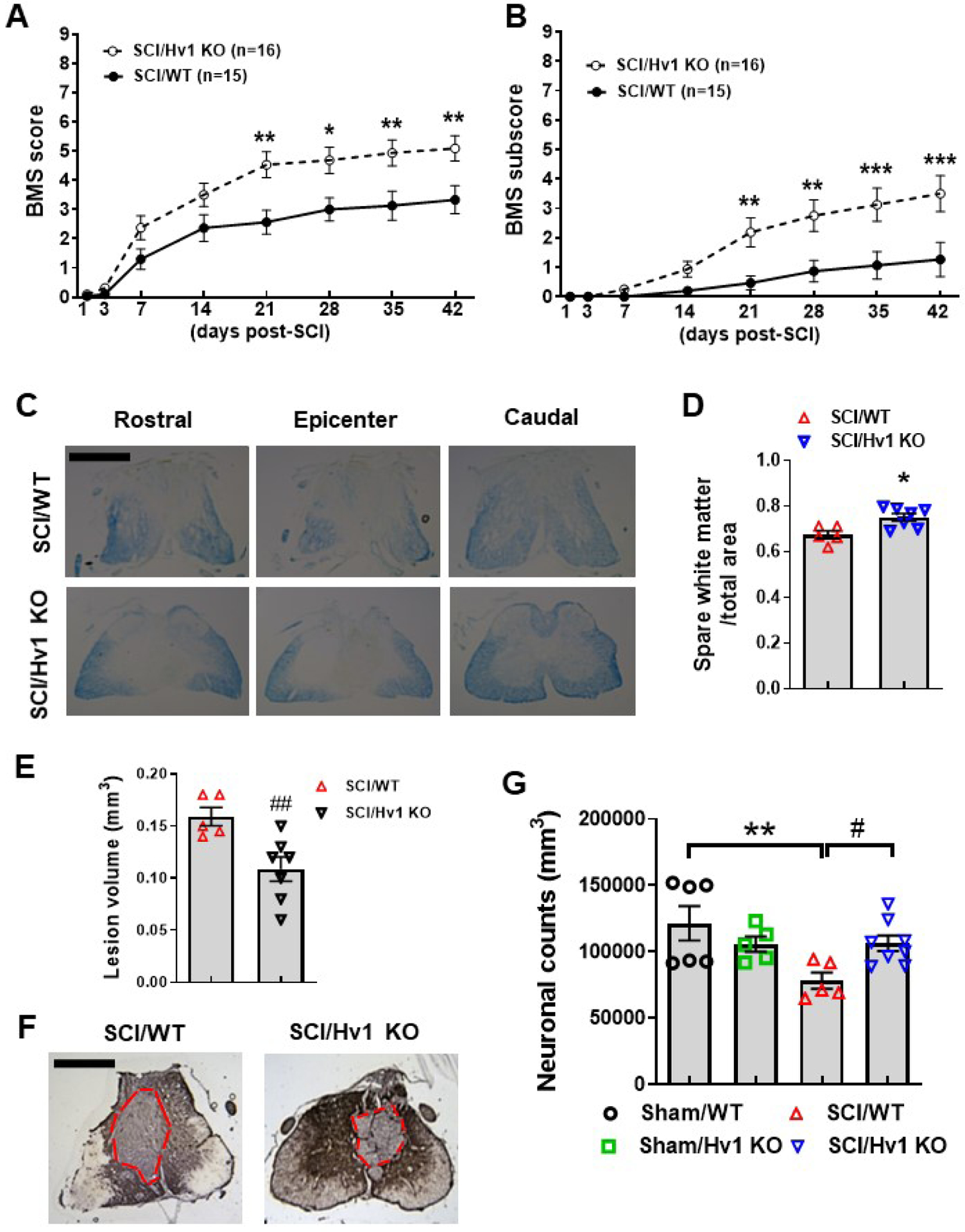
(A-B) Genetic deletion of Hv1 in Hv1 KO mice increased BMS scores (A) and subscore (B). N=15 (SCI/WT) and 16 (SCI/Hv1 KO) mice. *p<0.05, **p<0.01, ***p<0.001 vs SCI/WT. Two-way ANOVA with repeated measurement following by Sidak’s multiple comparisons test. (C-D) Spared whiter matter (SWM) at 6 weeks post-injury was assessed on Luxol Fast Blue (LFB) stained coronal sections. Representative images encompassed the rostral (R, 0.8 mm) to caudal (C, 0.8 mm) extent of the lesion from WT and Hv1 KO group. N=5 (SCI/WT) and 7 (SCI/Hv1 KO) mice/group. *p<0.05 with Mann Whitney test. Scale bar=500 μm. (E-F) Lesion volume was quantified by outlining the unstained or missing tissue on the injury core using unbiased stereology. Representative images with red dashed lines indicated the epicenter from SCI/WT and SCI/Hv1 KO group. N=5 (SCI/WT) and 7 (SCI/Hv1 KO) mice/group. ##p<0.01 with Unpaired t test. Scale bar=500 μm. (G) Unbiased stereology was performed to assess neuronal counts. N=6 (Sham/WT), 5 (Sham/Hv1 KO), 5 (SCI/WT), and 8 (SCI/Hv1 KO) mice/group. **p<0.001 vs. Sham/WT; #p<0.05 vs SCI/WT. Two-way ANOVA following Student-Newman-Keuls multiple comparisons test.
Histological staining with Solvent Blue showed less myelin damage in the SCI/Hv1 KO group compared to SCI/WT, with significantly higher volume of spared white matter (p<0.05, Fig. 9C–D). SCI/Hv1 KO mice had significantly smaller lesion volumes compared to SCI/WT control as evidenced by GFAP/DAB staining (p<0.01, Fig. 9E–F). To further confirm the extent of neuroprotection that global knockout of Hv1 has in traumatic SCI, we examined neuronal loss using unbiased stereological counting. Significantly fewer neurons were observed in WT mice at 6 w post-injury (p=0.002 vs Sham/WT), but not between SCI and Sham mice of Hv1 KO groups. Moreover, SCI/Hv1 KO mice had significantly greater preservation of neurons in the gray matter compared to SCI/WT control (p=0.026 vs SCI/WT), suggesting robust neuroprotection (Fig. 9G). Taken together, our results demonstrate that the severity of acute tissue acidosis is associated with long-term recovery and the progression of neurodegeneration after SCI.
4. Discussion
In the present study, we explored the role of the pH-sensitive ion channel Hv1 in tissue acidosis caused by traumatic injury of the spinal cord. Decreased extracellular pH in the peri-injury area was observed for the first 7 days in a contusion-based traumatic SCI model. High lactate concentrations were also evident, along with increased ROS levels in the epicenter region. We discovered the voltage-gated proton channel Hv1, a known regulator of acid (i.e., H+) extrusion in the microglia, was significantly upregulated in the same timespan as tissue acidosis and remained elevated for weeks after SCI. Depletion of the Hv1 gene by global knockout led to alleviation of spinal cord acidosis and reduced neuroinflammation during the acute period, resulting in enhanced functional recovery and attenuation of tissue damage.
Although SCI-induced tissue acidosis was first reported as early as the 1970’s with many studies replicating the increase of lactate concentrations in the extracellular microenvironment (Anderson et al., 1980; Anderson et al., 1976; Falconer et al., 1996; Farooque et al., 1997; Vink et al., 1987; Vink et al., 1989; Zaks, 1976), our mechanistic understanding has been limited. The discovery of acid-sensing ion channels ASIC and TRPV on neurons of the central and peripheral nervous system offered a new perspective to the field of localized acidosis (Caterina et al., 2000; Waldmann et al., 1997). Blocking ASIC channels with specific pharmacological inhibitors was found to confer neuroprotective effects in spinal cord injury, leading to alleviation of secondary injury and better functional outcome in rats. However, contrasting findings have also been reported (Hu et al., 2011; Koehn et al., 2016). Tissue acidosis is associated with nociceptive responses and enhanced excitability of sensory neurons in the dorsal root ganglia, and grey matter of the spinal cord after inflammation (Deval et al., 2010; Neelands et al., 2005; Wu et al., 2004). Controversially, a few studies also point to the effectiveness of moderate acidosis (pH 6.5) in curtailing glutamate excitotoxicity (Giffard et al., 1990; Tang et al., 1990). No matter how complex the effects of tissue acidosis on neuronal activity in the spinal cord, all of these studies demonstrate increased hydrogen (H+) concentrations after injury. Our initial findings of decreased pH, elevated lactate concentration, and increased oxidative stress in the injury site during the first week of SCI are consistent with earlier studies of acidosis and inflammation (Farooque et al., 1997; Hasse et al., 2000; Hu et al., 2011; Koller et al., 1984; Vink et al., 1989). However, instead of focusing on the downstream mechanisms underlying the neuronal response to acidotoxicity, we sought to uncover the potential source of extracellular H+ responsible for activating those acid-sensing channels. In doing so, we identified a proton extrusion pathway exclusively expressed on microglia that becomes hyperactive, or dysregulated, for weeks following SCI. We believe that microglial Hv1 may lie upstream of neuronal ASIC signaling and contribute to neuronal acidification and death. Future studies will be required to conclusively link Hv1 activity to ASIC activation to confirm whether microglia can communicate with neurons via the extracellular release of protons during homeostasis and if those downstream signaling pathways can be tempered after injury to ameliorate acidotoxicity.
We are the first to report evidence for the involvement of Hv1 in the modulation of pathological acidosis in a clinically-relevant in vivo model of spinal cord injury. Tissue pH was measured in situ using a micro electrode. In the past, similar in situ readings were recorded in experimental autoimmune encephalomyelitis (EAE) studies, a well-developed rodent model of multiple sclerosis (de Ceglia et al., 2015; Friese et al., 2007). But due to the technical limitations and caveats of contusion-induced traumatic injury, all prior pH measurements for SCI studies have been taken from extracellular fluids [e.g., Cerebrospinal fluid (CSF)] via microdialysis and HPLC (Anderson et al., 1976; Delheimer et al., 1980; Farooque et al., 1997; Lamid, 1983). One of the constraints of our methodology is the inability to take readings directly from the epicenter of the injury site. Pilot data for this study showed that probing the spinal cord of anesthetized mice caused severe hemorrhaging upon entrance of the electrode. Conversely, probing the spinal cord without perfusion of the blood vessels, or placing the probe directly within the epicenter where injury-induced hemorrhage is prominent during the acute stage yielded results that suggested an absence of acidosis. Both of these situations are due to the existence of blood, which has a significantly higher pH (varied between 7.20 and 7.43 in C57BL/6 mice anesthetized with isoflurane) (Constantinides et al., 2011) than spinal cord tissue (range of 6.80 to 6.98 in current study). Thus, we could only make inferences to the pH level of the epicenter via recordings taken from the peri-injury sites. However, we believe taking repeated measurements from freshly dissected peri-injury sites after perfusion with ice-cold saline is a more accurate and reliable readout for determination of localized acidosis in the spinal cord parenchyma.
It is important to emphasize that a difference in pH from ~6.8 to ~6.7 as seen between genotypes after injury, reflects a ~2-fold increase in the concentration of hydrogen ions at the injury site. Due to the logarithmic scale, small changes in tissue pH level can have significant biological consequences. It was previously reported that activation of a single Hv1 channel can allow up to 100,000 hydrogen ions across the membrane each second (DeCoursey and Hosler, 2014). Indeed, the data indisputably show higher pH in spinal cord tissue at baseline in Hv1 KO mice compared to WT control. Interestingly, no change between genotypes was observed in brain tissue pH of naïve mice (data not shown). Ostensibly this reflects a region-specific functionality of the Hv1 proton channel in microglia. There is not much known regarding how alterations in homeostatic pH alter the CNS environment and its response to trauma. Our data suggests that it has no obvious impact on motor function at baseline, and may in fact provide prophylactic protection by increasing the threshold to reach a pathological pH level after injury. While we agree that the SCI-induced decrease in pH is proportional within genotype groups, it stands to reason that the absolute pH value is more critical for CNS injury severity and recovery than the relative decrease itself. It is clear that the Hv1 proton channel represents only one of several known acidosis-related secondary injury mechanisms, including the ubiquitously expressed Na+/H+ exchangers (NHEs). However, few have been described in the context of spinal cord injury or directly involve microglia.
Using the NanoString Neuroinflammation panel to identify the transcriptional targets of Hv1 activity after SCI, our group was able to characterize the impact that genetic deletion had on Hv1-responsive genes. As shown in our findings, as many as 160 genes were significantly modified at baseline levels by Hv1 KO. The majority of these genes encode proteins involved with immune response, cytokine and inflammatory signaling and microglia function. These findings indicate that Hv1 ablation via knockout mutation could distinctly alter cellular function in the immune system. However, the magnitude of modification, as shown by fold change, is comparatively smaller than after SCI. These findings are consistent with prior studies that observed reduced oxidation and attenuation of immune cell signaling activation in Hv1 KO phenotypes, but no distinct impairment of bacterial clearance after phorbol 12-myristate 13-acetate (PMA) stimulation (Ramsey et al., 2009). On the other hand, findings by Sasaki et al showed an autoimmune disorder phenotype in germline Hv1 KO mice, featuring increased number of T-cells and spontaneous splenomegaly at 6 months of age (Sasaki et al., 2013). Based on these findings and our own results, discrete transcriptional changes in genes related to immune function are evidently present, but it does not seem to affect the survival rate of Hv1 KO mice in natural aging, nor does it impair the body’s immune system in combating disease infection. In line with our hypothesis, the majority of genes regulated by Hv1 KO were related to microglia function, cytokine signaling, and immune response. Several key pro-inflammatory genes were further validated with qPCR, confirming the reductions seen in injured KO mice. These findings stand in contrast to that reported by Wu et al in ischemic stroke (Wu et al., 2012). The authors found that Hv1 KO led to a reduction in ROS generation, but not pro-inflammatory cytokines, in cultured microglia after oxygen glucose deprivation (OGD), an in vitro model of ischemic stroke. However, the mechanism and severity of OGD and traumatic SCI differ greatly, not the least of which is the absence of neurons, vasculature, and other glia cells which also play important roles in secondary injury. Only one other study has reported the impact of Hv1 in SCI (Li et al., 2020). The authors reported that Hv1 deficiency resulted in stronger M2 phenotypes and lower expression levels of pro-inflammatory cytokines. This inflammatory profile was also confirmed by our own findings. However, whereas Li and colleagues only assessed the outcome of Hv1 KO with lesion volume and demyelination measurements, our study was able to determine functional outcome with BMS and more extensive stereological quantification of neuronal death.
Since Hv1 has been shown to be critical for phagocytosis and respiratory burst activity in neutrophils, we reasoned that it may also play a role in other myeloid cells of the immune system (Ramsey et al., 2009; Ratanayotha et al., 2019). Microglia activation and myeloid cell infiltration are hallmarks of the secondary injury phase after SCI (Pukos et al., 2019; Zhou et al., 2014). At the level of cellular function, we found that CD11b+CD45int microglia proliferation and CD11b+CD45hi leukocyte infiltration were lower in injured KO mice. Consistent with our understanding of Hv1 and its essential role in oxidative burst, microglia in SCI/Hv1 KO mice exhibited blunted induction of CD68, NOX2, and ROS. Although similar changes were seen in the CD45hiCD11b+ myeloid population, overall numbers paled in comparison to the tissue-resident microglia.
It is now accepted that microglia have been shown to have critical roles in SCI pathogenesis. Moreover, the temporal pattern of microglia responses suggest these resident immune cells can contribute to both injury and repair depending on the injury phase (Bellver-Landete et al., 2019). Pharmacological depletion of microglia was recently shown to reduce cytokine expression and alleviate neuropathic pain after sciatic nerve injury (Lee et al., 2018). Although pH levels were not examined in either of these studies, it is reasonable to assume upon our findings that the absence of microglia during the acute phase of injury would reduce extracellular H+ and ROS after injury. The relationship between spinal cord acidosis and chronic pain warrants further investigation. Whether or not microglia Hv1 play a critical role in the induction of extracellular acidosis after SCI is intriguing for future study.
In conclusion, our study examined the induction and molecular mechanism of pathological acidosis after SCI, revealing a novel regulatory role for the Hv1 proton channel. We demonstrated that Hv1 activity, while essential for phagocyte function and intracellular pH regulation, can become dysregulated following traumatic SCI, resulting in the extracellular release of protons and ROS. These findings establish a direct causal link between inflammation and acidosis. Targeting this ion channel by genetic deletion led to attenuation of these pathological processes in the acute stage and enhanced long-term recovery. Therefore, early intervention aimed at inhibiting Hv1 activity may mitigate tissue acidosis and alleviate the damage caused by spinal cord trauma.
Highlights.
Spinal cord acidosis and elevated NOX2 and ROS persist up to one week after SCI
SCI causes a sustained increase in Hv1 mRNA expression up to one-month post-injury
Genetic ablation of the voltage-dependent proton channel Hv1 reduced acidosis and ROS
Hv1 deficiency reduced microglia proliferation and leukocyte infiltration after SCI
Hv1 deficiency limited tissue damage and improved functional recovery after SCI
Acknowledgements
The work was supported by the National Institutes of Health Grants R01 NS110825 (JW and LW), R01 NS094527 (JW), RF1 NS110637 (JW), and K99 NS116032 (RMR). We would like to thank Dr. Zhuofan Lei, Ms. Lulu Liu, Ms. Jordan Carter, and Mr. Niaz Khan for assistance with the Hv1 antibody testing, tissue sectioning, and the cytometer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors have no conflicts of interest to declare.
References
- Anderson DK, Means ED, Waters TR, 1980. Spinal cord energy metabolism in normal and postlaminectomy cats. J Neurosurg 52, 387–391. [DOI] [PubMed] [Google Scholar]
- Anderson DK, Prockop LD, Means ED, Hartley LE, 1976. Cerebrospinal fluid lactate and electrolyte levels following experimental spinal cord injury. J Neurosurg 44, 715–722. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG, 2006. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23, 635–659. [DOI] [PubMed] [Google Scholar]
- Bellver-Landete V, Bretheau F, Mailhot B, Vallieres N, Lessard M, Janelle ME, Vernoux N, Tremblay ME, Fuehrmann T, Shoichet MS, Lacroix S, 2019. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun 10, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, et al. , 2000. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313. [DOI] [PubMed] [Google Scholar]
- Chesler M, 2003. Regulation and modulation of pH in the brain. Physiological reviews 83, 1183–1221. [DOI] [PubMed] [Google Scholar]
- Clark RA, 2016. Editorial: Proton pathway paradox: Hv1 H+ channel sustains neutrophil Nox2 activity, yet suppresses HOCl formation. Journal of leukocyte biology 99, 1–4. [DOI] [PubMed] [Google Scholar]
- Clausen T, Khaldi A, Zauner A, Reinert M, Doppenberg E, Menzel M, Soukup J, Alves OL, Bullock MR, 2005. Cerebral acid-base homeostasis after severe traumatic brain injury. J Neurosurg 103, 597–607. [DOI] [PubMed] [Google Scholar]
- Cohen J, 1992. A power primer. Psychological bulletin 112, 155–159. [DOI] [PubMed] [Google Scholar]
- Constantinides C, Mean R, Janssen BJ, 2011. Effects of isoflurane anesthesia on the cardiovascular function of the C57BL/6 mouse. ILAR J 52, e21–31. [PMC free article] [PubMed] [Google Scholar]
- de Ceglia R, Chaabane L, Biffi E, Bergamaschi A, Ferrigno G, Amadio S, Del Carro U, Mazzocchi N, Comi G, Bianchi V, Taverna S, Forti L, D’Adamo P, Martino G, Menegon A, Muzio L, 2015. Down-sizing of neuronal network activity and density of presynaptic terminals by pathological acidosis are efficiently prevented by Diminazene Aceturate. Brain Behav Immun 45, 263–276. [DOI] [PubMed] [Google Scholar]
- DeCoursey TE, 2015. The Voltage-Gated Proton Channel: A Riddle, Wrapped in a Mystery, inside an Enigma. Biochemistry 54, 3250–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Hosler J, 2014. Philosophy of voltage-gated proton channels. J R Soc Interface 11, 20130799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delheimer SC, Anderson RE, Sundt TM Jr., 1980. The measurement of spinal cord tissue pH using a diffusible, lipid-soluble, pH-sensitive fluorescent indicator. J Neurochem 34, 1514–1519. [DOI] [PubMed] [Google Scholar]
- Deval E, Gasull X, Noel J, Salinas M, Baron A, Diochot S, Lingueglia E, 2010. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacology & therapeutics 128, 549–558. [DOI] [PubMed] [Google Scholar]
- Dodge JC, Treleaven CM, Fidler JA, Tamsett TJ, Bao C, Searles M, Taksir TV, Misra K, Sidman RL, Cheng SH, Shihabuddin LS, 2013. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci U S A 110, 10812–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Chemaly A, Nunes P, Jimaja W, Castelbou C, Demaurex N, 2014. Hv1 proton channels differentially regulate the pH of neutrophil and macrophage phagosomes by sustaining the production of phagosomal ROS that inhibit the delivery of vacuolar ATPases. Journal of leukocyte biology 95, 827–839. [DOI] [PubMed] [Google Scholar]
- Falconer JC, Liu SJ, Abbe RA, Narayana PA, 1996. Time dependence of N-acetyl-aspartate, lactate, and pyruvate concentrations following spinal cord injury. J Neurochem 66, 717–722. [DOI] [PubMed] [Google Scholar]
- Farooque M, Hillered L, Holtz A, Olsson Y, 1997. Effects of moderate hypothermia on extracellular lactic acid and amino acids after severe compression injury of rat spinal cord. J Neurotrauma 14, 63–69. [DOI] [PubMed] [Google Scholar]
- Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L, 2007. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med 13, 1483–1489. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Monyer H, Christine CW, Choi DW, 1990. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res 506, 339–342. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Zygun DA, Johnston AJ, Steiner LA, Al-Rawi PG, Chatfield D, Shepherd E, Kirkpatrick PJ, Hutchinson PJ, Menon DK, 2004. Extracellular Brain pH and Outcome following Severe Traumatic Brain Injury. J Neurotrauma 21, 678–684. [DOI] [PubMed] [Google Scholar]
- Hasse W, Weidtmann A, Voeltz P, 2000. [Lactic acidosis: a complication of spinal cord injury in multiple trauma]. Unfallchirurg 103, 495–498. [DOI] [PubMed] [Google Scholar]
- Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, Szeto GL, Wu J, Stoica BA, Faden AI, Loane DJ, 2020. Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. J Neurosci 40, 2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Duan B, Wang D, Yu Y, Li W, Luo H, Lu P, Lin J, Zhu G, Wan Q, Feng H, 2011. Role of acid-sensing ion channel 1a in the secondary damage of traumatic spinal cord injury. Ann Surg 254, 353–362. [DOI] [PubMed] [Google Scholar]
- Jalalvand E, Robertson B, Tostivint H, Wallen P, Grillner S, 2016. The Spinal Cord Has an Intrinsic System for the Control of pH. Curr Biol 26, 1346–1351. [DOI] [PubMed] [Google Scholar]
- Koehn LM, Noor NM, Dong Q, Er SY, Rash LD, King GF, Dziegielewska KM, Saunders NR, Habgood MD, 2016. Selective inhibition of ASIC1a confers functional and morphological neuroprotection following traumatic spinal cord injury. F1000Res 5, 1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller ME, Breivik H, Greider P, Jones DJ, Smith RB, 1984. Synergistic effect of acidosis and succinylcholine-induced hyperkalemia in spinal cord transected rats. Acta Anaesthesiol Scand 28, 87–90. [DOI] [PubMed] [Google Scholar]
- Lamid S, 1983. Ascorbic acid and methenamine mandelate on the urinary pH of spinal cord injury patients. J Urol 129, 845–846. [DOI] [PubMed] [Google Scholar]
- Lee S, Shi XQ, Fan A, West B, Zhang J, 2018. Targeting macrophage and microglia activation with colony stimulating factor 1 receptor inhibitor is an effective strategy to treat injury-triggered neuropathic pain. Molecular pain 14, 1744806918764979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Ward R, Dong G, Ergul A, O’Connor P, 2019. Neurovascular protection in voltage-gated proton channel Hv1 knock-out rats after ischemic stroke: interaction with Na(+) /H(+) exchanger-1 antagonism. Physiological reports 7, e14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu R, Yu Z, He D, Zong W, Wang M, Xie M, Wang W, Luo X, 2020. Microglial Hv1 exacerbates secondary damage after spinal cord injury in mice. Biochem Biophys Res Commun. [DOI] [PubMed] [Google Scholar]
- Marmarou A, Holdaway R, Ward JD, Yoshida K, Choi SC, Muizelaar JP, Young HF, 1993. Traumatic brain tissue acidosis: experimental and clinical studies. Acta Neurochir Suppl (Wien) 57, 160–164. [DOI] [PubMed] [Google Scholar]
- Matyas JJ, O’Driscoll CM, Yu L, Coll-Miro M, Daugherty S, Renn CL, Faden AI, Dorsey SG, Wu J, 2017. Truncated TrkB.T1-Mediated Astrocyte Dysfunction Contributes to Impaired Motor Function and Neuropathic Pain after Spinal Cord Injury. J Neurosci 37, 3956–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musset B, Morgan D, Cherny VV, MacGlashan DW Jr., Thomas LL, Rios E, DeCoursey TE, 2008. A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc Natl Acad Sci U S A 105, 11020–11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelands TR, Jarvis MF, Han P, Faltynek CR, Surowy CS, 2005. Acidification of rat TRPV1 alters the kinetics of capsaicin responses. Molecular pain 1, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukos N, Goodus MT, Sahinkaya FR, McTigue DM, 2019. Myelin status and oligodendrocyte lineage cells over time after spinal cord injury: What do we know and what still needs to be unwrapped? Glia 67, 2178–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Ruchti E, Kaczmarek JS, Clapham DE, 2009. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc Natl Acad Sci U S A 106, 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratanayotha A, Kawai T, Okamura Y, 2019. Real-time functional analysis of Hv1 channel in neutrophils: a new approach from zebrafish model. Am J Physiol Regul Integr Comp Physiol 316, R819–R831. [DOI] [PubMed] [Google Scholar]
- Ritzel RM, Doran SJ, Glaser EP, Meadows VE, Faden AI, Stoica BA, Loane DJ, 2019. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging 77, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro HA, Carson JH, 2004. pH microdomains in oligodendrocytes. J Biol Chem 279, 37115–37123. [DOI] [PubMed] [Google Scholar]
- Ruffin VA, Salameh AI, Boron WF, Parker MD, 2014. Intracellular pH regulation by acid-base transporters in mammalian neurons. Front Physiol 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirzhanov B, Li Y, Coll-Miro M, Matyas JJ, He J, Kumar A, Ward N, Yu J, Faden AI, Wu J, 2019a. Inhibition of NOX2 signaling limits pain-related behavior and improves motor function in male mice after spinal cord injury: Participation of IL-10/miR-155 pathways. Brain Behav Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirzhanov B, Matyas J, Coll-Miro M, Yu LL, Faden AI, Stoica BA, Wu J, 2019b. Inhibition of microRNA-711 limits angiopoietin-1 and Akt changes, tissue damage, and motor dysfunction after contusive spinal cord injury in mice. Cell death & disease 10, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Tojo A, Okochi Y, Miyawaki N, Kamimura D, Yamaguchi A, Murakami M, Okamura Y, 2013. Autoimmune disorder phenotypes in Hvcn1-deficient mice. Biochem J 450, 295–301. [DOI] [PubMed] [Google Scholar]
- Seredenina T, Demaurex N, Krause KH, 2015. Voltage-Gated Proton Channels as Novel Drug Targets: From NADPH Oxidase Regulation to Sperm Biology. Antioxidants & redox signaling 23, 490–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RP, 2006. Acidotoxicity trumps excitotoxicity in ischemic brain. Arch Neurol 63, 1368–1371. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Stys PK, 2010. Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol Med 16, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CM, Dichter M, Morad M, 1990. Modulation of the N-methyl-D-aspartate channel by extracellular H+. Proc Natl Acad Sci U S A 87, 6445–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian DS, Li CY, Qin C, Murugan M, Wu LJ, Liu JL, 2016. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J Neurochem 139, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, McIntosh TK, Weiner MW, Faden AI, 1987. Effects of traumatic brain injury on cerebral high-energy phosphates and pH: a 31P magnetic resonance spectroscopy study. J Cereb Blood Flow Metab 7, 563–571. [DOI] [PubMed] [Google Scholar]
- Vink R, Noble LJ, Knoblach SM, Bendall MR, Faden AI, 1989. Metabolic changes in rabbit spinal cord after trauma: magnetic resonance spectroscopy studies. Ann Neurol 25, 26–31. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M, 1997. A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177. [DOI] [PubMed] [Google Scholar]
- Wu J, Sabirzhanov B, Stoica BA, Lipinski MM, Zhao Z, Zhao S, Ward N, Yang D, Faden AI, 2015. Ablation of the transcription factors E2F1–2 limits neuroinflammation and associated neurological deficits after contusive spinal cord injury. Cell Cycle 14, 3698–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhao Z, Kumar A, Lipinski MM, Loane DJ, Stoica BA, Faden AI, 2016. Endoplasmic Reticulum Stress and Disrupted Neurogenesis in the Brain Are Associated with Cognitive Impairment and Depressive-Like Behavior after Spinal Cord Injury. J Neurotrauma [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI, 2014. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci 34, 10989–11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, 2014. Voltage-gated proton channel HV1 in microglia. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 20, 599–609. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Duan B, Mei YD, Gao J, Chen JG, Zhuo M, Xu L, Wu M, Xu TL, 2004. Characterization of acid-sensing ion channels in dorsal horn neurons of rat spinal cord. J Biol Chem 279, 43716–43724. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE, 2012. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci 15, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaks IO, 1976. [Acid-base balance of spinal cord fluid in the post-resuscitation period]. Biull Eksp Biol Med 82, 1303–1305. [PubMed] [Google Scholar]
- Zhang B, Bailey WM, McVicar AL, Stewart AN, Veldhorst AK, Gensel JC, 2019. Reducing age-dependent monocyte-derived macrophage activation contributes to the therapeutic efficacy of NADPH oxidase inhibition in spinal cord injury. Brain Behav Immun 76, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, He X, Ren Y, 2014. Function of microglia and macrophages in secondary damage after spinal cord injury. Neural regeneration research 9, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygun DA, Steiner LA, Johnston AJ, Hutchinson PJ, Al-Rawi PG, Chatfield D, Kirkpatrick PJ, Menon DK, Gupta AK, 2004. Hyperglycemia and brain tissue pH after traumatic brain injury. Neurosurgery 55, 877–881; discussion 882. [DOI] [PubMed] [Google Scholar]