

Abstract
G protein-coupled receptor 68 (GPR68) responds to extracellular protons, thus called the proton-sensing G protein-coupled receptor (GPCR), leading to activation of the phospholipase C-β (PLCβ)/calcium (Ca2+) pathway or the adenylyl cyclase (AC)/cyclic AMP (cAMP) pathway. We recently found that whole body deletion of Gpr68 (Gpr68−/− mice) reduced the number of B lymphocytes with age and during hematopoietic regeneration, such as in response to fluorouracil (5-FU) administration. This prompted us to characterize the hematopoietic stem cell (HSC) phenotype in Gpr68−/− mice. Despite high level of Gpr68 protein expression on HSC in bone marrow (BM), the pool size of HSC was unaltered in Gpr68−/− mice either under steady state or upon stress, including aging and 5-FU treatment. HSC from Gpr68−/− mice exhibited comparable cellular features, such as cell cycle quiescence and cell survival. HSC from Gpr68−/− mice also exhibited comparable competitiveness after serial transplantation. Surprisingly, cytosolic Ca2+ accumulation was increased in HSC from Gpr68−/− mice. In contrast, cAMP levels were reduced in hematopoietic stem and progenitor cells (HSPC) from Gpr68−/− mice. Intriguingly, we found high level of Gpr68 protein expression on non-hematopoietic cells in BM, especially endothelial cells that function as HSC niche. In addition, expression of other proton-sensing GPCR was upregulated in HSPC from Gpr68−/− mice. Our studies suggest that Gpr68−/− mice display insignificant phenotype on HSC biology, possibly due to the function of Gpr68 in non-hematopoietic cells and/or the compensatory effects from other proton-sensing GPCR.
Keywords: Proton-sensing G protein-coupled receptors, G protein-coupled receptor 68, hematopoietic stem cells
1. INTRODUCTION
Recently, a group of G protein-coupled receptors (GPCR), including G protein-coupled receptor 4 (GPR4, also known as GPR19), G protein-coupled receptor 65 (GPR65, also known as T-cell death-associated gene 8), G protein-coupled receptor 68 (GPR68, also known as ovarian cancer G protein-coupled receptor 1) and G protein-coupled receptor 132 (GPR132, also known as G2 accumulation protein), have been demonstrated to respond to extracellular acidosis, i.e. protons[1–3]. These proton-sensing GPCR sense neutral to mildly acidic pH (i.e. 7.6 to 6.0) through several histidine residues on their extracellular domains[1–4]. After protonation, these histidine residues may change the structural conformation of the receptors, leading to activation of downstream signaling pathways. GPR68 is reported to couple with Gq/11, leading to activation of the phospholipase C-β (PLCβ)/calcium (Ca2+) pathway[1]. GPR4, GPR65 and GPR68 are shown to couple with Gs, leading to activation of the adenylyl cyclase (AC)/cyclic AMP (cAMP) pathway[1, 5, 6].
Growing evidence implicate these proton-sensing GPCR in pleotropic physiological and/or pathological processes, including ischemic heart disease, inflammation, insulin secretion, bone formation and tumorigenesis[7–12]. In addition to response to extracellular ligands, overexpression of GPCR is thought to increase basal level activation of downstream signaling pathways as well[13]. Indeed, dysregulation of these proton-sensing GPCR is observed in various pathological conditions, such as inflammation and cancer[14–16]. Understanding the function and the signaling pathways related to these proton-sensing GPCR merits novel therapies. However, genetic models reveal that deletion of Gpr4, Gpr65 and Gpr132 leads to lethality or severe phenotypes[17–19], limiting the application of agonists and/or antagonists targeting these GPCR in clinic due to potential side effects. In contrast, whole body genetic deletion of Gpr68, the Gpr68 knockout (i.e. Gpr68−/−) mice, results in only marginal phenotype[20]. In addition, tumorigenesis of melanoma and prostate cancer is reduced in Gpr68−/− mice, which requires myeloid-derived cells[20, 21]. These studies suggest that Gpr68 may play a role in suppressing immunosurveillance. These lines of evidence implicate GPR68 as a potential therapeutic target for anti-cancer therapy with less side effects.
We have found that GPR68 expression is upregulated in response to lenalidomide in cell lines derived from patients with Myelodysplastic Syndromes (MDS)[22]. Upregulation of GPR68 activates a Ca2+/calpain proapoptotic pathway in MDS cells[22]. In contrast, the Gpr68−/− mice display no striking phenotype under steady state[20], indicating that Gpr68 may be dispensable for normal hematopoiesis under steady state. Recently, we have found reduced frequency and/or number of B lymphocytes in peripheral blood (PB), bone marrow (BM) and spleens from Gpr68−/− mice under stressed conditions, such as aging and hematopoietic regeneration after fluorouracil (5-FU) treatment[23]. However, the function of Gpr68 in hematopoietic stem cells (HSC) remains unclear.
In this study, we characterized the HSC phenotype in Gpr68−/− mice under steady state and upon stresses, including aging and 5-FU treatment. Our results suggest that whole body deletion of Gpr68 does not lead to significant changes in the homeostasis or function of HSC.
2. MATERIAL AND METHODS
2.1. Mice
Gpr68−/− mice in C57Bl/6 genetic background were generated in Dr. Yan Xu’s lab[20, 21]. CD45.1+ B6.SJLPtprca Pep3b/BoyJ (BoyJ) mice were purchased from the Jackson Laboratory. All mice were bred, housed and handled in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of University of South Carolina. BM cells were harvested from tibia, femur and pelvic bones as previously described[24, 25]. PB was collected from retro orbital plexus. 5-FU (50mg/kg, Sigma, F6627) was injected intraperitoneally to WT and Gpr68−/− mice.
2.2. Flow cytometry
To analyze the cell surface immunophenotype of lineage+ cells and hematopoietic stem and progenitor cell (HSPC), BM cells were stained with biotin conjugated lineage markers (CD11b, Gr1, Ter119, CD3, B220, Mouse Hematopoietic Lineage Biotin Panel, 88–7774-75, eBiosciences), followed by staining with streptavidin (eBioscience, 48–4317-82), Sca-1 (eBioscience, 25–5981-81), c-Kit (eBioscience, 47–1171-82), CD48 (BioLegend, 103420), CD150 (eBioscience, 17–1501-81), CD127 (eBioscience, 15–1271-81), CD34 (eBioscience, 50–0341-82), CD16/32 (eBioscience, 45–0161-80). To monitor CD45.2 chimerism after cBMT, PB samples were treated with 1 × red blood cell (RBC) lysis buffer at 37°C for 15 minutes, washed and incubated with antibodies of CD45.1 (eBioscience, 25–0453-82) and CD45.2 (eBioscience, 47–0454-82). Otherwise, BM cells were treated with 1 × RBC lysis buffer at room temperature for 10 minutes, washed and stained for HSPC in combination with CD45.1 and CD45.2. To determine quiescence of HSPC, BM cells were treated with 1 × RBC lysis buffer at room temperature for 10 minutes, washed and stained for HSPC in combination with Hoechst 33342 (561908, BD Pharmingen™) and Pyronin Y (Sigma, P9172). To determine HSPC cell death, BM cells were treated with 1 × RBC lysis buffer at room temperature for 10 minutes, washed and stained for HSPC in combination with Annexin V (eBioscience, 88–8005-74). To measure the expression of Gpr68 protein, PB and BM cells were treated with 1 × RBC lysis buffer at room temperature for 10 minutes, washed and stained for HSPC or non-hematopoietic cells, including antibodies of CD45 (eBiosceince, 12–9459-42), and Ter119 (eBioscience, 25–5921-82) and CD31 (eBioscience, 48–0311-82), in combination with Gpr68 antibody (Alomone Labs, AGR-042). The cells were washed again and stained with secondary antibody (Jackson ImmunoResearch, 111–096-144). Analysis was performed using NovoCyte Flow Cytometer with NovoExpress software.
2.3. Competitive bone marrow transplantation (cBMT)
For primary cBMT, 3 × 106 BM mononuclear cells (MNC) from WT or Gpr68−/− mice were mixed with 3 × 106 BM MNCs from BoyJ mice, and then transplanted into lethally-irradiated (11.75 Gy) recipient BoyJ mice. For secondary cBMT, BM MNC were harvested from the primary recipient mice, followed by transplantation into lethally-irradiated BoyJ mice.
2.4. Colony forming cell (CFC) assay
BM cells from WT or Gpr68−/− mice were treated with 1 × RBC lysis buffer at room temperature for 10 minutes. 1 × 105 BM MNC were plated in methylcellulose (StemCell Technologies, 03444) for CFC assay. The colonies were counted 7 days after plating. Colonies were then collected and plated for secondary and tertiary replating with 1 × 105 cells.
2.5. Measurement of cytosolic Ca2+ levels
5 × 106 BM cells from WT or Gpr68−/− mice were treated with 1 × RBC lysis buffer at room temperature for 10 minutes, washed and resuspended in 300μL of 1 × PBS and 100μL of Fluo-4 Direct™ Calcium Assay Buffer (Molecular Probes, F10471). The cells were then incubated at 37°C for 30 minutes before measurement of cytosolic Ca2+ levels.
2.6. Measurement of cytosolic cAMP levels
BM cells were harvested from WT and Gpr68−/− mice. Lineage− cells were enriched with the EasySep™ Mouse Hematopoietic Progenitor Cell Enrichment Kit (StemCell Technologies, 19756). Cytosolic cAMP levels were measured in lineage− cells according to the instruction of the manufacture (Biovision, K371–100).
2.7. RT-qPCR
Total RNA was prepared with the Quick-RNA MiniPrep (Zymo research, R1055). Reverse transcription was performed with the SuperScript VILO cDNA Synthesis Kit (Invitrogen, 11754050). Quantitative PCR was performed with Taqman Master Mix (Life Technologies, 4324018). Probes included mGapdh (Applied Biosystems, Cat 4331182, Assay ID Mm99999915_g1), mGpr4 (Applied Biosystems, Cat 4448892, Assay ID Mm00558777_s1), mGpr65 (Applied Biosystems, Cat 4331182, Assay ID Mm02619732_s1), mGpr132 (Applied Biosystems, Cat 4331182, Assay ID Mm02620285_s1).
2.8. Statistical analysis
Results are shown as mean ± s.e.m. Student’s t-test was used for all the results with GraphPad Prism (v7, GraphPad).
3. RESULTS AND DISCUSSION
3.1. Gpr68 protein is expressed at high level on HSC.
Previous studies have shown that Gpr68−/− mice do not display a striking phenotype on peripheral hematopoietic output at young age (2-month old) and under steady state[20]. We have recently observed reduced frequency and/or number of B lymphocytes in PB, BM and spleens from Gpr68−/− mice upon aging (12-month old) and in response to 5-FU injection[23]. In addition, we have observed increased expression of Gpr68 protein on B cells and myeloid cells in BM from old (20-month old) than young (5-month old) mice as well as in hematopoietic stem and progenitor cells (HSPC) in response to 5-FU[23]. These preliminary studies prompt us to examine a potential function of Gpr68 in HSC biology. We first examined Gpr68 protein expression on cell surface of HSPC in BM from C57Bl6 mice with flow cytometry (5-month old, Figure 1A and Supplemental Figure 1). The mean fluorescence intensity (MFI) of Gpr68 protein was much higher in long-term HSC (LT-HSC, lineage−, Sca-1+, c-Kit+, CD34− and CD135−, MFI 4.8 × 104 ± 0.6 × 104) than other HSPC subpopulations (Figure 1B), including short-term HSC (ST-HSC, lineage−, Sca-1+, c-Kit+, CD34+, and CD135−, MFI 0.9 × 104 ± 0.2 × 104, P = 0.001), multipotential progenitor cells (MPP, lineage−, Sca-1+, c-Kit+, CD34+ and CD135+, MFI 1.5 × 104 ± 0.3 × 104, P = 0.0037), common myeloid progenitor cells (CMP, lineage−, Sca-1−, c-Kit+, CD34+ and CD16/32−, MFI 0.4 × 104 ± 0.07 × 104, P = 0.0004), megakaryocyte erythroid progenitor cells (MEP, lineage−, Sca-1−, c-Kit+, CD34− and CD16/32−, MFI 0.6 × 104 ± 0.04 × 104, P = 0.0005), granulocyte monocyte progenitor cells (GMP, lineage−, Sca-1−, c-Kit+, CD34+ and CD16/32+, MFI 0.3 × 104 ± 0.07 × 104, P = 0.0004), lymphoid-primed multipotent progenitor cells (LMPP, lineage−, Sca-1+, c-Kit+ and CD135+, MFI 2.0 × 104 ± 0.5 × 104, P = 0.014) and common lymphoid progenitor cells (CLP, lineage−, Sca-1low, c-Kitlow and CD127+, MFI 1.3 × 104 ± 0.2 × 104, P = 0.0015). In addition, Gpr68 protein was expressed at comparable levels on LT-HSC and lineage+ cells in PB (MFI 5.0 × 104 ± 0.7 × 104 for lineage+ PB, P = 0.86, Figure 1B), indicating that Gpr68 protein was expressed at high level on HSC.
Figure 1. The pool size of HSC is not changed in Gpr68−/− mice.
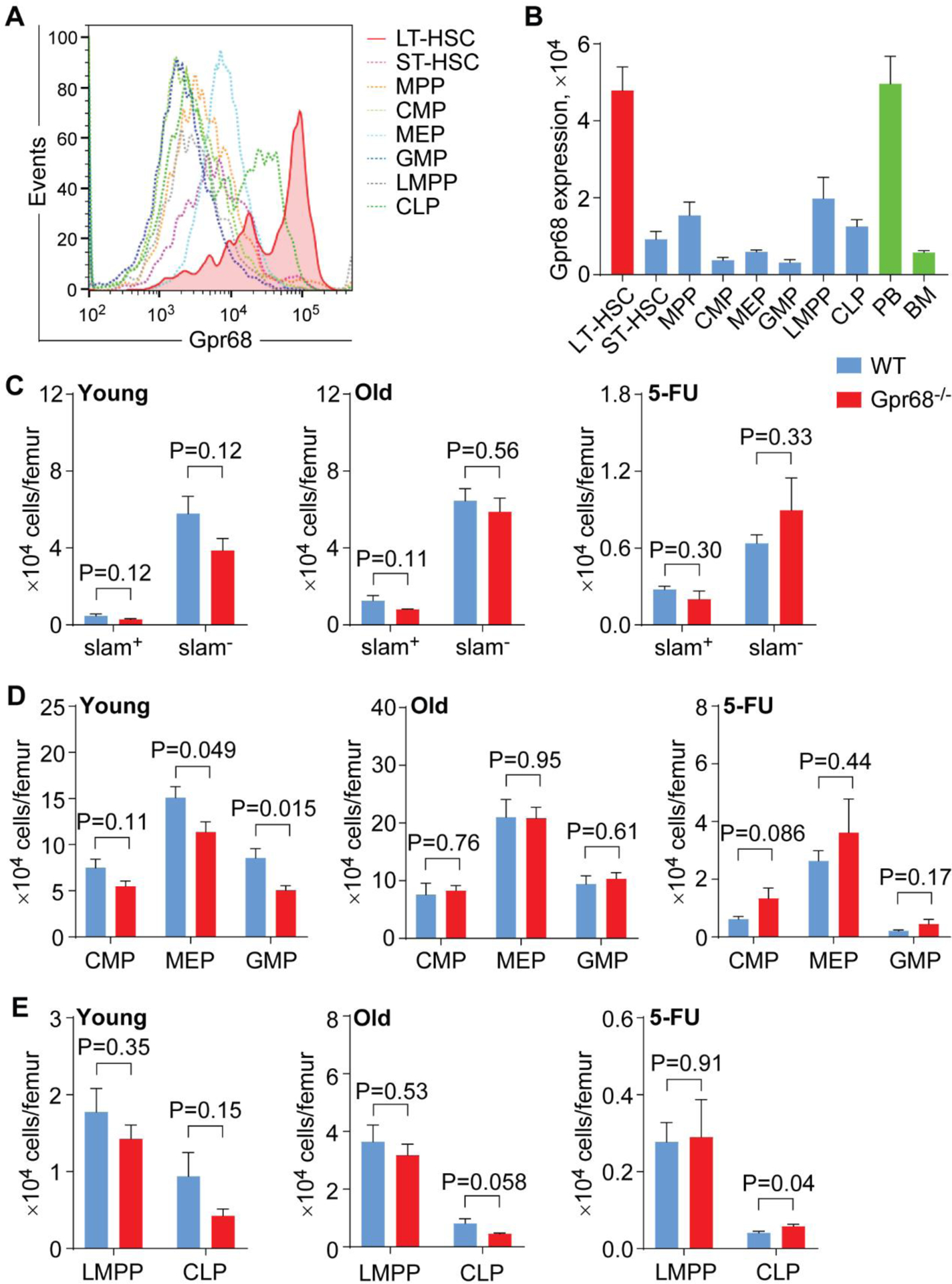
(A)Representative flow cytometric analysis of Gpr68 expression on HSPC in BM from C57Bl6 mice (5-month old). (B) Gpr68 protein expression on HSPC in BM and lineage+ cells in PB and BM from C57Bl6 mice (5-month old, n=4). (C-E) Numbers of HSPC in BM from WT and Gpr68−/− mice at young age (4~5 months old, n=5), upon aging (10~11 months old, n=5) and after 5-FU injection (8-month old, n=5).
3.2. The pool size of HSC is not changed in Gpr68−/− mice.
We next examined the numbers of HSPC in BM from WT and Gpr68−/− mice at young age (4~5 months old), upon aging (10~11 months old) and in response to 5-FU injection (8 months old). We found similar numbers of slam+ cells (lineage−, Sca-1+, c-Kit+, CD48− and CD150+) that were enriched for LT-HSC from WT and Gpr68−/− mice at young age (0.47 ± 0.10 × 104 cells/femur for WT group and 0.29 ± 0.04 × 104 cells/femur for Gpr68−/− group, P = 0.12, Figure 1C and Supplemental Figure 2), upon aging (1.3 ± 0.25 × 104 cells/femur for WT group and 0.81 ± 0.02 × 104 cells/femur for Gpr68−/− group, P = 0.11, Figure 1C) and post 5-FU injection (0.28 ± 0.02 × 104 cells/femur for WT group and 0.20 ± 0.06 × 104 cells/femur for Gpr68−/− group, P = 0.30, Figure 1C). We found fewer MEP and GMP cells from young Gpr68−/− than WT mice (15.1 ± 1.2 × 104 MEP cells/femur for WT group and 11.4 ± 1.1 × 104 MEP cells/femur for Gpr68−/− group, P = 0.049; 8.5 ± 1.0 × 104 GMP cells/femur for WT group and 5.1 ± 0.51 × 104 GMP cells/femur for Gpr68−/− group, P = 0.015, Figure 1D and Supplemental Figure 2). However, this did not correlate with alterations in hematopoietic output in PB[20]. Intriguingly, the numbers of MEP and GMP cells were comparable in WT and Gpr68−/− mice with age (21.0 ± 3.1 × 104 MEP cells/femur for WT group and 20.8 ± 1.9 × 104 MEP cells/femur for Gpr68−/− group, P = 0.95; 9.4 ± 1.4 × 104 GMP cells/femur for WT group and 10.4 ± 1.0 × 104 GMP cells/femur for Gpr68−/− group, P = 0.61, Figure 1D) and post 5-FU injection (2.6 ± 0.35 × 104 MEP cells/femur for WT group and 3.6 ± 1.2 × 104 MEP cells/femur for Gpr68−/− group, P = 0.45; 0.21 ± 0.03 × 104 GMP cells/femur for WT group and 0.45 ± 0.16 × 104 GMP cells/femur for Gpr68−/− group, P = 0.18, Figure 1D), indicating enhanced myelopoiesis in Gpr68−/− mice upon stress. This might contribute to increased myeloid output in PB from Gpr68−/− mice with age and after 5-FU treatment[23]. We didn’t observe a dramatic change in other HSPC subpopulations, including slam− cells (lineage−, Sca-1+, c-Kit+, CD48− and CD150+) that were enriched for MPP, myeloid or lymphoid progenitor cells, from WT or Gpr68−/− mice at young or old age or after 5-FU stress (Figure 1C–E and Supplemental Figure 2). These results suggest that despite high level of Gpr68 protein expression on HSC, whole body deletion of Gpr68 does not change the pool size of HSC either under steady state or upon stress.
3.3. Cellular features are not altered in HSC from Gpr68−/− mice.
HSC are in a quiescent cell cycle status, which is critical for HSC homeostasis[26]. To determine whether Gpr68 regulates HSC quiescence, we examined cell cycle status by staining slam+ cells with Pyronin Y and Hoechst 33342 (Figure 2A). As expected, slam+ cells from WT mice were primarily in G0 phase (quiescent cells), and rarely in G1 or S/G2/M phases (cycling cells, Figure 2A,B). The proportion of quiescent cells was similar in slam+ cells from WT and Gpr68−/− mice at young age (4~5 months old, 47.6% ± 2.4% for WT group and 46.7% ± 4.8% for Gpr68−/− group, P = 0.86, Figure 2A,B), upon aging (10~11 months old, 61.7% ± 4.7% for WT group and 59.0% ± 2.8% for Gpr68−/− group, P = 0.64, Figure 2B) and post 5-FU injection (8months old, 88.3% ± 3.3% for WT group and 81.8% ± 6.0% for Gpr68−/− group, P = 0.37, Figure 2B). To determine whether Gpr68 regulates HSPC survival, apoptosis was analyzed based on Annexin V staining. The frequencies of Annexin V+ cells were similar in slam+ cells (17.9% ± 0.6% for WT group and 19.5% ± 7.6% for Gpr68−/− group, P = 0.84, Figure 2C,D) as well as other HSPC subpopulations and BM lineage+ cells from WT and Gpr68−/− mice (4~5 months old, Figure 2C,D and Supplemental Figure 3A). Intriguingly, the frequencies of Annexin V+ cells were slightly increased in slam+ cells and BM lineage+ cells but not other HSPC subpopulations from Gpr68−/− than WT mice after 5-FU injection (17.8% ± 0.7% for WT slam+ cells and 24.2% ± 3.0% for Gpr68−/− slam+ cells, P = 0.069; 38.7% ± 1.4% for WT BM cells and 48.4% ± 1.4% for Gpr68−/− BM cells, P = 0.0011, 8-month old, Figure 2E and Supplemental Figure 3B). These results indicate slightly increased apoptosis due to loss of Gpr68 upon stress, a possible mechanism that could explain slightly reduced number of white blood cells (WBC) in PB from Gpr68−/− mice after 5-FU injection[23]. To determine whether Gpr68 is required for progenitor cell function, BM mononuclear cells (MNC) from WT and Gpr68−/− mice were plated in methylcellulose for colony formation. At younger age (4~5 months old), Gpr68-deficient BM cells formed less colonies than WT BM cells after primary plating (339 ± 19 colonies for WT group and 263 ± 10 colonies for Gpr68−/− group, P = 0.023, Figure 2F), which was consistent with reduced numbers of progenitor cells from young Gpr68−/− mice (Figure 1 and Supplemental Figure 2). However, similar numbers of colonies were formed after secondary (270 ± 20 colonies for WT group and 254 ± 18 colonies for Gpr68−/− group, P = 0.60) and tertiary (12 ± 2 colonies for WT group and 7 ± 3 colonies for Gpr68−/− group, P = 0.21) replating by WT and Gpr68−/− BM cells (Figure 2F). These suggest that loss of Gpr68 does not change self-renewal of HSC. At older age (10~11 months old), BM cells from WT and Gpr68−/− mice formed similar numbers of colonies after primary (366 ± 27 colonies for WT group and 363 ± 19 colonies for Gpr68−/− group, P = 0.94) and secondary (222 ± 16 colonies for WT group and 253 ± 8 colonies for Gpr68−/− group, P = 0.15) replating (Supplemental Figure 3C), which was consistent with comparable numbers of progenitor cells from old WT and Gpr68−/− mice (Figure 1 and Supplemental Figure 2). These results suggest that whole body deletion of Gpr68 does not change cell cycle quiescence, survival or self-renewal of HSC.
Figure 2. Cellular features are not altered in HSC from Gpr68−/− mice.

(A)Representative flow cytometric analysis of cell cycle status in slam+ cells from WT and Gpr68−/− mice (4~5 months old). (B) Proportion of quiescent (G0) and cycling (G1, S, G2, M) cells in slam+ cells from WT and Gpr68−/− mice at young age (4~5 months old, n=5, left panel), upon aging (10~11 months old, n=5, middle panel) and post 5-FU injection (8 months old, n=5, right panel). (C) Representative flow cytometric analysis of Annexin V in slam+ cells from WT and Gpr68−/− mice (4~5 months old). (D-E) Frequency of Annexin V+ cells in slam+ cells from WT and Gpr68−/− mice at young age (4~5 months old, n=3, D) and post 5-FU injection (8 months old, n=5, E). (F) Numbers of colonies formed by BM MNC from WT and Gpr68−/− mice after primary (1°), secondary (2°) and tertiary (3°) replating (4~5 months old, n=3).
3.4. HSC from WT and Gpr68−/− mice exhibit comparable competitiveness.
To examine the effect of Gpr68 on HSC function, we performed cBMT. Briefly, BM cells from WT or Gpr68−/− mice (6-week old, CD45.2+) were mixed with the competitor BM cells from BoyJ mice (CD45.1+) at 1:1 ratio, followed by transplantation into lethally irradiated syngeneic BoyJ mice. Donor-derived hematopoietic reconstitution was determined by measuring cell surface immunophenotype (i.e. CD45.2) on hematopoietic cells in PB and BM from recipient mice (Figure 3A). 4-month after primary cBMT, the proportion of CD45.2+ cells was comparable in PB from recipients of WT and Gpr68−/− groups (50.1% ± 1.9% for WT group and 44.4% ± 2.7% for Gpr68−/− group, P = 0.13, Figure 3B,C). Consistently, similar proportion of CD45.2+ cells was observed in BM HSPC from recipients of WT and Gpr68−/− groups (Figure 3D–F). The BM cells were then harvested from primary recipients for secondary cBMT (Figure 3A). 4-month after secondary cBMT, the proportion of CD45.2+ cells was still comparable in PB from recipients of WT and Gpr68−/− groups (58.0% ± 3.9% for WT group and 65.4% ± 3.4% for Gpr68−/− group, P = 0.19, Figure 3C). In addition, we performed cBMT with BM cells from old WT and Gpr68−/− mice (10~11 months old, Figure 3A). Comparable proportion of CD45.2+ cells was also found in PB from recipients of WT and Gpr68−/− groups (55.1% ± 1.3% for WT group and 53.0% ± 6.4% for Gpr68−/− group, P = 0.76, Figure 3G). These results reveal that HSC from WT and Gpr68−/− mice exhibit similar competitiveness, suggesting that whole body deletion of Gpr68 does not change HSC function.
Figure 3. HSC from WT and Gpr68−/− mice exhibit comparable competitiveness.
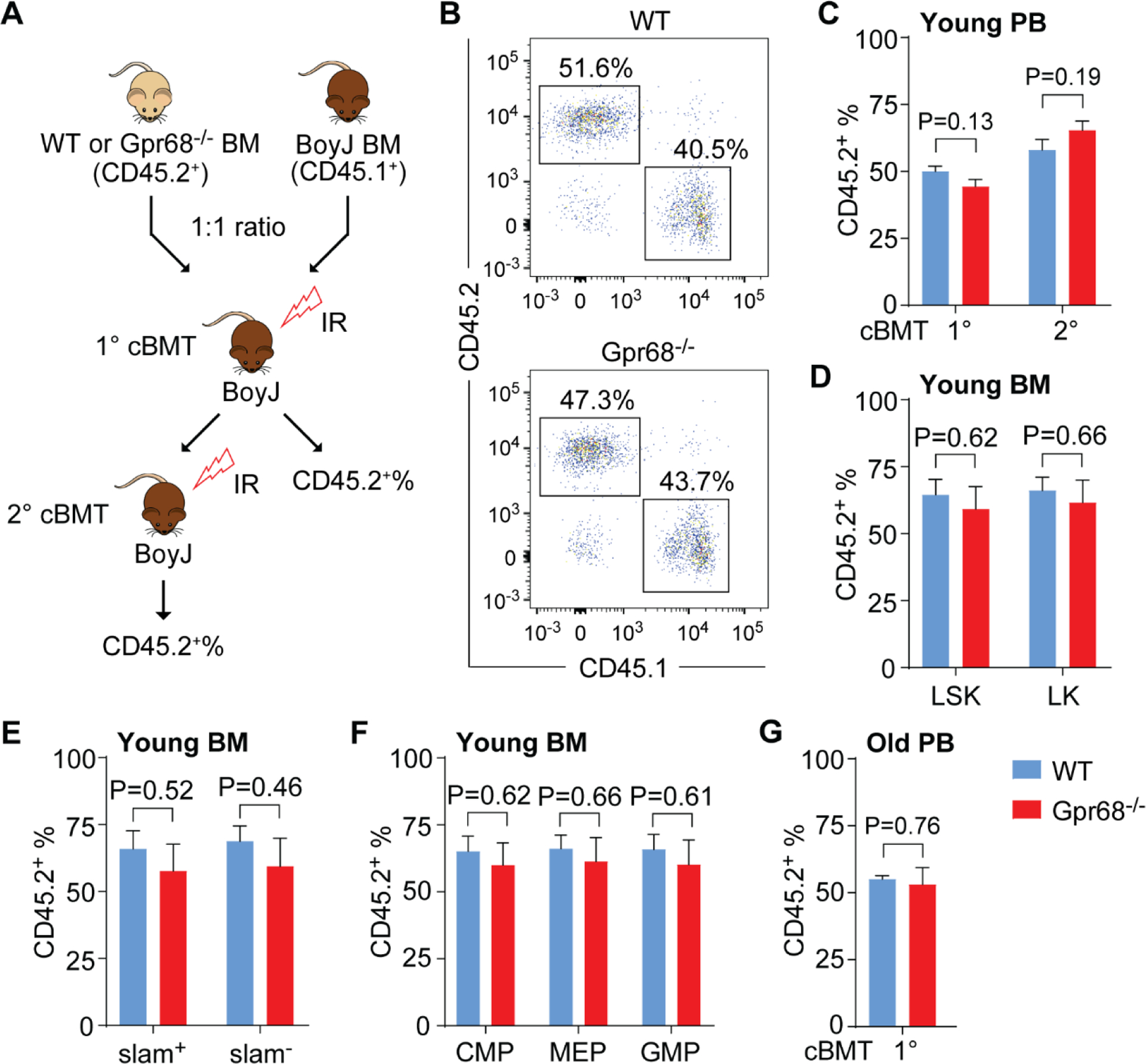
(A)Schematics of experimental approach for primary (1°) and secondary (2°) cBMT. (B) Representative flow cytometric analysis of donor-derived (CD45.2+) and competitor-derived (CD45.1+) hematopoietic cells in PB from recipients receiving BM cells from WT or Gpr68−/− mice (6 weeks old). (C) Frequency of CD45.2+ cells in PB from primary (1°) and secondary (2°) recipients receiving BM cells from WT or Gpr68−/− mice (6 weeks old, n=4~5). (D-F) Frequency of CD45.2+ cells in BM HSPC subpopulations from primary recipients receiving BM cells from WT or Gpr68−/− mice (6 weeks old, n=4). (G) Frequency of CD45.2+ cells in PB from primary (1°) recipients receiving BM cells from WT or Gpr68−/− mice at older age (10~11 months old, n=6).
3.5. Signaling pathways in HSPC from Gpr68−/− mice.
In response to extracellular ligand, such as protons, GPR68 associates with Gq/11, leading to activation of the PLCβ/Ca2+ pathway[1]. We next examined whether loss of Gpr68 resulted in alterations of downstream signaling pathways by measuring cytosolic Ca2+ accumulation. Unexpectedly, cytosolic Ca2+ levels were higher in slam+ cells (MFI 3.3 × 105 ± 0.1 × 105 for WT group and 4.6 × 105 ± 0.3 × 105 for Gpr68−/− group, P = 0.0034) as well as most of the HSPC subpopulations and lineage+ BM cells from young Gpr68−/− than WT mice (4~5 months old, Figure 4A,B and Supplemental Figure 4A), possibly due to compensation from the effects of Gpr68 on non-hematopoietic cells or other proton-sensing GPCR family members. Intriguingly, cytosolic Ca2+ levels were comparable in slam+ cells (MFI 3.8 × 105 ± 0.7 × 105 for WT group and 4.1 × 105 ± 0.6 × 105 for Gpr68−/− group, P = 0.70) as well as other HSPC and lineage+ BM cells from old WT and Gpr68−/− mice (10~11 months old, Figure 4B and Supplemental Figure 4B). In addition to the PLCβ/Ca2+ pathway, Gpr68 was also reported to activate the AC/cAMP pathway[6]. We found lower levels of cAMP in lineage− BM cells from Gpr68−/− than WT mice (2.1 ± 0.1 for WT group and 1.3 ± 0.02 for Gpr68−/− group, P = 0.0095, 4~5 months old, Figure 4C), indicating that Gpr68 may activate the AC/cAMP pathway in HSPC.
Figure 4. Signaling pathways in HSPC from Gpr68−/− mice.
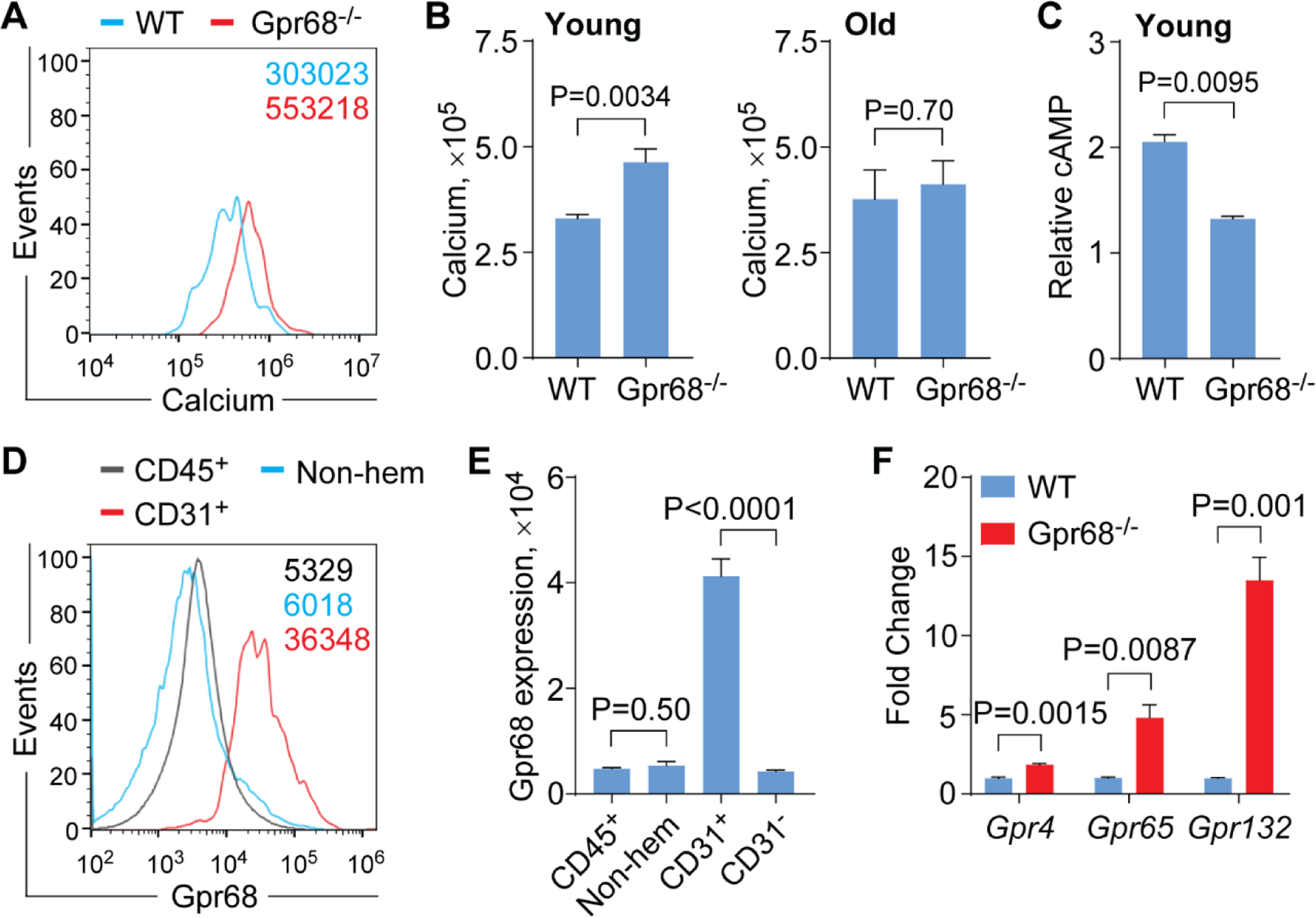
(A)Representative flow cytometric analysis of cytosolic Ca2+ levels in slam+ cells from WT and Gpr68−/− mice (4~5 months old). (B) Cytosolic Ca2+ levels in slam+ cells from WT and Gpr68−/− mice at young age (4~5 months old, n=5, left panel) and upon aging (10~11 months old, n=5, right panel). (C) Cytosolic cAMP levels in lineage- cells from WT and Gpr68−/− mice (4~5 months old, n=2). (D) Representative flow cytometric analysis of Gpr68 expression on hematopoietic cells (CD45+), non-hematopoietic cells (Non-hem) and CD31+ cells in BM from C57Bl6 mice (5-month old). (E) Gpr68 protein expression on hematopoietic cells (CD45+), non-hematopoietic cells (Non-hem), CD31+ and CD31- cells in BM from C57Bl6 mice (5-month old, n=4). (F) Relative mRNA levels of Gpr4, Gpr65 and Gpr132 in LSK cells from WT and Gpr68−/− mice (7-month old, n=3).
In addition to the function of Gpr68 in immune cells[27], Gpr68 is also shown to be expressed on structural cells, such as airway smooth muscle cells and epithelial cells, and regulate immune response[28]. To gain insights into possible mechanism of insignificant HSC phenotype and elevated cytosolic Ca2+ accumulation, we measured the expression of Gpr68 on non-hematopoietic cells in BM from C57Bl6 mice (5-month old). Gpr68 was expressed at similar levels on non-hematopoietic cells that were negative for CD45− and Ter119− compared to hematopoietic cells that was positive for CD45+ (MFI 0.5 × 104 ± 0.02 × 104 for non-hematopoietic cells and 0.5 × 104 ± 0.08 × 104 for hematopoietic cells, P = 0.50, Figure 4D,E and Supplemental Figure 4C). Among non-hematopoietic cells, Gpr68 was expressed at much higher levels in CD31+, a marker for vascular endothelial cells[29], than CD31− cells (4.1 × 104 ± 0.3 × 104 for CD31+ cells and 0.4 × 104 ± 0.02 × 104 for CD31− cells, P < 0.0001, Figure 4D,E and Supplemental Figure 4C). Elevated cytosolic Ca2+ accumulation could as well be due to compensation from other proton-sensing GPCR family members. Intriguingly, the transcript levels of Gpr4, Gpr65 and Gpr132 were significantly upregulated in LSK cells from Gpr68−/− than WT mice (fold change 1.8 ± 0.07 for Gpr4, P = 0.0015; 4.8 ± 0.8 for Gpr65, P = 0.0087; 13.5 ± 1.4 for Gpr132, P = 0.001; Figure 4F).
In the present study, we found that Gpr68 protein was expressed at high level on HSC. However, we failed to observe a dramatic change in the number, cellular features or function of HSC from Gpr68−/− mice under steady state or upon stress. This could be due to potential function of Gpr68 in non-hematopoietic cells. Indeed, we observed high level of Gpr68 expression on CD31+ cells from BM. CD31 is a marker for vascular endothelial cells, which act as a critical component of the HSC niche, maintaining the function of HSC in BM[29, 30]. These results suggest a potential function of Gpr68 in HSC niche, which needs further study, such as conditional deletion of Gpr68 in hematopoietic cells or endothelial cells. In addition, we explored potential compensation from other proton-sensing GPCR family members. Gpr65 has been shown to suppress the production of hematopoietic cells during embryogenesis in mouse and zebrafish via downregulating the expression of transcription factor Gata2[31]. In addition to protons, some proton-sensing GPCR also respond to other ligands. For example, Gpr132 responds to free fatty acids, leading to enhancement of embryonic and adult hematopoiesis in mouse and zebrafish[32]. BM cells from Gpr132−/− mice showed functional defects after competitive transplantation, indicating an indispensable role of Gpr132 in maintaining HSC function[32]. Intriguingly, we found increased expression of both Gpr65 and Gpr132 in LSK cells from Gpr68−/− mice. Given that Gpr65 suppresses while Gpr132 promotes hematopoiesis, we hypothesize that compensated expression of Gpr132 may play a role in maintaining the homeostasis and function of Gpr68-deficient HSC, which needs further study.
It is believed that overexpression of GPCR will increase basal level activation of downstream signaling pathways[13]. This prompts us to hypothesize that compensated expression of Gpr65 and Gpr132 may also activate the corresponding signaling pathways in Gpr68-deficient HSC. However, we observed reduced levels of cAMP in lineage− BM cells, indicating reduced AC/cAMP pathway. This is in line with loss of Gpr68 function instead of overexpression/activation of Gpr65. Gpr132 is shown to activate the Ca2+ pathway in macrophages that reside in tumor microenvironment in response to lactate[33]. Tumor cells produce large amount of lactate due to metabolic reprogramming from oxidative phosphorylation to glycolysis, leading to elevation of lactate levels in the extracellular space[34, 35]. Likewise, HSC predominantly depend on glycolysis to fuel energy[36, 37], which also leads to elevated extracellular lactate levels. We hypothesize that Gpr132 on Gpr68-deficient HSC activates the Ca2+ pathway either due to overexpression or in response to extracellular lactate, which maintains the normal function of HSC.
In summary, our studies suggest that whole body deletion of Gpr68 leads to insignificant phenotype on HSC biology, possibly due to the function of Gpr68 in non-hematopoietic cells or compensation from other proton-sensing GPCR, such as Gpr132. Our studies also implicate that inhibiting the function of Gpr68 even for longer time may not cause significant defects on HSC homeostasis and function. Therefore, Gpr68 could be a promising therapeutic target.
Supplementary Material
Highlights.
Despite high level of Gpr68 protein expression on HSC, the pool size of HSC is not changed in Gpr68−/− mice.
HSC from Gpr68−/− mice exhibit comparable cellular features and function.
HSC from Gpr68−/− mice exhibit increased cytosolic Ca2+ accumulation but reduced cAMP levels.
Gpr68 protein is expressed on non-hematopoietic cells, including components of HSC niche.
Compensated expression of other proton-sensing GPCR in HSC from Gpr68−/− mice.
ACKNOWLEDGEMENT
We thank the animal facility of University of South Carolina (USC), Center for Colon Cancer Research (supported by NIH 5 P30 GM103336) of USC, and the Instrumentation Resource Facility at USC School of Medicine for their help with mouse work. We acknowledge Dr. Xiang-ming Zha for providing the Gpr68−/− mice.
FUNDING
This work was supported by NIH (R01CA218076), NIH COBRE 1P20GM109091-01, St. Baldrick’s Foundation and Aplastic Anemia & MDS International Foundation, NSF (1736150) and USC SPARC Graduate Research Grant.
ABBREVIATION
- GPR68
G protein-coupled receptor 68
- PLCβ
Phospholipase C-β
- AC
Adenylyl cyclase
- MDS
Myelodysplastic syndromes
- HSC
Hematopoietic stem cells
- WT
Wild type
- PB
Peripheral blood
- BM
Bone marrow
- GPR4
G protein-coupled receptor 4
- GPR65
G protein-coupled receptor 65
- GPR132
G protein-coupled receptor 132
- KO
Knockout
- cBMT
Competitive bone marrow transplantation
- HSPC
Hematopoietic stem and progenitor cells
- WBC
White blood cells
- MNC
Mononuclear cells
- LSK
Lin−, Sca-1+, c-Kit+
- LK
Lin−, Sca-1−, c-Kit+
- LT-HSC
long-term HSC
- MPP
multipotential progenitor cells
- CMP
common myeloid progenitor cells
- MEP
megakaryocyte erythroid progenitor cells
- GMP
granulocyte monocyte progenitor cells
- LMPP
lymphoid primed multipotential progenitor cells
- CLP
common lymphoid progenitor cells
Footnotes
DECLARATIONS
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Ludwig MG, et al. , Proton-sensing G-protein-coupled receptors. Nature, 2003. 425(6953): p. 93–8. [DOI] [PubMed] [Google Scholar]
- 2.Wang JQ, et al. , TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J Biol Chem, 2004. 279(44): p. 45626–33. [DOI] [PubMed] [Google Scholar]
- 3.Seuwen K, Ludwig MG, and Wolf RM, Receptors for protons or lipid messengers or both? J Recept Signal Transduct Res, 2006. 26(5–6): p. 599–610. [DOI] [PubMed] [Google Scholar]
- 4.Liu JP, et al. , Each one of certain histidine residues in G-protein-coupled receptor GPR4 is critical for extracellular proton-induced stimulation of multiple G-protein-signaling pathways. Pharmacol Res, 2010. 61(6): p. 499–505. [DOI] [PubMed] [Google Scholar]
- 5.Radu CG, et al. , Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci U S A, 2005. 102(5): p. 1632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang XP, et al. , Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature, 2015. 527(7579): p. 477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell JL, et al. , Regulated expression of pH sensing G Protein-coupled receptor-68 identified through chemical biology defines a new drug target for ischemic heart disease. ACS Chem Biol, 2012. 7(6): p. 1077–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okajima F, Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal, 2013. 25(11): p. 2263–71. [DOI] [PubMed] [Google Scholar]
- 9.Mogi C, Nakakura T, and Okajima F, Role of extracellular proton-sensing OGR1 in regulation of insulin secretion and pancreatic beta-cell functions. Endocr J, 2014. 61(2): p. 101–10. [DOI] [PubMed] [Google Scholar]
- 10.Krieger NS, et al. , Increased bone density in mice lacking the proton receptor OGR1. Kidney Int, 2016. 89(3): p. 565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Justus CR, Dong L, and Yang LV, Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front Physiol, 2013. 4: p. 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damaghi M, Wojtkowiak JW, and Gillies RJ, pH sensing and regulation in cancer. Front Physiol, 2013. 4: p. 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Ligt RA, Kourounakis AP, and AP IJ, Inverse agonism at G protein-coupled receptors: (patho)physiological relevance and implications for drug discovery. Br J Pharmacol, 2000. 130(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellone RD, et al. , Inhibition of tumor cell migration and metastasis by the proton-sensing GPR4 receptor. Cancer Lett, 2011. 312(2): p. 197–208. [DOI] [PubMed] [Google Scholar]
- 15.Justus CR, et al. , Contextual tumor suppressor function of T cell death-associated gene 8 (TDAG8) in hematological malignancies. J Transl Med, 2017. 15(1): p. 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh LS, et al. , Ovarian cancer G protein-coupled receptor 1, a new metastasis suppressor gene in prostate cancer. J Natl Cancer Inst, 2007. 99(17): p. 1313–27. [DOI] [PubMed] [Google Scholar]
- 17.Yang LV, et al. , Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol Cell Biol, 2007. 27(4): p. 1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lassen KG, et al. , Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity, 2016. 44(6): p. 1392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le LQ, et al. , Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity, 2001. 14(5): p. 561–71. [DOI] [PubMed] [Google Scholar]
- 20.Li H, et al. , Abnormalities in osteoclastogenesis and decreased tumorigenesis in mice deficient for ovarian cancer G protein-coupled receptor 1. PLoS One, 2009. 4(5): p. e5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan L, et al. , Role of OGR1 in myeloid-derived cells in prostate cancer. Oncogene, 2014. 33(2): p. 157–64. [DOI] [PubMed] [Google Scholar]
- 22.Fang J, et al. , A calcium- and calpain-dependent pathway determines the response to lenalidomide in myelodysplastic syndromes. Nat Med, 2016. 22(7): p. 727–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He X, et al. , G protein-coupled receptor 68 increases the number of B lymphocytes. Am J Blood Res, 2020. 10(2): p. 15–21. [PMC free article] [PubMed] [Google Scholar]
- 24.Fang J, et al. , TRAF6 Mediates Basal Activation of NF-kappaB Necessary for Hematopoietic Stem Cell Homeostasis. Cell Rep, 2018. 22(5): p. 1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, et al. , Osteoblast Sorting and Intracellular Staining of CXCL12. Bio Protoc, 2018. 8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura-Ishizu A, Takizawa H, and Suda T, The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development, 2014. 141(24): p. 4656–66. [DOI] [PubMed] [Google Scholar]
- 27.Aoki H, et al. , Proton-sensing ovarian cancer G protein-coupled receptor 1 on dendritic cells is required for airway responses in a murine asthma model. PLoS One, 2013. 8(11): p. e79985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aoki H, Mogi C, and Okajima F, Ionotropic and metabotropic proton-sensing receptors involved in airway inflammation in allergic asthma. Mediators Inflamm, 2014. 2014: p. 712962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusumbe AP, et al. , Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature, 2016. 532(7599): p. 380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu C, et al. , Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun, 2018. 9(1): p. 2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X, et al. , GATA Factor-G-Protein-Coupled Receptor Circuit Suppresses Hematopoiesis. Stem Cell Reports, 2016. 6(3): p. 368–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lahvic JL, et al. , Specific oxylipins enhance vertebrate hematopoiesis via the receptor GPR132. Proc Natl Acad Sci U S A, 2018. 115(37): p. 9252–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen P, et al. , Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A, 2017. 114(3): p. 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warburg O, On the origin of cancer cells. Science, 1956. 123(3191): p. 309–14. [DOI] [PubMed] [Google Scholar]
- 35.Warburg O, On respiratory impairment in cancer cells. Science, 1956. 124(3215): p. 269–70. [PubMed] [Google Scholar]
- 36.Kohli L and Passegue E, Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Biol, 2014. 24(8): p. 479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takubo K, et al. , Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell, 2013. 12(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.