

Abstract
Background & Aims:
In autoimmune hepatitis (AIH) imbalance between Treg and Th17-cells has been linked to low levels of CD39, an ectoenzyme that hydrolyses ATP ultimately generating immunosuppressive adenosine. Upregulation of CD39 results from activation of aryl-hydrocarbon-receptor (AHR), which mediates toxin responses to modulate T-cell immunity. Upon binding to exogenous or endogenous ligands, AHR dimerizes with the aryl-hydrocarbon-receptor-nuclear-translocator (ARNT) or other ‘non-canonical’ binding factors like oestrogen-receptor-α (Erα) to modulate gene transcription. The AHR/ARNT complex is in turn regulated by hypoxia-inducible-factor-1α (HIF-1α) and the aryl-hydrocarbon receptor-repressor (AHRR). In this study we investigated whether altered AHR signalling underlies defective CD39 expression and function in AIH Treg and Th17-cells, therefore contributing to regulatory/effector cell imbalance.
Methods:
Treg and Th17-cells, obtained from the peripheral blood of 49 AIH patients and 21 healthy subjects (HS), were tested for response to AHR endogenous and exogenous ligands.
Results:
When compared to HS, AIH Treg and Th17-cells displayed impaired response to AHR activation, as reflected by impaired upregulation of CD39, delayed increase in ectoenzymatic activity and defective Treg suppressive function. These impairments resulted, at least in part, from heightened levels of AHRR and Erα in Treg and high HIF-1α in Th17-cells and were reverted upon molecular blockade. Importantly, in AIH Treg, the binding affinity of AHR was higher for Erα than ARNT.
Conclusions:
In AIH, high levels of AHRR and HIF-1α inhibit AHR signalling in Treg and Th17-cells. AHR non-canonical binding to Erα further amplifies lack of effective CD39 upregulation. Blockade of these inhibitory and/or non-canonical activation pathways represents a potential therapeutic approach to restore CD39 and immunohomeostasis in AIH.
Keywords: ectoenzyme, AHR, HIF-1α, AIH
Graphical Abstract
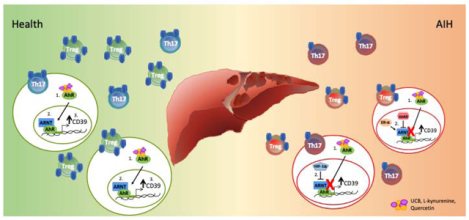
Lay summary
In patients with autoimmune hepatitis imbalance between Treg and Th17 effector lymphocytes is linked to dysfunction of aryl hydrocarbon receptor pathway, resulting from aberrant inhibition or non-canonical activation. These alterations lead to impaired Treg and Th17-cell ability to upregulate CD39, an ectoenzyme key to immunoregulation, and in defective Treg ability to suppress. Blockade of excessive aryl hydrocarbon receptor inhibition or non-canonical activation might represent a novel therapeutic strategy to control inflammation while restoring Treg/Th17 immune balance in autoimmune hepatitis.
Introduction
Autoimmune hepatitis type-1 (AIH-1) is an organ-specific autoimmune disorder characterized by hypergammaglobulinemia, positivity for anti-nuclear (ANA) and/or anti-smooth muscle antibodies (SMA) and evidence of interface hepatitis on histology(1, 2).
In AIH, dysfunction of regulatory T-cells (Treg) co-exists with exacerbated effector T-helper-type-17 (Th17)-cell immunity(3–7). Others and we have previously shown that peripheral blood and liver-derived Treg from AIH patients are functionally impaired and fail to induce infectious tolerance, when co-cultured with effector cells(3–5, 8, 9); whereas effector CD4 lymphocytes are resistant to Treg control, therefore also perpetuating liver pathogenicity(6, 10).
We have previously reported that both Treg and Th17-cells obtained from AIH patients express low levels and impaired ectoenzymatic activity of CD39(10, 11). This is an ectonucleotidase key to the maintenance of immune homeostasis because it hydrolyses pro-inflammatory ATP and ADP into AMP, which is subsequently catalysed into immunosuppressive adenosine by CD73, an ectoenzyme that works in tandem with CD39(12, 13). As a consequence of low CD39 levels and ectoenzymatic activity, Treg from AIH patients display decreased ability to control IL-17 production by CD4 effectors and are phenotypically and functionally unstable, when challenged with pro-inflammatory stimuli(11). Further, decreased CD39 levels and perturbed activity render Th17-cells refractory to immunoregulation and less prone to acquire immunosuppressive properties(10).
Upregulation of CD39 is mediated - at least in part - upon engagement of aryl-hydrocarbon-receptor (AHR)(14) that mediates toxin and xenobiotic responses to regulate Treg and effector T-cell immunity(15, 16). We have recently shown that AHR engagement with endogenous ligands like unconjugated bilirubin (UCB) leads to upregulation of CD39 by healthy control Th17-cells(17). After binding to exogenous and endogenous ligands, AHR translocates to the nucleus where it binds to the aryl-hydrocarbon-receptor-nuclear-translocator (ARNT) to regulate various genes including xenobiotic metabolizing enzymes like CYP1A1; the ATP binding cassette family of drug transporters ABCB1 and ABCC4; as well as cytokines like IL10, IL17 and IL22(18). Additional studies have indicated that AHR can modulate gene expression upon non-canonical interactions with the oestrogen-receptor-alpha (Erα) or the Kruppel-like-factor-6 (KLF6)(19–21). Whether CD39 expression and activity can be effectively upregulated and maintained upon AHR interaction with the non-canonical binding partners Erα and/or KLF6 remains unknown.
AHR-mediated immune responses are regulated by the aryl-hydrocarbon-receptor-repressor (AHRR) that binds to ARNT to interfere with the transcriptional activity of the AHR/ARNT complex(22); and by the hypoxia-inducible-factor-1-alpha (HIF-1α), which - like AHR - also binds to ARNT and is upregulated in chronic inflammatory conditions(23).
In this study we tested whether impaired CD39 expression and function in AIH-derived Treg and Th17-cells result from alterations in AHR levels and/or signalling pathway.
We report that, compared to health, AIH Treg and Th17-cells display dysfunctional AHR signalling. This dysfunction derives from excessive upregulation of AHRR and Erα in Treg and from heightened levels of HIF-1α in Th17-cells. Both these alterations result in disruption of Treg and Th17-cell purinergic activity and function in AIH.
Methods
Subjects
Forty-nine patients with AIH-1 were studied. A liver biopsy performed at the time of, or close to diagnosis, showed presence of histopathological features of interface hepatitis in all patients. At the time of study, twenty-two patients were ANA positive and twenty-six were SMA positive. Six of the forty-nine patients were negative for both ANA and SMA autoantibodies. In two patients, ANA and SMA levels were unknown at the time of study. Thirty-one patients were females. Thirty-seven patients were studied during remission (i.e. normal AST levels) while on immunosuppressive treatment; whereas twelve patients were studied during active disease. In five patients with active disease, blood was collected at the time of or close to diagnosis, before immunosuppression was started; while the remaining seven patients were studied during an episode of relapse. Twenty-seven patients were studied in multiple occasions, during active disease and at remission, following normalization of AST. Demographic and laboratory data of AIH patients are summarized in Supplementary Table 1. Patients were treated with prednisone (2.5 mg-20 mg/day) either alone or in combination with azathioprine (25–150 mg/day). Six patients were on prednisone and mycophenolate (1,000–2,000 mg/day) when studied.
Twenty-one healthy subjects (median age 40 years, range 24–55; 16 females) served as normal controls. The study received IRB approval at Beth Israel Deaconess Medical Center, Boston, MA. Written consent was obtained from all study participants prior to inclusion in the study.
Cell separation
Peripheral blood mononuclear cells (PBMCs) were obtained as previously reported(17). Mononuclear cell viability, determined by Trypan blue exclusion, exceeded 98%. Details on cell purification, polarization and culture are provided in the Supplemental Material. Information on flow cytometry, qPCR analysis, thin layer chromatography, chromatin immunoprecipitation, immunoprecipitation, immunoblotting and immunohistochemistry is also included in the Supplemental Material.
Erα, ARNT, KLF6, AHRR, CD39 and HIF-1α silencing
Treg and Th17-cell silencing of Erα, ARNT, KLF6, AHRR, CD39 (Treg) and HIF-1α (Th17-cells) was as previously reported(24). Further information is provided in the Supplemental Material.
Suppression assay
Treg ability to suppress CD4+CD25−-cell proliferation was assessed in co-culture experiments, in which untreated, UCB, L-Kynurenine or quercetin-treated Treg were added at a ratio of 1/8 to CD4+CD25− target cells(5). Additional details are given in the Supplemental Material.
Statistics
Results are expressed as mean + SEM. Normality of variable distribution was assessed by Kolmogorov-Smirnov goodness-of-fit test. Comparisons were performed using parametric (paired or unpaired Student’s t test) or non-parametric (Wilcoxon signed-rank or Mann-Whitney test) tests according to data distribution. One-way ANOVA or Kruskal-Wallis test, followed by Tukey’s or Dunn’s multiple comparison tests, were used when comparing more than two sets of data. P≤0.05 was considered significant; P≤0.1 indicated a trend to significance. Statistical analysis was performed using GraphPad Prism, version 7.0a (GraphPad Software, San Diego, CA).
Results
AIH Treg and Th17-cells display defective response to AHR ligation
Our previous studies showed defective CD39 expression and function in both Treg and Th17-cells derived from AIH patients(10, 11). To determine whether these impairments result from dysfunctional AHR/ARNT signalling, we tested the response of Treg and Th17-cells, differentiated from the peripheral blood of AIH patients and healthy controls to defined AHR endogenous (i.e. UCB, biliverdin, L-Kynurenine and 2-(1’H-Indole-3′-carbonyl)-thiazole-4-carboxylic acid, ITE) and exogenous (i.e. quercetin and benzo[a]pyrene) ligands. Phenotype validation after differentiation confirmed that Treg were CD4+CD25highCD127low and FOXP3+ while Th17-cells expressed RORC, IL23R and CCR6 and produced IL17 (Supplementary Fig. 1A–B). The expression of CYP1A1 - one of the main genes controlled by the AHR/ARNT complex - and CD39 was used as readout. Analysis of CYP1A1 and CD39 mRNA levels was carried out considering untreated Treg and Th17-cells as internal controls, to assess variations in the response to AHR ligands as compared to baseline levels.
We note that Treg and Th17-cells differentiated from CD4 lymphocytes of AIH patients were defective in the ability to upregulate CYP1A1 upon exposure to UCB, L-Kynurenine and quercetin (Fig. 1A–B); whereas no differences were noted in the levels of CYP1A1 upon stimulation in the presence of biliverdin, ITE and benzo[a]pyrene (Fig. 1A–B). Therefore, in all subsequent experiments, AHR activation was induced upon cell exposure to UCB, L-Kynurenine and quercetin.
Figure 1. In AIH Treg and Th17-cells display defective response to AHR activation.
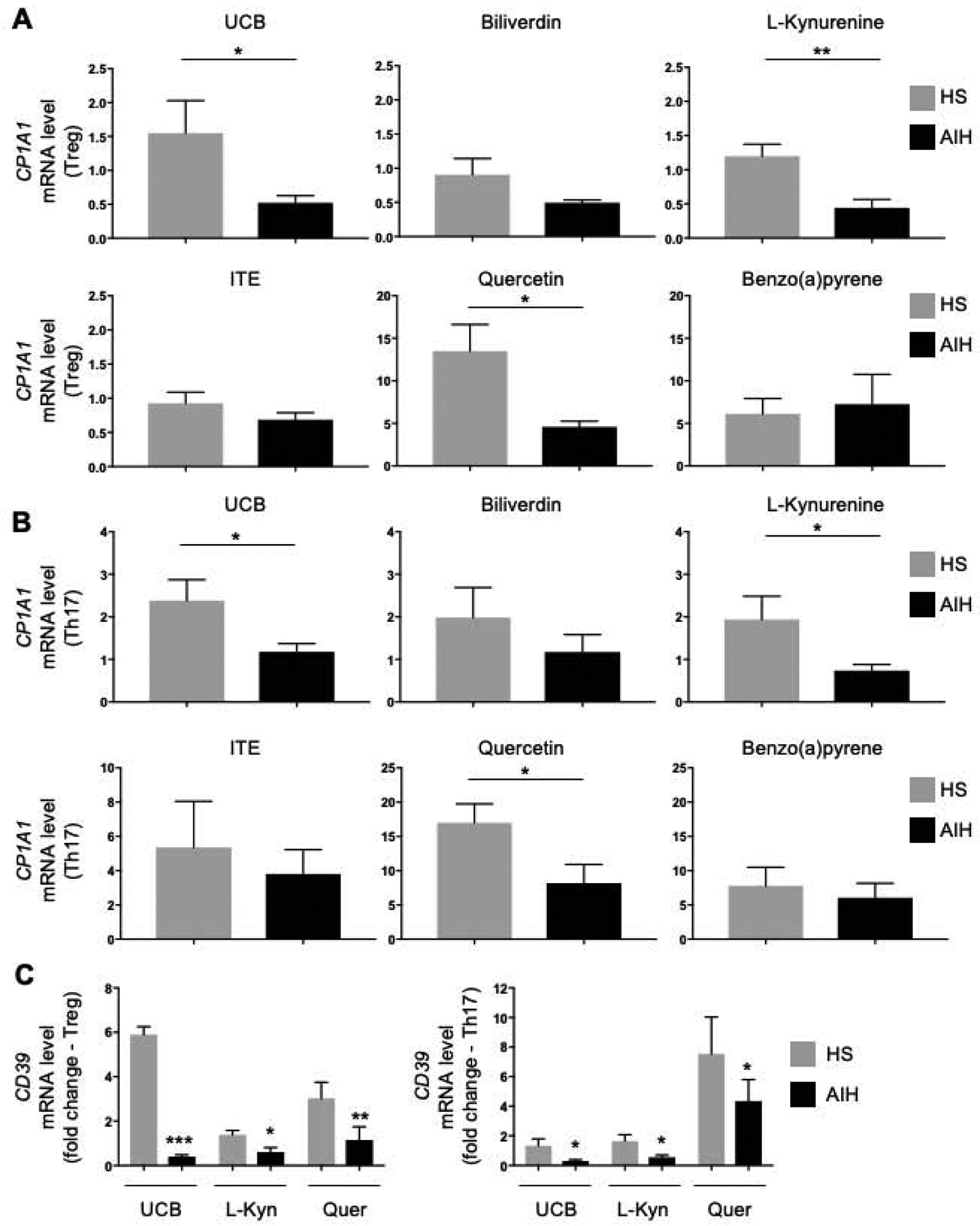
Mean+SEM CYP1A1 mRNA levels in (A) Treg and (B) Th17-cells exposed to UCB, biliverdin, L-Kynurenine, ITE, quercetin and benzo(a)pyrene (HS, n=6; AIH, n=7). Treg and Th17-cells from AIH patients failed to effectively upregulate CYP1A1 mRNA upon exposure to UCB, L-Kynurenine and quercetin. (C) Mean+SEM CD39 mRNA levels in Treg and Th17-cells obtained from HS (n=6) and AIH patients (n=6) and exposed to UCB, L-Kynurenine and quercetin. *P≤0.05; **P≤0.01; ***P≤0.001 (unpaired t test or Mann-Whitney test).
Akin to CYP1A1, Treg and Th17-cells from AIH patients failed to effectively upregulate CD39 in the presence of UCB, L-Kynurenine and quercetin (Fig. 1C). Comparable results were obtained when measuring CYP1A1 and CD39 mRNA levels in immunomagnetically purified Treg and Th17-cells following exposure to UCB, L-Kynurenine and quercetin (Supplementary Fig. 2A–C).
Analysis of ectoenzymatic activity revealed that L-Kynurenine boosted ADPase activity of healthy subject Treg and Th17-cells at earlier time points after exposure to 14C-labelled ADP, when compared to Treg and Th17-cells obtained from AIH patients (Fig. 2A–B and Supplementary Fig. 3A–B). Quercetin boosted the ADPase activity of healthy subject and AIH Treg and that of healthy subject Th17-cells forty minutes after exposure to 14C-labelled ADP. At the same time point, L-Kynurenine and quercetin increased Treg AMPase activity in healthy subjects and AIH patients respectively (Fig. 2C–D and Supplementary Fig. 3A–B). No changes in ADPase and AMPase activity were noted following Treg and Th17-cell exposure to UCB in both healthy subjects and AIH patients (Fig. 2A–D and Supplementary Fig. 3A–B).
Figure 2. In AIH Treg and Th17-cells display defective ADPase and AMPase activity upon exposure to AHR ligands.
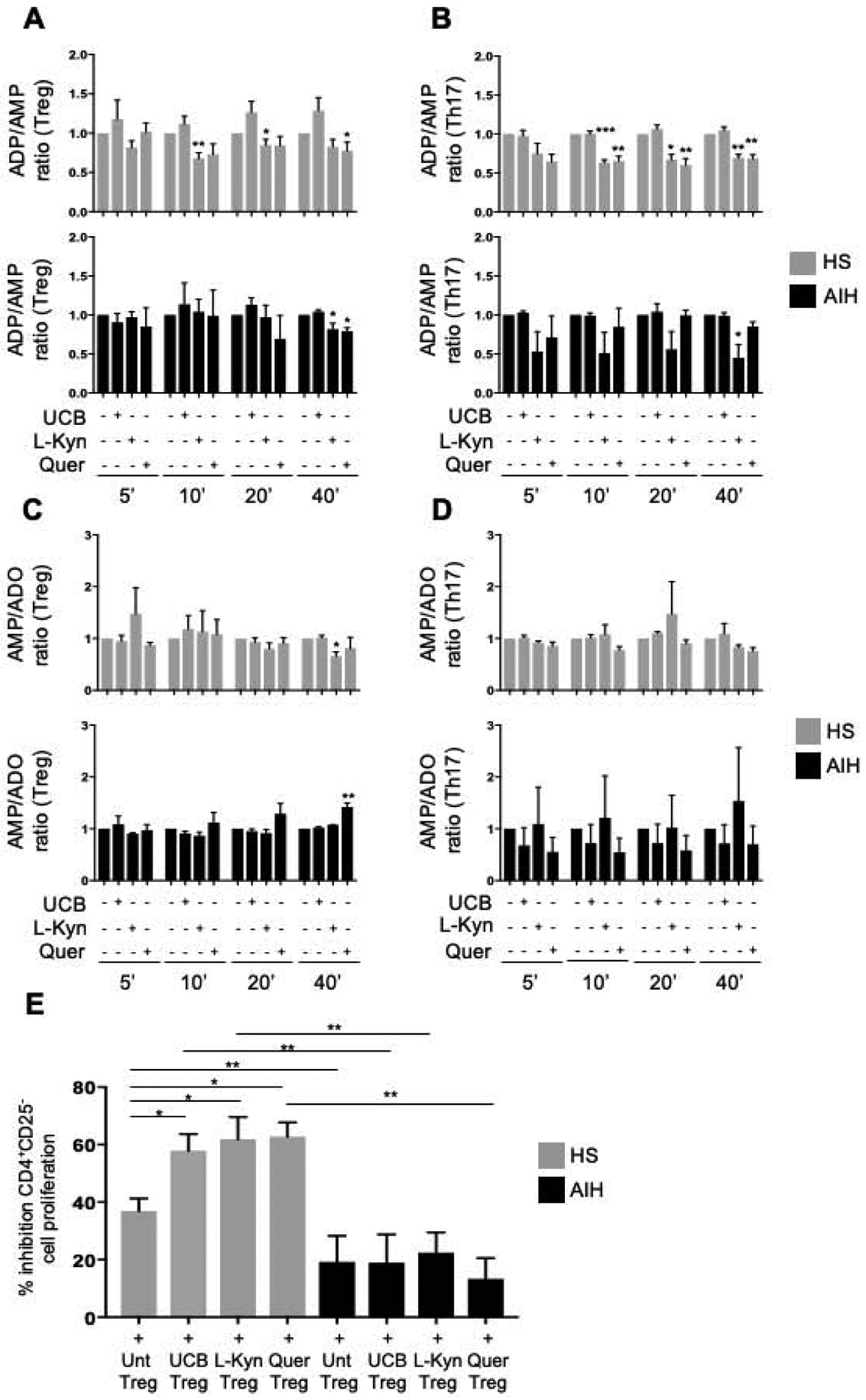
Mean+SEM (A-B) ADP/AMP ratio and (C-D) AMP/adenosine ratio in untreated, UCB, L-Kynurenine and quercetin-treated Treg and Th17-cells 5, 10, 20, 40 minutes after addition of 14C-labelled ADP (HS, n=4; AIH, n=4). (E) Mean+SEM % inhibition of CD4+CD25−-cell proliferation in the presence of untreated or treated Treg is shown (HS, n=8; AIH, n=10). *P≤0.05; **P≤0.01; ***P≤0.001 (one-way ANOVA).
When testing suppressive function, exposure to UCB, L-Kynurenine and quercetin boosted Treg ability to suppress CD4+CD25−-cell proliferation in healthy subjects but not in AIH Treg (Fig. 2E). Treg suppressive function in AIH remained lower than HS in untreated, UCB, L-Kynurenine and quercetin-treated Treg (Fig. 2E). Importantly, silencing of CD39 significantly reduced Treg ability to suppress in the presence of UCB, L-Kynurenine and quercetin in healthy controls but not in AIH patients (Supplementary Fig. 4A–B).
Overall, these data indicate that Treg and Th17-cells derived from AIH patients are defective in their response to AHR ligation, as reflected by impaired CYP1A1 and CD39 upregulation, delayed increase in ectoenzymatic activity and, in the case of Treg, inability to ameliorate suppressive function upon exposure to UCB, L-Kynurenine and quercetin.
Aberrant upregulation of AHR inhibitory and non-canonical binding factors in Treg and Th17-cells from AIH patients
AHR classically binds to ARNT to regulate various downstream genes, including CD39(14). Here we defined whether impaired response to AHR activation in AIH, results from defective AHR/ARNT levels or from aberrant expression of AHR non-canonical binding partners or inhibitory factors. To this end, Treg and Th17-cells differentiated or purified from the peripheral blood of healthy controls and AIH patients were tested for expression of Erα and KLF6 - two AHR alternative binding partners; for AHRR, which binds to ARNT interfering with the transcriptional activity of the AHR/ARNT complex(22); and for HIF-1α, which inhibits AHR and downstream signalling(23, 24). mRNA levels of AHR and ARNT were similar in Treg and Th17-cells differentiated from healthy controls and AIH patients (Fig. 3A–B). When analysing Erα and KLF6, we noted marked increase in these two factors in Treg from AIH patients, when compared to healthy controls (Fig. 3C–D). Comparable results were obtained when testing AHR, ARNT, Erα and KLF6 mRNA levels in purified Treg and Th17-cells (Supplementary Fig. 5A–D). Notably, polarized and freshly isolated Treg and Th17-cells from AIH patients expressed higher levels of AHRR (Fig. 3E and Supplementary Fig. 5E) and HIF-1α (Fig. 3F and Supplementary Fig. 5F) respectively; these data suggesting that dysfunctional AHR signalling in AIH might result from aberrant upregulation of AHR non-canonical binding partners, as well as from increase in inhibitory factors, namely AHRR in Treg and HIF-1α in Th17-cells.
Figure 3. Altered expression of AHR non-canonical binding partners and inhibitors in AIH Treg and Th17-cells.
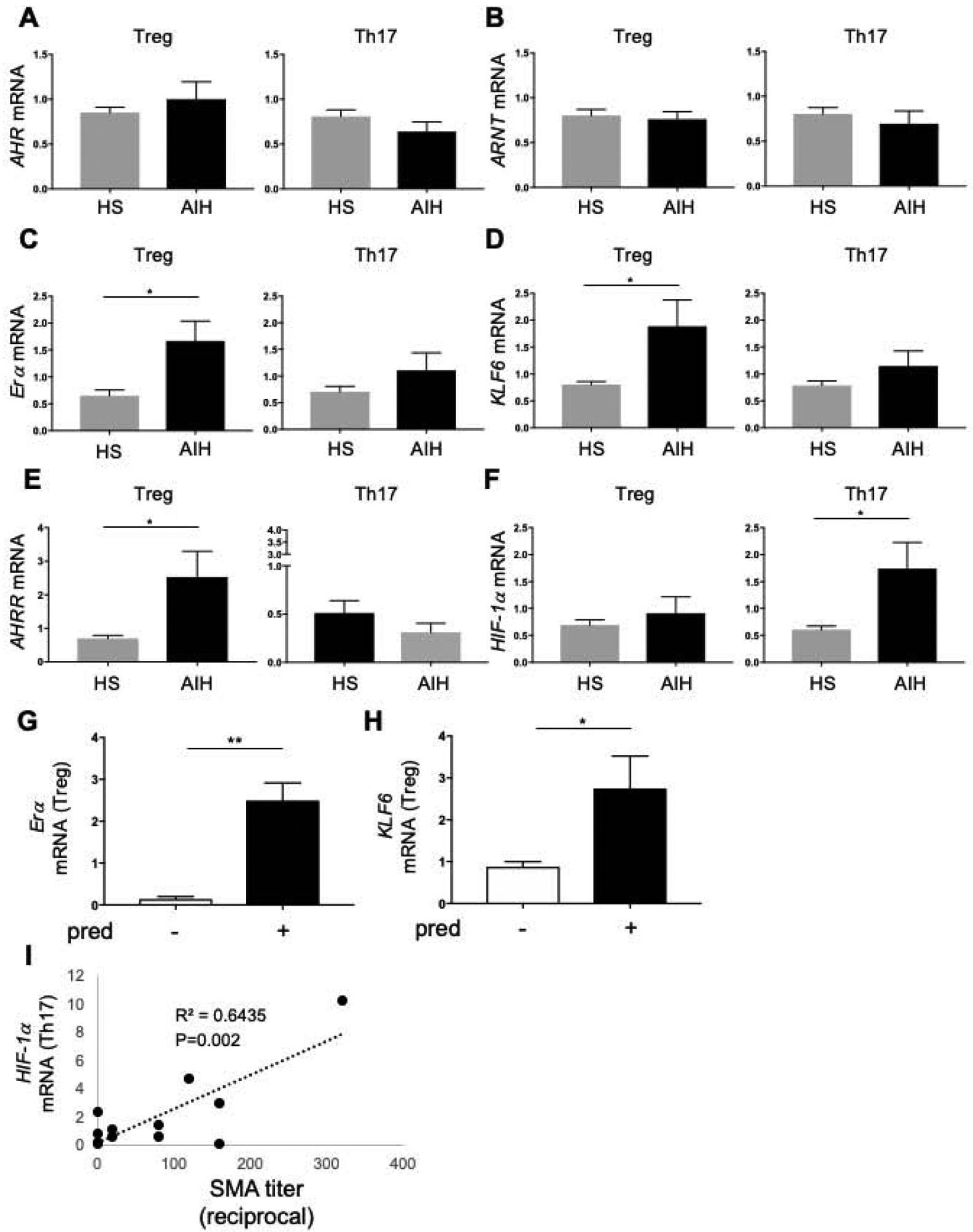
Mean+SEM (A) AHR, (B) ARNT, (C) Erα, (D) KLF6, (E) AHRR and (F) HIF-1α mRNA levels in Treg and Th17-cells from HS (n=13) and AIH patients (n=13). (G-H) Patients on prednisone treatment (n=7) display higher Erα and KLF6 mRNA levels than patients who were not under prednisone treatment (n=6). Mean+SEM is shown. *P≤0.05; **P≤0.01 (unpaired t test or Mann-Whitney test). (I) Positive correlation between HIF-1α mRNA levels with SMA autoantibody titre in AIH Th17-cells.
In 5 AIH patients, response of Treg and Th17-cells to AHR ligands was also evaluated in samples obtained at the time of (or close to) diagnosis before the immunosuppression was started and later while on immunosuppressive treatment. Th17-cells derived from AIH patients on immunosuppressive treatment displayed heightened CYP1A1 and CD39 mRNA levels following exposure to UCB and quercetin, when compared to Th17-cells obtained from AIH patients before immunosuppression was started (Supplementary Fig. 6A–B).
Levels of Erα and KLF6 mRNA were higher in Treg obtained from patients on prednisone treatment (Fig. 3G–H) and did not differ between females and males (Supplementary Fig. 7A–B) Importantly, levels of HIF-1a in Th17 cells were positively correlated with SMA autoantibody titre (Fig. 3I). When comparing Treg and Th17-cells obtained from AIH patients before and after immunosuppressive treatment was started, we noted higher Erα mRNA levels in patients on immunosuppressive treatment (Supplementary Fig. 7C).
Levels of Erα and HIF-1α were also tested by flow cytometry in CD45RA+FOXP3low ‘resting’ Treg, CD45RA−FOXP3high ‘activated’ Treg as well as in CD45RA−FOXP3low ‘non-suppressive’ Treg(25). In AIH samples, all three Treg subsets displayed higher Erα (Supplementary Fig. 8A–C) and HIF-1α (Supplementary Fig. 9A–C) MFI when compared to health.
Silencing of AHRR and Erα boosts CD39 levels in AIH-derived Treg
We determined whether silencing of the non-canonical AHR binding partners Erα and KLF6 or the inhibitory factor AHRR could impact CD39 expression in Treg from AIH patients and healthy controls. Differentiated Treg were exposed to UCB, L-Kynurenine or quercetin in the absence or presence of siRNA specific for ARNT, Erα, KLF6 or AHRR. At variance with healthy controls, silencing of AIH-derived Treg with Erα or AHRR siRNA resulted in increased frequencies of CD39+-cells upon exposure to UCB (Fig. 4A and Supplementary Fig. 10A–B); increase in CD39+-cells was also noted upon AIH Treg exposure to quercetin in the presence of Erα siRNA treatment (Fig. 4A). These effects were not observed when Treg from healthy controls were analysed (Fig. 4A and Supplementary Fig. 10A). Similar effects were noted also when considering CD39 expression at the mRNA level (Supplementary Fig. 10C).
Figure 4. Silencing of Erα and AHRR increases Treg response to UCB and quercetin in AIH.
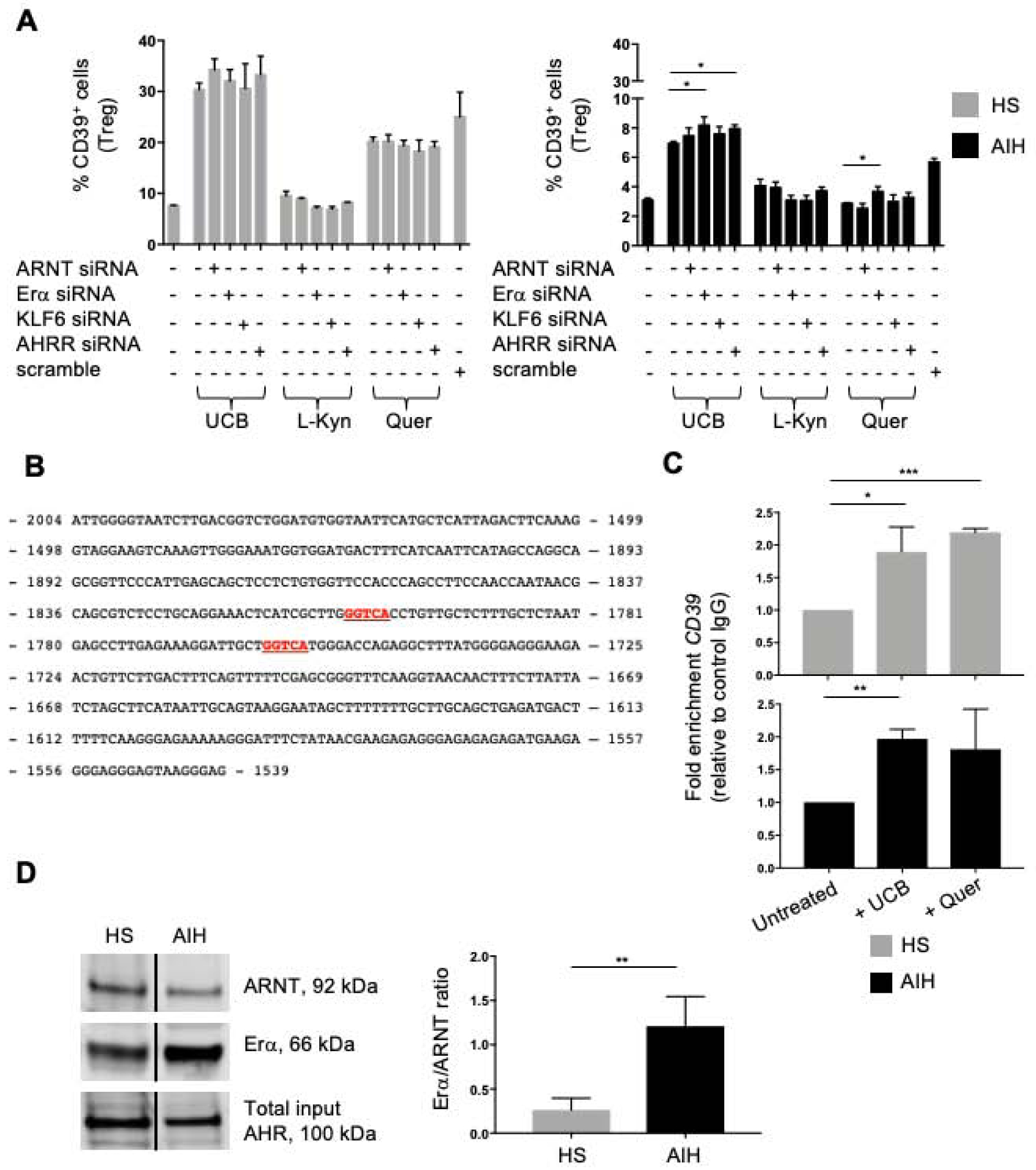
(A) Mean+SEM CD39+-cell frequency within untreated, siRNA (or scramble)-treated Treg before and after AHR ligand exposure (HS, n=10; AIH, n=14). (B) Putative Erα binding sites (in red) within CD39 promoter. (C) Mean+SEM fold enrichment in CD39 (CHiP) in untreated and ligand-treated Treg (HS, n=3; AIH, n=3). (D) Immunoblot (cropped) of ARNT and Erα in AHR-immunoprecipitated Treg exposed to UCB. AHR expression in input samples is represented. Mean+SEM ARNT/Erα ratio is shown (HS, n=4; AIH, n=4). *P≤0.05; **P≤0.01; ***P≤0.001 (one-way ANOVA and unpaired t test).
Exposure of AIH and healthy donor-derived Treg to L-Kynurenine in the presence of ARNT, Erα and KLF6 specific siRNA left unchanged the frequency of CD39+ lymphocytes (Fig. 4A), as well as CD39 mRNA levels (Supplementary Fig. 10C) in Treg.
Importantly, heightened AHRR and Erα expression was also present in liver infiltrating lymphocytes - along with CD3 and FOXP3 positivity and scanty CD39+-cells - in AIH biopsies, when compared to control liver sections (Supplementary Fig. 11A–B).
We then tested whether the impaired ability to upregulate CD39 upon AHR/Erα interaction derives from altered Erα binding to the CD39 promoter. To this end, we performed sequence analysis of the CD39 promoter region and identified putative binding sites for Erα (Fig. 4B). Chromatin immunoprecipitation analysis revealed that in both healthy control and AIH Treg Erα binds to the CD39 promoter, as reflected by enrichment in CD39 expression, following Treg exposure to UCB (Fig. 4C) and, in the case of healthy control Treg, quercetin (Fig. 4C).
To define whether AIH Treg impairment to effectively upregulate CD39 in the presence of high Erα levels results from altered interactions between AHR and Erα, we performed Treg-cell lysate immunoprecipitation for AHR and then measured ARNT and Erα expression in immunoprecipitated samples by immunoblotting. As depicted in Fig. 4D, Erα was present in larger amount than ARNT in AHR immunoprecipitated samples obtained from AIH Treg.
Overall the data presented here indicate that, in AIH Treg, blockade of AHRR and Erα increases the frequency of CD39+-cells and that, in these cells, AHR binds to Erα with greater affinity than ARNT.
Blockade of HIF-1α restores CD39 levels in AIH Th17 cells
We have shown that AIH Th17-cells display heightened HIF-1α levels (Fig. 3F and Supplementary Fig. 5F); therefore, we postulated that hypoxia and HIF-1α, could negatively impact AHR signalling, and consequently CD39 levels, in this T-cell subset. To this end, we measured HIF-1α levels in Th17-cells differentiated from circulating CD4-cells of healthy blood donors and AIH patients under normoxic conditions and after exposure to 2% O2. We found increased HIF-1α mRNA levels upon Th17-cell exposure to hypoxia in both controls and AIH patients (Fig. 5A). Levels of HIF-1α were higher in AIH than healthy control-derived Th17-cells both under normoxic (P=0.02) and hypoxic (P=0.038) conditions. Exposure of Th17-cells to hypoxia left AHR mRNA levels unchanged in Th17-cells from both controls and patients (Fig. 5B).
Figure 5. . Hypoxia impairs Th17-cell response to AHR activation in AIH.
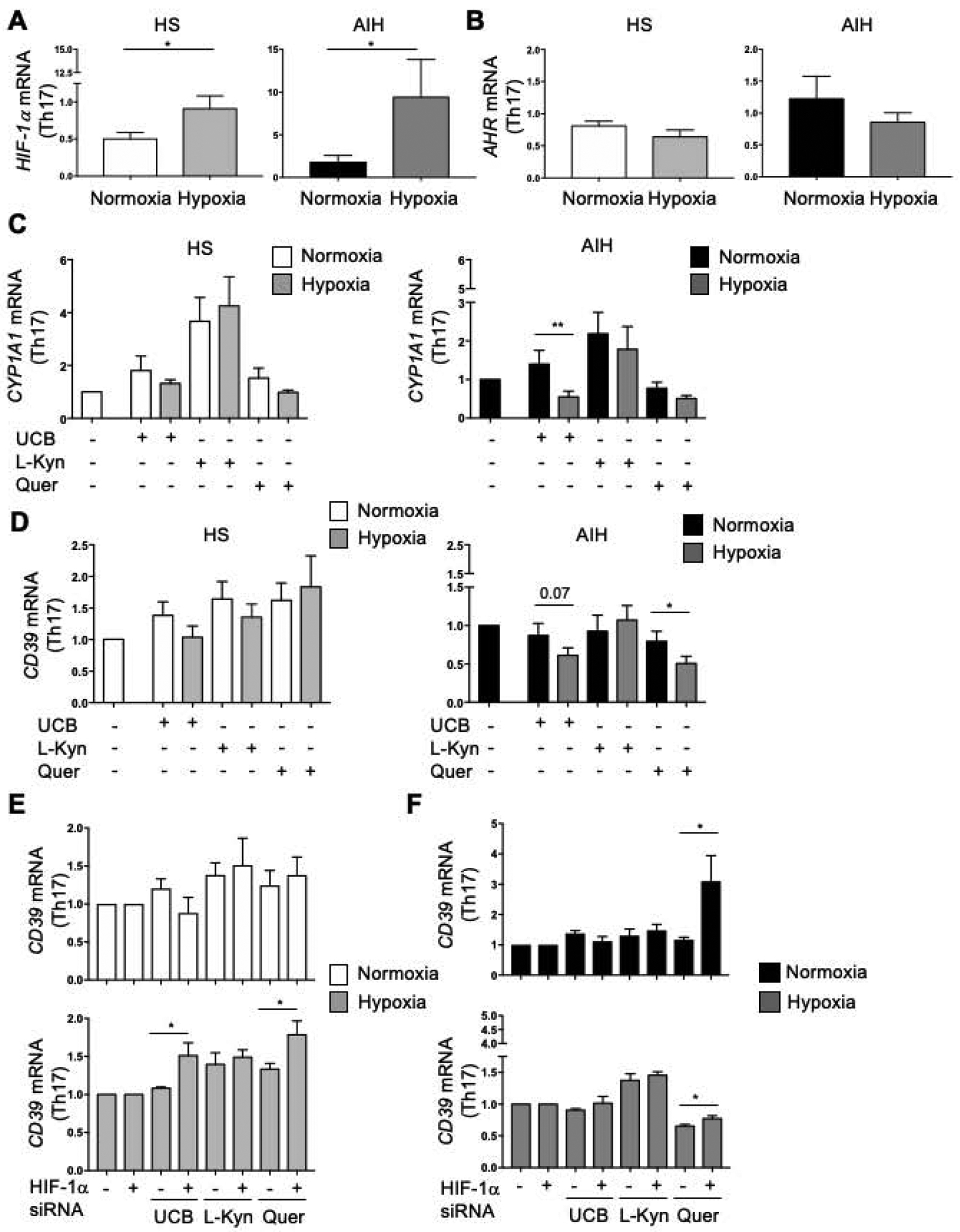
Mean+SEM (A) HIF-1α and (B) AHR mRNA levels in Th17-cells under normoxia and hypoxia (HS, n=14; AIH, n=12). (C) Mean+SEM CYP1A1 mRNA levels of untreated and AHR ligand-treated Th17-cells under normoxia and hypoxia (HS, n=8; AIH, n=11). (D) Mean+SEM CD39 mRNA levels of untreated and AHR ligand-treated Th17-cells under normoxia and hypoxia (HS, n=7; AIH, n=9). (E-F) Mean+SEM CD39 mRNA levels of untreated and AHR-ligand-treated Th17-cells under normoxia and hypoxia, in the absence or presence of HIF-1α siRNA (HS, n=5; AIH, n=5). *P≤0.05; **P≤0.01 (unpaired t test or Mann-Whitney test; one-way ANOVA).
When testing whether hypoxia impacted AHR signalling, we found that exposure of AIH-derived Th17-cells to UCB, under hypoxic conditions, decreased CYP1A1 mRNA levels (Fig. 5C). Hypoxia did not impact CYP1A1 levels in AIH and healthy control Th17-cells exposed to L-Kynurenine and quercetin (Fig. 5C).
When assessing CD39 expression, we noted decrease in CD39 mRNA levels upon exposure of UCB and quercetin-treated Th17-cells to hypoxia in AIH but not in healthy controls (Fig. 5D).
Treatment with HIF-1α siRNA resulted in an increase in CD39 mRNA levels in healthy blood donor Th17-cells treated with UCB and quercetin and exposed to hypoxic conditions (Fig. 5E). In AIH, treatment with HIF-1α siRNA boosted CD39 mRNA levels in Th17-cells exposed to quercetin both under normoxic and hypoxic conditions (Fig. 5F).
Collectively, these data show that, in AIH, hypoxia exerts inhibitory effects on AHR signalling, this being reverted, at least in part, upon exposure to HIF-1α molecular silencing.
Discussion
This study reports differential AHR alterations in Treg and Th17-cells obtained from patients with AIH; these consist of heightened levels of the inhibiting factors AHRR in Treg and HIF-1α in Th17-cells; and in upregulated levels of the non-canonical AHR binding partner Erα in Treg. All these alterations result in impaired upregulation of CD39 in Treg and Th17-cells and in impaired functionality of the Treg subset. Impaired CD39 expression and upregulation might impact Treg function by favouring phenotypic instability upon pro-inflammatory challenge(11) and limits Th17-cell ability to undergo regulation and acquire immunosuppressive properties(10).
Our data support the evidence that AHR signalling alterations involve both the regulatory and effector T-cell arm in AIH, as shown by inability of both polarized and freshly purified Treg and Th17-cells to upregulate CYP1A1 - one of the main genes regulated by the AHR/ARNT complex - upon stimulation with endogenous ligands or metabolites like UCB and L-Kynurenine; and exogenous ligands like quercetin. Inability to upregulate CYP1A1 is reflected by a markedly impaired defect in increasing CD39 levels by both T-cell types. When considering the Treg subset, it appears that high levels of AHRR impact the response of these cells to AHR ligation in the presence of UCB, suggesting an intrinsic defect of Treg in responding to immunoregulation in the AIH context. Further, aberrantly high levels of Erα, likely induced by prednisone treatment, might also exacerbate inability of Treg to effectively respond to AHR stimulation; lack of CD39 upregulation and amelioration of Treg suppressive properties upon AHR activation reflects this functional impairment. Increases in Treg functionality, as noted following treatment with AHR ligands in health, depend - at least to some extent - on CD39, as shown by our silencing experiments. Lack of an effect of CD39 blockade in AIH Treg might derive from low levels of CD39 at baseline and impaired Treg ability to adequately respond to AHR ligation.
Amongst the AHR non-canonical binding factors, it appears that Erα is the most relevant for impacting CD39 levels and function since molecular blockade of this factor results in partial restoration of CD39 in AIH Treg. Blockade of KLF6 - the tumour repressor that, like Erα, serves as non-canonical binding partner for AHR and is also upregulated in AIH Treg - does not result in effective CD39 upregulation in these cells. These data suggest that the alterations of AHR signalling in AIH Treg are induced by aberrantly high inhibition, as reflected by increase in AHRR expression, and could be further exacerbated by immunosuppression with prednisone (Fig. 6), the dose of which strongly correlates with the levels of the non-canonical binding partner Erα. The heightened Erα levels noted in Treg obtained from patients on immunosuppressive treatment further corroborate this clinical correlation.
Figure 6. AHR dysfunctional signalling in AIH.
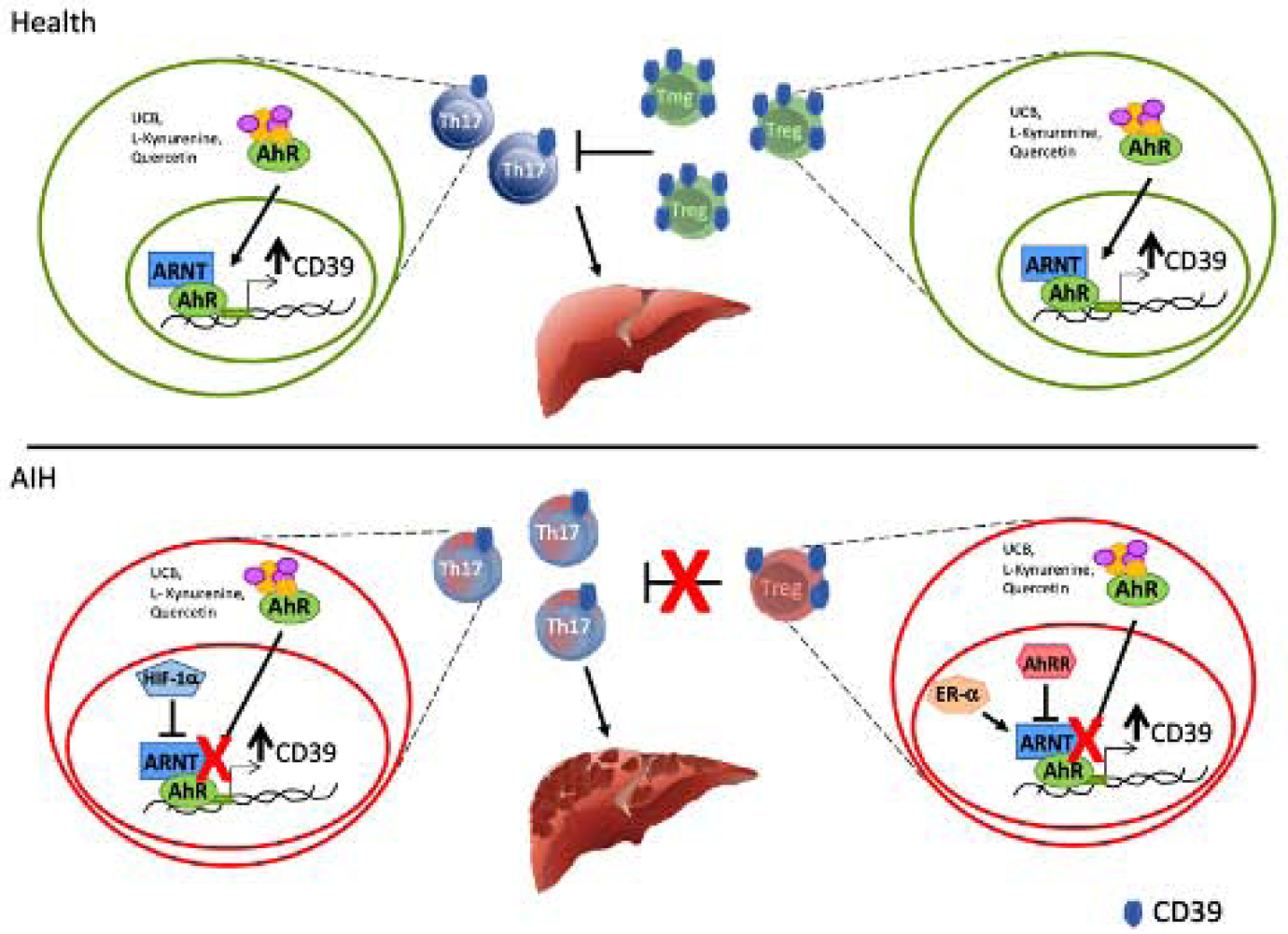
In health, Treg and Th17-cell ability to upregulate CD39 upon exposure to AHR endogenous and exogenous ligands, results from effective interactions between AHR and ARNT. In AIH Treg and Th17-cells, increase in the levels of the AHR inhibitors AHRR (Treg) and HIF-1α (Th17) results in altered AHR signalling and consequent inability to effectively upregulate CD39. In AIH Treg, AHR signalling is further impacted because of heightened expression of the AHR non-canonical binding partner Erα, the expression of which directly correlates with steroid dose.
Importantly, high numbers of AHRR+ and Erα+ positive lymphocytes are also noted amongst liver infiltrating CD3+ and FOXP3+-cells.
Chromatin immunoprecipitation data indicate that Erα does bind to the CD39 promoter, suggesting that lack of effective CD39 upregulation in AIH Treg might derive from altered AHR and Erα interactions, rather than inability of Erα to bind to the CD39 promoter. Immunoprecipitation studies showing that, in AIH Treg, AHR binds to Erα with greater affinity than ARNT support this postulate. Evidence that Erα might impact immunoregulatory/inhibitory pathways was provided by previous work, showing that Erα functions as a negative regulator of PD-L1 in the cancer setting(26). Further, oestrogens were reported to induce expression of pro-inflammatory cytokines by LPS stimulated macrophages via Erα signalling(27).
Our data also indicate that, differently from Treg, Th17-cells obtained from AIH patients upregulate HIF-1α both under normoxic and hypoxic conditions.
Elevated HIF-1α levels are likely to derive from protracted hypoxia generated from chronic inflammation. Importantly, like we previously noted in the setting of IBD(24), upregulation of HIF-1α inhibits AHR signalling (Figure 6). This is reflected by downregulation of CD39 in AIH Th17-cells exposed to hypoxic conditions, without AHR expression being impacted. Similar to what observed in IBD(24), silencing of HIF-1α results in a partial restoration of CD39 levels under hypoxic conditions. Hypoxia is known to regulate T-cell immunity although the impact might differ in acute versus chronic inflammation. Previous studies have shown that during acute inflammation hypoxia has beneficial effects, as marked by increases in FOXP3+ Treg(28). In the chronic inflammatory setting, however, hypoxia has been reported to inhibit the differentiation of T-regulatory-type-1 (Tr1)-cells(23), a regulatory subset predominant during chronic inflammation(29, 30); and limits the acquisition of regulatory properties by Th17-cells while boosting their pathogenic potential(31).
Previous work in the context of ethanol-induced liver disease and non-alcoholic fatty liver disease indicated HIF-1α as a pivotal factor in determining liver tissue damage(32–34). Additional work confirmed HIF role in the mechanism leading to liver fibrosis in cholestatic liver disease and demonstrated nuclear accumulation of HIF-1α in liver macrophages of patients with primary biliary cirrhosis and primary sclerosing cholangitis(35).
Interestingly, further analysis of the Treg subset based on the expression of CD45RA and FOXP3, revealed that, in AIH, ‘resting’, ‘activated’ and ‘non-suppressive’ Treg all display increased Erα and HIF-1α levels, suggesting that alterations in AHR signalling pathway are present in all Treg subpopulations in this setting.
Importantly, defects in AHR signalling in AIH Treg and Th17-cells are more evident following cell stimulation in the presence of UCB and L-Kynurenine, two AHR endogenous ligands with well-known immunosuppressive properties(36–41); and in the presence of quercetin, a polyphenol that impairs T-cell proliferation, while modulating the pro-inflammatory properties of dendritic cells(42). The lack of an effective Treg and Th17-cell response to these AHR ligands might compromise immune homeostasis in AIH by interfering with AHR control over immunoregulatory genes and pathways, like CD39 and the purinergic signalling. The reasons for this selective impairment in AIH Treg and Th17-cell response to UCB, L-Kynurenine and quercetin remain unclear although differences in the structure or chemical composition of AHR ligands might result in changes in the type and extent of the T-cell response elicited(43). In this regard, we did not observe alterations in the response of Treg and Th17-cells to benzo[a]pyrene, which is found in air pollution and cigarette smoke, the latter being previously indicated as a possible trigger for the recruitment of mucosal effector lymphocytes in patients with both autoimmune liver disease and inflammatory bowel disease(44). The different clinical setting and lymphocyte compartments studied might account for these dissimilarities.
Another important implication of our findings relates to immunotherapy. The evidence that silencing of AHRR, HIF-1α or Erα could restore CD39 levels in AIH Treg and Th17-cells along with Treg function postulates a role for therapeutic approaches based on the use of AHRR, Erα or HIF-1α antagonists/silencers for the treatment of AIH. This would enable re-establishing the levels of CD39 and, consequently, the immune homeostatic balance between regulatory and effector cells in this autoimmune condition. The prednisone-linked upregulation of Erα, for which AHR has preferential binding in AIH, suggests a potential side effect of this treatment that, despite restraining inflammation, might be involved in perpetuating, and perhaps even exacerbating, the immunoregulatory defects already present in AIH.
An additional strategy that might be considered to correct these alterations would be to bypass AHR disrupted signalling and rather focus on boosting CD39 per se. Our recent data obtained in the preclinical IBD setting propose the use of recombinant apyrase enzymes with ectoenzymatic activity comparable to that of CD39 to control inflammation in colitis while boosting the immunoregulatory properties of Treg and Tr1-cells(45).
In conclusion, our findings propose that immune imbalance between Treg and Th17-cells in AIH derives from alterations of AHR signalling that result in defective ability to upregulate CD39. Interference with AHR dysfunctional signalling or boosting downstream CD39-driven pathways would represent valuable therapeutic options to halt inflammation while correcting altered immunoregulatory mechanisms.
Supplementary Material
Highlights.
In AIH Treg and Th17 display dysfunctional aryl-hydrocarbon-receptor (AhR) pathway
This results in defective upregulation of CD39, a key immunoregulatory ectoenzyme
Altered AhR pathway results from increased inhibitory factors
Binding of AhR to estrogen-receptor-alpha amplifies defective CD39 upregulation
Blockade of this aberrant pathways would restore CD39 and immunotolerance in AIH
Financial support:
This work has been supported by the National Institute of Health (R01 DK108894 to M.S.L. and R21 CA164970 to S.C.R.); AASLD Pilot Research Award (to M.S.L.); Seed Grant Award (Department of Anesthesia, Critical Care & Pain Medicine to M.S.L.); the Department of Defense Award W81XWH-16-0464 (to S.C.R.); Postdoctoral Fellowship from CAPES, Brazil (88881.171857/2018-01 to L.A.F.); Scholarship Dr. Ludmily Sedlarovey-Rabanovej and Nadacia Tatra banky (to B.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
Nothing to disclose.
References
Author names in bold designate shared co-first authorship
- 1.Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS. Autoimmune hepatitis. Lancet 2013;382:1433–1444. [DOI] [PubMed] [Google Scholar]
- 2.Kapila N, Higa JT, Longhi MS, Robson SC. Autoimmune Hepatitis: Clinical Review with Insights into the Purinergic Mechanism of Disease. J Clin Transl Hepatol 2013;1:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol 2004;41:31–37. [DOI] [PubMed] [Google Scholar]
- 4.Longhi MS, Ma Y, Mitry RR, Bogdanos DP, Heneghan M, Cheeseman P, et al. Effect of CD4+ CD25+ regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J Autoimmun 2005;25:63–71. [DOI] [PubMed] [Google Scholar]
- 5.Longhi MS, Hussain MJ, Mitry RR, Arora SK, Mieli-Vergani G, Vergani D, et al. Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol 2006;176:4484–4491. [DOI] [PubMed] [Google Scholar]
- 6.Liberal R, Grant CR, Holder BS, Ma Y, Mieli-Vergani G, Vergani D, et al. The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin-9/tim-3 pathway. Hepatology 2012;56:677–686. [DOI] [PubMed] [Google Scholar]
- 7.Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One 2011;6:e18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longhi MS, Mitry RR, Samyn M, Scalori A, Hussain MJ, Quaglia A, et al. Vigorous activation of monocytes in juvenile autoimmune liver disease escapes the control of regulatory T-cells. Hepatology 2009;50:130–142. [DOI] [PubMed] [Google Scholar]
- 9.Ferri S, Longhi MS, De Molo C, Lalanne C, Muratori P, Granito A, et al. A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology 2010;52:999–1007. [DOI] [PubMed] [Google Scholar]
- 10.Liberal R, Grant CR, Ma Y, Csizmadia E, Jiang ZG, Heneghan MA, et al. CD39 mediated regulation of Th17-cell effector function is impaired in juvenile autoimmune liver disease. J Autoimmun 2016;72:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC, et al. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology 2014;59:1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vuerich M, Robson SC, Longhi MS. Ectonucleotidases in Intestinal and Hepatic Inflammation. Front Immunol 2019;10:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev 2017;276:121–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol 2010;11:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008;453:65–71. [DOI] [PubMed] [Google Scholar]
- 16.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008;453:106–109. [DOI] [PubMed] [Google Scholar]
- 17.Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez-Vazquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018;48:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang Z, Lu M, Kim SS, Park JS. Potential role of HSP90 in mediating the interactions between estrogen receptor (ER) and aryl hydrocarbon receptor (AhR) signaling pathways. Toxicol Lett 2014;226:6–13. [DOI] [PubMed] [Google Scholar]
- 20.Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol 2006;72:267–279. [DOI] [PubMed] [Google Scholar]
- 21.Wilson SR, Joshi AD, Elferink CJ. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J Pharmacol Exp Ther 2013;345:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev 1999;13:20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med 2015;21:638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie A, Robles RJ, Mukherjee S, Zhang H, Feldbrugge L, Csizmadia E, et al. HIF-1alpha-induced xenobiotic transporters promote Th17 responses in Crohn’s disease. J Autoimmun 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009;30:899–911. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Shen Y, Zhu X, Lv R, Li S, Zhang Z, et al. ERalpha is a negative regulator of PD-L1 gene transcription in breast cancer. Biochem Biophys Res Commun 2018;505:157–161. [DOI] [PubMed] [Google Scholar]
- 27.Calippe B, Douin-Echinard V, Delpy L, Laffargue M, Lelu K, Krust A, et al. 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. J Immunol 2010;185:1169–1176. [DOI] [PubMed] [Google Scholar]
- 28.Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A 2012;109:E2784–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol 2014;380:39–68. [DOI] [PubMed] [Google Scholar]
- 30.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 2006;212:28–50. [DOI] [PubMed] [Google Scholar]
- 31.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011;146:772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambade A, Satishchandran A, Szabo G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and miR-122-mediated HIF-1alpha activation. Sci Rep 2016;6:21340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satishchandran A, Ambade A, Rao S, Hsueh YC, Iracheta-Vellve A, Tornai D, et al. MicroRNA 122, Regulated by GRLH2, Protects Livers of Mice and Patients From Ethanol-Induced Liver Disease. Gastroenterology 2018;154:238–252 e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mesarwi OA, Shin MK, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY. Hepatocyte Hypoxia Inducible Factor-1 Mediates the Development of Liver Fibrosis in a Mouse Model of Nonalcoholic Fatty Liver Disease. PLoS One 2016;11:e0168572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Copple BL, Kaska S, Wentling C. Hypoxia-inducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J Pharmacol Exp Ther 2012;341:307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Li P, Lu J, Xiong W, Oger J, Tetzlaff W, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol 2008;181:1887–1897. [DOI] [PubMed] [Google Scholar]
- 37.Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, Otterbein LE, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J 2007;21:3450–3457. [DOI] [PubMed] [Google Scholar]
- 38.Rocuts F, Zhang X, Yan J, Yue Y, Thomas M, Bach FH, et al. Bilirubin promotes de novo generation of T regulatory cells. Cell Transplant 2010;19:443–451. [DOI] [PubMed] [Google Scholar]
- 39.Vetvicka V, Sima P, Miler I, Bilej M. The immunosuppressive effects of bilirubin. Folia Microbiol (Praha) 1991;36:112–119. [DOI] [PubMed] [Google Scholar]
- 40.Allegri G, Bertazzo A, Biasiolo M, Costa CV, Ragazzi E. Kynurenine pathway enzymes in different species of animals. Adv Exp Med Biol 2003;527:455–463. [DOI] [PubMed] [Google Scholar]
- 41.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 2010;185:3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez de Medina F, Vera B, Galvez J, Zarzuelo A. Effect of quercitrin on the early stages of hapten induced colonic inflammation in the rat. Life Sci 2002;70:3097–3108. [DOI] [PubMed] [Google Scholar]
- 43.O’Driscoll CA, Gallo ME, Fechner JH, Schauer JJ, Mezrich JD. Real-world PM extracts differentially enhance Th17 differentiation and activate the aryl hydrocarbon receptor (AHR). Toxicology 2019;414:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liaskou E, Karikoski M, Reynolds GM, Lalor PF, Weston CJ, Pullen N, et al. Regulation of mucosal addressin cell adhesion molecule 1 expression in human and mice by vascular adhesion protein 1 amine oxidase activity. Hepatology 2011;53:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robles RJ, Mukherjee S, Vuerich M, Xie A, Harshe R, Cowan PJ, et al. Modulation of CD39 and exogenous APT102 correct immune dysfunction in experimental colitis and Crohn’s disease. J Crohns Colitis 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.