

Abstract
The most common complications in patients with type-2 diabetes are hyperglycemia and hyperlipidemia that can lead to cardiovascular disease. Alleviation of these complications constitutes the major therapeutic approach for the treatment of diabetes mellitus. Agonists of peroxisome proliferator-activated receptor (PPAR) alpha and PPARγ are used for the treatment of hyperlipidemia and hyperglycemia, respectively. PPARs belong to the nuclear receptors super-family and regulate fatty acid metabolism. PPARα ligands, such as fibrates, reduce circulating triglyceride levels, and PPARγ agonists, such as thiazolidinediones, improve insulin sensitivity. Dual-PPARα/γ agonists (glitazars) were developed to combine the beneficial effects of PPARα and PPARγ agonism. Although they improved metabolic parameters, they paradoxically aggravated congestive heart failure in patients with type-2 diabetes via mechanisms that remain elusive. Many of the glitazars, such as muraglitazar, tesaglitazar, and aleglitazar, were abandoned in phase-III clinical trials. The objective of this review article pertains to the understanding of how combined PPARα and PPARγ activation, which successfully targets the major complications of diabetes, causes cardiac dysfunction. Furthermore, it aims to suggest interventions that will maintain the beneficial effects of dual PPARα/γ agonism and alleviate adverse cardiac outcomes in diabetes.
Keywords: glitazars, dual PPARα/γ agonists, cardiovascular effects, diabetes, heart failure
INTRODUCTION
Diabetes is the seventh leading cause of death worldwide.1 It is classified into two major forms, type 1 and 2. Type-2 diabetes (T2D) melitus affects the majority of the diabetic population, and it is associated with obesity and insulin resistance combined with insufficient compensatory insulin secretion.2 For patients in whom the recommended lifestyle changes3 are not adequate to control the complications of the disease, pharmacotherapy is required to preserve glycemic control. Besides the noninsulin antihyperglycemic drugs, such as biguanides, sulfonylureas, meglitinides, glitazones alpha-glucosidase inhibitors, glucagon-like peptide-1, dipeptidyl peptidase 4 inhibitors, and sodium/glucose cotransporter 2 inhibitors, drugs that regulate both glucose and lipid metabolism have been generated. The dual-PPARα/γ coactivators or “glitazars” combine the insulin-sensitizing effect of PPARγ agonists with the lipid- and triglyceride-lowering effects of PPARα agonists, which are additionally accompanied by an increase in the high-density lipoprotein (HDL)-cholesterol levels (Fig. 1).4 The dual actions of these agents come in varying degrees of PPARα and PPARγ activation, depending on their chemical structure (Fig. 2). Some of the glitazars, when compared with the single PPARγ agonists, which are also used in antidiabetic treatment, were more efficient in lowering hyperglycemia and hyperlipidemia.5,6 However, although glitazars improved clinical efficacy on glucose control and lipid metabolism, they were discontinued because of major cardiovascular events, such as stroke, myocardial infarction (MI), peripheral edema, and heart failure (Fig. 1 and Table 1).7–10 This review aims to summarize the basic and clinical research findings to date on glitazars and contribute to the understanding of the mechanisms that involve PPARα and PPARγ activation and underlie their cardiovascular adverse effects.
FIGURE 1.
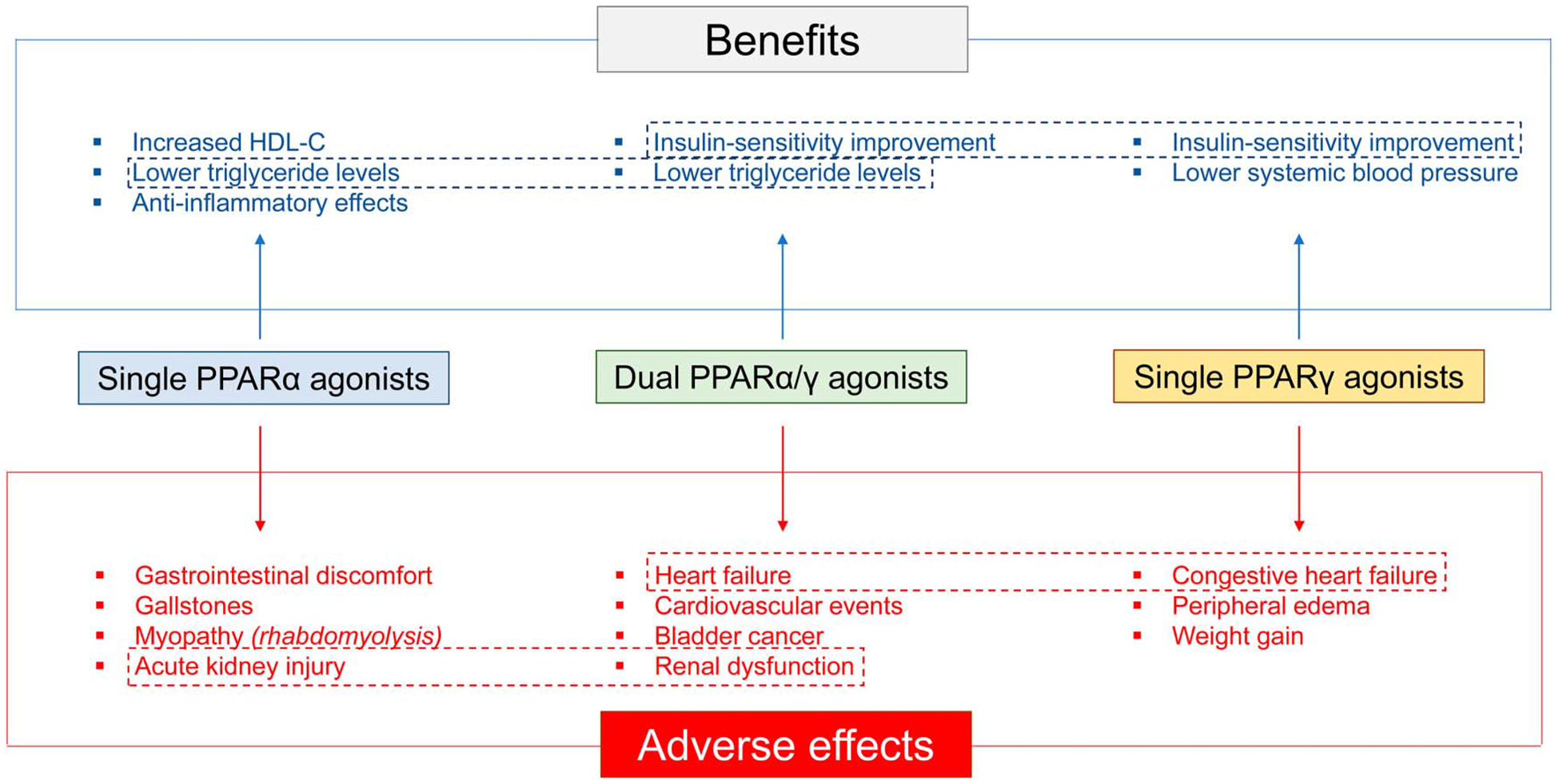
Summary of benefits and adverse effects of single PPARα or PPARγ agonists and dual PPARα/γ agonists. Common effects of single and dual agonists are shown in frames.
FIGURE 2.
Chemical structures of glitazars.
TABLE 1.
Dual PPARα/γ agonists That Have Been Tested in Clinical Trials
| Substance | Company | Disease | Discontinuation | Notes |
|---|---|---|---|---|
| Farglitazar | GlaxoSmithKline (London) | T2D | 2003 | Discontinued due to the evidence of edema |
| Ragaglitazar | Novo Nordisk (Denmark) | T2D | 2004 | Discontinued due to weight gain, anemia and bladder tumors in rodents |
| TAK-559/Imiglitazar | Takeda (Japan) | T2D | 2004 | Discontinued in phase III following abnormalities in liver enzymes |
| Muraglitazar (PARGLUVA) | Bristol-Myers Squibb (United States of America) | T2D | 2006 | Approved then discontinued due to CV events (heart failure) |
| Tesaglitazar (GALIDA) | AstraZeneca (United Kingdom) | T1D, T2D, Metabolic disorders | 2006 | Discontinued in phase III trials due to increased creatinine levels and decreased glomerular filtration rate |
| Naveglitazar (LY519818) | Eli Lilly (United States of America) | Cardiovascular disease, T2D, dyslipidemia | 2006 | Discontinued due to adverse preclinical findings in rodents |
| Sipoglitazar | Takeda (Japan) | T2D | 2006 | Discontinued due to safety concerns |
| Cevoglitazar | Novartis (Switzerland) | T2D, obesity | 2008 | Discontinued due to insufficient positive benefits |
| AVE-0847 | Sanofi-Aventis (France) | T2D | 2008 | Termination of development due to reprioritization of product portfolio |
| Aleglitazar | Hoffman-La-Roche (Switzerland) | T2D | 2013 | Discontinued in phase III trials due to gastrointestinal bleeding, bone fractures, heart failure |
| Chiglitazar | Shenzhen Chipscreen, (China) | T2D | - | Safe and effective treatment for patients with T2D according to study results presented at the American Diabetes Association Scientific Sessions in 2019 |
| Saroglitazar (LIPAGLYN) | Zydus Cadila (India) | T2D, dyslipidemia | - | Approved for use in India by the Drug Controller General of India |
T1D, Type-1 diabetes; T2D, Type-2 diabetes.
Clinical Applications of PPAR Agonists PPARα Agonists
Several studies and clinical trials on PPARα agonists (fibrates) have also attempted to investigate their benefits versus adverse effects. The FIELD study showed that fibrates reduce total cardiovascular events, specifically reducing nonfatal MIs and revascularizations. This decrease however was not significant in the lowering of the primary outcome of coronary events in the general population.11 Another study showed that long-term use of fenofibrate reduced microvascular-associated complications and was generally well-tolerated, specifically in patients with T2D. However, it had no significant effect on mortality or on other adverse cardiovascular outcomes.12 The positive effect of fibrates has been suggested to be mediated by increased expression of HDL proteins (AI, AII), reverse cholesterol transport factors (ABCA1, SR-BI), lipid-remodeling factors (lipoprotein lipase), and anti-inflammatory functions. It has also been suggested that fibrates alter the composition of the atheromatic plaque in a way that leads to regression of atherosclerosis.13,14 Although the magnitude of the benefits of fibrates is intermediate, it can have high clinical significance in high-risk individuals and in those with combined dyslipidemia. Despite their partially beneficial role, fibrates are accompanied by several other adverse effects due to non–lipid-related differential regulation on homocysteine, creatinine, and hemoglobin A1c (HbA1c). Specifically, fibrates have been shown to increase homocysteine with gemfibrozil, increasing it less than fenofibrate.15 The ACCORD lipid trial studied whether a coadministration of fibrates and statins (another lipid-lowering drug) is more efficient in reducing the risk of cardiovascular events by either of the drugs used alone. The outcome of the study indicated that combination therapy could not effectively reduce cardiovascular disease in most high-risk adults with T2D. In addition, the researchers noted that men may experience a more robust benefit from the combination therapy, whereas women on combined fibrates and statins therapy seemed to have more cardiovascular problems than those on statins alone.16 Furthermore, the combination of fibrates and statins has also been reported to increase the incidence of rhabdomyolysis.17 However, a recently developed fibrate, permafibrate (K-877), when coadministered with statins showed milder side effects.18,19 Further research on this drug is currently in phase-III clinical trials (PROMINENT, NCT03071692, started in 2017).
PPARγ Agonists
Most of the antidiabetic drugs that activate PPARγ exhibit cardiovascular risk in a direct or indirect manner. The thiazolidinediones (TZDs), such as pioglitazone and rosiglitazone, have been associated with fluid retention and increased risk for cardiac dysfunction and heart failure.20–22 A meta-analysis of randomized trials and the PROactive study showed that although diabetic patients treated with pioglitazone had lower all-cause mortality, they had a higher heart failure incidence.23,24 Rosiglitazone has been associated with even higher risk of heart failure and other cardiovascular events25,26 and MI.27 In contrast, the RECORD trial showed that rosiglitazone treatment increased the risk for heart failure but did not reveal any association with MIs.28,29 In accordance to the RECORD trial, the AHA/ACCF Science Advisory in 2010, upon reevaluation of the cardiovascular effects of TZDs, acknowledged that the association of rosiglitazone with increased risk for heart failure still remains uncertain30. Afterward, in 2013, rosiglitazone was reapproved without any restrictions by the United States Food and Drug Administration.
Dual PPARα/γ Agonists
The effort to develop PPAR agonists without adverse effects led to the development of dual agonists (glitazars) that activate both PPARα and PPARγ, thus combining the lipid-lowering effects of PPARα with the insulin-sensitizing effects of PPARγ. Depending on their molecular structure, these molecules exert varying degrees of PPARα and PPARγ activation. Although glitazars lower plasma glucose and triglycerides,4 they have been associated with ischemic events31 and congestive heart failure.32 A precautionary statement for an approved dual agonist, saroglitazar, was issued for patients with diabetes and congestive heart failure.32 Other glitazars, such as tesaglitazar, aleglitazar, muraglitazar, and cevoglitazar have been discontinued due to adverse side effects, including heart failure and myocardial ischemia.31
The first dual agonist was farglitazar, which was discontinued in phase-III clinical trials due to evidence of edema.33 Farglitazar had also been tested in phase-II clinical trials for the treatment of hepatic fibrosis but failed to show significant therapeutic effects.34 The retraction of farglitazar was followed by discontinuation of ragaglitazar,35 imiglitazar, and tesaglitazar trials,36 due to carcinogenicity in rodent toxicity models, elevated creatinine levels in serum, and cardiac dysfunction, respectively. Imiglitazar treatment increased HDL cholesterol levels. This was associated with higher apolipoprotein A-I levels, larger HDL particles, and suppression of small-density HDL particles. Imiglitazar decreased plasma triglycerides and apolipoprotein B-100 levels. However, evidence of liver enzyme abnormalities led to its withdrawal from phase-III clinical trials.37,38
Tesaglitazar decreases hyperglycemia and dyslipidemia9 and prevents atherosclerosis in mice with low-density lipoprotein (LDL) receptor deficiency.21 Later, two series of phase-III clinical trials, named GALLANT and GALEX, tested tesaglitazar versus common antidiabetic drugs, such as metformin, pioglitazone, and placebos. All studies were terminated due to side effects, such as congestive heart failure and renal toxicity, that occurred despite tesaglitazar’s strong antihyperlipidemic and antihyperglycemic effects.21,39–42
Similarly, muraglitazar development was discontinued due to higher rates of all-cause mortality compared with pioglitazone. Higher mortality was associated with edema, cardiac dysfunction, stroke, heart failure, and increased weight gain rate.22,43 A double-blind randomized clinical trial that compared muraglitazar and pioglitazone showed that muraglitazar decreased hemoglobin A1c (HbA1c), cholesterol, apolipoprotein B, and triglycerides (P < 0.0001) and increased HDL-C.43
Muraglitazar and tesaglitazar failure was followed by the development of glitazars with slightly modified chemical structures. However, all alternatives were eventually abandoned due to various cardiovascular and other adverse effects. Naveglitazar, a dual PPARα/γ agonist with γ-dominance, was also discontinued due to carcinogenic effects in the urinary bladder of rats, which were preceded by urothelial hypertrophy, inflammation, and changes in urinary composition.44 Three more dual agonists, sipoglitazar, cevoglitazar, and AVE-0847, were also discontinued due to adverse effects.
Another attempt with aleglitazar portrayed a protective role of the drug in mouse cardiomyocytes exposed to high glucose levels in vitro.45 Aleglitazar protects the heart against ischemia–reperfusion injury by suppressing myocardial apoptosis and decreasing infarct size.46 However, aleglitazar was discontinued in phase-III trials due to other side effects, including severe gastrointestinal bleeding and bone fractures.47
Another PPARα/γ agonist, CG301269, also had beneficial metabolic effects in animal studies with rodents.48 Furthermore, administration of CG301269 in a mouse model of myocardial ischemia/reperfusion did not exacerbate heart failure. Conversely, another dual PPARα/γ agonist, LY510929, which treated hyperlipidemia and hyperglycemia in rats, caused left ventricular hypertrophy after treatment for 2 weeks.49
The most recently developed dual PPARα/γ agonists are chiglitazar and saroglitazar. Specifically, chiglitazar is a PPARα/γ/δ panagonist that improves insulin sensitivity and glucose tolerance, and reduces the homeostatic model assessment index, plasma triglycerides, total cholesterol, low-density lipoprotein cholesterol levels, alanine gluconeogenesis, and hepatic glycogen levels in monosodium glutamate (MSG) obese rats.50 Two phase-III trials concluded that chiglitazar is a safe treatment option against T2D (NCT02121717), although adverse events, such as weight gain, edema, and hypoglycemia, were reported.
Saroglitazar is the only glitazar still in clinical development. It is a dual PPARα/γ agonist with significant lowering effects on serum lipids and glucose. A study with 23 male and 11 female patients with T2D and dyslipidemia, who were treated with saroglitazar, showed significant reduction in total cholesterol, LDL cholesterol, and plasma triglycerides.4 Saroglitazar incurs moderate PPARγ activation and more robust activation of PPARα,51 even in comparison with the PPARα activator, WY14,643.52 A recent study showed that saroglitazar diminishes hypertriglyceridemia and improves insulin sensitivity and β-pancreatic cell function by alleviating glucolipotoxicity, possibly through direct PPARγ activation in patients with T2D and high blood triglyceride levels.7 The substance was approved in June 2013 for clinical use in India.53 Saroglitazar has never been associated with cardiovascular complications, although its product information contains a precautionary statement for its use in patients with T2D and congestive heart failure. These findings combined with those from older studies for other dual PPARα/γ agonists presented the high efficiency of dual PPARα/γ agonists against hyperglycemia and hyperlipidemia.54–56
Molecular Mechanisms of Cardiac Toxicity by Single or Dual PPARα–PPARγ Activation
Since the beginning of clinical development for both single PPAR agonists and dual PPARα/γ agonists, there have been concerns about their potential association with cardiovascular complications.57 Therefore, elucidation of the mechanisms that underlie adverse outcomes is warranted. Accordingly, various studies have focused on the molecular basis of these unexplained effects.
PPARs are nuclear receptors that act as transcriptional factors by forming heterodimers with retinoid X receptors and binding to cis-acting DNA elements (PPREs). Endogenous ligands, such as steroids, retinoids, cholesterol metabolites, and dietary lipids, can activate PPARs.58 PPARs transcriptional activity can be increased or decreased depending on interaction with either coactivators, including the CBP/p300, the p160/SRC family (SRC-2/GRIP1/TIF2, SRC-3/Pcip/rac3/ACTR/AIB1/TRAM-1), and the PPARγ coactivator 1 (PGC-1) family (PGC-1α, PGC-1β)59 or corepressors, such as nuclear receptor corepressor protein (NCoR)60 and the silencing mediator of retinoid and thyroid hormone receptors (SMRT).61 There are three PPAR isoforms: PPAR-α (NR1C1), PPAR-γ (NR1C3), and PPAR-β/δ (NR1C2).62
PPARα
PPARα is predominantly expressed in tissues with high capacity for fatty acid oxidation (FAO), including the heart, liver, macrophages, muscle, and kidneys,63 and regulates the expression of proteins that are involved in this process.64 PPARα ligands, such as fibrates, lower plasma triglyceride levels and increase HDL levels.65
PPARα regulates the expression of genes that encode for proteins that are involved in FAO, such as cluster of differentiation (Cd)36, carnitine palmitoyl transferase I (Cpt1), diacylglycerol acyltransferase (Dgat), malonyl-CoA decarboxylase (Mcd), and fatty acid-binding protein (Fabp).66 Mice with global deletion of PPARα have lower levels of FAO, higher glucose oxidation, and increased hepatic lipid content.67 Cardiomyocyte PPARα is activated by fatty acids released from intracellular triglyceride stores to be catabolized for mitochondrial ATP synthesis.68,69 Accordingly, overexpression of PPARα increases FAO and decreases glucose oxidation.64 Constitutive cardiomyocyte-specific expression of PPARα (αMHC-PPARα) increases oxidation rate, as shown by increased palmitate turnover from triglyceride stores.70 Interestingly, along with increased lipid oxidation, constitutive cardiomyocyte PPARα expression leads to cardiac lipid accumulation.64 This is likely accounted for by increased fatty acid uptake through CD36,71 which also stimulates a positive-feedback PPARα activation loop that is constituted by phosphorylation of PPARα by GSK3α.72 The ventricular hypertrophy and dysfunction phenotype of these mice resembles diabetic cardiomyopathy because they have profound accumulation of intramyocardial triglycerides after short-term fasting.64 It has been shown that PPARα cooperates synergistically with KLF15 to induce gene expression,73 whereas binding of Sirtuin (SIRT) 1 on PPARα has the opposite effect.74 In conclusion, PPARα plays a central role in stimulating fatty acid uptake and oxidation.
Various factors activate PPARα expression, such as glucocorticoids,75 farnesoid X receptor (FXR),76 AMP-activated protein kinase (AMPK),77–79 estrogen-related receptor (EER)α,80 retinoic acid,81 retinoid X receptor (RxR),82 phorbol-12-myristate-13-acetate,83 branched chain amino acids,84 nitrate,85 heat shock factor-1,86 diabetes,64,87 and exercise training.88 On the other hand, cardiac PPARα expression is reduced during heart failure,89 MI,90 hypoxia,91,92 endotoxemia,93,94 and polymicrobial sepsis.95 The details of the mechanisms that lead to this reduction remain elusive. The JNK signaling pathway has been associated with reduced cardiac PPARα gene expression.96 Many factors, such as IL-1β,97 IL-6,97 PPARδ,98,99 NF-κ?,100 glucose,101,102 insulin,103 Akt,104 c-Myc,105 the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway,106 periostin,107 reactive oxygen species,100 growth hormone,108 androgens,109 and angiotensin II,110 have been reported to downregulate Ppara expression.
Pharmacologic activation or constitutive cardiomyocyte expression of PPARα causes intramyocardial lipid accumulation and cardiac dysfunction64,111 accompanied by the activation of protein kinase C signaling and β-adrenergic receptor desensitization.112 Adrenergic signaling attenuation may also be accounted for by phosphodiesterase 1C, which is stimulated by PPARα.113 PPARα−/− mice have decreased myocardial fatty acid metabolism.114–116 Despite the importance of FAO for cardiac function,117 these mice have normal cardiac function at baseline according to some studies.116,118,119 This suggests the activation of other proteins that may substitute PPARα in stimulating FAO. Conversely, other studies showed that PPARα−/− mice have reduced cardiac function at baseline, which was attributed to fibrosis,115,120 abnormal mitochondrial structure, and oxidative stress.121,122 Following metabolic stress, such as starvation, exposure to high temperature, and high workload, PPARα−/− mice had lower levels of cardiac ATP115,119 and lower contractile performance.119 Stimulation of β1-adrenergic receptors by isoproterenol reduced positive inotropic effect.120 Short-term starvation67,118 and CPT1 inhibition114 caused hepatic and cardiac lipid accumulation and hypoglycemia.
These studies indicate that PPARα is important for activation of cardiac FAO and inhibition of glucose utilization. It is possible that PPARα−/− mice do not develop cardiac dysfunction at baseline because of either compensatory activation of other PPAR isoforms or higher glucose utilization.118 Nevertheless, activation of PPARγ may act as an insulin-sensitizing process that promotes glucose uptake. However, this compensation is not sufficient when prolonged during myocardial stress.
PPARγ
PPARγ is expressed mainly in adipose tissue. It has a pivotal role in adipogenesis, thermogenesis, and fatty acid and triglyceride storage.63,123,124 TZDs, recognized as “glitazones,” are PPARγ ligands, which act as insulin sensitizers that lower plasma glucose.125 However, until today, there is a lot of controversy about the association of PPARγ agonists with cardiovascular risk and whether they can cause heart failure via direct actions on the heart or indirectly via increased salt and water retention.20
There is a broad range of transcriptional factors and physiological stimuli that upregulate PPARγ expression, such as C/EBPs,126,127 estrogen,128 Krüppel-like factor (KLF)5,129 KLF15,130 MEK/ERK signaling,131 c-Fos132 TGF-β,133 Smad1,134 p38 kinase, early growth-response factor-1 (Egr-1),135 FGF21,136,137 ghrelin,138 polyunsaturated fatty acids,102,139,140 the orphan nuclear receptor RORα,141 the zinc-finger protein Zfp423,142 and vitamin E.143 Transcriptional factors and stimuli that suppress PPARγ expression are lipopolysacharides,144,145 JNK,146–148 TNFα,149–152 IL-11,151 CCAAT/enhancer-binding protein homologous protein (CHOP),153 retinoic acid,81,154 estrogen receptor (ER)-α,155 KLF2,146,156 KLF7,157 the JAK/STAT pathway,106,126,127 interferon-gamma,144,158 leptin,159 angiotensin II,110 fasting,160 and androgens.161
Both gain and loss of PPARγ function mouse models have been generated. Mice with cardiomyocyte-specific constitutive PPARγ1 expression have increased cardiac lipid accumulation, abnormal mitochondrial structure, and dilated cardiomyopathy.124 This mouse model has also been described as a model of arrhythmia and sudden death, which is attributed to prolongation of the QRS and QT intervals, polymorphic ventricular tachycardia, and ventricular fibrillation.162 The level of PPARγ overexpression seems to influence the timing and severity of the phenotype.124
Although constitutive expression of cardiomyocyte PPARγ is clearly cardiotoxic, ablation of PPARγ does not seem to be protective either. Mice with global PPARγ deletion are lethal, and the embryos have cardiac abnormalities caused by placental defects.163 Cardiomyocyte-specific PPAγ−/− mice develop cardiac hypertrophy with preserved systolic cardiac function, which is not associated with altered cardiac metabolism.164–166 Hypertrophy has been attributed to elevated NF-κB expression,164 macrophage infiltration,165 and cardiomyocyte elongation.167 Cardiomyocyte-specific PPARγ−/− mice have increased oxidative damage, mitochondrial abnormalities, and die from dilated cardiomyopathy.168 Interestingly, PPARγ ablation was not accompanied by changes in the expression profile of PPARγ target genes. This implies that compensatory mechanisms, likely via the activation of other PPAR isoforms, are activated when PPARγ is blocked. Inducible cardiomyocyte-specific PPARγ deletion decreases the expression of FAO-related genes and proteins and lowers fatty acid utilization, whereas glucose utilization does not change.169 Thus, although activation of PPARγ has been attributed to cardiotoxicity, its inhibition also compromises cardiomyocyte biology and leads to cardiac dysfunction.
Combined Activation of PPARα and PPARγ
Controversial conclusions have been reported about the effects of dual PPARα/γ agonists on atherosclerotic plaque formation. Despite findings suggesting that treatment with a dual PPARα/γ agonist (tesaglitazar) stabilizes atherosclerotic plaque in diabetic LDLr−/− mice,170 an experimental model of insulin resistance and dyslipidemia showed increased macrophage-rich plaque formation in apolipoprotein E–knockout mice receiving the dual PPARα/γ agonist (S)-3-(4-(2-carbazol(phenoxazin)-9-yl-ethoxy)phenyl)-2-ethoxypropionic acid (compound 3q).171 The latter was associated with higher vascular expression of P-selectin, MCP-1, VCAM-1, and CD36, which are markers of endothelial activation and inflammation associated with plaque complexity.171 These findings provided a potential framework for investigation on whether dual α/γ agonists are associated with high risk for cardiovascular adverse effects, when used by individuals with insulin resistance.22
A study in rats showed that daily treatment with suprapharmacological doses of the PPARα/γ agonist LY510929 increases heart weight, left ventricular lumen volume, and wall thickness in male and female rats on a dose-dependent manner. This study identified plasma aminoterminal proatrial natriuretic peptide (NTproANP) levels as a potential biomarker of the progression of PPARα/γ agonist–induced left ventricular hypertrophy in rats.49
A possible explanation for the observed heart failure pertains to glucolipotoxicity that is driven by the combined increase of PPARγ-driven insulin sensitization and glucose uptake in the setting of higher PPARα-induced activation of fatty acid uptake and oxidation.172 This hypothesis corroborated previous studies showing that PPARγ can induce cardiac FAO-related gene expression124 without causing cardiac dysfunction when PPARα is inhibited.173,174
Newer studies124,173,174 indicated that combined PPARα and PPARγ activation compromise cardiac function. Constitutive cardiomyocyte-specific overexpression of PPARγ causes intramyocardial lipid accumulation and cardiac dysfunction124 characterized by ventricular tachycardia and fibrillation.162 Interestingly, cardiac dysfunction seems to occur when both cardiomyocyte PPARα and PPARγ are present. Along these lines, constitutive PPARγ expression in cardiomyocytes of PPARα-knockout mice did not result in cardiac dysfunction, despite the increased myocardial lipid content.173 Pharmacologic activation of PPARγ with TZDs promotes cardiomyocyte hypertrophy in vitro.175 However, it has also been proposed that the cardiotoxic effect of rosiglitazone may be independent to cardiomyocyte PPARγ activation as shown in mice with cardiomyocyte-specific deletion of PPARγ, which develop cardiac hypertrophy that is further exacerbated by treatment with rosiglitazone.164,176 Availability of fatty acids that follows lipoprotein-derived or intracellular triglyceride hydrolysis is a critical component for the activation of PPARs.68,177
PPARγ-mediated cardiac toxicity occurs primarily in the presence of PPARα. Therefore, cardiac dysfunction of mice that overexpress cardiomyocyte PPARγ may be due to fatty acid–mediated PPARα activation, which does not occur in the aMHC-Pparg;Ppara–/– mice.173 Nevertheless, there are mixed results on PPARγ-driven activation of PPARα with some studies showing that rosiglitazone increases PPARα expression,178 although others showing reduced expression of PPARα-target genes.179 Along these lines, previous studies have shown that activation of either PPARα or PPARγ in cardiomyocytes is associated with accumulation of acyl-carnitines and diacylglycerols in cardiomyocytes that lead to cardiac dysfunction,112,173 although cardiac acyl-carnitine content was reduced in the aMHC-Pparg;Ppara–/– mice.173
In accordance with those studies, a recent one focusing on tesaglitazar has confirmed that cardiac dysfunction is associated with alterations in cardiac content of triglycerides, acyl-carnitines, diacylglycerols, and phosphatidic acid.180 Combined treatment with tesaglitazar and resveratrol, which blocked the cardiotoxic effect of tesaglitazar, restored normal levels of acyl-carnitines, phosphatidic acid, and phosphatidylcholine.180 In a similar context, activation of different PPAR isoforms seems to alter representation of lipid species in total lipidome. Specifically, rats treated with a PPARα agonist had higher hepatic saturated fatty acids and monounsaturated fatty acids and lower ω−6 and ω−3 polyunsaturated fatty acids.181 On the other hand, treatment with a PPARγ agonist resulted in lower plasma triglycerides and phospholipids, higher hepatic phospholipids, lower total cardiac fatty acids and saturated fatty acids, and higher cardiac ω−6 polyunsaturated fatty acids.181 Thus, the modulation of relative activation of PPARα and PPARγ by different dual PPARα/γ agonists may result in differential organ lipid content that may eventually protect or compromise organ function.
Combined PPARα/γ activation by tesaglitazar suppresses gene expression of SIRT1 and activation of PGC1α, which eventually leads to lower mitochondrial abundance.180 Inhibition of PGC1α expression also occurs in aMHC-Pparg when treated with a PPARα agonist.173 PGC-1α is the main coactivator for PPARs that serves as a scaffold, recruiting regulatory proteins for chromatin remodeling and transcription activation.182–184 PGC1a controls FAO-related gene expression185 and mitochondrial biogenesis.186 Cardiac PGC1α gene expression is decreased in rodents and humans with heart failure.187 Similarly, PGC1α-knockout mice develop cardiac dysfunction,185 and they aggravate heart failure following pressure overload.188 Surprisingly, overexpression of cardiomyocyte PGC1α also leads to cardiac dysfunction.186 The mechanism behind the toxic effect of increased levels of PGC1α is attributed to the impairment of mitochondrial biogenesis and function.186 On the other hand, short-term PGC1α overexpression in cardiomyocytes improves mitochondrial biogenesis and oxidative respiration and cardiac function. Thus, dilated cardiomyopathy of the αMHC-PGC1α transgenic mice is most likely due to the profound and long-term increase of PGC1α expression. Nevertheless, the same study186 reported that transgenic lines with constitutive but not extremely high expression of PGC1α have normal cardiac function. Thus, the level of PGC1α activity seems to be a critical factor for the protective or aggravating role of this protein.
The activity of PGC1α is regulated by posttranslational mechanisms, including phosphorylation and acetylation. Acetylation of PGC1α is mainly controlled by the enzymatic activities of the acetyltransferase (general control of amino acid synthesis) GCN5 and the deacetylase SIRT1. A recent study showed that the dual PPARα/γ agonist (tesaglitazar) reduces cardiac SIRT1 expression and increases PGC1α acetylation. SIRT1 inhibition has been previously linked with cardiac dysfunction in several forms of cardiac stress. Resveratrol, which activates PPARγ,189 SIRT1, and PGC1α,190 maintained the beneficial antihyperlipidemic and antihyperglycemic effects of tesaglitazar and simultaneously negated the cardiotoxic effect of dual PPARα/γ activation while increasing mitochondrial abundance.180 Conversely, treatment of PPARα−/− mice with tesaglitazar did not change PGC1α and SIRT1 expression and had a less profound effect in cardiac function compared with C57BL/6 mice that were also treated with tesaglitazar.180 Thus, PPARα activation seems to account primarily for the cardiotoxic effects of dual PPARα/γ activation. Future studies on the role of PPARα in stimulating transcriptional and posttranslational repressors of PGC1α and SIRT1 would be useful to delineate the pathways that mediate the cardiotoxic effects of PPARα activation by single or dual agonists. Several factors that suppress PGC1α expression have been described so far in different tissues, including the histone methyltransferase Smyd1,191 hypoxia inducible factor,192 p160 myb binding protein193, and various miRs, such as miR-23a,194 miR-696,195 miR-211,196 miR-130b,197 miR-494,198 miR-29a,199 miR-485–5p, miR-485–3p,200 and miR-30b.201 Accordingly, the role of combined activation of PAPRα and PPARγ in stimulating suppressors of SIRT1 or altering NAD+/NADH ratio is another potential area for investigation that can delineate how dual PPARα/γ activation suppresses SIRT1 expression.180
Administration of rosiglitazone in mice with low levels of cardiac PPARα expression increases PGC1α expression,174 whereas PPARγ activation in LDLr−/− mice fed with high-fat diet, which increases cardiac PPARα levels,202,203 reduces PGC1α expression,179 and causes cardiac hypertrophy. It has also been shown that treatment of mice with tesaglitazar increases PPARδ expression,180 which regulates cardiac FAO.204 PPARδ increases expression of UCP3 in the heart204 and skeletal muscle.205,206 Cardiomyocyte-specific overexpression of PPARδ increases glucose utilization and glycogen content without affecting fatty acid utilization, lipid content, and cardiac function.207 Inducible cardiomyocyte-specific expression of constitutively active PPARδ also increases glucose utilization.208 However, these mice have increased cardiac FAO, decreased glycogen content, and increased mitochondrial biogenesis without oxidative stress.208 Thus, the level of activation of PPARδ seems to define the effect of this transcriptional factor independently from its expression levels.
These observations signify the importance of future studies focusing on the interplay between all three PPAR isoforms and their role in leading to PGC1α inhibition, when combined activation occurs. Furthermore, studies on potential cardioprotection by PPARα inhibition or activation of the SIRT1-PGC1α signaling in diabetes animal models that will be subjected to glitazars treatment are warranted.
CONCLUSIONS
Modulation of PPAR signaling has attracted a lot of interest for discovery and development of drugs for the treatment of diabetes. Single agonists of PPARα (fibrates) have been used as an adjunct therapy in the treatment of hypertriglyceridemia and hypercholesterolemia.65 In addition, single PPARγ agonists (TZDs) have been used to treat hyperglycemia.125 To alleviate toxicity of single PPAR agonists, dual PPARα/γ agonists (glitazars) were developed with the expectation to be an effective treatment for diabetic patients. Although these drugs were successful in improving insulin sensitivity and treating metabolic conditions associated with diabetes, most of glitazars have been suspended due to adverse effects on the urothelial, renal, and cardiovascular system (Fig. 3). Various studies have identified effects of combined PPARα and PPARγ activation on SIRT1 and PGC1γ expression and signaling. Interestingly, activation of the SIRT1-PGC1α metabolic network by resveratrol ameliorates the toxic effects of combined PPARα/γ activation.180
FIGURE 3.

Cardiovascular-related beneficial and toxic effects of glitazars. Figures were produced using Servier medical art (http://www.servier.com).
Up to date, saroglitazar seems to be the only dual PPARα/γ agonist with antihyperlipidemic and antihyperglycemic action that does not have serious adverse effects. However, it is accompanied by a warning and precautionary statement for use by patients at high risk for heart failure.4 Investigation on whether saroglitazar is less toxic due to milder effects on SIRT1-PGC1α signaling and mitochondrial biology remains to be pursued. Additional studies are needed to explore whether combined use of glitazars with resveratrol or other SIRT1 activators will reduce the adverse effects of glitazars in humans. Also, further studies are necessary to elucidate whether combined PPARγ activation and PPARα inhibition can alleviate toxicity that has been speculated for the use of single PPARγ agonists, such as TZDs.
The goal of the present review article pertains primarily to understanding of the biological mechanisms that underlie cardiac toxicity of combined activation of PPARα and PPARγ, which materialized in several clinical applications of the dual PPARα/γ agonists. Undoubtedly, in the era of the use of sodium/glucose cotransporter 2 inhibitors and metformin, development of dual PPAR agonists is not of highest priority for the pharma industry. However, single PPARα or PPARγ agonists are still used. Importantly, in several cases, activation of a PPAR isoform may induce the expression of others, which eventually can resemble the situation of dual PPAR agonists, especially in the context of T2D that incurs aberrant load of tissues with lipids that can activate PPARs. Therefore, elucidation of the mechanisms, which underlie toxicity of single or multi-PPAR agonists, holds significant potential for the improvement of safety of existing treatments. Thus, understanding of critical pathophysiology aspects of dual PPARα/γ activation is important from the scientific point of view, as well as for the drug development industry.
Acknowledgments
Supported by the National Heart Lung and Blood Institute of the National Institutes of Health (Bethesda, MD) (HL130218, HL151924—K.D.), the PA Cure, and the “Stavros Niarchos Foundation” Research Training Program in Clinical & Experimental Medicine organized by the World Hellenic Biomedical Association-WHBA, Inc (C.K.).
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.World Health Organization. Diabetes. 2018. Available at: www.who.int/news-room/fact-sheets/detail/diabetes. Accessed October 30, 2018.
- 2.Centers for Disease Control and Prevention. Diabetes. 2019; Available at: www.cdc.gov/diabetes. Accessed January 20, 2020.
- 3.Nathan DM, Buse JB, Davidson MB, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2009;32:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatterjee S, Majumder A, Ray S. Observational study of effects of Saroglitazar on glycaemic and lipid parameters on Indian patients with type 2 diabetes. Sci Rep. 2015;5:7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oakes ND, Thalén P, Hultstrand T, et al. Tesaglitazar, a dual PPAR {alpha}/{gamma} agonist, ameliorates glucose and lipid intolerance in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R938–R946. [DOI] [PubMed] [Google Scholar]
- 6.Chakrabarti R, Vikramadithyan RK, Misra P, et al. Ragaglitazar: a novel PPAR alpha PPAR gamma agonist with potent lipid-lowering and insulin-sensitizing efficacy in animal models. Br J Pharmacol. 2003;140:527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain N, Bhansali S, Kurpad AV, et al. Effect of a dual PPAR alpha/gamma agonist on insulin sensitivity in patients of type 2 diabetes with hypertriglyceridemia- randomized double-blind placebo-controlled trial. Sci Rep. 2019;9:19017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balachandran K Dual PPAR alpha/gamma agonists: continuing cardiac concerns. Indian J Endocrinol Metab. 2019;23:586–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balakumar P, Mahadevan N, Sambathkumar R. A contemporary overview of PPARalpha/gamma dual agonists for the management of diabetic dyslipidemia. Curr Mol Pharmacol. 2019;12:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munigoti SP, Harinarayan CV. Role of Glitazars in atherogenic dyslipidemia and diabetes: two birds with one stone? Indian J Endocrinol Metab. 2014;18:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. [DOI] [PubMed] [Google Scholar]
- 12.Saha SA, Arora RR. Fibrates in the prevention of cardiovascular disease in patients with type 2 diabetes mellitus-a pooled meta-analysis of randomized placebo-controlled clinical trials. Int J Cardiol. 2010;141:157–166. [DOI] [PubMed] [Google Scholar]
- 13.Corti R, Osende J, Hutter R, et al. Fenofibrate induces plaque regression in hypercholesterolemic atherosclerotic rabbits: in vivo demonstration by high-resolution MRI. Atherosclerosis. 2007;190:106–113. [DOI] [PubMed] [Google Scholar]
- 14.Jeanpierre E, Tourneau T, Zawadzki C, et al. Beneficial effects of fenofibrate on plaque thrombogenicity and plaque stability in atherosclerotic rabbits. Cardiovasc Pathol. 2009;18:140–147. [DOI] [PubMed] [Google Scholar]
- 15.Dierkes J, Luley C, Westphal S. Effect of lipid-lowering and anti-hypertensive drugs on plasma homocysteine levels. Vasc Health Risk Manag. 2007;3:99–108. [PMC free article] [PubMed] [Google Scholar]
- 16.Elam M, Lovato L, Ginsberg H. The ACCORD-Lipid study: implications for treatment of dyslipidemia in Type 2 diabetes mellitus. Clin Lipidol. 2011;6:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marais GE, Larson KK. Rhabdomyolysis and acute renal failure induced by combination lovastatin and gemfibrozil therapy. Ann Intern Med. 1990;112:228–230. [DOI] [PubMed] [Google Scholar]
- 18.Yamashita S, Masuda D, Matsuzawa Y. Pemafibrate, a new selective PPARalpha modulator: drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr Atheroscler Rep 2020;22:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fruchart JC. Pemafibrate (K-877), a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc Diabetol. 2017;16:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seferovic PM, Petrie MC, Filippatos GS, et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2018;20:853–872. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein BJ, Rosenstock J, Anzalone D, et al. Effect of tesaglitazar, a dual PPAR alpha/gamma agonist, on glucose and lipid abnormalities in patients with type 2 diabetes: a 12-week dose-ranging trial. Curr Med Res Opin. 2006;22:2575–2590. [DOI] [PubMed] [Google Scholar]
- 22.Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581–2586. [DOI] [PubMed] [Google Scholar]
- 23.Erdmann E, Charbonnel B, Wilcox RG, et al. Pioglitazone use and heart failure in patients with type 2 diabetes and preexisting cardiovascular disease: data from the PROactive study (PROactive 08). Diabetes Care. 2007;30:2773–2778. [DOI] [PubMed] [Google Scholar]
- 24.Lincoff AM, Wolski K, Nicholls SJ, et al. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188. [DOI] [PubMed] [Google Scholar]
- 25.Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, et al. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. JAMA. 2010;304:411–418. [DOI] [PubMed] [Google Scholar]
- 26.Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Circulation. 2003;108:2941–2948. [DOI] [PubMed] [Google Scholar]
- 27.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. [DOI] [PubMed] [Google Scholar]
- 28.Home PD, Pocock SJ, Beck-Nielsen H, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373:2125–2135. [DOI] [PubMed] [Google Scholar]
- 29.Mahaffey KW, Hafley G, Dickerson S, et al. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J. 2013;166:240.e1. [DOI] [PubMed] [Google Scholar]
- 30.Kaul S, Bolger AF, Herrington D, et al. Thiazolidinedione drugs and cardiovascular risks: a science advisory from the American heart association and American College of Cardiology Foundation. Circulation. 2010;121:1868–1877. [DOI] [PubMed] [Google Scholar]
- 31.Lincoff AM, Tardif JC, Schwartz GG, et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA. 2014;311:1515–1525. [DOI] [PubMed] [Google Scholar]
- 32.Discovery Z Lipaglyn Product Information; 2013.
- 33.Henke BR, Blanchard SG, Brackeen MF, et al. N-(2-Benzoylphenyl)-L-tyrosine PPARgamma agonists. 1. Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J Med Chem. 1998;41:5020–5036. [DOI] [PubMed] [Google Scholar]
- 34.McHutchison J, Goodman Z, Patel K, et al. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology. 2010;138:1365–1372. [DOI] [PubMed] [Google Scholar]
- 35.Lohray BB, Lohray VB, Bajji AC, et al. (−)3-[4-[2-(Phenoxazin-10-yl) ethoxy]phenyl]-2-ethoxypropanoic acid [(−)DRF 2725]: a dual PPAR agonist with potent antihyperglycemic and lipid modulating activity. J Med Chem. 2001;44:2675–2678. [DOI] [PubMed] [Google Scholar]
- 36.Hegarty BD, Furler SM, Oakes ND, et al. Peroxisome proliferator-activated receptor (PPAR) activation induces tissue-specific effects on fatty acid uptake and metabolism in vivo-a study using the novel PPARalpha/gamma agonist tesaglitazar. Endocrinology. 2004;145:3158–3164. [DOI] [PubMed] [Google Scholar]
- 37.Imoto H, Matsumoto M, Odaka H, et al. Studies on non-thiazolidinedione antidiabetic agents. 3. Preparation and biological activity of the metabolites of TAK-559. Chem Pharm Bull. 2004;52:120–124. [DOI] [PubMed] [Google Scholar]
- 38.Sakamoto J, Kimura H, Moriyama S, et al. A novel oxyiminoalkanoic acid derivative, TAK-559, activates human peroxisome proliferator-activated receptor subtypes. Eur J Pharmacol. 2004;495:17–26. [DOI] [PubMed] [Google Scholar]
- 39.Ratner RE, Parikh S, Tou C. Efficacy, safety and tolerability of tesaglitazar when added to the therapeutic regimen of poorly controlled insulin-treated patients with type 2 diabetes. Diab Vasc Dis Res. 2007;4:214–221. [DOI] [PubMed] [Google Scholar]
- 40.Göke B, Gause-Nilsson I, Persson A. The effects of tesaglitazar as add-on treatment to metformin in patients with poorly controlled type 2 diabetes. Diab Vasc Dis Res. 2007;4:204–213. [DOI] [PubMed] [Google Scholar]
- 41.Wilding JP, Gause-Nilsson I, Persson A. Tesaglitazar, as add-on therapy to sulphonylurea, dose-dependently improves glucose and lipid abnormalities in patients with type 2 diabetes. Diab Vasc Dis Res. 2007;4:194–203. [DOI] [PubMed] [Google Scholar]
- 42.Bays H, McElhattan J, Bryzinski BS. A double-blind, randomised trial of tesaglitazar versus pioglitazone in patients with type 2 diabetes mellitus. Diab Vasc Dis Res. 2007;4:181–193. [DOI] [PubMed] [Google Scholar]
- 43.Kendall DM, Rubin CJ, Mohideen P, et al. Improvement of glycemic control, triglycerides, and HDL cholesterol levels with muraglitazar, a dual (alpha/gamma) peroxisome proliferator-activated receptor activator, in patients with type 2 diabetes inadequately controlled with metformin monotherapy: a double-blind, randomized, pioglitazone-comparative study. Diabetes Care. 2006;29:1016–1023. [DOI] [PubMed] [Google Scholar]
- 44.Long GG, Reynolds VL, Lopez-Martinez A, et al. Urothelial carcino-genesis in the urinary bladder of rats treated with naveglitazar, a gamma-dominant PPAR alpha/gamma agonist: lack of evidence for urolithiasis as an inciting event. Toxicol Pathol. 2008;36:218–231. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Chen H, Birnbaum Y, et al. Aleglitazar, a dual peroxisome proliferator-activated receptor-alpha and -gamma agonist, protects cardiomyocytes against the adverse effects of hyperglycaemia. Diab Vasc Dis Res. 2017;14:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian J, Chen H, Birnbaum Y, et al. Aleglitazar, a balanced dual PPARalpha and -gamma agonist, protects the heart against ischemiareperfusion injury. Cardiovasc Drugs Ther. 2016;30:129–141. [DOI] [PubMed] [Google Scholar]
- 47.Bopst M, Atzpodien EA. Non-clinical safety evaluation and risk assessment to human of aleglitazar, a dual PPAR alpha/gamma agonist, and its major human metabolite. Regul Toxicol Pharmacol. 2017;86:107–116. [DOI] [PubMed] [Google Scholar]
- 48.Jeong HW, Lee JW, Kim WS, et al. A newly identified CG301269 improves lipid and glucose metabolism without body weight gain through activation of peroxisome proliferator-activated receptor alpha and gamma. Diabetes. 2011;60:496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engle SK, Solter PF, Credille KM, et al. Detection of left ventricular hypertrophy in rats administered a peroxisome proliferator-activated receptor alpha/gamma dual agonist using natriuretic peptides and imaging. Toxicol Sci. 2010;114:183–192. [DOI] [PubMed] [Google Scholar]
- 50.Li PP, Shan S, Chen YT, et al. The PPARalpha/gamma dual agonist chiglitazar improves insulin resistance and dyslipidemia in MSG obese rats. Br J Pharmacol. 2006;148:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agrawal R The first approved agent in the Glitazar’s Class: Saroglitazar. Curr Drug Targets. 2014;15:151–155. [DOI] [PubMed] [Google Scholar]
- 52.Jain MR, Giri SR, Trivedi C, et al. Saroglitazar, a novel PPARalpha/gamma agonist with predominant PPARalpha activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol Res Perspect. 2015;3:e00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma A, Amarnath S, Kushwah DS. Saroglitazar, a novel cardiometabolic agent for diabetic dyslipidemia—a Review. J Young Pharm. 2014;7:2–6. [Google Scholar]
- 54.Stirban AO, Andjelkovic M, Heise T, et al. Aleglitazar, a dual peroxisome proliferator-activated receptor-alpha/gamma agonist, improves insulin sensitivity, glucose control and lipid levels in people with type 2 diabetes: findings from a randomized, double-blind trial. Diabetes Obes Metab. 2016;18:711–715. [DOI] [PubMed] [Google Scholar]
- 55.Harrity T, Farrelly D, Tieman A, et al. Muraglitazar, a novel dual (alpha/gamma) peroxisome proliferator-activated receptor activator, improves diabetes and other metabolic abnormalities and preserves beta-cell function in db/db mice. Diabetes. 2006;55:240–248. [PubMed] [Google Scholar]
- 56.Wallenius K, Kjellstedt A, Thalén P, et al. The PPAR alpha/gamma agonist, tesaglitazar, improves insulin mediated Switching of tissue glucose and free fatty acid utilization in vivo in the obese Zucker rat. PPAR Res. 2013;2013:305347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balakumar P, Rose M, Ganti SS, et al. PPAR dual agonists: are they opening Pandora’s Box? Pharmacol Res. 2007;56:91–98. [DOI] [PubMed] [Google Scholar]
- 58.Pol CJ, Lieu M, Drosatos K. PPARs: protectors or opponents of myocardial function? PPAR Res. 2015;2015:835985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gollamudi R, Gupta D, Goel S, et al. Novel orphan nuclear receptorscoregulator interactions controlling anti-cancer drug metabolism. Curr Drug Metab. 2008;9:611–613. [DOI] [PubMed] [Google Scholar]
- 60.Hörlein AJ, Näär AM, Heinzel T, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor corepressor. Nature. 1995;377:397–404. [DOI] [PubMed] [Google Scholar]
- 61.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. [DOI] [PubMed] [Google Scholar]
- 62.Poulsen L, Siersbæk M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23:631–639. [DOI] [PubMed] [Google Scholar]
- 63.Tyagi S, Gupta P, Saini AS, et al. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fruchart JC. Peroxisome proliferator-activated receptor-alpha (PPARalpha): at the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis. 2009;205:1–8. [DOI] [PubMed] [Google Scholar]
- 66.Lopaschuk GD, Ussher JR, Folmes CD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. [DOI] [PubMed] [Google Scholar]
- 67.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lahey R, Wang X, Carley AN, et al. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation. 2014;130:1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haemmerle G, Moustafa T, Woelkart G, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med. 2011;17:1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Banke NH, Wende AR, Leone TC, et al. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ Res. 2010;107:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem. 1998;273:23786–23792. [DOI] [PubMed] [Google Scholar]
- 72.Nakamura M, Liu T, Husain S, et al. Glycogen synthase kinase-3alpha promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab. 2019;29:1119–1134 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prosdocimo DA, John JE, Zhang L, et al. KLF15 and PPARalpha cooperate to regulate cardiomyocyte lipid gene expression and oxidation. PPAR Res. 2015;2015:201625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oka S, Zhai P, Yamamoto T, et al. Peroxisome proliferator activated receptor-alpha association with silent information regulator 1 suppresses cardiac fatty acid metabolism in the failing heart. Circ Heart Fail. 2015;8:1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lemberger T, Staels B, Saladin R, et al. Regulation of the peroxisome proliferator-activated receptor alpha gene by glucocorticoids. J Biol Chem. 1994;269:24527–24530. [PubMed] [Google Scholar]
- 76.Pineda Torra I, Claudel T, Duval C, et al. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–272. [DOI] [PubMed] [Google Scholar]
- 77.Lee WJ, Kim M, Park HS, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun. 2006;340:291–295. [DOI] [PubMed] [Google Scholar]
- 78.Meng RS, Pei ZH, Yin R, et al. Adenosine monophosphate-activated protein kinase inhibits cardiac hypertrophy through reactivating peroxisome proliferator-activated receptor-alpha signaling pathway. Eur J Pharmacol. 2009;620:63–70. [DOI] [PubMed] [Google Scholar]
- 79.Ravnskjaer K, Boergesen M, Dalgaard LT, et al. Glucose-induced repression of PPARalpha gene expression in pancreatic beta-cells involves PP2A activation and AMPK inactivation. J Mol Endocrinol. 2006;36:289–299. [DOI] [PubMed] [Google Scholar]
- 80.Huss JM, Torra IP, Staels B, et al. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Valmaseda A, Carmona MC, Barberá MJ, et al. Opposite regulation of PPAR-alpha and -gamma gene expression by both their ligands and retinoic acid in brown adipocytes. Mol Cell Endocrinol. 1999;154:101–109. [DOI] [PubMed] [Google Scholar]
- 82.Beigneux AP, Moser AH, Shigenaga JK, et al. The acute phase response is associated with retinoid X receptor repression in rodent liver. J Biol Chem. 2000;275:16390–16399. [DOI] [PubMed] [Google Scholar]
- 83.Yaacob NS, Norazmi MN, Gibson GG, et al. The transcription of the peroxisome proliferator-activated receptor alpha gene is regulated by protein kinase C. Toxicol Lett. 2001;125:133–141. [DOI] [PubMed] [Google Scholar]
- 84.Li Y, Xiong Z, Yan W, et al. Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-alpha pathway-dependent fatty acid oxidation. Theranostics. 2020;10:5623–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Horscroft JA, O’Brien KA, Clark AD, et al. Inorganic nitrate, hypoxia, and the regulation of cardiac mitochondrial respiration-probing the role of PPARalpha. FASEB J. 2019;33:7563–7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vallanat B, Anderson SP, Brown-Borg HM, et al. Analysis of the heat shock response in mouse liver reveals transcriptional dependence on the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha). BMC Genomics. 2010;11:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drosatos K, Pollak NM, Pol CJ, et al. Cardiac myocyte KLF5 regulates Ppara expression and cardiac function. Circ Res. 2016;118:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iemitsu M, Miyauchi T, Maeda S, et al. Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol. 2002;283:H1750–H1760. [DOI] [PubMed] [Google Scholar]
- 89.Karbowska J, Kochan Z, Smole[notdef]nski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett. 2003;8:49–53. [PubMed] [Google Scholar]
- 90.Masamura K, Tanaka N, Yoshida M, et al. Myocardial metabolic regulation through peroxisome proliferator-activated receptor alpha after myocardial infarction. Exp Clin Cardiol. 2003;8:61–66. [PMC free article] [PubMed] [Google Scholar]
- 91.Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001;166:7543–7548. [DOI] [PubMed] [Google Scholar]
- 92.Razeghi P, Young ME, Abbasi S, et al. Hypoxia in vivo decreases peroxisome proliferator-activated receptor alpha-regulated gene expression in rat heart. Biochem Biophys Res Commun. 2001;287:5–10. [DOI] [PubMed] [Google Scholar]
- 93.Feingold K, Kim MS, Shigenaga J, et al. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;286:E201–E207. [DOI] [PubMed] [Google Scholar]
- 94.Maitra U, Chang S, Singh N, et al. Molecular mechanism underlying the suppression of lipid oxidation during endotoxemia. Mol Immunol. 2009;47:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kokkinaki D, Hoffman M, Kalliora C, et al. Chemically synthesized Secoisolariciresinol diglucoside (LGM2605) improves mitochondrial function in cardiac myocytes and alleviates septic cardiomyopathy. J Mol Cell Cardiol. 2019;127:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Drosatos K, Drosatos-Tampakaki Z, Khan R, et al. Inhibition of C-JUNN-terminal kinase increases cardiac PPAR{alpha} expression and fatty acid oxidation and prevents LPS-induced heart dysfunction. J Biol Chem. 2011;286:36331–36339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Parmentier JH, Schohn H, Bronner M, et al. Regulation of CYP4A1 and peroxisome proliferator-activated receptor alpha expression by interleukin-1beta, interleukin-6, and dexamethasone in cultured fetal rat hepatocytes. Biochem Pharmacol. 1997;54:889–898. [DOI] [PubMed] [Google Scholar]
- 98.Chu R, Lin Y, Rao MS, et al. Cloning and identification of rat deoxyuridine triphosphatase as an inhibitor of peroxisome proliferator-activated receptor alpha. J Biol Chem. 1996;271:27670–27676. [DOI] [PubMed] [Google Scholar]
- 99.Shi Y, Hon M, Evans RM. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc Natl Acad Sci USA. 2002;99:2613–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cabrero A, Alegret M, Sanchez RM, et al. Increased reactive oxygen species production down-regulates peroxisome proliferator-activated alpha pathway in C2C12 skeletal muscle cells. J Biol Chem. 2002;277:10100–10107. [DOI] [PubMed] [Google Scholar]
- 101.Roduit R, Morin J, Massé F, et al. Glucose down-regulates the expression of the peroxisome proliferator-activated receptor-alpha gene in the pancreatic beta -cell. J Biol Chem. 2000;275:35799–35806. [DOI] [PubMed] [Google Scholar]
- 102.Joly E, Roduit R, Peyot ML, et al. Glucose represses PPARalpha gene expression via AMP-activated protein kinase but not via p38 mitogen-activated protein kinase in the pancreatic beta-cell. J Diabetes. 2009;1:263–272. [DOI] [PubMed] [Google Scholar]
- 103.Panadero M, Vidal H, Herrera E, et al. Nutritionally induced changes in the peroxisome proliferator-activated receptor-alpha gene expression in liver of suckling rats are dependent on insulinaemia. Arch Biochem Biophys. 2001;394:182–188. [DOI] [PubMed] [Google Scholar]
- 104.Cook SA, Matsui T, Li L, et al. Transcriptional effects of chronic Akt activation in the heart. J Biol Chem. 2002;277:22528–22533. [DOI] [PubMed] [Google Scholar]
- 105.Riu E, Ferre T, Mas A, et al. Overexpression of c-myc in diabetic mice restores altered expression of the transcription factor genes that regulate liver metabolism. Biochem J. 2002;368:931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou YC, Waxman DJ. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain. J Biol Chem. 1999;274:29874–29882. [DOI] [PubMed] [Google Scholar]
- 107.Bian X, Su X, Wang Y, et al. Periostin contributes to renal and cardiac dysfunction in rats with chronic kidney disease: reduction of PPARalpha. Biochimie. 2019;160:172–182. [DOI] [PubMed] [Google Scholar]
- 108.Carlsson L, Lindén D, Jalouli M, et al. Effects of fatty acids and growth hormone on liver fatty acid binding protein and PPARalpha in rat liver. Am J Physiol Endocrinol Metab. 2001;281:E772–E781. [DOI] [PubMed] [Google Scholar]
- 109.Collett GP, Betts AM, Johnson MI, et al. Peroxisome proliferator-activated receptor alpha is an androgen-responsive gene in human prostate and is highly expressed in prostatic adenocarcinoma. Clin Cancer Res. 2000;6:3241–3248. [PubMed] [Google Scholar]
- 110.Tham DM, Martin-McNulty B, Wang YX, et al. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics. 2002;11:21–30. [DOI] [PubMed] [Google Scholar]
- 111.Zungu M, Young ME, Stanley WC, et al. Chronic treatment with the peroxisome proliferator-activated receptor alpha agonist Wy-14,643 attenuates myocardial respiratory capacity and contractile function. Mol Cell Biochem. 2009;330:55–62. [DOI] [PubMed] [Google Scholar]
- 112.Drosatos K, Bharadwaj KG, Lymperopoulos A, et al. Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metab. 2011;300:E489–E499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shete V, Liu N, Jia Y, et al. Mouse cardiac Pde1C is a direct transcriptional target of Pparalpha. Int J Mol Sci. 2018;19:3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Djouadi F, Weinheimer CJ, Saffitz JE, et al. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor alpha- deficient mice. J Clin Invest. 1998;102:1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Watanabe K, Fujii H, Takahashi T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem. 2000;275:22293–22299. [DOI] [PubMed] [Google Scholar]
- 116.Campbell FM, Kozak R, Wagner A, et al. A role for peroxisome proliferator-activated receptor alpha (PPARalpha ) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098–4103. [DOI] [PubMed] [Google Scholar]
- 117.Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. 2016;118:1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu J, Wang P, He L, et al. Cardiomyocyte-Restricted deletion of PPARbeta/delta in PPARalpha-null mice causes impaired mitochondrial biogenesis and defense, but No further depression of myocardial fatty acid oxidation. PPAR Res. 2011;2011:372854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Luptak I, Balschi JA, Xing Y, et al. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112:2339–2346. [DOI] [PubMed] [Google Scholar]
- 120.Loichot C, Jesel L, Tesse A, et al. Deletion of peroxisome proliferator-activated receptor-alpha induces an alteration of cardiac functions. Am J Physiol Heart Circ Physiol. 2006;291:H161–H166. [DOI] [PubMed] [Google Scholar]
- 121.Guellich A, Damy T, Lecarpentier Y, et al. Role of oxidative stress in cardiac dysfunction of PPARalpha−/− mice. Am J Physiol Heart Circ Physiol. 2007;293:H93–H102. [DOI] [PubMed] [Google Scholar]
- 122.Guellich A, Damy T, Conti M, et al. Tempol prevents cardiac oxidative damage and left ventricular dysfunction in the PPAR-alpha KO mouse. Am J Physiol Heart Circ Physiol. 2013;304:H1505–H1512. [DOI] [PubMed] [Google Scholar]
- 123.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. [DOI] [PubMed] [Google Scholar]
- 124.Son NH, Park TS, Yamashita H, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heikkinen S, Auwerx J, Argmann CA. PPARgamma in human and mouse physiology. Biochim Biophys Acta. 2007;1771:999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clarke SL, Robinson CE, Gimble JM. CAAT/enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor gamma 2 promoter. Biochem Biophys Res Commun. 1997;240:99–103. [DOI] [PubMed] [Google Scholar]
- 127.Wu Z, Rosen ED, Brun R, et al. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell. 1999;3:151–158. [DOI] [PubMed] [Google Scholar]
- 128.Wang X, Kilgore MW. Signal cross-talk between estrogen receptor alpha and beta and the peroxisome proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7 breast cancer cells. Mol Cell Endocrinol. 2002;194:123–133. [DOI] [PubMed] [Google Scholar]
- 129.Oishi Y, Manabe I, Tobe K, et al. Krüppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27–39. [DOI] [PubMed] [Google Scholar]
- 130.Mori T, Sakaue H, Iguchi H, et al. Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005;280:12867–12875. [DOI] [PubMed] [Google Scholar]
- 131.Prusty D, Park BH, Davis KE, et al. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J Biol Chem. 2002;277:46226–46232. [DOI] [PubMed] [Google Scholar]
- 132.Xiao H, Leblanc SE, Wu Q, et al. Chromatin accessibility and transcription factor binding at the PPARgamma2 promoter during adipogenesis is protein kinase A-dependent. J Cell Physiol. 2011;226:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kintscher U, Wakino S, Bruemmer D, et al. TGF-beta(1) induces peroxisome proliferator-activated receptor gamma1 and gamma2 expression in human THP-1 monocytes. Biochem Biophys Res Commun. 2002;297:794–799. [DOI] [PubMed] [Google Scholar]
- 134.Hata K, Nishimura R, Ikeda F, et al. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol Biol Cell. 2003;14:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fu M, Zhang J, Lin Y, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor gamma (PPAR gamma) gene expression by transforming growth factor beta in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (AP1) and Smads. Biochem J. 2003;370:1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cai G, Liu J, Wang M, et al. Mutual promotion of FGF21 and PPARgamma attenuates hypoxia-induced pulmonary hypertension. Exp Biol Med. 2019;244:252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu J, Cai G, Li M, et al. Fibroblast growth factor 21 attenuates hypoxia-induced pulmonary hypertension by upregulating PPARgamma expression and suppressing inflammatory cytokine levels. Biochem Biophys Res Commun. 2018;504:478–484. [DOI] [PubMed] [Google Scholar]
- 138.Wang Q, Sui X, Chen R, et al. Ghrelin ameliorates angiotensin II-induced myocardial fibrosis by upregulating peroxisome proliferator-activated receptor gamma in young male rats. Biomed Res Int. 2018;2018:9897581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chambrier C, Bastard JP, Rieusset J, et al. Eicosapentaenoic acid induces mRNA expression of peroxisome proliferator-activated receptor gamma. Obes Res. 2002;10:518–525. [DOI] [PubMed] [Google Scholar]
- 140.Oster RT, Tishinsky JM, Yuan Z, et al. Docosahexaenoic acid increases cellular adiponectin mRNA and secreted adiponectin protein, as well as PPARgamma mRNA, in 3T3-L1 adipocytes. Appl Physiol Nutr Metab. 2010;35:783–789. [DOI] [PubMed] [Google Scholar]
- 141.Sundvold H, Lien S. Identification of a novel peroxisome proliferator-activated receptor (PPAR) gamma promoter in man and transactivation by the nuclear receptor RORalpha1. Biochem Biophys Res Commun. 2001;287:383–390. [DOI] [PubMed] [Google Scholar]
- 142.Gupta RK, Arany Z, Seale P, et al. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464:619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Landrier JF, Gouranton E, El Yazidi C, et al. Adiponectin expression is induced by vitamin E via a peroxisome proliferator-activated receptor gamma-dependent mechanism. Endocrinology. 2009;150:5318–5325. [DOI] [PubMed] [Google Scholar]
- 144.Welch JS, Ricote M, Akiyama TE, et al. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc Natl Acad Sci USA. 2003;100:6712–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jennewein C, von Knethen A, Schmid T, et al. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA destabilization. J Biol Chem. 2010;285:11846–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lee J, Lee J, Jung E, et al. Ultraviolet A regulates adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells via up-regulation of Kruppel-like factor 2. J Biol Chem. 2010;285:32647–32656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang Q, Chen C, Wu S, et al. Advanced glycation end products down-regulates peroxisome proliferator-activated receptor gamma expression in cultured rabbit chondrocyte through MAPK pathway. Eur J Pharmacol. 2010;649:108–114. [DOI] [PubMed] [Google Scholar]
- 148.Lee J, Jung E, Lee J, et al. Anti-adipogenesis by 6-thioinosine is mediated by downregulation of PPAR gamma through JNK-dependent up-regulation of iNOS. Cell Mol Life Sci. 2010;67:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang B, Berger J, Hu E, et al. Negative regulation of peroxisome proliferator-activated receptor-gamma gene expression contributes to the antiadipogenic effects of tumor necrosis factor-alpha. Mol Endocrinol. 1996;10:1457–1466. [DOI] [PubMed] [Google Scholar]
- 150.Xing H, Northrop JP, Grove JR, et al. TNF alpha-mediated inhibition and reversal of adipocyte differentiation is accompanied by suppressed expression of PPARgamma without effects on Pref-1 expression. Endocrinology. 1997;138:2776–2783. [DOI] [PubMed] [Google Scholar]
- 151.Meng L, Zhou J, Sasano H, et al. Tumor necrosis factor alpha and interleukin 11 secreted by malignant breast epithelial cells inhibit adipocyte differentiation by selectively down-regulating CCAAT/enhancer binding protein alpha and peroxisome proliferator-activated receptor gamma: mechanism of desmoplastic reaction. Cancer Res. 2001;61:2250–2255. [PubMed] [Google Scholar]
- 152.Kurebayashi S, Sumitani S, Kasayama S, et al. TNF-alpha inhibits 3T3-L1 adipocyte differentiation without downregulating the expression of C/EBPbeta and delta. Endocr J. 2001;48:249–253. [DOI] [PubMed] [Google Scholar]
- 153.Park SH, Choi HJ, Yang H, et al. Repression of peroxisome proliferator-activated receptor gamma by mucosal ribotoxic insult-activated CCAAT/enhancer-binding protein homologous protein. J Immunol. 2010;185:5522–5530. [DOI] [PubMed] [Google Scholar]
- 154.Lobo GP, Amengual J, Li HN, et al. Beta,beta-carotene decreases peroxisome proliferator receptor gamma activity and reduces lipid storage capacity of adipocytes in a beta,beta-carotene oxygenase 1-dependent manner. J Biol Chem. 2010;285:27891–27899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xiao J, Wang NL, Sun B, et al. Estrogen receptor mediates the effects of pseudoprotodiocsin on adipogenesis in 3T3-L1 cells. Am J Physiol Cell Physiol. 2010;299:C128–C138. [DOI] [PubMed] [Google Scholar]
- 156.Banerjee SS, Feinberg MW, Watanabe M, et al. The Krüppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem. 2003;278:2581–2584. [DOI] [PubMed] [Google Scholar]
- 157.Kawamura Y, Tanaka Y, Kawamori R, et al. Overexpression of Kruppel-like factor 7 regulates adipocytokine gene expressions in human adipocytes and inhibits glucose-induced insulin secretion in pancreatic beta-cell line. Mol Endocrinol. 2006;20:844–856. [DOI] [PubMed] [Google Scholar]
- 158.Waite KJ, Floyd ZE, Arbour-Reily P, et al. Interferon-gamma-induced regulation of peroxisome proliferator-activated receptor gamma and STATs in adipocytes. J Biol Chem. 2001;276:7062–7068. [DOI] [PubMed] [Google Scholar]
- 159.Zhou Y, Jia X, Qin J, et al. Leptin inhibits PPARgamma gene expression in hepatic stellate cells in the mouse model of liver damage. Mol Cell Endocrinol. 2010;323:193–200. [DOI] [PubMed] [Google Scholar]
- 160.Escher P, Braissant O, Basu-Modak S, et al. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142:4195–4202. [DOI] [PubMed] [Google Scholar]
- 161.Kajita K, Ishizuka T, Mune T, et al. Dehydroepiandrosterone down-regulates the expression of peroxisome proliferator-activated receptor gamma in adipocytes. Endocrinology. 2003;144:253–259. [DOI] [PubMed] [Google Scholar]
- 162.Morrow JP, Katchman A, Son NH, et al. Mice with cardiac overexpression of peroxisome proliferator-activated receptor gamma have impaired Repolarization and Spontaneous fatal ventricular arrhythmias. Circulation. 2011;124:2812–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Barak Y, Nelson MC, Ong ES, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. [DOI] [PubMed] [Google Scholar]
- 164.Duan SZ, Ivashchenko CY, Russell MW, et al. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372–379. [DOI] [PubMed] [Google Scholar]
- 165.Caglayan E, Stauber B, Collins AR, et al. Differential roles of cardiomyocyte and macrophage peroxisome proliferator-activated receptor gamma in cardiac fibrosis. Diabetes. 2008;57:2470–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Barbieri M, Di Filippo C, Esposito A, et al. Effects of PPARs agonists on cardiac metabolism in littermate and cardiomyocyte-specific PPAR-gamma-knockout (CM-PGKO) mice. PLoS One. 2012;7:e35999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hinrichs S, Heger J, Schreckenberg R, et al. Controlling cardiomyocyte length: the role of renin and PPAR-{gamma}. Cardiovasc Res. 2011;89:344–352. [DOI] [PubMed] [Google Scholar]
- 168.Ding G, Fu M, Qin Q, et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res. 2007;76:269–279. [DOI] [PubMed] [Google Scholar]
- 169.Luo J, Wu S, Liu J, et al. Conditional PPARgamma knockout from cardiomyocytes of adult mice impairs myocardial fatty acid utilization and cardiac function. Am J Transl Res. 2010;3:61–72. [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang BC, Li XK, Che WL, et al. [Peroxisome proliferator-activated receptor alpha/gamma agonist tesaglitazar stabilizes atherosclerotic plaque in diabetic low density lipoprotein receptor knockout mice]. Zhonghua Xin Xue Guan Bing Za Zhi. 2013;41:143–149. [PubMed] [Google Scholar]
- 171.Calkin AC, Allen TJ, Lassila M, et al. Increased atherosclerosis following treatment with a dual PPAR agonist in the ApoE knockout mouse. Atherosclerosis. 2007;195:17–22. [DOI] [PubMed] [Google Scholar]
- 172.Nolan CJ, Ruderman NB, Kahn SE, et al. Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes. 2015;64:673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Son NH, Yu S, Tuinei J, et al. PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARalpha deficiency despite increases in fatty acid oxidation. J Clin Invest. 2010;120:3443–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Drosatos K, Khan RS, Trent CM, et al. Peroxisome proliferator-activated receptor-gamma activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pharaon LF, El-Orabi NF, Kunhi M, et al. Rosiglitazone promotes cardiac hypertrophy and alters chromatin remodeling in isolated cardiomyocytes. Toxicol Lett. 2017;280:151–158. [DOI] [PubMed] [Google Scholar]
- 176.He H, Tao H, Xiong H, et al. Rosiglitazone causes cardiotoxicity via peroxisome proliferator-activated receptor gamma-independent mitochondrial oxidative stress in mouse hearts. Toxicol Sci. 2014;138:468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ziouzenkova O, Perrey S, Asatryan L, et al. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: evidence for an antiinflammatory role for lipoprotein lipase. Proc Natl Acad Sci USA. 2003;100:2730–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lee HH, Yeh CH, Chen YT, et al. Effects of metformin on rosiglitazone-induced cardiac hypertrophy in mice. Biol Pharm Bull. 2010;33:1506–1510. [DOI] [PubMed] [Google Scholar]
- 179.Verschuren L, Wielinga PY, Kelder T, et al. A systems biology approach to understand the pathophysiological mechanisms of cardiac pathological hypertrophy associated with rosiglitazone. BMC Med Genomics. 2014;7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kalliora C, Kyriazis ID, Oka SI, et al. Dual peroxisome-proliferator-activated-receptor-alpha/gamma activation inhibits SIRT1-PGC1alpha axis and causes cardiac dysfunction. JCI Insight 2019;5:e129556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Strand E, Lysne V, Grinna ML, et al. Short-term activation of peroxisome proliferator-activated receptors alpha and gamma induces tissue-specific effects on lipid metabolism and fatty acid composition in male wistar rats. PPAR Res. 2019;2019:8047627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. [DOI] [PubMed] [Google Scholar]
- 183.Wallberg AE, Yamamura S, Malik S, et al. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1alpha. Mol Cell. 2003;12:1137–1149. [DOI] [PubMed] [Google Scholar]
- 184.Puigserver P, Adelmant G, Wu Z, et al. Activation of PPARgamma coactivator-1 through transcription factor docking. Science. 1999;286:1368–1371. [DOI] [PubMed] [Google Scholar]
- 185.Arany Z, He H, Lin J, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. [DOI] [PubMed] [Google Scholar]
- 186.Lehman JJ, Barger PM, Kovacs A, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sihag S, Cresci S, Li AY, et al. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Arany Z, Novikov M, Chin S, et al. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci USA. 2006;103:10086–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hoseini A, Namazi G, Farrokhian A, et al. The effects of resveratrol on metabolic status in patients with type 2 diabetes mellitus and coronary heart disease. Food Funct. 2019;10:6042–6051. [DOI] [PubMed] [Google Scholar]
- 190.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. [DOI] [PubMed] [Google Scholar]
- 191.Warren JS, Tracy CM, Miller MR, et al. Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc Natl Acad Sci USA. 2018;115:E7871–E7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.LaGory EL, Wu C, Taniguchi CM, et al. Suppression of PGC-1alpha is critical for Reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep. 2015;12:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fan M, Rhee J, St-Pierre J, et al. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sun LY, Wang N, Ban T, et al. MicroRNA-23a mediates mitochondrial compromise in estrogen deficiency-induced concentric remodeling via targeting PGC-1alpha. J Mol Cell Cardiol. 2014;75:1–11. [DOI] [PubMed] [Google Scholar]
- 195.Aoi W, Naito Y, Mizushima K, et al. The microRNA miR-696 regulates PGC-1{alpha} in mouse skeletal muscle in response to physical activity. Am J Physiol Endocrinol Metab. 2010;298:E799–E806. [DOI] [PubMed] [Google Scholar]
- 196.Sahoo A, Lee B, Boniface K, et al. MicroRNA-211 regulates oxidative phosphorylation and energy metabolism in human vitiligo. J Invest Dermatol. 2017;137:1965–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jiang S, Teague AM, Tryggestad JB, et al. Role of microRNA-130b in placental PGC-1alpha/TFAM mitochondrial biogenesis pathway. Biochem Biophys Res Commun. 2017;487:607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lemecha M, Morino K, Imamura T, et al. MiR-494–3p regulates mitochondrial biogenesis and thermogenesis through PGC1-alpha signalling in beige adipocytes. Sci Rep. 2018;8:15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Caravia XM, Fanjul V, Oliver E, et al. The microRNA-29/PGC1alpha regulatory axis is critical for metabolic control of cardiac function. PLoS Biol. 2018;16:e2006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lou C, Xiao M, Cheng S, et al. MiR-485–3p and miR-485–5p suppress breast cancer cell metastasis by inhibiting PGC-1alpha expression. Cell Death Dis. 2016;7:e2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhang Q, Ma XF, Dong MZ, et al. MiR-30b-5p regulates the lipid metabolism by targeting PPARGC1A in Huh-7 cell line. Lipids Health Dis. 2020;19:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Li Y, Cheng L, Qin Q, et al. High-fat feeding in cardiomyocyte-restricted PPARdelta knockout mice leads to cardiac overexpression of lipid metabolic genes but fails to rescue cardiac phenotypes. J Mol Cell Cardiol. 2009;47:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cole MA, Abd Jamil AH, Heather LC, et al. On the pivotal role of PPARalpha in adaptation of the heart to hypoxia and why fat in the diet increases hypoxic injury. FASEB J. 2016;30:2684–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cheng L, Ding G, Qin Q, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. [DOI] [PubMed] [Google Scholar]
- 205.Chevillotte E, Rieusset J, Roques M, et al. The regulation of uncoupling protein-2 gene expression by omega-6 polyunsaturated fatty acids in human skeletal muscle cells involves multiple pathways, including the nuclear receptor peroxisome proliferator-activated receptor beta. J Biol Chem. 2001;276:10853–10860. [DOI] [PubMed] [Google Scholar]
- 206.Dressel U, Allen TL, Pippal JB, et al. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol. 2003;17:2477–2493. [DOI] [PubMed] [Google Scholar]
- 207.Burkart EM, Sambandam N, Han X, et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Liu J, Wang P, Luo J, et al. Peroxisome proliferator-activated receptor beta/delta activation in adult hearts facilitates mitochondrial function and cardiac performance under pressure-overload condition. Hypertension. 2011;57:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]