

Abstract
Background:
Lipoprotein (a) [Lp(a)] is a risk factor for coronary heart disease and calcific aortic valve disease. We determined the relationships of Lp(a) with prevalence and progression of coronary artery calcification (CAC), mitral annular calcification (MAC), and thoracic aortic calcification (TAC) in a multi-ethnic cohort of middle to older-aged adults.
Methods:
This analysis included 6705 Multi-Ethnic Study of Atherosclerosis participants. Lp(a) was measured with a turbidimetric immunoassay. CAC, MAC, and TAC were assessed by cardiac computed tomography both at baseline and once during follow-up.
Results:
In adjusted relative risk regression cross-sectional analysis, a Lp(a) level ≥50 mg/dL was associated with a 22% higher prevalence of MAC (relative risk (RR)=1.22, 95% confidence interval (CI) 1.00, 1.49). No significant associations were observed for prevalent CAC or TAC. In adjusted prospective analyses, participants with Lp(a) ≥50 mg/dL were at significantly higher risk for rapid CAC progression (median follow-up=8.9 years), defined as ≥100 units/year, compared to those with lower Lp(a) levels (RR=1.67, 95% CI=1.23, 2.27). The association between higher Lp(a) levels and incident CHD was no longer significant after adjusting for CAC progression. No significant associations were observed for MAC or TAC progression (median follow-up=2.6 years).
Conclusions:
Higher Lp(a) levels are associated with more rapid CAC progression. Additional study is needed to better understand how this relationship can further improve the ability of Lp(a) to enhance cardiovascular disease risk prediction.
Summary
Lipoprotein (a) [Lp(a)] is a risk factor for calcified aortic valvular disease; however, studies assessing associations between Lp(a) and coronary or other extra-coronary measures of calcification are limited. In adjusted prospective analyses, participants with Lp(a) ≥50 mg/dL were at significantly higher risk for rapid coronary artery calcium progression (median follow-up=8.9 years), defined as ≥100 units/year, compared to those with lower Lp(a) levels (RR=1.67, 95% CI=1.23, 2.27). The association between higher Lp(a) levels and incident coronary heart disease was no longer significant after adjusting for CAC progression. Additional study is needed to better understand how this relationship can further improve the ability of Lp(a) to enhance cardiovascular disease risk prediction.
Introduction
Lipoprotein (a) [Lp(a)] is a cholesterol-rich LDL sub-particle consisting of a molecule of apolipoprotein B100 covalently linked with a molecule of apolipoprotein(a).1 Lp(a) activates adhesion of monocytes and migration of macrophage foam cells into the arterial wall, and elevated levels increase risk for development of coronary heart disease (CHD).1–4 Prior study in selected populations has suggested that higher Lp(a) levels are also associated with severe stenosis of the aortic and mitral valves, possibly due to increased valvular calcification.5 Higher Lp(a) levels are already known to predict more prevalent and more severe calcific aortic valve disease (CAVD).6 And yet, to our knowledge, no study has examined whether an association exists between elevated Lp(a) and mitral annular calcification (MAC). Like CAVD, MAC also occurs in the left heart, has similar risk factors for development, and is associated with an increased risk of coronary heart disease, stroke, atrial fibrillation, and overall mortality.7–10
Aortic and mitral valve calcification are detected on cardiac computed tomography (CT). Cardiac CT detects arterial calcification as well in the same imaging protocol without additional radiation. Similar to left-sided cardiac valve calcification, the presence of coronary artery calcification (CAC) and thoracic aortic calcification (TAC) are also each associated with an increased risk of incident CHD.11–12 Despite the etiologic role of Lp(a) in atherosclerotic development, the strong correlation between and similar underlying risk factor profile for left-sided heart valves and CAC, Lp(a) has not been consistently associated with CAC.13–16 No prior studies have yet reported whether a relationship exists between Lp(a) and TAC. We determined the association of Lp(a) with the presence, incidence, and progression of CAC, MAC and TAC.
Methods
Study Population
The Multi-Ethnic Study of Atherosclerosis (MESA) is a National Heart, Lung, and Blood Institute–funded multicenter community-based study. The study recruited 6814 adults aged 45–84 years and free of clinically recognized cardiovascular disease to undergo baseline examination between 2000 and 2002.17 The study participants self-identified with one of four race/ethnic groups: non-Hispanic white, African-American, Hispanic, and Chinese. Follow-up visits 2, 3, 4, and 5 were conducted between the years 2002 to 2012. Institutional review boards at each site approved the study, and all participants gave informed consent.
Lipoprotein (a)
Lp(a) mass concentration was measured with a latex-enhanced turbidimetric immunoassay (Denka Seiken, Tokyo, Japan) at visit 1 and methods have been reported previously.6, 18 The active reagent (R2) contains a suspension of latex particles coated with anti-Lp(a) antibodies. Following incubation with serum, agglutination is detected by a change in absorbance at a wavelength of 700 nm, which is proportional to the mass, based on a five-level calibration. This assay employed five independent calibrators to control for apo(a) size heterogeneity, and the total imprecision was found to be <5%. At a concentration of 4 mg/dL, the imprecision was 6%.
Coronary artery, Mitral annular, and Thoracic aortic calcification
CAC, MAC, and TAC were assessed by cardiac computed tomography (CT) using either cardiac-gated electron-beam CT or multi-detector CT systems, depending on the study site, and the average effective radiation dose per scan in millisieverts (mSv) ranged from 0.6 to 5.6 mSv.19 Per study protocol, all participants underwent a cardiac CT scan at baseline, a random half at exam 2 and the other half at exam 3. CAC, MAC, and TAC were all evaluated at these visits. Additionally, all participants underwent a cardiac CT at exam 5, but only CAC was assessed on this scan. Scoring methods have been described previously.20
CAC was defined by the baseline presence of calcium (Agatston score >0) in any of the coronary arteries. Phantom-adjusted Agatston scores for baseline and follow-up measurements were used to compute the change in CAC per year. Incident CAC was defined as a CAC score>0 on any follow-up cardiac CT examination amongst participants with a CAC score=0 at baseline. CAC progression was defined by the presence of >0 U of change per year and further categorized according to rate of change (1–100 U change per year and >100 U change per year). An increase in CAC >100 U per year has previously been demonstrated to be associated with a significantly higher risk of incident CHD.21 Participants without evidence of progression were defined as ≤0 U change per year (e.g., no change or reduction in CAC score).
MAC was defined by the presence of calcium (Agatston score> 0) on the mitral valve annulus. Phantom-adjusted Agatston scores for baseline and follow-up measurements were used to compute the change in MAC per year.22 Incident MAC was defined as a MAC score>0 on follow-up cardiac CT examination amongst participants with a MAC score=0 at baseline. Progression in MAC was defined by the presence of >0 units of change per year.23
TAC was defined as the presence of calcium (Agatston score >0) in the visualized thoracic aorta from each participant’s computed tomographic scan—from the aortic annulus, above the aortic valve, to the lower edge of the pulmonary artery bifurcation (ascending aorta), and from the lower edge of the pulmonary bifurcation to the cardiac apex (descending aorta). The TAC score quantification method has been previously described.24 Incident TAC was defined as a TAC score>0 on follow-up cardiac CT examination amongst participants with a TAC score=0 at baseline. Progression in TAC was defined by the presence of >0 units of change per year.
Coronary heart disease (CHD)
Incident CHD was defined as the first occurrence of any of the following: myocardial infarction (n=101), resuscitated cardiac arrest (n=17), CHD death (n=45), or definite angina (n=109). Definite angina was defined as symptoms of typical chest pain and physician diagnosis of angina followed by coronary artery bypass grafting and percutaneous coronary intervention (PTCA), evidence of ischemia by stress tests or resting ECG, or ≥70% obstruction on coronary angiography.
Covariates
Standardized questionnaires were used at baseline to obtain demographic information, level of education, annual household income, physical activity, smoking history (never/former/current), family history of coronary heart disease (CHD), and medication usage, including statin, anti-hypertensive, or anti-diabetic use. Physical activity was recorded as participant-reported number of intentional exercise metabolic equivalent (MET)-minutes per week. Total and high-density lipoprotein (HDL) cholesterol, high-sensitivity C-reactive protein (hs-CRP), serum creatinine, and plasma glucose were measured from fasting blood samples. Estimated glomerular filtration rate (eGFR) was calculated using the CKD-EPI creatinine equation.25 Hypertension was defined as a self-report of physician diagnosis and use of an anti-hypertensive medication, or SBP ≥140, or DBP ≥90 mmHg. Diabetes was defined as a fasting glucose >125 mg/dl or use of anti-diabetic medications.
Statistical Analysis
Participants with Lp(a) measurement, CAC, MAC, & TAC scores on baseline cardiac CT examination, and complete covariate data at the baseline examination were included in the cross-sectional analysis. Individuals with an additional evaluation of CAC, MAC, & TAC on follow-up cardiac CT examination were included in the prospective analysis. The exam 5 value was used for participants with two follow-up CAC scores. Baseline characteristics were compared according to Lp(a) levels (≥50 mg/dL vs. <50 mg/dL). Categorical variables were reported as frequency and percentage while continuous variables were reported as mean ± standard deviation. Statistical significance for categorical variables was tested using the chi-square method and the analysis of variance procedure for continuous variables.
Relative risk regression models were run to calculate adjusted relative risks and 95% confidence intervals for Lp(a) levels with the risk of CAC, MAC, and TAC. Prevalence ratios and the risk for incidence and progression of each of these measures, as defined above, according to Lp(a) levels were determined. Multinomial logistic regression was used for CAC progression. Models were run using the clinically recognized cut-points: Lp(a) ≥30 mg/dL and Lp(a) ≥50 mg/dL. Multivariable models were constructed as follows: model 1 adjusted for age, sex, race, education, and clinic site; model 2 adjusted for model 1 covariates with the addition of clinical factors associated with each of the endpoints: cigarette smoking, BMI, diabetes, hypertension, family history of coronary heart disease, total and HDL cholesterol, eGFR, hs-CRP, statin use and aspirin use. Statistical significance was defined as p <0.05. Stata Version 16.1 (StataCorp, College Station, TX) was used for all analyses.
We examined potential mediation effects of CAC progression on the association between Lp(a) and CHD. Cox regression analysis was performed to test for association between Lp(a) and the primary outcome of CHD, adjusting for the same set of covariates as in previous models, with and without CAC progression variable. In this analysis, Lp(a) was categorized as a dichotomous variable (≥50 mg/dL vs. <50 mg/dL), while CAC progression was analyzed as a continuous variable (U change per year prior to occurrence of CHD event). A formal mediation analysis was performed to estimate the direct and indirect effects between Lp(a) and CHD, following the methods proposed by Huang and Yang (2017), using the R software (version 3.5.2).26, 27
Results
Figure 1 shows the flow of included participants. A total of 6705 participants were included in the cross-sectional analysis (mean age=62 years; 53% female; 38% white, 28% African-American 22% Hispanic, and 12% Chinese). Table 1 shows that participants with Lp(a) levels ≥50 mg/dL were more likely to be female, African-American, have a family history of CHD, have a higher BMI, have a higher SBP and DBP, have hypertension, have a higher total and HDL cholesterol, have diabetes, have a higher CRP, and report using both statin and anti-hypertensive medications.
Figure 1:
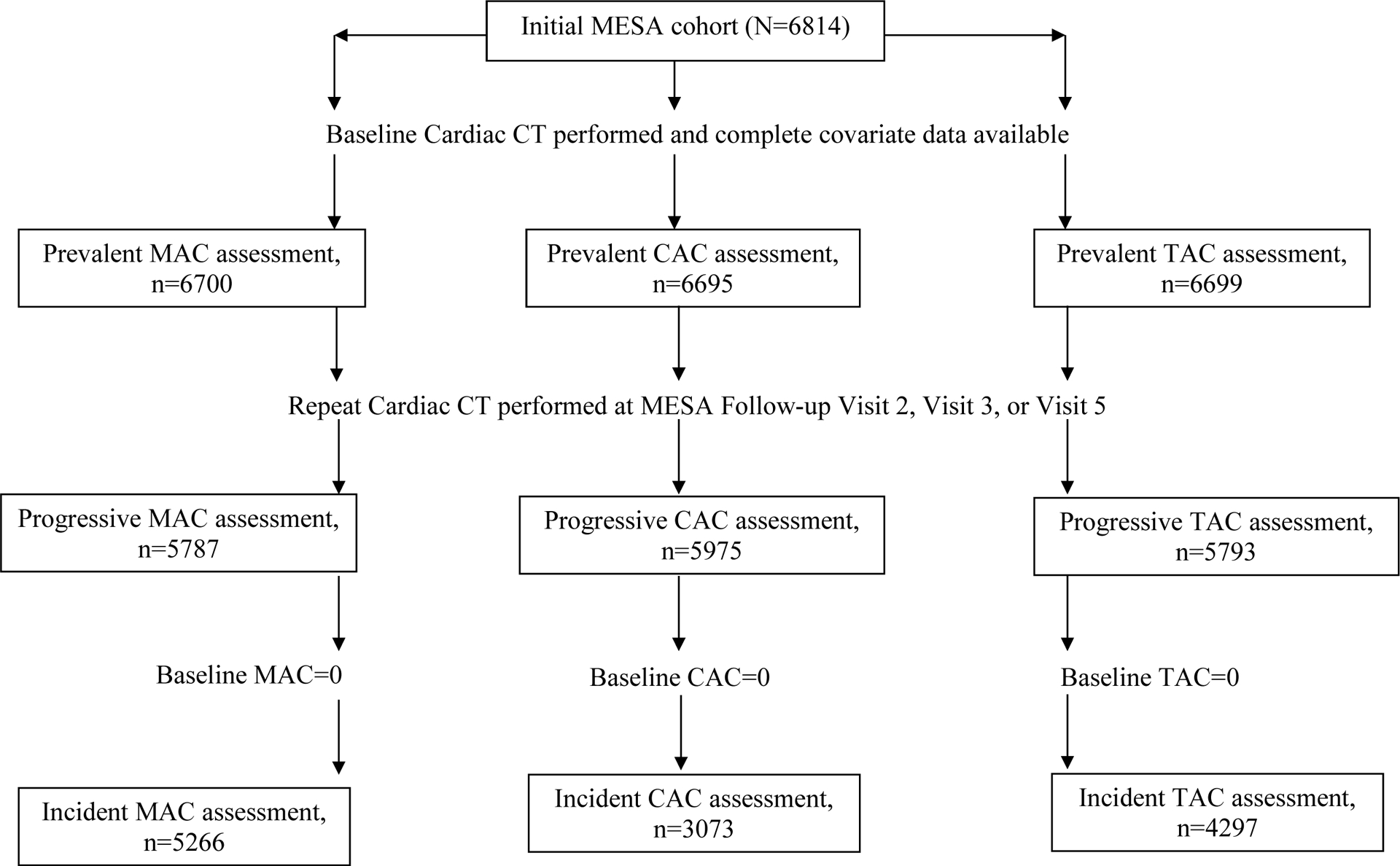
Flowchart of participants included in the analysis for CAC, MAC, and TAC
Table 1.
Baseline characteristics of MESA participants according to Lp(a) levels*
| Characteristic | Lp(a) <50 mg/dL (n=5359) | Lp(a) ≥50 mg/dL (n=1346) | p-value† |
|---|---|---|---|
| Age | 62.0 (10.3) | 62.4 (10.0) | 0.27 |
| Male, % | 2614 (48.8%) | 550 (40.9%) | <0.001 |
| Race, % | |||
| White | 2169 (40.5%) | 413 (30.7%) | <0.001 |
| Chinese | 718 (13.4%) | 75 (5.6%) | |
| African-American | 1207 (22.5%) | 641 (47.6%) | |
| Hispanic | 1265 (23.6%) | 217 (16.1%) | |
| Education, % | |||
| High school or less | 1946 | 482 | 0.65 |
| Some college | 1503 | 394 | |
| College or more | 1893 | 464 | |
| Body mass index, kg/m2 | 28.2 (5.4) | 28.8 (5.7) | <0.001 |
| Family history of MI, | 2106 (41.8%) | 583 (46.3%) | 0.004 |
| Hypertension, % | 2328 (43.4%) | 670 (49.8%) | <0.001 |
| Systolic blood pressure, mmHg | 126.0 (21.3) | 128.2 (22.1) | 0.001 |
| Diastolic blood pressure, mmHg | 71.7 (10.2) | 72.4 (10.4) | 0.023 |
| Total cholesterol, mg/dL | 191.7 (35.1) | 203.1 (36.6) | <0.001 |
| HDL cholesterol, mg/dL | 114.8 (30.8) | 126.1 (32.1) | <0.001 |
| Glucose, mg/dL | 97.5 (30.4) | 96.6 (29.4) | 0.32 |
| Diabetes, % | 649 (12.1%) | 193 (14.4%) | 0.007 |
| Smoking status, % | |||
| Never | 2688 (50.3%) | 675 (50.4%) | 0.17 |
| Former | 1980 (37.1%) | 472 (35.2%) | |
| Current | 675 (12.6%) | 193 (14.4%) | |
| hs-CRP, mg/dL | 3.7 (5.8) | 4.2 (6.4) | 0.008 |
| eGFR | 81.3 (18.5) | 80.7 (18.3) | 0.22 |
| Anti-hypertensive use, % | 1927 (36.0%) | 560 (41.6%) | <0.001 |
| Statin use, % | 1078 (18.9%) | 267 (27.0%) | <0.001 |
| Aspirin use, % | 1669 (31.2%) | 449 (33.4%) | 0.12 |
| Coronary artery calcium score | 140.9 (394.4) | 164.8 (499.7) | 0.060 |
| Mitral annular calcium score | 48.8 (438.0) | 48.7 (386.7) | 1.00 |
| Thoracic aortic calcium score | 220.3 (871.5) | 204.2 (836.3) | 0.54 |
MI=myocardial infarction; HDL=high-density lipoprotein
Continuous variables are expressed as mean (SD). Categorical variables are N (percent).
Comparisons were made between Lp(a)<50 mg/dL and Lp(a) ≥50 mg/dL. Fisher’s exact used to make statistical comparison.
The numbers of participants with CAC, MAC, and TAC at the baseline examination were 3342 (49.8%), 631 (9.4%), and 1810 (27.0%) respectively. Table 2 shows the prevalence of CAC, MAC, and TAC according to Lp(a) levels. After adjustment for model 2 variables, participants with Lp(a) levels ≥50 mg/dL had more prevalent MAC compared to those with lower levels (prevalence ratio=1.22; 95% confidence interval (CI) 1.00, 1.49). Higher Lp(a) levels were not associated with a higher prevalence of CAC or TAC.
Table 2.
Associations of Lp(a) levels with prevalence and incidence of CAC, MAC, and TAC*
| Prevalent CAC | Incident CAC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Events/# at risk | Model 1† PR (95% CI) | p-value | Model 2‡ PR (95% CI) | p-value | Events/# at risk | Model 1 RR (95% CI) | p-value | Model 2 RR (95% CI) | p-value | |
| Lp(a) level | ||||||||||
| <30 mg/dL | 2269/4484 | 1.00 | 1.00 | 700/2024 | 1.00 | 1.00 | ||||
| ≥30 mg/dL | 1073/2221 | 1.03 (0.99, 1.08) | 0.12 | 1.01 (0.97, 1.05) | 0.67 | 378/1049 | 1.07 (0.96, 1.18) | 0.24 | 1.06 (0.95, 1.18) | 0.30 |
| <50 mg/dL | 2683/5359 | 1.00 | 1.00 | 839/2449 | 1.00 | 1.00 | ||||
| ≥50 mg/dL | 659/1346 | 1.04 (0.99, 1.09) | 0.15 | 1.01 (0.96, 1.06) | 0.66 | 239/624 | 1.13 (1.01, 1.27) | 0.035 | 1.11 (0.99, 1.25) | 0.07 |
| Prevalent MAC | Incident MAC | |||||||||
| Events/# at risk | Model 1 PR (95% CI) | p-value | Model 2 PR (95% CI) | p-value | Events/# at risk | Model 1 RR (95% CI) | p-value | Model 2 RR (95% CI) | p-value | |
| Lp(a) level | ||||||||||
| <30 mg/dL | 418/4480 | 1.00 | 1.00 | 152/3537 | 1.00 | 1.00 | ||||
| ≥30 mg/dL | 213/2220 | 1.10 (0.92, 1.31) | 0.28 | 1.06 (0.87, 1.29) | 0.57 | 85/1729 | 1.32 (0.89, 1.97) | 0.17 | 1.40 (0.82, 2.39) | 0.21 |
| <50 mg/dL | 487/5355 | 1.00 | 1.00 | 184/4242 | 1.00 | 1.00 | ||||
| ≥50 mg/dL | 144/1345 | 1.26 (1.05, 1.51) | 0.013 | 1.22 (1.00, 1.49) | 0.049 | 53/1024 | 1.33 (0.86, 2.06) | 0.20 | 1.47 (0.70, 3.08) | 0.31 |
| Prevalent TAC | Incident TAC | |||||||||
| Events/# at risk | Model 1 PR (95% CI) | p-value | Model 2 PR (95% CI) | p-value | Events/# at risk | Model 1 RR (95% CI) | p-value | Model 2 RR (95% CI) | p-value | |
| Lp(a) level | ||||||||||
| <30 mg/dL | 1241/4479 | 1.00 | 1.00 | 333/2852 | 1.00 | 1.00 | ||||
| ≥30 mg/dL | 569/2220 | 1.02 (0.95, 1.10) | 0.52 | 1.01 (0.94, 1.09) | 0.71 | 151/1445 | 0.94 (0.76, 1.15) | 0.53 | 0.84 (0.68, 1.04) | 0.12 |
| <50 mg/dL | 1445/5354 | 1.00 | 1.00 | 393/3450 | 1.00 | 1.00 | ||||
| ≥50 mg/dL | 365/1345 | 1.04 (0.96, 1.12) | 0.31 | 1.04 (0.96, 1.13) | 0.33 | 91/847 | 0.94 (0.74, 1.20) | 0.61 | 0.82 (0.64, 1.06) | 0.14 |
CAC=coronary artery calcification, MAC=mitral annular calcification, PR=prevalence ratio, RR=relative risk, TAC=thoracic aortic calcification
Results of multivariable relative risk regression models
Model 1 adjusted for age, sex, race, education, and clinic site
Model 2 adjusted for Model 1 + cigarette smoking, body mass index, diabetes, hypertension, family history of coronary heart disease, total cholesterol, HDL cholesterol, eGFR, hs-CRP, statin use, and aspirin use
For our analysis of incident calcification outcomes, there were 3073, 5266, and 4297 participants without evidence of baseline CAC, MAC, and TAC, respectively, who underwent a follow-up cardiac CT. Median follow-up was 8.9 years for CAC and 2.6 years for MAC and TAC. Of these participants, 1078 (35.1%), 237 (4.5%), and 393 (11.3%) developed new CAC, MAC, and TAC, respectively, on the follow-up examination. Adjusting for model 1 variables, an Lp(a) level ≥50 mg/dL was associated with a 13% higher risk of incident CAC compared to those with lower levels (relative risk (RR)=1.13; 95% CI 1.01, 1.27). After additional adjustment for model 2 variables, associations were attenuated and of borderline significance (RR=1.11; 95% CI 0.99, 1.25). No significant associations were observed for Lp(a) levels and risk of incident MAC or TAC (Table 2).
Table 3 reports the risk of CAC progression associated with Lp(a) levels. Compared to participants without evidence of CAC progression (≤0 U/year), Lp(a) levels ≥30 mg/dL and ≥50 mg/dL were each associated with a higher risk of rapid CAC progression (>100 U/year). Higher Lp(a) levels were not associated with an increased risk of either MAC or TAC progression (Table 4).
Table 3.
Associations of Lp(a) levels with CAC progression*
| CAC progression | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1–100 U/year | >100 U/year | |||||||||
| Events/# at risk | Model 1† RR (95% CI) | p-value | Model 2‡ RR (95% CI) | p-value | Events/# at risk | Model 1 RR (95% CI) | p-value | Model 2 RR (95% CI) | p-value | |
| Lp(a) level | ||||||||||
| <30 mg/dL | 2289/4017 | 1.00 | 1.00 | 255/4017 | 1.00 | 1.00 | ||||
| ≥30 mg/dL | 1069/1958 | 1.07 (0.93, 1.23) | 0.34 | 1.00 (0.86, 1.16) | 0.97 | 146/1958 | 1.50 (1.16, 1.92) | 0.002 | 1.48 (1.12, 1.95) | 0.005 |
| <50 mg/dL | 2716/4794 | 1.00 | 1.00 | 297/4794 | 1.00 | 1.00 | ||||
| ≥50 mg/dL | 642/1181 | 1.05 (0.92, 1.19) | 0.49 | 1.00 (0.87, 1.15) | 0.98 | 104/1181 | 1.81 (1.38, 2.39) | <0.001 | 1.67 (1.23, 2.27) | 0.001 |
CAC=coronary artery calcification, RR=relative risk
Results of multivariable relative risk regression models
Model 1 adjusted for age, sex, race, education, and clinic site
Model 2 adjusted for Model 1 + cigarette smoking, body mass index, diabetes, hypertension, family history of coronary heart disease, total cholesterol, HDL cholesterol, eGFR, hs-CRP, statin use, and aspirin use
Table 4.
Associations of Lp(a) levels with MAC and TAC progression*
| MAC progression (>0 U/year) | TAC progression (>0 U/year) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Events/# at risk | Model 1† RR (95% CI) | p-value | Model 2 RR (95% CI) | p-value | Events/# at risk | Model 1 RR (95% CI) | p-value | Model 2 RR (95% CI) | p-value | |
| Lp(a) level | ||||||||||
| <30 mg/dL | 388/3882 | 1.00 | 1.00 | 1115/3888 | 1.00 | 1.00 | ||||
| ≥30 mg/dL | 198/1905 | 1.12 (0.93, 1.35) | 0.23 | 1.13 (0.92, 1.39) | 0.24 | 487/1905 | 0.97 (0.89, 1.06) | 0.57 | 0.96 (0.88, 1.05) | 0.37 |
| <50 mg/dL | 463/4644 | 1.00 | 1.00 | 1302/4650 | 1.00 | 1.00 | ||||
| ≥50 mg/dL | 123/1143 | 1.11 (0.90, 1.37) | 0.31 | 1.14 (0.90, 1.45) | 0.28 | 300/1143 | 0.97 (0.88, 1.08) | 0.58 | 0.96 (0.87, 1.06) | 0.45 |
MAC=mitral annular calcification, RR=relative risk, TAC=thoracic aortic calcification
Results of multivariable relative risk regression models
Model 1 adjusted for age, sex, race, education, and clinic site
Model 2 adjusted for Model 1 + cigarette smoking, body mass index, diabetes, hypertension, family history of coronary heart disease, total cholesterol, HDL cholesterol, eGFR, hs-CRP, statin use, and aspirin use
A significant association was observed between the binary Lp(a) category (≥50 mg/dL vs. <50 mg/dL) and risk of CHD (hazard ratio (HR)= 1.25; 95% CI 1.01, 1.55) after adjusting for age, sex, race, education, clinic site, cigarette smoking, BMI, diabetes, hypertension, family history of coronary heart disease, total and HDL cholesterol, eGFR, hs-CRP, and aspirin use. However, when CAC progression score was added to the model, Lp(a) was no longer significant (HR=1.13; 95% CI 0.87, 1.46), while CAC progression score was significantly associated with risk of CHD (HR=1.26 per SD increment; 95% CI 1.19, 1.34). In the mediation analysis, we assumed a biological pathway from Lp(a) → CAC progression → CHD, and that all confounders have been considered in our analysis. We estimated a direct effect between Lp(a) and CHD (HR = 1.13, 95% CI: (0.87, 1.46), p = 0.35), and marginally significant indirect effect through CAC progression (HR = 1.02, 95% CI: (1.00, 1.03), p = 0.07).
Discussion
Participants with higher Lp(a) levels were at a substantially higher risk for rapid CAC progression. In addition, we found that the association between high Lp(a) levels and incident CHD was no longer significant after adjusting for CAC progression. While no relationship was observed for Lp(a) levels and prevalence of CAC, a greater risk of incident CAC approached statistical significance. Although our findings regarding relationships between Lp(a) levels and either MAC or TAC did not demonstrate meaningful associations, our data are novel in that we evaluated the impact of Lp(a) on progression of calcification in vascular beds which has not been extensively studied in a large population-based cohort free of cardiovascular disease.
Lp(a) adheres to multiple components of the extracellular matrix in the arterial wall and promotes atherogenesis by transporting cholesterol and oxidized phospholipids into the subendothelial arterial space.26, 27 Calcium deposition in the arterial wall occurs as the atherosclerotic lesion continues to develop.28 Prior pathologic studies have documented a close correlation between coronary calcium and atherosclerotic plaque.29–32 Since Lp(a) may be reflective of underlying atherosclerotic burden, it would follow that individuals with higher Lp(a) levels would have a greater CAC prevalence. Lp(a), however, was not associated with prevalent CAC in this study and this finding is consistent with other population-based cohort studies.6, 15, 16, 33 Additionally, genetic variations in Lp(a) genetic locus have not been associated with the presence of CAC either.34
Although the published literature does not suggest a strong relationship between Lp(a) and CAC, nearly all of these studies were cross-sectional. A longitudinal study involving over 2500 healthy Korean adults undergoing sequential cardiac CT scans 4 years apart found Lp(a) levels ≥50 mg/dL were associated with a 33% higher risk of any CAC progression (>0 U/year).35 Our study not only extends these findings to a multi-ethnic cohort with a longer follow-up but also suggests the relationship may be particularly relevant for rapid CAC progression. The contrast in the association of Lp(a) with prevalent CAC as opposed to CAC progression may be due to higher Lp(a) levels being more indicative of atherosclerotic disease activity rather than coronary atherosclerotic burden. Lp(a) has been previously demonstrated to preferentially accumulate at sites of endothelial injury, where plaque formation occurs, compared with LDL as a result of its greater capacity to bind to exposed fibrin or glycosaminoglycans.36 Calcification is known to occur during the healing of these plaques and CAC progression might correlate more with progression of noncalcified plaque than pathologically stable calcified plaque. The intimal calcification that forms in response to the healing plaque might then propagate a positive feedback loop of further inflammation and calcification.37, 38 CAC progression is, therefore, an increasingly recognized indicator of disease activity and can identify a subset of individuals at high risk for future CHD events.21, 39
Since CAC and TAC share similar risk factors, we expected associations between Lp(a) levels and TAC to be similar to those reported for Lp(a) levels and CAC.24 Despite an assumed shared pathophysiology with CAC, higher Lp(a) levels did not increase risk for any TAC outcome. Indeed, the disparate results for CAC and TAC may be a due to a variety of etiologic factors. Most coronary artery smooth muscle cells derive from a distinct embryological origin than those smooth muscle cells found in the thoracic aorta.40, 41 In addition, CAC is only seen in the intimal layer whereas TAC occurs in both the intima and media.42 Calcification in the coronary arteries is a result of the healing response to atherosclerotic plaque inflammation.43 Medial calcification, however, is more reflective of aging and metabolic processes such as diabetes and renal disease rather than atherosclerosis.43, 44 Finally, shear stress, a phenomenon associated with atherosclerosis development, may be lower in the ascending thoracic aorta as it is devoid of arterial branches, unlike the coronary arteries.45
Considering that mitral and aortic valves experience similar blood flow hemodynamics and share many of the same clinical risk factors for valvular calcification, we also anticipated that Lp(a) levels would be associated with MAC development in a similar fashion to that reported for CAVD.6, 7 Higher Lp(a) levels were associated with a higher prevalence of MAC; however, no associations were noted for MAC incidence or progression. Genetic factors may help explain the seemingly paradoxical findings for associations of Lp(a) with MAC compared to CAVD. A large genome-wide association study was performed in participants across multiple cohorts to better understand genetic contributions to valvular calcification.34 In this study, genetic variation in the LPA locus rs10455872 was associated with CAVD across multiple ethnic groups and the relationship was mediated by Lp(a) levels. Similar associations were not seen for genetic variations in the LPA locus and MAC.
An important strength of this study is the recruitment of a racially/ethnically diverse population spread across 6 U.S. cities and who were without prior clinical CVD at the time of enrollment. Additionally, CAC, MAC, and TAC were all assessed with serial CT scan measurements and potential confounders that were also well characterized in MESA. Our study also has some limitations. Although no significant associations for Lp(a) and both MAC and TAC were found in prospective analyses, the confidence intervals were wide and median follow-up was only 2.6 years. This timeframe was likely too short to definitively assess prospective associations of Lp(a) with these extra-coronary measures of calcification. Calcification in the aortic arch and in the abdominal aorta were not considered in the assessment of TAC as chest CT imaging studies in MESA were performed for CAC screening purposes only. We had limited power to perform stratified analyses. We also acknowledge the limitation of our mediation analysis which heavily relies on model assumptions, including the validity of the causal pathway between Lp(a), CAC progression, and CHD, and no other confounders for the Lp(a)-CAC progression, CAC progression-CHD, and Lp(a)-CHD relationship that were not included in our model. These assumptions are not all verifiable. Finally, there was a selection bias for CAC progression. Because these participants had follow-up visits at MESA Exam 5, the resulting cohort was comprised of healthier individuals—though this would have skewed our findings toward the null.
In conclusion, higher Lp(a) levels were associated with CAC progression and the relationship appeared to be strongest in those participants experiencing a rapid annual increase (>100 U/year). More importantly, the association between higher Lp(a) levels and incident CHD in this cohort was no longer significant after adjusting for CAC progression, although Lp(a) has been previously shown as an important biomarker for CHD. Current guidelines consider an elevated Lp(a) (≥50 mg/dL) to be a risk-enhancing factor for the development of cardiovascular disease and should prompt either the initiation of statin therapy or further risk-stratification with CAC testing in appropriate individuals.46 Our findings suggest that additional study is needed to better understand how this relationship of high Lp(a) levels with rapid CAC progression can improve the ability of Lp(a) to enhance cardiovascular disease risk prediction. Further studies are also needed to better target which individuals with high Lp(a) levels are more likely to experience rapid CAC progression and determine whether more aggressive primary prevention measures lead to slower CAC progression and, ultimately, a reduced cardiovascular risk.
Acknowledgments:
The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesanhlbi.org.
Funding sources: This research was supported by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169 and by grant R01-HL-127659 from the National Heart, Lung, and Blood Institute, and by grants UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420 from NCATS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have no conflicts of interest to disclose.
References
- 1.) Berglund L, Ramakrishnan R. Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler Thromb Vasc Biol 2004;24:2219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.) Dangas G, Mehran R, Harpel PC, Sharma SK, Marcovina SM, Dube G, Ambrose JA, Fallon JT. Lipoprotein (a) and inflammation in human coronary atheroma: association with the severity of clinical presentation. J Am Coll Cardiol 1998;32:2035–2042. [DOI] [PubMed] [Google Scholar]
- 3.) Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein (a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation 2008;117:176–184. [DOI] [PubMed] [Google Scholar]
- 4.) Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009;302:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.) Hojo Y, Kumakura H, Kanai H, Iwasaki T, Ichikawa S, Kurabayashi M. Lipoprotein(a) is a risk factor for aortic and mitral valvular stenosis in peripheral arterial disease. Eur Heart J Cardiovasc Imaging 2016;17:492–497. [DOI] [PubMed] [Google Scholar]
- 6.) Cao J, Steffen BT, Budoff M, Post WS, Thanassoulis G, Kestenbaum B, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol 2016;36:1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.) Abramowitz Y, Jilaihawi H, Chakravarty T, Mack MJ, Makkar RR. Mitral annulus calcification. J Am Coll Cardiol 2015;66:1934–1941. [DOI] [PubMed] [Google Scholar]
- 8.) Fox CS, Vasan RS, Parise H, Levy D, O’Donnell CJ, D’Agostino RB, et al. Mitral annular calcification predicts cardiovascular morbidity and mortality: the Framingham Heart Study. Circulation 2003; 107:1492–1496. [DOI] [PubMed] [Google Scholar]
- 9.) Kizer JR, Wiebers DO, Whisnant JP, Galloway JM, Welty TK, Lee ET, et al. Mitral annular calcification, aortic valve sclerosis, and incident stroke in adults free of clinical cardiovascular disease: the Strong Heart Study. Stroke 2005; 36:2533–2537. [DOI] [PubMed] [Google Scholar]
- 10.) Fox CS, Parise H, Vasan RS, Levy D, O’Donnell CJ, D’Agostino RB, et al. Mitral annular calcification is a predictor for incident atrial fibrillation. Atherosclerosis 2004; 173:291–294. [DOI] [PubMed] [Google Scholar]
- 11.) Silverman MG, Blaha MJ, Krumholz HM, Budoff MJ, Blankstein R, Sibley CT, et al. Impact of coronary artery calcium on coronary heart disease events in individuals at the extremes of traditional risk factor burden: the multi-ethnic study of atherosclerosis. Eur Heart J 2014;35:2232–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.) Budoff MJ, Nasir K, Katz R, Takasu J, Carr JJ, Wong ND, et al. Thoracic aortic calcification and coronary heart disease events: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2011;215:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.) Yeboah J, McClelland RL, Polonsky TS, Burke GL, Sibley CT, O’Leary D, et al. Comparison of novel risk markers for improvement in cardiovascular risk assessment in intermediate-risk individuals. JAMA 2012;308:788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.) Hamirani YS, Nasir K, Blumenthal RS, Takasu J, Shavelle D, Kronmal R, et al. Relation of mitral annular calcium and coronary calcium (from the multiethnic study of atherosclerosis [MESA]). Am J Cardiol 2011;107:1291–1294. [DOI] [PubMed] [Google Scholar]
- 15.) Guerra R, Yu Z, Marcovina S, Peshock R, Cohen JC, Hobbs HH. Lipoprotein(a) and apolipoprotein(a) isoforms: no association with coronary artery calcification in the Dallas Heart Study. Circulation 2005;111:1471–1479. [DOI] [PubMed] [Google Scholar]
- 16.) Erbel R, Lehmann N, Churzidse S, Mohlenkamp S, Moebus S, Mahabadi AA, et al. Gender-specific association of coronary artery calcium and lipoprotein parameters: The Heinz Nixdorf Recall Study. Atherosclerosis 2013;229:531–540. [DOI] [PubMed] [Google Scholar]
- 17.) Bild DE, Bluemke DA, Burke GL, Detrano R, Diez-Roux AV Folsom AR, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol 2002;156:871–81. [DOI] [PubMed] [Google Scholar]
- 18.) Guan W, Cao J, Steffen BT, Post WS, Stein JH, Tattersall MC, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: The Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.) Carr JJ, Nelson JC, Wong ND, McNitt-Gray M, Arad Y, Jacobs DR Jr, et al. Calcified coronary artery plaque measurement with cardiac CT in population-based studies: standardized protocol of Multi-Ethnic Study of Atherosclerosis (MESA) and Coronary Artery Risk Development in Young Adults (CARDIA) study. Radiology 2005;234:35–43. [DOI] [PubMed] [Google Scholar]
- 20.) Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol 1990;15:827–832. [DOI] [PubMed] [Google Scholar]
- 21.) Budoff MJ, Young R, Lopez VA, Kronmal RA, Nasir K, Blumenthal RS, et al. Progression of coronary calcium and incident coronary heart disease events: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol 2013;61:1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.) Budoff MJ, Takasu J, Katz R, Mao S, Shavelle DM, O’Brien KD, et al. Reproducibility of CT measurements of aortic valve calcification, mitral annulus calcification, and aortic wall calcification in the multi-ethnic study of atherosclerosis. Acad Radiol 2006;13:166–172. [DOI] [PubMed] [Google Scholar]
- 23.) O’Neal WT, Efird JT, Qureshi WT, Nazarian S, Alonso A, Heckbert SR, Soliman EZ. Mitral annular calcification and incident atrial fibrillation in the Multi-Ethnic Study of Atherosclerosis Europace 2015;17:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.) Wong ND, Gransar H, Shaw L, Polk D, Moon JH, Miranda-Peats R, et al. Thoracic aortic calcium versus coronary artery calcium for the prediction of coronary heart disease and cardiovascular disease events. J Am Coll Cardiol Imaging 2009;2:319– 326. [DOI] [PubMed] [Google Scholar]
- 25.) Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.) Huang YT, Yang HI. Causal mediation analysis of survival outcome with multiple mediators. Epidemiology (Cambridge, Mass.) 2017;28:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.) R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2018. https://www.R-project.org/. [Google Scholar]
- 28.) Bihari-Varga M, Gruber E, Rotheneder M, Zechner R, Kostner GM. Interaction of lipoprotein Lp(a) and low density lipoprotein with glycosaminoglycans from human aorta. Arteriosclerosis 1988;8:851–857. [DOI] [PubMed] [Google Scholar]
- 29.) Anuurad E, Boffa MB, Koschinsky ML, Berglund L. Lipoprotein(a): A unique risk factor for cardiovascular disease. Clin Lab Med 2006;26:751–772. [DOI] [PubMed] [Google Scholar]
- 30.) Doherty TM, Asotra K, Fitzpatrick LA, Qiao JH, Wilkin DJ, Detrano RC, et al. Calcification in atherosclerosis: bone biology and chronic inflammation at the arterial crossroads. Proc Natl Acad Sci U S A 2003;100:11201–11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.) Blankenhorn SR, Stern D. Calcification of the coronary arteries. Am J Roentgenol 1959;81:772–777. [PubMed] [Google Scholar]
- 32.) Eggen D, Strong J, McGill H. Coronary calcification: relationship to clinically significant coronary lesions and race, sex and topographic distribution. Circulation 1965;32:948–955. [DOI] [PubMed] [Google Scholar]
- 33.) McCarthy JH, Palmer FJ. Incidence and significance of coronary artery calcification. Br Heart J 1974;36:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.) Rumberger JA, Simons DB, Fitzpatrick LA, Sheedy PF, Schwartz RS. Coronary artery calcium areas by electron beam computed tomography and coronary atherosclerotic plaque area: a histopathologic correlative study. Circulation 1995;92:2157–2162. [DOI] [PubMed] [Google Scholar]
- 35.) Kullo IJ, Bailey KR, Bielak LF, Sheedy PF, Klee GG, Kardia SL, et al. Lack of association between lipoprotein(a) and coronary artery calcification in the Genetic Epidemiology Network of Arteriopathy (GENOA) Study. Mayo Clin Proc 2004;79:1258–1263. [DOI] [PubMed] [Google Scholar]
- 36.) Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med 2013;368:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.) Guerra R, Yu Z, Marcovina S, Peshock R, Cohen JC, Hobbs HH. Lipoprotein(a) and apolipoprotein(a) isoforms: No association with coronary artery calcifications in The Dallas Heart Study. Circulation 2005;111:1471–1479. [DOI] [PubMed] [Google Scholar]
- 38.) Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res 2016;57:1953–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.) Beckman JA, Ganz J, Creager MA, Ganz P, Kinlay S. Relationship of clinical presentation and calcification of culprit coronary artery stenosis. Arterioscler Thromb Vasc Biol 2001;21:1618–1622. [DOI] [PubMed] [Google Scholar]
- 40.) Nadra I, Mason JC, Philippidis P, Florey O, Smythe CDW, McCarthy GM, et al. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ Res 2005;96:1248–1256. [DOI] [PubMed] [Google Scholar]
- 41.) Lehmann N, Erbel R, Mahabadi AA, Rauwolf M, Mohlenkamp S, Moebus S, et al. Value of progression of coronary artery calcification for risk prediction of coronary and cardiovascular events: Result of the HNR Study (Heinz Nixdorf Recall). Circulation 2018;137:665–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.) Leroux-Berger M, Queguiner I, Maciel TT, Ho A, Relaix F, Kempf H. Pathologic calcification of adult vascular smooth muscle cells differs on their crest or mesodermal embryonic origin. J Bone Miner Res 2011;26:1543–1553. [DOI] [PubMed] [Google Scholar]
- 43.) Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development 2000;127:1607–1616. [DOI] [PubMed] [Google Scholar]
- 44.) Demer LL, Tintut Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008;117:2938–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.) Doherty TM, Fitzpatrick LA, Inoue D, Qiao JH, Fishbein MC, Detrano RC, et al. Molecular, endocrine, and genetic mechanisms of arterial calcification. Endocr Rev 2004;25:629–672. [DOI] [PubMed] [Google Scholar]
- 46.) Iribarren C, Sidney S, Sternfeld B, Browner WS. Calcification of the aortic arch: risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. JAMA 2000;283:2810–2815. [DOI] [PubMed] [Google Scholar]
- 47.) Chiu J-J, Chien S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol Rev 2011;91:327–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.) Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; ePub ahead of print. [Google Scholar]