

Abstract
Background:
Chronic renovascular disease (RVD) can lead to a progressive loss of renal function, and current treatments are inefficient. We designed a fusion of vascular endothelial growth factor (VEGF) conjugated to an elastin-like polypeptide (ELP) carrier protein with an N-terminal kidney-targeting peptide (KTP). We tested the hypothesis that KTP-ELP-VEGF therapy will effectively recover renal function with an improved targeting profile. Further, we aimed to elucidate potential mechanisms driving renal recovery.
Methods:
Unilateral RVD was induced in 14 pigs. Six weeks later renal blood flow (RBF) and glomerular filtration rate (GFR) were quantified by multidetector-CT imaging. Pigs then received a single intrarenal injection of KTP-ELP-VEGF or vehicle. CT-quantification of renal hemodynamics were repeated 4 weeks later, and then pigs were euthanized. Ex-vivo renal microvascular (MV) density and media-to-lumen ratio, macrophage infiltration, and fibrosis were quantified. In parallel, THP-1 human monocytes were differentiated to naive macrophages (M0) or inflammatory macrophages (M1) and incubated with VEGF, KTP-ELP, KTP-ELP-VEGF, or control media. The mRNA expression of macrophage polarization and angiogenic markers were quantified (qPCR).
Results:
Intra-renal KTP-ELP-VEGF improved RBF, GFR, and MV density; attenuated MV media-to-lumen ratio and renal fibrosis compared to placebo, accompanied by augmented renal M2 macrophages. In vitro, exposure to VEGF/KTP-ELP-VEGF shifted M0 macrophages to a pro-angiogenic M2 phenotype while M1s were non-responsive to VEGF treatment.
Conclusions:
Our results support the efficacy of a new renal-specific biologic construct in recovering renal function and suggest that VEGF may directly influence macrophage phenotype as a possible mechanism to improve MV integrity and function in the stenotic kidney.
Keywords: renovascular disease, vascular endothelial growth factor, macrophage, angiogenesis, microcirculation, drug-delivery technology
Introduction
Renovascular disease (RVD), most often caused by atherosclerotic renal artery stenosis[1], is a leading cause of secondary hypertension. It may lead to and exacerbate the progression of chronic kidney disease (CKD)[2, 1], and significantly increases cardiovascular risk[3]. Progressive damage and loss of the renal microvasculature is a major pathological feature in chronic renal diseases that correlates with the progression of renal injury[4, 5], irrespective of the etiology. We and others have shown that renal microvascular (MV) rarefaction develops in the setting of blunted angiogenic signaling and reduced availability of renal VEGF[6-9]. We also showed in a swine model of unilateral RVD that intrarenal replenishment of VEGF stimulates angiogenesis, leading to improved renal MV integrity and endothelial function, demonstrated by the improved stenotic kidney renal blood flow (RBF), glomerular filtration rate (GFR), fibrosis, and attenuated hypertension[8]. These data support a prominent role for VEGF in the kidney and a pathophysiological link between MV damage and renal injury.
In subsequent studies[10-12], we aimed to refine VEGF therapy by addressing the limited circulating time of this cytokine and offsetting the possibility of VEGF binding outside the kidney. To address this, we used drug delivery technology in the form of elastin-like polypeptide (ELP). ELP is an inert, non-immunogenic peptide that may be conjugated to virtually any therapeutic cargo or tissue-targeting agents[13-15]. We developed an ELP-VEGF construct and successfully showed that single ELP-VEGF therapy in RVD improved RBF, GFR, MV density, and hypertension more effectively than free VEGF[10, 11].
We aimed to extend those studies by refining ELP-VEGF to improve renal specificity. Using a phage display screen in mice, Pasqualini et al. identified a cyclic peptide of seven amino acids that naturally accumulates in the kidney 10 times more than in comparator organs[16]. We fused the kidney targeting peptide (KTP) to the ELP-VEGF construct and showed in multiple animal models that these constructs display greater deposition in the kidneys and prolonged tissue half-life relative to untargeted ELP constructs[17, 18]. Furthermore we showed that VEGF fused to KTP-ELP retains its biological activity in vitro[18]. These studies support the use of KTP as a potential means to improve delivery of ELP-VEGF therapy. However, the renal therapeutic efficacy of the KTP-ELP-VEGF construct has not yet been evaluated.
Based on these exciting data we hypothesize that the novel KTP-ELP-VEGF construct will improve renal injury in RVD without compromising efficacy. Furthermore, we recently showed that ELP-VEGF in a model of CKD improved renal health partly by inducing a shift in renal macrophage phenotype in favor of M2s[12], thus reducing renal inflammation and promoting repair. How modulation of renal macrophage phenotype is mechanistically related to recovery of renal function following VEGF therapy is still unknown. Thus, the model of unilateral RVD offers the opportunity to extend our previous work[12] and test the hypothesis that long-term protective effects of KTP-ELP-VEGF therapy are mediated by a direct effect of VEGF to induce macrophages to an anti-inflammatory, pro-angiogenic phenotype, leading the recovery of renal MV architecture and function.
Materials and Methods
In vivo studies
The Institutional Animal Care and Use Committee at the University of Mississippi Medical Center approved all studies. Twenty-one juvenile intact male and female pigs (sus scrofa domesticus) used for this study were pooled, similarly distributed, and observed for a total of 10 weeks. In 14 pigs, RVD was induced by unilateral renal artery stenosis, as described[19, 20, 7, 21, 22, 8, 23, 10]. The remaining 7 pigs underwent sham surgery and were kept as normal controls.
Six weeks following induction of RVD or sham, pigs underwent contrast enhanced multidetector computed tomography (MDCT) scans to quantify RBF, regional perfusion, and GFR, as previously described[7, 8, 24, 10]. After scanning, KTP-ELP-VEGF (100 μg/kg) or vehicle (saline) was injected into the stenotic kidney. Four weeks later, at the 10-week timepoint, all pigs underwent repeated MDCT-derived quantification of renal function to determine the responses to treatment. Two days after the 10-week in vivo studies, pigs were euthanized with IV phenobarbital (100 mg/kg). Urine was collected to measure blood urea nitrogen (BUN) and kidneys were collected for ex vivo studies.
Quantification of renal microvascular density: Micro CT studies
Excised kidneys were perfused with a silicon polymer contrast (Microfil MV122) and scanned on a benchtop high-resolution Micro-CT (SkyScan 1076, Bruker Biospin Corp., MA) at 0.3° increments with a resolution of 9 μm. The Analyze™ software package was used to generate 3D reconstructions and measure cortical and medullary MV density as previously described[20, 25, 21, 23]. To further localize the effects of VEGF, MV density was quantified for all microvessel diameters (0-500 μm) as well as incrementally smaller subgroups (0-200 μm and 0-80 μm).
Histology
Mid-hilar kidney tissue was fixed in 10% formalin, embedded in paraffin, and cut to 5 μm sections. Slides were then stained with hematoxylin and eosin and Masson’s trichrome to quantify MV media-to-lumen ratio and renal fibrosis[23]. Measurement of media to lumen ratio and quantification of fibrosis was performed using ImageJ (National Institutes of Health (NIH), Bethesda, MD) as previously described[26, 12, 27].
Immunohistochemistry
Mid-hilar kidney sections from each animal were stained for indoleamine-2,3-dioxygenase (IDO1, Invitrogen, 711778, 1/50) and mannose receptor-c type 1 (MRC1, LSBio, LS-B9805, 1/50) to identify M1 and M2 macrophages as previously described[26, 12]. Slides were counter-stained with the nuclear label DAPI. Nucleated IDO1+ (M1) and MRC1+ (M2) macrophages were quantified in 15 randomly captured high power images per slide.
In vitro studies
Frozen THP-1 human monocytes (ATCC, TIB-202) were reconstituted in culture media consisting of RPMI 1640 + glutamine and hepes (Gibco, 22400-089) containing 10% fetal bovine serum and penicillin/streptomycin (100 U/mL, Gibco, 15070-063). Cells were incubated at 19% O2 and 5% CO2 in a water jacketed incubator until 80-90% confluence in T75 flasks before passaging 1:4 per supplier instructions. Cells in passage 4-7 were used in all experiments. The majority of the cells used in this experiment were between passages 4 and 5. More passages were needed for just three samples to provide sufficient RNA for analysis. Monocytes were seeded in 6-well plates at 1.2*106 live cells per well. All plates were then incubated for 48 hours in culture media containing 100 nM phorbol 12-myristate-13-acetate (PMA) to induce terminal differentiation to naïve M0 macrophages[28]. Plates were then divided into two groups, receiving normal culture media or media containing 100 ng/mL lipopolysaccharide (LPS) and 20 ng/mL interferon gamma (IFNγ) for 48 hours to induce differentiation of M1 macrophages[29]. Plates containing M0 or M1 macrophages were finally cultured for 48 hours in normal media or media containing 100 nM VEGF, KTP-ELP, or KTP-ELP-VEGF. This concentration was chosen based on previous work assessing effective dose of these constructs to stimulate human endothelial and tubular cells in vitro[18]. Following this step, media was removed from all wells and cells were lysed to perform RNA isolation.
Real-time polymerase chain reaction (qPCR)
Total RNA was isolated from cell culture using the PureLink RNA Mini Kit (Invitrogen, 12183018A). 500 ng of RNA from each sample was used to generate cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, K1621). qPCR was performed using Hot Start Taq 2x Master Mix (New England BioLabs, M0496L). Samples were probed using primer pairs for Beta Actin (Hs01060665_g1), VEGF (Hs00900055_m1), KDR/Flk-1 (Hs00911700_m1), Flt-1 (Hs01052961_m1), IDO1 (Hs00984148_m1), CD163 (Hs00174705_m1), and sFlt-1 (5’-3’ TGG GGA GGG GAG GAT GTT AG, 3’-5’ TAA GGG AGG TGC GTT GAA CC).
Statistical analysis
Results were expressed as mean±SEM. Statistical comparisons within each group were performed by ANOVA followed by Fisher’s post-test and between groups with factorial ANOVA followed by Tukey’s post-hoc test. Six and 10-week comparisons with baseline values were performed by repeated measures ANOVA with the Bonferroni adjustment for multiple pairwise comparisons. Statistical significance was accepted for p<0.05.
Results
General Characteristics
Body weight was similar between all groups and sexes at the 6 and 10-week timepoints (Table 1a-b). The degree of renal artery stenosis, quantified by renal angiography, remained stable throughout the study at approximately 70% (Table 1a-b), which is hemodynamically significant[30]. Prior to treatment at 6 weeks, blood pressure was significantly and similarly elevated in all pigs compared to normal. Four weeks following treatment, there was no change in blood pressure in the RVD+KTP-ELP-VEGF group while untreated RVD showed a significant rise compared to 6 weeks (Table 1b).
Table 1a:
General characteristics of each group at 6 weeks (immediately prior to treatment, n=7/group).
| Normal | RVD | RVD+KTP-ELP-VEGF | |
|---|---|---|---|
| Body weight (kg) | 45.6 ± 1.1 | 44.8 ± 1.4 | 46.6 ± 2.5 |
| Mean arterial pressure (mm Hg) | 111.7 ± 2.1 | 131.8 ± 8.0* | 132.4 ± 6.7* |
| Renal artery stenosis (%) | 0 | 70 ± 7.4* | 71.3 ± 3.4* |
| Cortical perfusion (mL/min/cc tissue) | 3.9 ± 0.3 | 3.2 ± 0.6* | 3.1 ± 0.1* |
| Medullary perfusion (mL/min/cc tissue) | 1.8 ± 0.3 | 1.9 ± 0.6 | 1.85 ±0.4 |
| RVR (mm Hg/mL per min) | 0.31 ± 0.05 | 0.60 ± 0.12* | 0.66 ± 0.07* |
| Renal volume (cc) | 124.7 ± 8.3 | 69.3 ± 6.4* | 82.0 ± 3.4*† |
Values are expressed as mean ± SEM. Renal vascular resistance (RVR) was quantified as mean systolic blood pressure divided by renal blood flow.
p<0.05 vs normal
p<0.05 vs untreated RVD
Table 1b:
General characteristics of each group at 10 weeks (4 weeks post-treatment, n=6-7/group).
| Normal | RVD | RVD+KTP-ELP-VEGF | |
|---|---|---|---|
| Body weight (kg) | 54.9 ± 1.9^ | 54.4 ± 2.1^ | 57.7 ± 2.5^ |
| Mean arterial pressure (mm Hg) | 110.2 ± 3.7 | 145.3 ± 7.0*^ | 135.1 ± 6.4* |
| Cortical perfusion (mL/min/cc tissue) | 3.8 ± 0.4 | 3.4 ± 0.6 | 3.5 ± 0.3^ |
| Medullary perfusion (mL/min/cc tissue) | 2.0 ± 0.3 | 1.9 ± 0.6 | 1.9 ± 0.2 |
| RVR (mm Hg/mL per min) | 0.33 ± 0.07 | 0.54 ± 0.12* | 0.52 ± 0.04*^ |
| Renal volume (cc) | 136.2 ± 12.1 | 76.8 ± 11.2* | 100.6 ± 5.5*†^ |
| Blood urea nitrogen (mg/dL) | 4.4 ± 0.7 | 7.5 ± 0.9* | 5.3 ± 0.8† |
Values are expressed as mean ± SEM. Renal vascular resistance (RVR) was quantified as mean systolic blood pressure divided by renal blood flow. The degree of renal artery stenosis in RVD and RVD+KTP-ELP-VEGF pigs was unchanged compare to 6 weeks.
p<0.05 vs normal
p<0.05 vs untreated RVD
p<0.05 vs same group at 6 weeks (Table 1a)
MDCT-derived renal hemodynamics
At 6 weeks of RVD, immediately prior to treatment, RBF was similarly and significantly reduced in both RVD (−65.5% vs normal, p<0.05) and RVD KTP-ELP-VEGF (−63.2% vs normal, p<0.05) compared to the sham-operated normal group (Figure 1). This was paralleled by a significant drop in GFR in RVD (−47.9% vs normal, p<0.05) and RVD KTP-ELP-VEGF (−50.7% vs normal, p<0.05) compared to normal.
Figure 1.
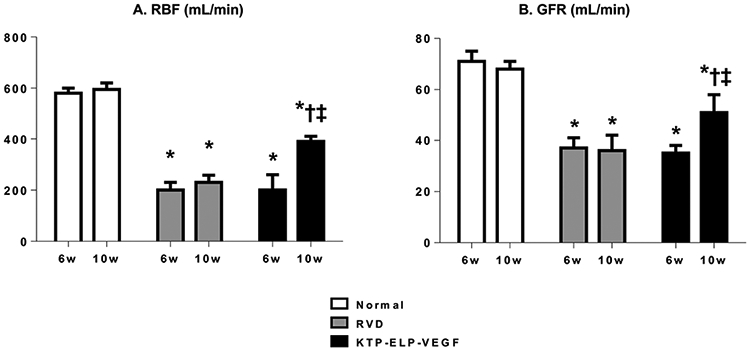
Single-kidney renal blood flow (RBF, A) and glomerular filtration rate (GFR, B) in normal, RVD, and RVD+KTP-ELP-VEGF pigs (stenotic kidney) at 6 and 10 weeks. Both RBF and GFR were significantly reduced in RVD groups at 6 weeks. No change was observed in untreated pigs at 10 weeks while a significant increase was observed in those receiving KTP-ELP-VEGF. n=6-7 per group. *p<0.05 vs. normal; † p<0.05 vs. RVD; ‡ p<0.05 vs. 6 weeks (Single way ANOVA, Tukey).
At 10 weeks, four weeks after treatment, BUN was significantly increased in untreated pigs but not those receiving KTP-ELP-VEGF (Table 1b). No significant change was observed in the untreated RVD compared to pre-treatment/placebo values in either RBF (+15% vs 6w, p=NS) or GFR (−2.7% vs 6w, p=NS), demonstrating stable disease. Conversely, KTP-ELP-VEGF treated pigs were found to have significantly improved RBF (+85.7% vs 6w, p<0.05) and GFR (+45.7% vs 6w p<0.05) compared to pre-treatment values (Figure 1).
Micro-CT quantification of renal MV density
MV rarefaction was observed in both cortex and medulla of the untreated RVD group (Figure 2a,b). This was found in association with an increase in media-to-lumen ratio indicating concomitant remodeling of remaining vasculature (Figure 2c). Notably, MV rarefaction was markedly improved in pigs receiving KTP-ELP-VEGF. This effect was much more pronounced in the cortex, particularly in small and medium sized microvessels (0-200). Improvements in MV density following KTP-ELP-VEGF were associated with reduced renal vascular resistance (RVR), restored cortical perfusion (Table 1b), and attenuation of vascular remodeling, indicated by reduced media-to-lumen ratio.
Figure 2.

Representative Micro-CT images of renal microvasculature (A.) and quantification of cortical and medullary microvascular (MV) density (B.), media-to-lumen ratio (C.) representative trichrome stained mid-hilar stenotic kidney sections (D.), and morphometric analysis (quantification of renal fibrosis, E.) from normal, RVD and RVD+KTP+ELP-VEGF pigs after 10 weeks of observation. MV density was markedly decreased in untreated RVD across all diameters assessed and was significantly increased but not normalized in pigs receiving KTP-ELP-VEGF. Consistent with this was increased media-to-lumen ratio in RVD which was attenuated by treatment, suggesting attenuated vascular remodeling. Significant fibrosis was observed in the all compartments of the untreated RVD kidneys, most notably in the perivascular and tubulointerstitial compartments (arrows). Fibrosis was significantly reduced with KTP-ELP-VEGF although limited residual tubulointerstitial fibrosis was noted (arrow). Scale bar represents 20 μm. n=6-7 per group. *p<0.05 vs. normal; † p<0.05 vs. RVD; ‡ p<0.05 vs. 6 weeks (Single way ANOVA, Tukey).
Renal morphology and macrophage infiltration
Significant fibrosis was observed in the all compartments of the untreated RVD kidneys, most prominently in the perivascular and tubulointerstitial compartments (Figure 2d). Notably, image analysis and quantification revealed dramatic overall reduction in fibrosis compared to untreated RVD (Figure 2e). M1 macrophage infiltration was significantly greater in untreated RVD compared to normal whereas KTP-ELP-VEGF treated pigs showed no difference from normal. KTP-ELP-VEGF treated pigs showed significantly increased renal infiltration of M2 macrophages compared to RVD or normal (Figure 3a-b).
Figure 3.

Representative immunostaining (A) and quantification (B) of M1 and M2 macrophages using the M1 marker indoleamine-2,3-dioxygenase (IDO1, red), the M2 marker mannose receptor c type-1 (MRC1, green), and the nuclear counterstain DAPI (blue) in normal, RVD, and RVD+KTP-ELP-VEGF pigs after 10 weeks of observation. Untreated RVD pigs showed significantly increased macrophage infiltration which was markedly skewed towards inflammatory M1 polarization. Pigs treated with KTP-ELP-VEGF showed similar overall macrophage infiltration which was shifted in favor of anti-inflammatory M2s. Scale bar represents 20 μm. n=6 per group. *p<0.05 vs. normal; † p<0.05 vs. RVD (Single way ANOVA, Tukey).
In vitro studies
Macrophage polarization
In a previous study, we showed that renal VEGF therapy induces M2 polarization and VEGF expression in renal macrophages[12]. Here, we aimed to determine the direct effects of VEGF on macrophage polarization and effector function using human macrophages in vitro. mRNA expression of IDO1 and CD163 was quantified by qPCR as these are accepted markers of M1 and M2 macrophage polarization respectively[31, 32]. As expected, IDO1 expression was significantly elevated in all M1 but no M0 macrophages, supporting the successful generation of an M1 phenotype (Figure 4a). CD163 expression was significantly increased in M0s treated with VEGF or KTP-ELP-VEGF relative to unstimulated M0 and increased in the M1+VEGF group compared to unstimulated M1s (Figure 4b).
Figure 4.
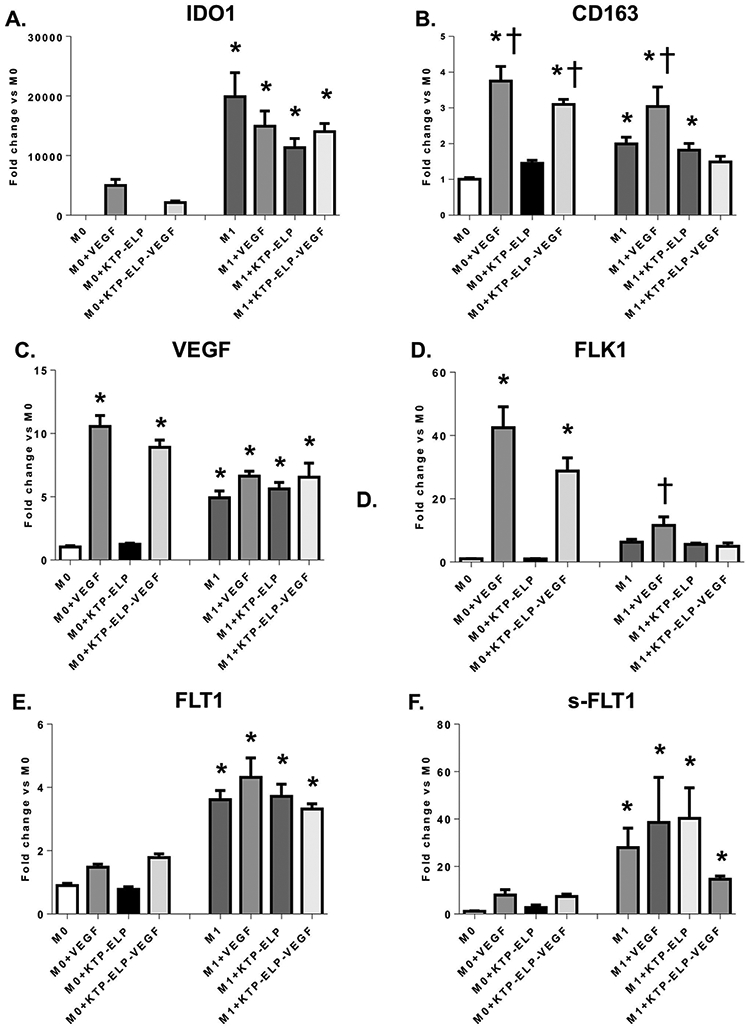
Quantitative real time polymerase chain reaction (qRT-PCR) performed on M0 and M1 macrophages incubated in VEGF, KTP-ELP, KTP-ELP-VEGF, or control media. The macrophage M1 marker (indoleamine-2,3-dioxygenase 1, IDO1, A) was significantly increased in all macrophages incubated in LPS and IFNγ supporting successful M1 polarization. The M2 marker (CD163, B) was dramatically elevated by VEGF or KTP-ELP-VEGF in M0 macrophages indicating that VEGF induces an M2 phenotype primarily through effects on naive macrophages. A significant increase in CD163 was seen in M1s treated with VEGF only, suggesting some phenotype switching from M1 to M2 may occur. M0s showed substantial increases in expression of VEGF (C.) and its receptor Flk-1 (D.) when stimulated with VEGF or KTP-ELP-VEGF, suggesting a feed-forward induction of a pro-angiogenic phenotype. In contrast M1 macrophages expressed moderate levels of VEGF and both membrane-bound (E.) and soluble (F.) forms of its receptor Flt-1 at baseline but showed no demonstrable response to VEGF therapy. n=6 per group. *p<0.05 vs. M0 † p<0.05 vs. M1 (Single way ANOVA, Tukey).
Macrophage angiogenic signaling
Naïve M0 macrophages demonstrated a robust increase in VEGF expression when exposed to either VEGF or KTP-ELP-VEGF but not KTP-ELP (Figure 4c). M1 macrophages exhibited a modest level of VEGF expression which was notably unaltered by VEGF stimulus. We subsequently measured expression of VEGF’s primary receptors Flk-1 and Flt-1. We found that Flk-1 (Figure 4d) mirrored VEGF expression in M0 macrophages, increasing dramatically in those treated with VEGF or KTP-ELP-VEGF (approximately 40-fold and 30-fold respectively compared to unstimulated M0). M1s showed a small but significant increase in Flk-1 expression only when treated with VEGF alone. Flt-1 and sFlt-1 expression (Figure 4e,f), in contrast, was significantly elevated only in M1s and again was unresponsive to VEGF stimulus.
Discussion
In a series of recent studies from our laboratories that developed and characterized the constructs, we demonstrate that addition of ELP to VEGF, KTP to ELP and KTP-ELP to VEGF increases renal targeting, prolongs its half-life, and significantly shifts its deposition from the liver to the kidney without interfering with VEGF’s biological activity or inducing any collateral effects in other organs such as the liver or the heart[17, 11, 18]. Herein we extended those studies by investigating the therapeutic efficacy in vivo and found that the KTP-ELP-VEGF construct effectively recovered stenotic kidney blood flow and filtration function in the swine model of RVD, which was accompanied by improved MV density, suppressed M1 and augmented M2 macrophage infiltration in the kidneys, and attenuated fibrosis. The renal functional improvements were comparable to those seen after ELP-VEGF treatment of RVD[10] and CKD[12] in our recent work, indicating that addition of the KTP moiety did not interfere with the efficacy of renal VEGF therapy, which extends our previous work[17, 18]. Thus, this novel drug-delivery technology and strategy may refine renal delivery of VEGF, which could have significant implications for clinical translation.
Owing in part to its high metabolic activity, MV rarefaction has been shown to occur in the cortex before the medulla, driving functional impairment and parenchymal atrophy[21]. MV recovery in the KTP-ELP-VEGF treated kidney was most pronounced in the cortex, particularly the small and medium sized vessels (0-200 μm), and accordingly associated with preferential improvement of cortical perfusion and a reduction in overall RVR, suggesting functional restoration of the renal MV architecture. Recovery of cortical blood flow was linked to increased renal volume, in line with normal animals, compared to no change in untreated RVD. This finding supports the notion that rescued glomerular function may have facilitated the expansion of active renal parenchyma. This cortical effect driving renal recovery is in agreement with our previous study showing that fluorescently labeled KTP-ELP-VEGF displays a clear preference for deposition in the cortex, notably binding to glomerular capillaries and tubular cells[18]. This may be attributable to the increased vascularity of the cortex relative to the medulla or, likely, a binding partner for KTP which is preferentially expressed in the cortical parenchyma and will be addressed in future studies. Regardless, by targeted glomerular deposition, KTP-ELP-VEGF may act locally to restore blood flow to under perfused glomeruli, thereby rescuing ischemic but recoverable parenchyma.
The current study also aimed to shed light into potential mechanisms of KTP-ELP-VEGF effects. Macrophages are well described players in the pathogenesis of renal injury. Yet more recently, it has been appreciated that macrophages play a pivotal role in renal regeneration as well[33, 34]. Shortly after renal injury, resident and infiltrating macrophages polarize to a pro-inflammatory M1 phenotype[35]. In the normal course of injury and recovery these M1 macrophages are progressively replaced by regenerative M2 macrophages over 2-3 weeks[35]. However, we have shown that this phenotype switch can fail to occur in chronic renal injury[26] leading to a persistent M1 response and pervasive inflammation which exacerbates parenchymal damage.
We recently showed that ELP-VEGF therapy in CKD results in a shift in the renal macrophage population from M1 to a predominantly M2 phenotype[12]. Herein we demonstrated a similar effect, as KTP-ELP-VEGF suppressed M1 macrophage polarization and induced a significant increase in renal M2 macrophage polarization in vivo compared to untreated RVD. However, the magnitude of this effect was less than that observed using ELP-VEGF in our CKD model[12]. Several factor may have played a role in these differences. First, the length of observation and model are different compared to[12]. Speculatively, differences in the magnitude of M2 increase may also be attributable to differences in the therapeutic constructs, with addition of KTP modifying macrophage binding. Alternatively, macrophage presence in the stenotic kidney may be improved by a healthier microenvironment after KTP-ELP-VEGF therapy (reflected by improved RBF and GFR) or may have been reduced in this model compared to established CKD due to the less advanced stage of renal injury. Fewer renal macrophages prior to treatment would naturally diminish the degree of the M2 phenotype shift observed after VEGF in the RVD model.
While the augmented M2 population and suppressed M1 infiltration was associated with significant MV and functional recovery in vivo, it was not certain whether this is driven directly by an effect of VEGF on macrophages. Rather, if VEGF induces renal recovery by some other means it is possible that the attenuated inflammatory milieu allows the M2 phenotype to emerge after the fact. Herein we aimed to address this uncertainty in vitro. Our results show that naive resting macrophages (M0) do, in fact, directly respond to stimulus from VEGF or KTP-ELP-VEGF by polarizing to an M2 phenotype, indicated by the increase in CD163 expression. No change in polarization state was seen in response to KTP-ELP, suggesting the effect was indeed mediated by the VEGF moiety. M1 macrophages were relatively insensitive to the polarizing effects of VEGF. These findings strongly indicate that the influx of M2 macrophages observed in vivo may occur secondary to an effect of VEGF on naïve M0 macrophages rather than by shifting the phenotype of M1s.
One perplexing finding is the apparent increase in CD163 in M1 macrophages following VEGF but not KTP-ELP-VEGF. Note that, compared to the untreated M1 control, all M1 groups showed a trend towards decrease in IDO1, however this was not significant. In the context of persistently high IDO1 expression, the elevated CD163 expression following free VEGF may suggest adoption of a mixed phenotype, supporting the now well-accepted notion that macrophage polarization is more akin to a spectrum, with the M1 and M2 phenotype being on opposite ends[35]. Experiments showing equivalent activity between VEGF and KTP-ELP-VEGF have been focused on endothelial and tubular cells function such as proliferation and tube formation[18]. It is well established that the effects of VEGF in these cells are primarily mediated through the Flk-1 receptor[36]. Thus, the possibility remains that addition of the KTP-ELP motif impedes binding of VEGF to the Flt-1 receptor, leading to limited effects on M1s marked by high Flt-1 expression. If true, this may contribute to therapeutic efficacy in vivo by shifting binding in favor of Flk-1 and away from Flt-1 and sFlt-1. Further studies may be needed to evaluate this phenomenon.
The shift towards M2 polarization following VEGF stimulus in M0s was associated with dramatically increased mRNA expression of VEGFA as well as its receptor Flk-1. This pronounced effect suggests a potential feed forward phenomenon wherein binding of the Flk-1 receptor by VEGF acts to increase the cell’s own VEGF production which further stimulates Flk-1 signaling. Taken together these results suggest that VEGF induces a M2 phenotype characterized by high angiogenic potential and support a potential role for these cells in prolonging the MV protective effects of a single transient dose of VEGF in RVD.
M2 macrophages do not have a monopoly on angiogenic signaling, as it has been documented that M1s may be a source of VEGF as well[37]. This is consistent with our in vitro results showing that under control conditions, M1s demonstrate markedly higher VEGF expression than M0s. However, in the presence of exogenous VEGF, its expression in M0s increased to levels much greater than that of M1s under any conditions. Notably, we observed that M0s favor expression of the Flk-1 receptor while M1s predominantly express Flt-1 suggesting that differences in receptor expression may explain the contrasting responses to VEGF. Flk-1 is known to mediate the majority of the angiogenic effects of VEGF as it exerts strong mitogenic and pro-survival signals in endothelial cells[38]. In contrast, Flt-1 has been shown to primarily suppress or modulate Flk-1 signaling[36]. Indeed, it has been shown that knockout of Flt-1 leads to unregulated Flk-1 activation and excessive angiogenesis [39]. This suppressive effect of Flt-1 activation may explain the apparent insensitivity of M1s to VEGF stimulus.
Interestingly, M1 macrophages in vitro showed very high expression of a soluble form of the Flt-1 receptor (sFlt-1). Created by alternative splicing of the Flt-1 gene[36], sFlt-1 retains the extracellular domain of Flt-1 along with its ability to avidly bind VEGF. However, it lacks transmembrane and intracellular motifs, allowing it to be released freely from cells where it can bind and trap free VEGF[36]. sFlt-1 produced by M1s may have acted to neutralize VEGF and KTP-ELP-VEGF, further explaining the relative insensitivity of M1s to VEGF. Elevated sFlt-1 levels have been shown to be detrimental in the kidney, causing glomerular injury and damage to the filtration barrier[40-42]. Given the extensive infiltration of M1 macrophages in chronic renal injury[26], it is possible that secretion of sFlt-1 by M1s may drive the suppression of VEGF signaling and subsequent MV injury in RVD. Furthermore, suppressing sFlt-1 production by modulating or suppressing renal macrophages may be a viable therapeutic strategy though additional studies will be needed to elucidate this in vivo.
Limitations and Opportunities
The use of human cell lines in controlled in vitro studies allows us to build upon previous work with swine in vivo [26, 12] and provide mechanistic insights to previous associations and support translatability to humans. However, validation of these effects using primary cell culture and in vivo studies specifically targeting macrophages (e.g. macrophage depletion[43, 44]) as well as extended characterization of M1 and M2 macrophages in the presence of a specific macrophage marker in the swine kidney would be necessary in future studies. The animals used in this study were juvenile pigs from both sexes, with RVD as the sole insult. Due to its gradually progressive course, RVD primarily presents in older individuals, with most patients being over 60[45]. Additionally, most RVD patients present at a relatively advanced stage with multiple co-morbid conditions[45]. Thus, studies using older animals with additional insults (e.g. diabetes, obesity[46-48]) will expand our findings and may contribute to their translation. Furthermore, although we showed that the KTP-ELP construct and the fusion of KTP-ELP-VEGF targets the kidney independent of the species tested [17, 18], additional studies in the swine model of RVD would be needed to characterize its biodistribution, the targeting profile, and safety, as we did with ELP-VEGF[10, 11]. Still, it is important to highlight that the current study supports the plasticity of ELP technology to be fused to different therapeutic agents[10, 11, 49] as well as the possibility of applying ELP-driven strategies in other diseases[49] and in other forms of renal disease[12, 50] offers the opportunity for more avenues of needed research towards new applications and for the progression of its potential for clinical translation.
In summary, our results demonstrate the importance of renal MV integrity in the pathophysiology of renal injury in the stenotic kidney and expand upon mechanisms of therapeutic angiogenesis as a viable treatment strategy for RVD and CKD. Herein we demonstrate the efficacy of a novel kidney-specific KTP-ELP-VEGF construct in recovering renal function. Furthermore, we show a novel potential mechanism of M1 macrophages in driving MV rarefaction and establish a direct role of VEGF in modulating macrophages to an angiogenic M2 phenotype, indicating that macrophage modulation may be a prominent mechanism of therapeutic angiogenesis to induce renal recovery.
Acknowledgments
Sources of Funding
This work was supported by grants R01HL095638, P01HL51971, P20GM104357, and R01HL121527 from the National Institutes of Health and grants IPA34170267, PRE34380314, and PRE34380274 from the American Heart Association.
Footnotes
Disclosures
G.L. Bidwell is the owner of Leflore Technologies LLC, a private company working to develop and commercialize ELP (elastin-like polypeptide)-based technologies in several disease areas. G.L. Bidwell and A.R. Chade are inventors on patents related to ELP technology.
Conflicts of Interest: None
Literature Cited
- 1.Herrmann SM, Textor SC. Renovascular Hypertension. Endocrinology and Metabolism Clinics of North America. 2019. 2019/December/01/;48(4):765–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalra PA, Guo H, Kausz AT, Gilbertson DT, Liu J, Chen SC, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney international. 2005. July;68(1):293–301. [DOI] [PubMed] [Google Scholar]
- 3.Investigators A, Wheatley K, Ives N, Gray R, Kalra PA, Moss JG, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med. 2009. November 12;361(20):1953–62. [DOI] [PubMed] [Google Scholar]
- 4.Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney international. 2015. February;87(2):297–307. [DOI] [PubMed] [Google Scholar]
- 5.Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, et al. Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. Journal of the American Society of Nephrology : JASN. 2015. April;26(4):817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leonard EC, Friedrich JL, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. American journal of physiology Renal physiology. 2008. December;295(6):F1648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chade AR, Kelsen S. Renal microvascular disease determines the responses to revascularization in experimental renovascular disease. Circulation Cardiovascular interventions. 2010. August;3(4):376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chade AR, Kelsen S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: a novel potential therapeutic approach. American journal of physiology Renal physiology. 2012. May 15;302(10):F1342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivaskandarajah GA, Jeansson M, Maezawa Y, Eremina V, Baelde HJ, Quaggin SE. Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 2012. November;61(11):2958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chade AR, Tullos NA, Harvey TW, Mahdi F, Bidwell GL 3rd., Renal Therapeutic Angiogenesis Using a Bioengineered Polymer-Stabilized Vascular Endothelial Growth Factor Construct. Journal of the American Society of Nephrology : JASN. 2016. June;27(6):1741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chade AR, Williams ML, Guise E, Vincent LJ, Harvey TW, Kuna M, et al. Systemic biopolymer-delivered vascular endothelial growth factor promotes therapeutic angiogenesis in experimental renovascular disease. Kidney international. 2018. April;93(4):842–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel J, Williams E, Williams M, Bidwell G, Chade A. Targeted VEGF (Vascular Endothelial Growth Factor) Therapy Induces Long-Term Renal Recovery in Chronic Kidney Disease via Macrophage Polarization. Hypertension. 2019;74:00–00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bidwell GL 3rd, Raucher D. Application of thermally responsive polypeptides directed against c-Myc transcriptional function for cancer therapy. Mol Cancer Ther. 2005. July;4(7):1076–85. [DOI] [PubMed] [Google Scholar]
- 14.Bidwell GL 3rd, Davis AN, Fokt I, Priebe W, Raucher D. A thermally targeted elastin-like polypeptide-doxorubicin conjugate overcomes drug resistance. Invest New Drugs. 2007. August;25(4):313–26. [DOI] [PubMed] [Google Scholar]
- 15.Bidwell GL 3rd, Fokt I, Priebe W, Raucher D. Development of elastin-like polypeptide for thermally targeted delivery of doxorubicin. Biochem Pharmacol. 2007. March 1;73(5):620–31. [DOI] [PubMed] [Google Scholar]
- 16.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996. March 28;380(6572):364–6. [DOI] [PubMed] [Google Scholar]
- 17.Bidwell GL 3rd, Mahdi F, Shao Q, Logue OC, Waller JP, Reese C, et al. A kidney-selective biopolymer for targeted drug delivery. American journal of physiology Renal physiology. 2017. January 1;312(1):F54–f64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahdi F, Chade AR, Bidwell GL 3rd. Utilizing a Kidney-Targeting Peptide to Improve Renal Deposition of a Pro-Angiogenic Protein Biopolymer. Pharmaceutics. 2019. October 18;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerman LO, Schwartz RS, Grande JP, Sheedy PF, Romero JC. Noninvasive evaluation of a novel swine model of renal artery stenosis. Journal of the American Society of Nephrology : JASN. 1999;10:1455–65. [DOI] [PubMed] [Google Scholar]
- 20.Chade AR, Rodriguez-Porcel M, Grande JP, Krier JD, Lerman A, Romero JC, et al. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation. 2002. August 27;106(9):1165–71. [DOI] [PubMed] [Google Scholar]
- 21.Iliescu R, Fernandez SR, Kelsen S, Maric C, Chade AR. Role of renal microcirculation in experimental renovascular disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2010. April;25(4):1079–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelsen S, Hall JE, Chade AR. Endothelin-A receptor blockade slows the progression of renal injury in experimental renovascular disease. American journal of physiology Renal physiology. 2011. July;301(1):F218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chade AR, Stewart NJ, Peavy PR. Disparate effects of single endothelin-A and -B receptor blocker therapy on the progression of renal injury in advanced renovascular disease. Kidney international. 2014. April;85(4):833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eirin A, Zhu XY, Li Z, Ebrahimi B, Zhang X, Tang H, et al. Endothelial outgrowth cells shift macrophage phenotype and improve kidney viability in swine renal artery stenosis. Arteriosclerosis, thrombosis, and vascular biology. 2013. May;33(5):1006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006. August;20(10):1706–8. [DOI] [PubMed] [Google Scholar]
- 26.Chade AR, Williams ML, Engel J, Guise E, Harvey TW. A translational model of chronic kidney disease in swine. American journal of physiology Renal physiology. 2018. August 1;315(2):F364–f73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guise E, Engel JE, Williams ML, Mahdi F, Bidwell GL 3rd, Chade AR. Biopolymer-delivered vascular endothelial growth factor improves renal outcomes following revascularization. American journal of physiology Renal physiology. 2019. May 1;316(5):F1016–f25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PloS one. 2010. January 13;5(1):e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wheeler KC, Jena MK, Pradhan BS, Nayak N, Das S, Hsu CD, et al. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PloS one. 2018;13(1):e0191040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrmann SM, Textor SC. Renovascular Hypertension. Endocrinol Metab Clin North Am. 2019. December;48(4):765–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapetanovic R, Fairbairn L, Beraldi D, Sester DP, Archibald AL, Tuggle CK, et al. Pig Bone Marrow-Derived Macrophages Resemble Human Macrophages in Their Response to Bacterial Lipopolysaccharide. The Journal of Immunology. 2012;188(7):3382–94. [DOI] [PubMed] [Google Scholar]
- 32.Schroder K, Irvine KM, Taylor MS, Bokil NJ, Le Cao KA, Masterman KA, et al. Conservation and divergence in Toll-like receptor 4-regulated gene expression in primary human versus mouse macrophages. Proc Natl Acad Sci U S A. 2012. April 17;109(16):E944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishida M, Okumura Y, Fujimoto S, Shiraishi I, Itoi T, Hamaoka K. Adoptive transfer of macrophages ameliorates renal fibrosis in mice. Biochemical and biophysical research communications. 2005. June 24;332(1):11–6. [DOI] [PubMed] [Google Scholar]
- 34.Ji L, Chen Y, Wang H, Zhang W, He L, Wu J, et al. Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int J Oncol. 2019. July;55(1):103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. Journal of immunology (Baltimore, Md : 1950). 2000. June 15; 164(12):6166–73. [DOI] [PubMed] [Google Scholar]
- 36.Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Romanian journal of morphology and embryology = Revue roumaine de morphologie et embryologie. 2018;59(2):455–67. [PubMed] [Google Scholar]
- 37.Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014. May;35(15):4477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cellular Signalling. 2007. 2007/October/01/;19(10):2003–12. [DOI] [PubMed] [Google Scholar]
- 39.Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am J Pathol. 2004. May;164(5):1531–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henao DE, Arias LF, Mathieson PW, Ni L, Welsh GI, Bueno JC, et al. Preeclamptic sera directly induce slit-diaphragm protein redistribution and alter podocyte barrier-forming capacity. Nephron Exp Nephrol. 2008;110(3):e73–81. [DOI] [PubMed] [Google Scholar]
- 41.Zhao S, Gu X, Groome LJ, Wang Y. Decreased nephrin and GLEPP-1, but increased VEGF, Flt-1, and nitrotyrosine, expressions in kidney tissue sections from women with preeclampsia. Reproductive sciences (Thousand Oaks, Calif). 2009. October;16(10):970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moghaddas Sani H, Zununi Vahed S, Ardalan M. Preeclampsia: A close look at renal dysfunction. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2019. January;109:408–16. [DOI] [PubMed] [Google Scholar]
- 43.D'Souza MJ, Oettinger CW, Shah A, Tipping PG, Huang XR, Milton GV. Macrophage depletion by albumin microencapsulated clodronate: attenuation of cytokine release in macrophage-dependent glomerulonephritis. Drug Dev Ind Pharm. 1999. May;25(5):591–6. [DOI] [PubMed] [Google Scholar]
- 44.Ferenbach DA, Sheldrake TA, Dhaliwal K, Kipari TM, Marson LP, Kluth DC, et al. Macrophage/monocyte depletion by clodronate, but not diphtheria toxin, improves renal ischemia/reperfusion injury in mice. Kidney international. 2012. October;82(8):928–33. [DOI] [PubMed] [Google Scholar]
- 45.Herrmann SM, Textor SC. Current Concepts in the Treatment of Renovascular Hypertension. American journal of hypertension. 2018;31(2):139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iliescu R, Chade AR. Progressive renal vascular proliferation and injury in obese Zucker rats. Microcirculation. 2010. May;17(4):250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Futrakul N, Futrakul P. Renal microvascular disease predicts renal function in diabetes. Ren Fail. 2012;34(1):126–9. [DOI] [PubMed] [Google Scholar]
- 48.Hall ME, do Carmo JM, da Silva AA, Juncos LA, Wang Z, Hall JE. Obesity, hypertension, and chronic kidney disease. Int J Nephrol Renovasc Dis. 2014;7:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eddy AC, Howell JA, Chapman H, Taylor E, Mahdi F, George EM, et al. Biopolymer-Delivered, Maternally Sequestered NF-κB (Nuclear Factor-κB) Inhibitory Peptide for Treatment of Preeclampsia. Hypertension. 2020. January;75(1): 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chade AR, Williams ML, Engel JE, Williams E, Bidwell GL 3rd. Molecular targeting of renal inflammation using drug delivery technology to inhibit NF-κB improves renal recovery in chronic kidney disease. American journal of physiology Renal physiology. 2020. July 1;319(1):F139–f48. [DOI] [PMC free article] [PubMed] [Google Scholar]