

Abstract
Rotenone, a mitochondrial complex I inhibitor, has been widely used to study the effects of mitochondrial dysfunction on dopaminergic neurons in the context of Parkinson’s disease. Although the deleterious effects of rotenone are well documented, we found that young adult Caenorhabditis elegans showed resistance to 24- and 48-hour rotenone exposures. To better understand the response to rotenone in C. elegans, we evaluated mitochondrial bioenergetic parameters after 24- and 48-hour exposures to 1μM or 5 μM rotenone. Results suggested upregulation of mitochondrial complexes II and V following rotenone exposure, without major changes in oxygen consumption or steady-state ATP levels after rotenone treatment at the tested concentrations. We found evidence that the glyoxylate pathway (an alternate pathway not present in higher metazoans) was induced by rotenone exposure; gene expression measurements showed increases in mRNA levels for two complex II subunits and for isocitrate lyase, the key glyoxylate pathway enzyme. Targeted metabolomics analyses showed alterations in the levels of organic acids, amino acids, and acylcarnitines, consistent with the metabolic restructuring of cellular bioenergetics pathways including activation of complex II, the glyoxylate pathway, glycolysis, and fatty acid oxidation. This expanded understanding of how C. elegans responds metabolically to complex I inhibition via multiple bioenergetic adaptations, including the glyoxylate pathway, will be useful in interrogating the effects of mitochondrial and bioenergetic stressors and toxicants.
Keywords: Caenorhabditis elegans (C. elegans), rotenone, complex I, metabolism, metabolomics, mitochondrial metabolism
1. Introduction
The pesticide rotenone was discovered to act via electron transport chain (ETC) complex I inhibition in the early 1960s (Lindahl and Öberg 1961). This mechanism of action became of particular interest to the Parkinson’s disease (PD) research community after it was discovered that there was significant complex I inhibition in parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and that postmortem substantia nigra samples from PD patients exhibited decreased complex I function (Langston et al. 1983; Schapira et al. 1990). Rotenone has since become a widely used tool in the study of PD, particularly after a study by Betarbet et al. (2000) showed that chronic, systemic infusion of this chemical can recapitulate hallmarks of parkinsonism (including substantia nigra neurodegeneration). Research into rotenone’s mode of action, particularly how systemic complex I inhibition can lead to such region-specific neurodegeneration, has helped unveil its effects on mitochondrial and neuronal health beyond inhibiting complex I. It is now known that rotenone exposure causes oxidative stress, ATP depletion, and cell death (Li et al. 2003; Sherer et al. 2003). This increase in oxidative stress results in damage to cellular macromolecules (Sanders and Greenamyre 2013). It has also been documented that rotenone increases calcium influx via the N-methyl-D-aspartate (NMDA) receptor (which is constantly active in dopaminergic neurons), leading to excitotoxicity; another study showed that a dopamine metabolite causes structural modification and oligomerization of α-synuclein (a protein that aggregates to form Lewy bodies in PD) following rotenone exposure (Costa et al. 2008; De Miranda et al. 2016; Follmer et al. 2015). Both of these studies help shed light on rotenone’s selectivity for dopaminergic neurons.
Another research focus for rotenone toxicity has been its effects on bioenergetics and metabolism, which are particularly interesting given the link between PD and bioenergetic alteration (Requejo-Aguilar and Bolaños 2016). Studies have revealed alterations in mitochondrial bioenergetic parameters, reporting decreases in oxygen consumption, increases in glycolysis, and alterations in the levels of organic and amino acids after rotenone exposure (Dranka et al. 2012; Giordano et al. 2012; Karlsson et al. 2016; Lei et al. 2014; Xu et al. 2011). Increases in lipid β-oxidation have also been reported (Worth et al. 2014).
Although the mitochondrial and neuronal alterations caused by rotenone exposure have been well documented in the scientific literature, we have observed instances in which the model organism Caenorhabditis elegans displays resistance to rotenone’s toxic effects. Previous work in our laboratory evaluated DNA damage levels and mitochondrial genome copy number in C. elegans following rotenone exposure; we were unable to detect significant alterations (Gonzalez-Hunt et al. 2014). We also did not detect significant levels of dopaminergic neurodegeneration after 24- or 48-hour rotenone exposures in young adult worms (unpublished observations), despite robust neurodegeneration at this lifestage with 6-hydroxydopamine and other chemical exposures (Gonzalez-Hunt et al. 2014; Smith et al. 2019). Both mtDNA damage and neurodegeneration have been reported in other systems after rotenone exposure (Gonzalez-Hunt and Sanders 2020; Sanders et al. 2014a; Sanders et al. 2014b). The goal of this study was to characterize the response to rotenone in C. elegans, focusing on functional mitochondrial and metabolic endpoints. We performed chemical metabolic challenges, measured oxygen consumption utilizing the Seahorse XFe24 Bioanalyzer, and carried out targeted metabolomics analyses. We saw alterations in amino acids, organic acids, and acylcarnitines similar to those previously reported for mitochondrial complex I mutants or after rotenone exposure in other model systems, but we did not see major alterations in oxygen consumption. Our data shows that there is an upregulation of complex II activity, fatty acid oxidation, and induction of the glyoxylate cycle (an alternative pathway in the citric acid cycle not present in higher metazoans (Holmes 1993; Kondrashov et al. 2006), although there may be a role for dietary glyoxylate (Strittmatter et al. 2014)). Overall, our results show that C. elegans’ response to rotenone involves multiple metabolic changes that might result in compensation for loss of complex I function. They also highlight the importance of considering the biology of C. elegans and how it might differ from other metazoans when performing experiments, particularly when the goal is to extrapolate results from lower to higher organisms.
2. Materials and Methods
2.1. C. elegans culture
This study utilizes C. elegans as our model organism; these nematodes have long been used to study mechanisms of toxicity for exogenous compounds, and they are increasingly being used to predict human responses to toxicologically relevant exposures (Leung et al. 2008). Synchronized populations of nematodes were obtained by harvesting eggs from gravid adults via hypochlorite treatment (Lewis and Fleming 1995). Worms were hatched for no more than 14 hours in K+ medium, referred to as “complete K-medium” in Boyd et al. (2009). Worm transfers were performed by washing nematodes off agar plates or treatment wells and rinsing (after centrifugation at 2200 g for 2 min) with K medium.
C. elegans strains used were N2 (Bristol), JK1107 glp-1(q224) III, PE255 glp-4(bn2), and RB766 icl-1(ok531). The N2, JK1107, and RB766 strains were obtained from the Caenorhabditis Genetics Center (University of Minnesota). The PE255 strain was generously provided by Cristina Lagido (University of Aberdeen, Aberdeen, UK).
For experiments with adults (all except larval growth assays), populations of C. elegans were grown on OP50-seeded K agar plates (Williams and Dusenbery 1988) at 25 °C until dosing; at this temperature, the glp-1 and glp-4 strains lose germline proliferation (Kodoyianni et al. 1992). For population maintenance and expansion purposes, N2 and RB766 were kept at 20 °C on OP50-seeded K agar plates; PE255 and JK1107 were maintained at 15 °C until ready for experiments due to their temperature-sensitive sterility.
2.2. Rotenone exposures in young adults
After liquid hatch in K+ medium, worms were grown at 25 °C for 48 hours and then transferred to K medium and allowed to clear their guts for 15 minutes. They were then exposed for 24 or 48 hours to rotenone (CAS Number 83-79-4, ≥ 95% purity, Sigma-Aldrich; dissolved in DMSO, final concentration 1%) in K+ medium and fed 2X concentrated UVC-inactivated UvrA-deficient E. coli; bacteria were exposed to 1000 J/m2 UVC with the UVLMS-38 EL Series Lamp emitting at 254 nm [UVP, LLC], as previously described (Meyer et al. 2010). Following the exposure, worms were washed, allowed to clear their guts for 15 minutes, and measured as described in the next sections. The rotenone concentrations were chosen based on a high dose that would cause no lethality but minimal growth inhibition for each strain. Published analyses of treated C. elegans have shown that the internal concentrations of many compounds are lower than the dose in the culture media; this is likely due to the presence of a collagen cuticle limiting uptake of ionized or bulky compounds (Maurer et al. 2018; Watanabe et al. 2005). Based on published data, we estimate that the doses chosen for this study result in comparable internal concentrations to those observed in human cell culture (Crone et al. 2015; Luz et al. 2016a; Naranmandura et al. 2011; Zheng et al. 2013). These doses likely result in internal concentrations orders of magnitude below what has been reported as leading to human fatalities, and more comparable to acute, low-dose exposures (Caboni et al. 2008; Rhee et al. 2016).
For oxygen consumption analysis with the Seahorse Bioanalyzer and targeted metabolomics, 4500 JK1107 (glp-1) nematodes were exposed to 0, 0.25, or 5 μM rotenone in a 25 cm2 vented cell culture flask. For gene expression measurements, 500 JK1107 (glp-1) worms/well were exposed to 0 or 5 μM rotenone in a 12-well plate. For the ATP measurements using the PE255 nematodes (metabolic inhibition assay), 4500 worms were exposed to 0, 0.25, or 1 μM rotenone in a 25 cm2 vented cell culture flask. PE255 nematodes were more sensitive to rotenone than JK1107 worms (data not shown), so we reduced the highest dose to avoid lethality; this increased sensitivity (compared to JK1107) has also been observed after arsenite and 5-fluoro-2'-deoxyuridine exposures (Luz et al. 2016a; Rooney et al. 2014). For growth assays, larval stage 1 (L1) nematodes were exposed to 0.25, 0.5, 1, 2.5, and 5 μM rotenone for 48 hours in a 96-well plate. For ATP measurements using the Promega kit, 2000 N2, JK1107 (glp-1), or RB766 (icl-1) nematodes/well were exposed to 0, 0.25, 1, or 5 μM rotenone in a 6-well plate.
2.3. Oxygen consumption analysis
We measured oxygen consumption rate (OCR) in age-matched young adult glp-1 mutant nematodes after 48 h rotenone exposure using an XFe24 Extracellular Flux Analyzer (Seahorse Bioscience). We then calculated the following parameters of mitochondrial performance: basal and maximal respiration, ATP-linked respiration, spare respiratory capacity, and proton leak as previously described (Luz et al. 2015a; Luz et al. 2015b). Following exposure, worms were pipetted into a 24-well utility plate as follows: 50 worms/well for carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) plate, and 60 worms/well for sodium azide + N,N'-Dicyclohexylcarbodiimide (DCCD) plate. Seven or eight wells were run per treatment (plus two wells as blanks) and final inhibitor concentrations per well were 25 μM FCCP, 20 μM DCCD and 10 mM sodium azide.
We calculated spare respiratory capacity by subtracting average basal OCR from average maximal respiration (induced by FCCP) for each well. To calculate ATP-linked respiration we subtracted the DCCD response from the basal OCR for each well. For proton leak, we subtracted the response to sodium azide from the response to DCCD for each well. All OCR measurements were normalized to total protein. The experiment was repeated twice.
2.4. Metabolic inhibition assay
This protocol was performed as described in Luz et al. (2016b). Age-matched young adult nematodes expressing luciferase were exposed to rotenone for 48 h as described above and incubated with various metabolic inhibitors. We then measured luminescence after the addition of luciferin as a way to assess the contribution of specific ETC complexes or metabolic pathways to steady-state ATP levels.
Fifty aged-matched young adult nematodes/well were added to a 96-well white plate. For each experiment, 4 wells were run for each rotenone dose and inhibitor combination (including unbuffered EPA water or 1% DMSO as vehicle controls for the inhibitors). The experiment was repeated at least three times for each inhibitor. Final inhibitor concentration, target and incubation time are listed in Table 1.
Table 1:
Inhibitor targets, final concentrations and incubation times as used in the mitochondrial inhibitor assay.
| Inhibitor | Target | Final Concentration |
Incubation Time (hr) |
|---|---|---|---|
| Rotenone | Complex I | 20 μM | 1 |
| Malonate | Complex II | 10 mM | 1 |
| Antimycin A | Complex III | 150 μM | 1 |
| Sodium azide | Complex IV | 250 μM | 1 |
| DCCD | ATP synthase | 20 μM | 1 |
| FCCP | Mitochondrial coupling | 25 μM | 1 |
| Perhexiline | Fatty acid oxidation | 100 μM | 1 |
| 2-DG | Glycolysis | 50 mM | 4.5 |
Following incubation, ATP levels were measured as previously described (Lagido et al. 2015; Lagido et al. 2008b; Luz et al. 2016b). After injection of luminescence buffer (140 mM Na2PO4, 30 mM citric acid [pH 6.5], 1% DMSO, 0.05% Triton X-100, 100 μM D-luciferin) into each well and a 3-minute incubation, we measured luminescence on a FLUOstar Optima plate reader (BMG Labtech). All values were normalized to green fluorescent protein (GFP).
2.5. Targeted gene expression measurements
Gene expression measurements were performed as described in Leung et al. (2013). The mRNA of approximately 500 worms was extracted using the RNeasy mini kit (QIAGEN) following rotenone exposure of age-matched young adults as described above. After extraction and quantification, mRNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression was measured by real-time PCR; primers used were as follows: cdc-42 forward - 5’-GAG AAA AAT GGG TGC CTG AA-3’, reverse - 5’-CTC GAG CAT TCC TGG ATC AT-3’ (111 bp, published in Leung et al. (2013)); pmp-3 forward – AAG ATG ATT GGC CGG ATG AT , reverse – GCA ACG AGA GCA ACT GAA CT (102 bp); tba-1 forward – TCA TCT CGC AGG TTG TGT CT, reverse – GGT AAG CCT TGT CAG CAG AG (159 bp); mev-1 forward – GTTGGACAGATCTACAAATCGGG reverse – TCTTGTTGCTCTTGTTCTGGC (100 bp); sdhA-1 forward – TCGCAGCTCAAGGAGGAATC reverse – ATGGCATCCTGATCTCCGAG (115 bp); and icl-1 forward – TGCTCATCCAGGATTGGTGC reverse – CTGAGCCAAGAGTCGAGGTATCCA (198 bp). Annealing temperature was 60 °C for all primer sets except the primers for icl-1 (59 °C). All samples were run in triplicate, and the experiment was repeated three times. Replicates were averaged and normalized to the expression levels of each housekeeping gene (cdc-42, pmp-3, tba-1). These values were then averaged and presented as fold change.
2.6. Growth assay
Growth from L1 was measured as previously described (Boyd et al. 2012). Briefly, worms were grown in 96 well plates in K+ medium, UvrA bacteria, and with 0, 0.25, 0.5, 1, 2.5, or 5 μM rotenone dissolved in DMSO (final concentration 1%) for 48 hours at 25°C. After 48 hours, worm size was measured as Time of Flight (TOF), utilizing a COPAS Biosort large particle flow cytometer (Union Biometrica, Holliston MA). The experiment was performed twice for glp-1, and three times for N2 and icl-1.
2.7. ATP measurements
ATP levels after rotenone exposure were measured for N2, and the glp-1 and icl-1 mutants using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). This protocol was adapted from previously published methods (Bailey et al. 2016; Todt et al. 2016). Following rotenone exposure of age-matched young adults as described above, 200 worms per well (at a concentration of 4 nematodes/μl in K medium) were added to a 96-well white plate. We then added 50 μl of CellTiter-Glo reagent and measured luminescence after 30 minutes incubating at room temperature. Twenty measurements were taken (60 minutes total measuring time, with a measurement occurring every 3 minutes). Three measurements were averaged and used for ATP calculations; these three were chosen based on luminescence having reached a steady, peak level. For N2s and icl-1, this was mostly the first three measurements. For glp-1, the peak luminescence varied but generally occurred within the first ten measurements. Four wells were run per dose and strain combination for each experiment; the experiment was repeated three times for glp-1, six times for icl-1 and N2. Luminescence data was normalized to average worm volume per treatment group as calculated using the WormSizer software (Moore et al. 2013). ATP is reported as the percent of the 0 μM rotenone control luminescence for each strain.
2.8. Targeted Metabolomics
This protocol was performed as described in Luz et al. (2016a). Following 24 and 48 hours of rotenone exposure on aged-matched young adults, nematodes were prepared for metabolomics analyses. After a rinse with cold PBS, worms were resuspended in 300 μl of an aqueous solution of 0.6% formic acid and flash-frozen in liquid nitrogen. Samples were then sonicated with five 30-second pulses at 20% power (Biologics, Inc.). A third of each sample was stored for protein analysis using the BCA protein assay kit (Thermo Fisher Scientific). The remaining sample was mixed 1:1 with acetonitrile and then aliquoted into two: one aliquot for quantifying amino acids and acylcarnitines, and another for organic acids. Amino acids and acylcarnitines were measured as described in An et al. (2004) and Wu et al. (2004). Organic acids were measured as described in Jensen et al. (2006). Metabolite concentrations were normalized to total protein. The experiment was performed twice, with 2-3 technical replicates per experiment.
2.9. Statistical analyses
Data were analyzed with JMP Pro for Mac (Version 14.0.0, SAS Institute Inc.) and GraphPad Prism (Version 7.05). One- or two-factor analysis of variance (ANOVA) was used to evaluate the effect of the rotenone exposures on all endpoints. The Tukey-Kramer honestly significant difference (HSD) test or Dunnett’s multiple comparisons test were used for posthoc analyses. A p-value of less than 0.05 was considered statistically significant.
3. Results
3.1. Rotenone exposure resulted in increased dependence on complex II and ATP synthase activity, and increased sensitivity to uncoupling-mediated ATP depletion
We first tested whether a 48h sublethal exposure to rotenone would result in functional metabolic changes in PE255 glp-4 young adult nematodes; to avoid overt toxicity (i.e. lethality) the highest dose of rotenone used on this worm strain was 1μM (see Materials and Methods). PE255 glp-4 lack germline proliferation at the temperature employed. As a result, since C. elegans somatic cells are invariant, any changes observed could be attributed only to changes in cellular metabolism, not changes in cell composition. We evaluated mitochondrial function in rotenone-exposed young adults by assaying different mitochondrial energy production pathways using a previously-described metabolic inhibition assay that permits detection of metabolic shifts (Lagido et al. 2008a; Luz et al. 2016b). This assay measures ATP levels in PE255 glp-4 nematodes transgenically expressing the luciferase enzyme by measuring luminescence after addition of luciferin plus a mitochondrial uncoupler (FCCP) or inhibitor of an ETC complex (rotenone, malonate, antimycin A, sodium azide, or DCCD), fatty acid oxidation (perhexiline), or glycolysis (2-deoxy-D-glucose, 2-DG). The principle of this assay is that following inhibitor treatment, larger reductions in ATP in a rotenone-exposed group than in a control group would indicate increased dependence on the ETC complex or metabolic pathway being inhibited. Conversely, lesser or no reductions in ATP following inhibitor treatment in a rotenone-exposed group compared to its control group, would indicate that the ETC complex or metabolic pathway being inhibited is playing a lesser role in energy production in the rotenone-exposed group.
After complex II inhibition with malonate, we observed larger ATP decreases in the 1 μM rotenone-exposed group than in the control group, indicating increased reliance on complex II function to maintain steady-state ATP levels after 48 h of 1 μM rotenone exposure (Fig. 1B). We also saw larger decreases in ATP in the 0.25 and 1 μM rotenone-exposed groups than in the control group after inhibition of the ATP synthase with DCCD, suggesting increased ATP synthase function after rotenone exposure (Fig. 1E). Finally, there was also a larger ATP decrease in the 1 μM rotenone-treated group compared to the control group after mitochondrial uncoupling with the protonophore FCCP, indicating an increased susceptibility to uncoupling-mediated ATP depletion (Fig. 1F). No differences were observed after inhibition with rotenone, antimycin A, sodium azide, perhexiline, or 2-DG.
Figure 1: Contribution of specific ETC complexes and metabolic pathways to ATP levels in rotenone-exposed PE255 C. elegans.
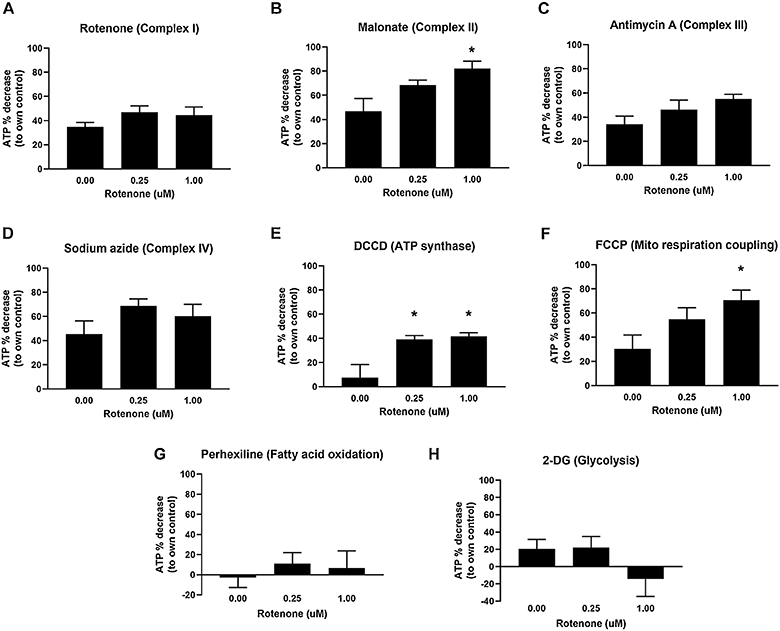
ATP decreases after exposure to malonate (B), DCCD (E), and FCCP (F) suggest increased reliance on Complex II and ATP synthase, as well as increased sensitivity to uncoupling-mediated ATP depletion, in rotenone-exposed nematodes. For malonate, one-way ANOVA p=0.0419*, Dunnett’s test vs control for 1 μM p=0.0279*. For DCCD, one-way ANOVA p=0.0218*, Dunnett’s test vs control for 0.25 μM p=0.0300*, for 1 μM p=0.0219*. For FCCP, one-way ANOVA p=0.0423*, Dunnett’s test vs control for 1 μM p=0.0261*. Bars ± SEM of three to six independent experiments.
These results suggest significant shifts in how the ETC is used to maintain ATP after 48 h of rotenone exposure. However, these shifts were only revealed after exposure to inhibitors or an uncoupler: we did not observe changes in ATP levels following the 48-hr rotenone treatments and subsequent incubation with vehicle controls (EPA water and 1% DMSO), which further suggests compensatory mechanisms are upregulated following exposure to rotenone to maintain steady-state ATP levels (Supplemental Figure 1).
3.2. Rotenone did not cause major alterations in oxygen consumption, but increased proton leak
We then tested whether the 48 h rotenone exposure affected mitochondrial respiration. For the reasons described above, we again employed a germline-deficient strain, glp-1 (Kodoyianni et al. 1992). The highest rotenone dose used (5 μM) did not cause overt toxicity. We first measured the OCR, both basally and following chemical challenges with DCCD (to permit calculation of ATP-linked oxygen consumption), FCCP (to assess spare respiratory capacity by uncoupling), and sodium azide (an inhibitor of ETC complex IV, to calculate non-mitochondrial oxygen consumption and proton leak (Benz and McLaughlin 1983; Fillingame 1975; Wharton and Tzagoloff 1967).
We did not observe significant changes to the basal OCR after rotenone exposure (Fig. 2). We also did not see any alterations due to rotenone exposure in ATP-linked respiration (basal OCR minus OCR after DCCD injection). Although spare respiratory capacity (FCCP-induced OCR minus basal OCR) appeared altered, these changes were not statistically significant. However, we did detect a substantial (>5-and 10-fold in low and high rotenone exposures, respectively) increase in proton leak (OCR after DCCD injection minus OCR after azide injection) in rotenone-exposed worms (Fig. 2).
Figure 2: Rotenone caused a statistically significant increase in proton leak, but not basal respiration, ATP-linked respiration or spare capacity.
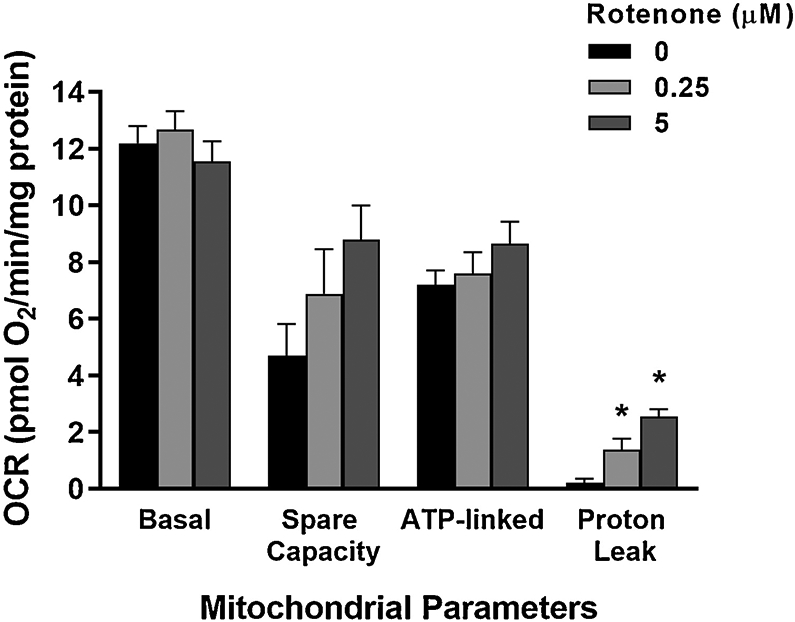
For proton leak, one-way ANOVA p<.0001*, Tukey-Kramer HSD vs control: p=0.0107* for 0.25 μM, p<.0001* for 5 μM). Bars ± SEM of duplicate independent experiments.
3.3. mRNA levels for complex II subunits and isocitrate lyase were elevated after rotenone exposure
The lack of effect on levels of ATP and oxygen consumption that we observed after 48 h rotenone exposure, coupled with evidence for functional metabolic shifts uncovered by the metabolic inhibition assay and increased proton leak, led us to hypothesize that the nematodes were able to compensate for complex I inhibition in our exposure paradigm by inducing the glyoxylate pathway, an alternate pathway present in C. elegans (Kahn and McFadden 1980). Induction of this pathway could allow the nematodes to maintain the functionality of the ETC and ATP levels, as described in the Discussion section. The glyoxylate pathway requires two exclusive enzymes, isocitrate lyase and malate synthase, which in C. elegans are both encoded by the gene icl-1 (Liu et al. 1995). To test this, we measured expression of the genes coding for isocitrate lyase (icl-1) and two complex II subunits, succinate dehydrogenase subunit A (a flavoprotein that is part of the hydrophilic head) and cytochrome b560 (part of the hydrophobic membrane anchor), sdha-1 and mev-1, respectively. mRNA levels for all three genes were elevated in young adult glp-1 worms after 48 h rotenone exposure (Figure 3).
Figure 3: Rotenone exposure induced isocitrate lyase (icl-1) and both complex two subunits (mev-1 and sdha-1).
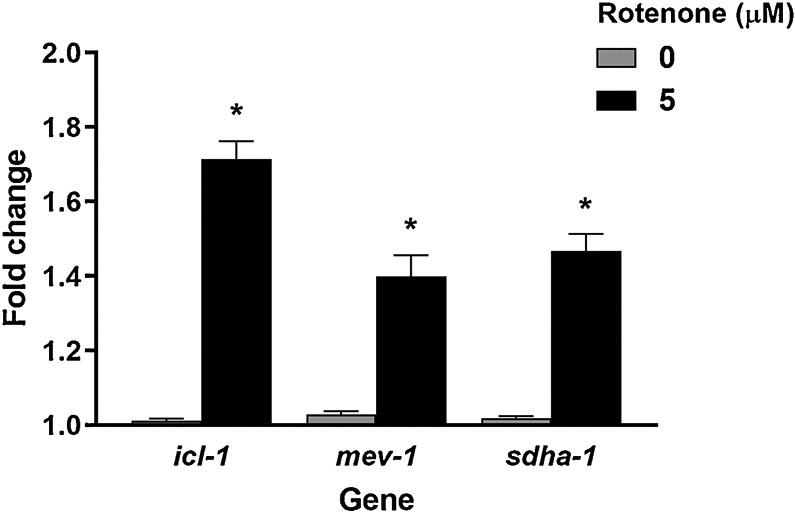
Two-tailed t-test p-value: icl-1 p=0.0041*, mev-1 0.0201*, sdha-1 0.0097*. Bars ± SEM of triplicate independent experiments.
To further test the functional importance of the glyoxylate pathway in maintaining ETC function after rotenone exposure, we measured total nematode growth from L1 in the presence of rotenone. For this growth assay, we measured the average nematode size (as time of flight, TOF) of the glyoxylate pathway mutant strain icl-1, the germline deficient strain glp-1, and the wild type N2 strain after a 48- hour exposure to rotenone. However, although the icl-1 strain appeared slightly more sensitive than N2 to rotenone-induced growth defects, these differences were not statistically significant (Figure 4). ATP levels were also measured in young adults after a 48 h exposure to rotenone in these strains, and no significant differences were observed among them (Supplemental Figure 2).
Figure 4: The glyoxylate mutant strain icl-1 did not exhibit additional sensitivity to rotenone exposure in growth assays.
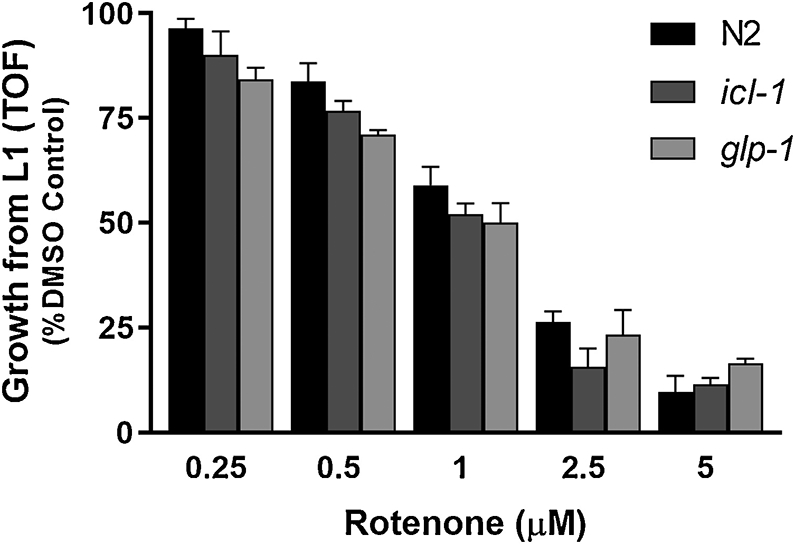
Bars ± SEM of 2-3 independent experiments.
3.4. Rotenone caused metabolomic shifts consistent with upregulated glyoxylate cycle, glycolysis, and fatty acid oxidation
Finally, we evaluated the effect of rotenone on young adult glp-1 C. elegans metabolism by measuring metabolite levels using mass spectrometry after exposure to 0.25 and 5 μM rotenone at 25°C. Changes were observed for all three metabolite categories analyzed: organic acids, amino acids, and acyl carnitines. For amino acids, we saw only one statistically significant change at 48h: a ~four-fold increase (normalized to control) in alanine levels after 5 μM rotenone exposure. There was also a trend towards increased levels of glycine (Figure 5). We also found changes in organic acids, with lactate, malate, and pyruvate increasing ~two- to seven-fold after the 5 μM exposure (Figure 6). These results are collectively consistent with an increase in glycolysis, complex II, and glyoxylate cycle activity. Moreover, we saw widespread ~two- to four-fold decreases in short, medium and long chain acylcarnitines (Figure 7), consistent with an increase in fatty acid β-oxidation. Finally, these changes in metabolite levels appeared to be incremental in a dose- and time- dependent manner, as we also evaluated these metabolites after 24-hour exposures to rotenone and observed similar, albeit smaller changes in alanine, malate, pyruvate, and acylcarnitines (Supplemental Figures 3 and 4).
Figure 5. Metabolomics analysis of amino acid levels in C. elegans after a 48h rotenone exposure.
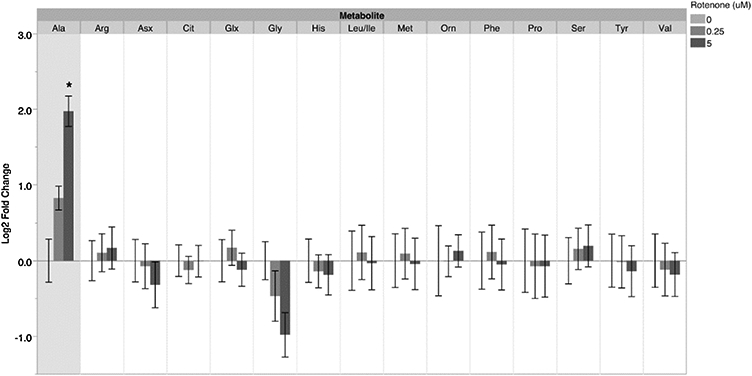
For alanine levels after the 5 μM rotenone exposure: one-way ANOVA p=0.0001*, Tukey-Kramer HSD vs control p=0.0001* for 5 μM. Bars ± SEM of three independent experimental replicates, with 2-3 technical replicates per experiment.
Figure 6. Metabolomics analysis of organic acid levels in C. elegans after a 48h rotenone exposure.
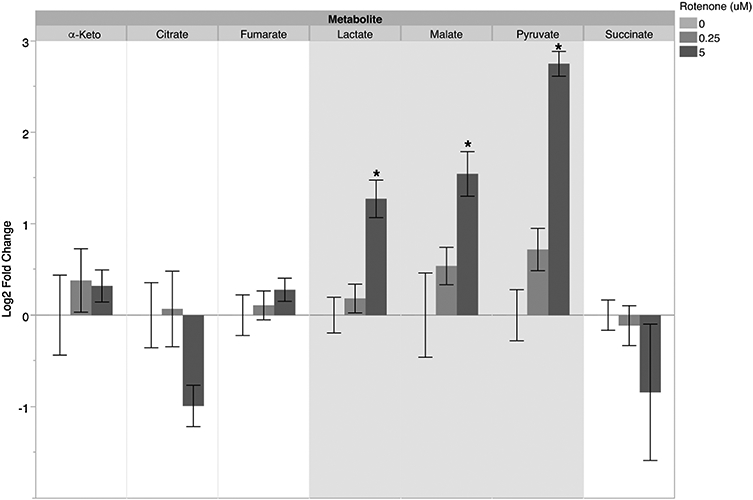
For malate levels after the 5 μM exposure: one-way ANOVA p=0.0165*, Tukey-Kramer HSD vs control p=0.0140*. For lactate levels after the 5 μM exposure: one-way ANOVA p=0.0008*, Tukey-Kramer HSD vs control p=0.0011*. For pyruvate levels after the 5 μM exposure: one-way ANOVA p<.0001*, Tukey-Kramer HSD vs control p<.0001*. Bars ± SEM of three independent experimental replicates, with 2-3 technical replicates per experiment.
Figure 7: Metabolomics analysis of acylcarnitine levels in C. elegans after a 48h rotenone exposure.

A, short chain acylcarnititnes. B, medium chain acylcarnititnes. C, long chain acylcarnititnes. Bars ± SEM of three independent experimental replicates, with 2-3 technical replicates per experiment. P-values for statistically significant changes are listed in Supplemental Table 1.
4. Discussion
Based on previous experiments with rotenone, we hypothesized that C. elegans activates a wide variety of metabolic pathways to deal with the effects of rotenone toxicity. Our results support that idea, as we found that rotenone caused several alterations in C. elegans bioenergetics. Some of these have been observed in vertebrates, but one, an induction in the glyoxylate cycle, does not appear to be possible in vertebrates.
Although there is not a precisely defined, published metabolic signature for the effect of rotenone in mammalian models, some patterns can be gleaned from the literature; overall, our results align with these patterns. Particularly, increases in lactate have been consistently reported (Karlsson et al. 2016; Lei et al. 2014; Xu et al. 2011). Xu et al. (2011) also saw an increase in alanine, while Lei et al. (2014) reported a decrease. An increase in pyruvate following rotenone exposure was also observed by Karlsson et al. (2016). It is worth noting that very similar results have been observed in complex I mutant nematodes (Falk et al. 2008; Grad and Lemire 2004; Morgan et al. 2015; Schrier Vergano et al. 2014; Zuryn et al. 2010), supporting the assumption that the primary, direct mode of action of rotenone is indeed complex I inhibition. The complex I subunit mutant gas-1 exhibits a decrease in complex I-dependent respiration, and an increase in complex II activity (Kayser et al. 2001; Pujol et al. 2013). Gas-1 also exhibits signs of disrupted pyruvate metabolism (with alterations in the levels of lactate, pyruvate, and alanine) and widespread changes in amino acid levels (Falk et al. 2008; Morgan et al. 2015; Schrier Vergano et al. 2014). Knockdown or mutations in the gene coding for another complex I subunit, nuo-1, also cause changes in lactate and pyruvate levels, impaired respiration, and increases in mRNA levels for genes involved in fatty acid β-oxidation, gluconeogenesis, and glycolysis (Grad and Lemire 2004; Zuryn et al. 2010).
Interestingly, several of these studies also presented evidence of upregulation of the glyoxylate cycle in complex I mutants (Falk et al. 2008; Morgan et al. 2015; Pujol et al. 2013; Zuryn et al. 2010). This pathway, which does not appear to be present in higher metazoans, employs acetyl-CoA to make malate and succinate. Isocitrate lyase, a key enzyme in this pathway, converts isocitrate produced by the citric acid cycle to glyoxylate and succinate; glyoxylate can then be transformed into malate, bypassing two NADH-producing steps in the citric acid cycle (Braeckman et al. ; Kondrashov et al. 2006). This might be adaptive in the context of complex I inhibition, as it would result in less complex I substrate and thus less reactive oxygen species (ROS) production in the context of rotenone-induced complex I inhibition (De Miranda et al. 2016). Inducing the glyoxylate pathway can then theoretically lower oxidative stress by reducing the need for complex I to catalyze electron transfer from NADH to ubiquinone, and as a result prevent complex I-mediated dysfunction (Morgan et al. 2015). The increase in proton leak might also be an adaptive response to oxidative stress, potentially decreasing ROS production by decreasing the degree of reduction of the ETC. An increase in succinate due to the glyoxylate cycle could also provide the substrate required for the complex II upregulation observed in complex I mutants, permitting at least partial maintenance of energy production despite a decrease in complex I activity in these mutants. While not statistically significant, we did observe a trend towards decreased succinate levels, again supporting increased Complex II activity. Thus, if induced, the glyoxylate pathway could allow mitochondria to maintain somewhat functional ETC and ATP levels, with increased reliance on complex II function. This idea is supported by reports of increased complex II activity in complex I mutants (Kayser et al. 2001; Pujol et al. 2013). Our mitochondrial respiration results support this hypothesis, as the nematodes do not display major alterations in OCR despite rotenone’s well-documented effects on oxygen consumption (De Miranda et al. 2016; Dranka et al. 2012; Giordano et al. 2012).
However, our attempts to characterize the importance of the glyoxylate pathway induction after rotenone exposure resulted in mixed results. We found evidence that rotenone-exposed worms had an increased reliance on complex II, and induced mRNA levels of the bifunctional glyoxylate pathway enzyme, icl-1. Glyoxylate pathway induction is also supported by our metabolite analysis. We observed an increase in malate in our metabolomics experiments, which has been previously reported after rotenone exposure (Xu et al. 2011) and in complex I mutants, where it was attributed to a possible increase in the glyoxylate cycle (Schrier Vergano et al. 2014). However, our experiments with the glyoxylate pathway mutant strain icl-1 evaluating growth and ATP levels did not convincingly show increased susceptibility to rotenone. We speculate that this lack of susceptibility might be due to other metabolic alterations occurring in the icl-1 mutant strain, allowing it to compensate for the lack of isocitrate lyase/malate synthase; however, this needs to be experimentally tested. It is also possible that the larval growth assay was uninformative because it was performed at a different lifestage, in which the role of the glyoxylate cycle may be different. Finally, in both cases, it is possible that the use of a mutant strain that has existed for generations without icl-1 led to compensatory adaptive changes; future experiments should employ RNAi. Nonetheless, overall, our data contributes to the growing evidence that the glyoxylate pathway, known as critically important in embryogenesis and larval development (Braeckman et al.), is also engaged in response to stress in adult nematodes—perhaps in particular stress which inhibits complex I.
Further evidence of metabolic restructuring in C. elegans after rotenone exposure was provided by the results of our metabolomics analyses. Our data indicate a disruption in pyruvate metabolism. Pyruvate can be converted into alanine and lactate; very high levels of pyruvate might explain the increases we see in both (Chandel 2015). Increased pyruvate and lactate would be consistent with inhibition of pyruvate dehydrogenase due to oxidative stress caused by rotenone (Samikkannu et al. 2003; Tabatabaie et al. 1996), and upregulation of glycolysis, an observation that has also been made in other metabolic studies on the effect of rotenone (Dranka et al. 2012; Giordano et al. 2012; Karlsson et al. 2016; Lei et al. 2014). We did not detect increased reliance on glycolysis (for ATP production) in our 2-DG inhibition experiment; however, again, this may have resulted from the utilization by worms of multiple adaptive pathways providing functional redundancy at the level of ATP maintenance. Another possibility is an increase in glutaminolysis, through which pyruvate and glutamate are converted into alanine and α-ketoglutarate. More α-ketoglutarate feeding into the citric acid cycle would result in more succinate, which could explain why we do not see a significant decrease in succinate levels despite the potential upregulation of complex II after rotenone exposure. Based on our experiments, we cannot unequivocally say which pathways are responsible for the alterations in the levels of these organic acids.
The widespread decreases in short, medium, and long chain acylcarnitines are consistent with increased fatty acid β-oxidation, an observation that has been made previously in cell culture after rotenone exposure (Worth et al. 2014). This is also supported by the evidence of complex II upregulation (a key component of fatty acid oxidation) and pyruvate and lactate accumulation, as increases in fatty acid oxidation could help replenish acetyl-CoA levels, which might be reduced due to increased pyruvate conversion to alanine and lactate, or due to inhibition of pyruvate dehydrogenase due to oxidative stress caused by rotenone (Samikkannu et al. 2003; Tabatabaie et al. 1996). We did not detect an alteration in reliance on fatty acid oxidation for ATP production in our perhexiline inhibition experiment; however, again, this could be the result of either redundant ATP-preserving pathways being induced. Alternatively, this could be attributed to the undetectable effect of perhexiline, potentially precluding observation of a change induced by rotenone.
Taken together, our results show that rotenone causes various alterations in metabolite levels in C. elegans at exposure levels that did not cause major changes in oxygen consumption or steady-state ATP. These changes are consistent with a multi-pathway metabolic restructuring that helps maintain oxidative phosphorylation and ATP production: we found evidence for concomitantly increased glycolysis and fatty acid oxidation—pathways that also function in compensatory fashion in mammals— as well as the glyoxylate pathway, which appears to be absent in mammals. Induction of the glyoxylate pathway could help the worms deal with complex I inhibition and maintain baseline levels of ATP and oxygen consumption. Other groups have seen mitochondrial dysfunction and toxicity after rotenone exposure in C. elegans, including decreased respiration and neurodegeneration (Chikka et al. 2016; Grad and Lemire 2004; Ray et al. 2014; Schouest et al. 2009; Ved et al. 2005; Zhou et al. 2013), and we have observed rotenone toxicity in older adults, which was reduced by exercise preconditioning (Hartman et al. 2018). This discrepancy may likely be due to the exposure protocol (exposure level, timecourse, lifestage, etc.) or the worm strain used. Indeed, when we expose adult worms to higher doses of rotenone for just an hour (as seen in the metabolic inhibition assay), we see decreases in ATP levels. We propose that these are in fact entirely consistent results; the longer, 48-hour exposure at a lower concentration likely allows the nematodes to “acclimate” to complex I inhibition by inducing the glyoxylate pathway. Similarly, even with the availability of an additional compensatory pathway (the glyoxylate pathway), high enough doses will eventually result in the same downstream results of mitochondrial inhibition. Thus, it is very likely that the exposure protocol used dictates whether or not rotenone toxicity can be compensated with metabolic changes.
It is important to note that these apparently compensatory metabolic shifts may not be entirely without cost. While we did not directly test susceptibility to a secondary stressor at the organismal level, we did find that rotenone-exposed worms had a decreased ability to maintain ATP levels upon acute exposure to the mitochondrial uncoupler FCCP. The mechanistic basis for this sensitivity is not clear; possibilities include the fact that the rotenone-exposed worms already have partially-uncoupled mitochondria (i.e., increased proton leak), or that substrate availability for the alternate, non-complex I pathways is limited. Whatever the mechanism, these results suggest that rotenone-acclimated worms may be more susceptible to certain subsequent stressors.
Finally, an important conclusion from this study is that nematode biology needs to be considered when using C. elegans to study complex I inhibitors. Nematodes have alternate metabolic pathways that increase their ability to compensate for such inhibition, and this compensation could confuse interpretation of results from toxicological studies (in particular in the context of environmentally-relevant long-term, low-dose experiments).
Supplementary Material
Highlights.
Rotenone exposure resulted in metabolic shifts in Caenorhabditis elegans, allowing the nematode to maintain ATP levels.
C. elegans increased dependence on complex II and ATP synthase activity after rotenone exposure, suggesting changes in how the electron transport chain is used to maintain ATP levels after Complex I inhibition.
Rotenone caused changes in metabolite levels consistent with upregulated glyoxylate cycle, glycolysis, and fatty acid oxidation.
Rotenone-exposed C. elegans displayed increased sensitivity to uncoupling-mediated ATP depletion, suggesting that these metabolic changes were not without trade-offs.
Acknowledgments
Funding
As a Duke Cancer Institute member, I acknowledge support from the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: P30 CA014236). This work was also supported by the National Institute of Environmental Health Sciences and National Institute of Health (R01ES017540 and R01ES028218, P42ES010356). Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington D and Newgard CB 2004. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med 10, 268–274. [DOI] [PubMed] [Google Scholar]
- Bailey DC, Todt CE, Orfield SE, Denney RD, Snapp IB, Negga R, Montgomery KM, Bailey AC, Pressley AS, Traynor WL and Fitsanakis VA 2016. Caenorhabditis elegans chronically exposed to a Mn/Zn ethylene-bis-dithiocarbamate fungicide show mitochondrial Complex I inhibition and increased reactive oxygen species. NeuroToxicology 56, 170–179. [DOI] [PubMed] [Google Scholar]
- Benz R and McLaughlin S 1983. The molecular mechanism of action of the proton ionophore FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazone). Biophysical Journal 41, 381–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV and Greenamyre JT 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Boyd WA, Smith MV and Freedman JH 2012. Caenorhabditis elegans as a model in developmental toxicology. Methods Mol Biol 889, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd WA, Smith MV, Kissling GE, Rice JR, Snyder DW, Portier CJ and Freedman JH 2009. Application of a Mathematical Model to Describe the Effects of Chlorpyrifos on Caenorhabditis elegans Development. PLoS ONE 4, e7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckman BP, Houthoofd K and Vanfleteren JR Intermediary metabolism In: T.C.e.R. Community (Ed), WormBook, WormBook. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboni P, Sarais G, Vargiu S, De Luca MA, Garau VL, Ibba A and Cabras P 2008. LC–MS– MS Determination of Rotenone, Deguelin, and Rotenolone in Human Serum. Chromatographia 68, 739–745. [Google Scholar]
- Chandel NS 2015. Navigating metabolism, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. [Google Scholar]
- Chikka MR, Anbalagan C, Dvorak K, Dombeck K and Prahlad V 2016. The Mitochondria-Regulated Immune Pathway Activated in the C. elegans Intestine Is Neuroprotective. Cell Rep 16, 2399–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Belcastro V, Tozzi A, Di Filippo M, Tantucci M, Siliquini S, Autuori A, Picconi B, Spillantini MG, Fedele E, Pittaluga A, Raiteri M and Calabresi P 2008. Electrophysiology and Pharmacology of Striatal Neuronal Dysfunction Induced by Mitochondrial Complex I Inhibition. The Journal of Neuroscience 28, 8040–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone B, Aschner M, Schwerdtle T, Karst U and Bornhorst J 2015. Elemental bioimaging of Cisplatin in Caenorhabditis elegans by LA-ICP-MS. Metallomics 7, 1189–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda BR, Van Houten B and Sanders LH 2016. Toxin-Mediated Complex I Inhibition and Parkinson’s Disease In: Buhlman LM (Ed), Mitochondrial Mechanisms of Degeneration and Repair in Parkinson's Disease, Springer International Publishing, Cham, pp. 115–137. [Google Scholar]
- Dranka BP, Zielonka J, Kanthasamy AG and Kalyanaraman B 2012. Alterations in bioenergetic function induced by Parkinson’s disease mimetic compounds: lack of correlation with superoxide generation. Journal of Neurochemistry 122, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Zhang Z, Rosenjack JR, Nissim I, Daikhin E, Nissim I, Sedensky MM, Yudkoff M and Morgan PG 2008. Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Molecular Genetics and Metabolism 93, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingame RH 1975. Identification of the dicyclohexylcarbodiimide-reactive protein component of the adenosine 5'-triphosphate energy-transducing system of Escherichia coli. Journal of Bacteriology 124, 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follmer C, Coelho-Cerqueira E, Yatabe-Franco DY, Araujo GD, Pinheiro AS, Domont GB and Eliezer D 2015. Oligomerization and Membrane-binding Properties of Covalent Adducts Formed by the Interaction of alpha-Synuclein with the Toxic Dopamine Metabolite 3,4-Dihydroxyphenylacetaldehyde (DOPAL). J Biol Chem 290, 27660–27679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S, Lee J, Darley-Usmar VM and Zhang J 2012. Distinct Effects of Rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on Cellular Bioenergetics and Cell Death. PLOS ONE 7, e44610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hunt CP, Leung MC, Bodhicharla RK, McKeever MG, Arrant AE, Margillo KM, Ryde IT, Cyr DD, Kosmaczewski SG, Hammarlund M and Meyer JN 2014. Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. PLoS One 9, e114459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hunt CP and Sanders LH 2020. DNA damage and repair in Parkinson's disease: Recent advances and new opportunities. J Neurosci Res. [DOI] [PubMed] [Google Scholar]
- Grad LI and Lemire BD 2004. Mitochondrial complex I mutations in Caenorhabditis elegans produce cytochrome c oxidase deficiency, oxidative stress and vitamin-responsive lactic acidosis. Human Molecular Genetics 13, 303–314. [DOI] [PubMed] [Google Scholar]
- Hartman JH, Smith LL, Gordon KL, Laranjeiro R, Driscoll M, Sherwood DR and Meyer JN 2018. Swimming Exercise and Transient Food Deprivation in Caenorhabditis elegans Promote Mitochondrial Maintenance and Protect Against Chemical-Induced Mitotoxicity. Scientific Reports 8, 8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RP 1993. The absence of glyxylate cycle enzymes in rodent and embryonic chick liver. Biochimica et Biophysica Acta (BBA) - General Subjects 1158, 47–51. [DOI] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ilkayeva O, Burgess S, Lu D, Ronnebaum SM, Odegaard M, Becker TC, Sherry AD and Newgard CB 2006. Compensatory Responses to Pyruvate Carboxylase Suppression in Islet β-Cells: PRESERVATION OF GLUCOSE-STIMULATED INSULIN SECRETION. Journal of Biological Chemistry 281, 22342–22351. [DOI] [PubMed] [Google Scholar]
- Kahn FR and McFadden BA 1980. Embryogenesis and the glyoxylate cycle. FEBS Lett 115, 312–314. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Ehinger JK, Piel S, Sjövall F, Henriksnäs J, Höglund U, Hansson MJ and Elmér E 2016. Changes in energy metabolism due to acute rotenone-induced mitochondrial complex I dysfunction – An in vivo large animal model. Mitochondrion 31, 56–62. [DOI] [PubMed] [Google Scholar]
- Kayser E-B, Morgan PG, Hoppel CL and Sedensky MM 2001. Mitochondrial Expression and Function of GAS-1 in Caenorhabditis elegans. Journal of Biological Chemistry 276, 20551–20558. [DOI] [PubMed] [Google Scholar]
- Kodoyianni V, Maine EM and Kimble J 1992. Molecular basis of loss-of-function mutations in the glp-1 gene of Caenorhabditis elegans. Molecular Biology of the Cell 3, 1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA, Koonin EV, Morgunov IG, Finogenova TV and Kondrashova MN 2006. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol Direct 1, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagido C, McLaggan D and Glover LA 2015. A Screenable In Vivo Assay for Mitochondrial Modulators Using Transgenic Bioluminescent Caenorhabditis elegans. Journal of visualized experiments: JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagido C, Pettitt J, Flett A and Glover LA 2008a. Bridging the phenotypic gap: real-time assessment of mitochondrial function and metabolism of the nematode Caenorhabditis elegans. BMC Physiol 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagido C, Pettitt J, Flett A and Glover LA 2008b. Bridging the phenotypic gap: Real-time assessment of mitochondrial function and metabolism of the nematode Caenorhabditis elegans. BMC Physiology 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston J, Ballard P, Tetrud J and Irwin I 1983. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. [DOI] [PubMed] [Google Scholar]
- Lei S, Zavala-Flores L, Garcia-Garcia A, Nandakumar R, Huang Y, Madayiputhiya N, Stanton RC, Dodds ED, Powers R and Franco R 2014. Alterations in Energy/Redox Metabolism Induced by Mitochondrial and Environmental Toxins: A Specific Role for Glucose-6-Phosphate-Dehydrogenase and the Pentose Phosphate Pathway in Paraquat Toxicity. ACS Chemical Biology 9, 2032–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung M, Rooney J, Ryde I, Bernal A, Bess A, Crocker T, Ji A and Meyer J 2013. Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacology and Toxicology 14, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung MC, Williams PL, Benedetto A, Au C, Helmcke KJ, Aschner M and Meyer JN 2008. Caenorhabditis elegans: an emerging model in biomedical and environmental toxicology. Toxicol Sci 106, 5–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA and Fleming JT 1995. Basic culture methods. Methods in cell biology 48, 3–29. [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA and Robinson JP 2003. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. Journal of Biological Chemistry 278, 8516–8525. [DOI] [PubMed] [Google Scholar]
- Lindahl PE and Öberg KE 1961. The effect of rotenone on respiration and its point of attack. Experimental Cell Research 23, 228–237. [DOI] [PubMed] [Google Scholar]
- Liu F, Thatcher JD, Barral JM and Epstein HF 1995. Bifunctional Glyoxylate Cycle Protein of Caenorhabditis elegans: A Developmentally Regulated Protein of Intestine and Muscle. Developmental Biology 169, 399–414. [DOI] [PubMed] [Google Scholar]
- Luz AL, Godebo TR, Bhatt DP, Ilkayeva OR, Maurer LL, Hirschey MD and Meyer JN 2016a. From the Cover: Arsenite Uncouples Mitochondrial Respiration and Induces a Warburg-like Effect in Caenorhabditis elegans. Toxicological Sciences 152, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz AL, Lagido C, Hirschey MD and Meyer JN 2016b. In Vivo Determination of Mitochondrial Function Using Luciferase-Expressing Caenorhabditis elegans: Contribution of Oxidative Phosphorylation, Glycolysis, and Fatty Acid Oxidation to Toxicant-Induced Dysfunction. Curr Protoc Toxicol 69, 25 28 21–25 28 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz AL, Rooney JP, Kubik LL, Gonzalez CP, Song DH and Meyer JN 2015a. Mitochondrial Morphology and Fundamental Parameters of the Mitochondrial Respiratory Chain Are Altered in Caenorhabditis elegans Strains Deficient in Mitochondrial Dynamics and Homeostasis Processes. PLOS ONE 10, e0130940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz AL, Smith LL, Rooney JP and Meyer JN 2015b. Seahorse Xfe24 Extracellular Flux Analyzer-Based Analysis of Cellular Respiration in Caenorhabditis elegans Current Protocols in Toxicology, John Wiley & Sons, Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer LL, Luz AL and Meyer JN 2018. Detection of Mitochondrial Toxicity of Environmental Pollutants Using Caenorhabditis elegans. Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants, pp. 655–689. [Google Scholar]
- Meyer JN, Lord CA, Yang XY, Turner EA, Badireddy AR, Marinakos SM, Chilkoti A, Wiesner MR and Auffan M 2010. Intracellular uptake and associated toxicity of silver nanoparticles in Caenorhabditis elegans. Aquatic Toxicology 100, 140–150. [DOI] [PubMed] [Google Scholar]
- Moore BT, Jordan JM and Baugh LR 2013. WormSizer: high-throughput analysis of nematode size and shape. PLoS One 8, e57142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan PG, Higdon R, Kolker N, Bauman AT, Ilkayeva O, Newgard CB, Kolker E, Steele LM and Sedensky MM 2015. Comparison of proteomic and metabolomic profiles of mutants of the mitochondrial respiratory chain in Caenorhabditis elegans. Mitochondrion 20, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranmandura H, Xu S, Sawata T, Hao WH, Liu H, Bu N, Ogra Y, Lou YJ and Suzuki N 2011. Mitochondria Are the Main Target Organelle for Trivalent Monomethylarsonous Acid (MMAIII)-Induced Cytotoxicity. Chemical Research in Toxicology 24, 1094–1103. [DOI] [PubMed] [Google Scholar]
- Pujol C, Bratic-Hench I, Sumakovic M, Hench J, Mourier A, Baumann L, Pavlenko V and Trifunovic A 2013. Succinate Dehydrogenase Upregulation Destabilize Complex I and Limits the Lifespan of gas-1 Mutant. PLOS ONE 8, e59493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Martinez BA, Berkowitz LA, Caldwell GA and Caldwell KA 2014. Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson's model. Cell Death Dis 5, e984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requejo-Aguilar R and Bolaños JP 2016. Mitochondrial control of cell bioenergetics in Parkinson's disease. Free Radical Biology and Medicine 100, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J, Yum H, Moon S, In S, Lee S and Seo J 2016. Rotenone Analysis by Liquid Chromatography–Tandem Mass Spectrometry with Information-Dependent Acquisition in a Fatal Case of Rotenone Poisoning with a Commercial Organic Insecticide Being Sold in Korea. Journal of Analytical Toxicology 40, 460–465. [DOI] [PubMed] [Google Scholar]
- Rooney JP, Luz AL, González-Hunt CP, Bodhicharla R, Ryde IT, Anbalagan C and Meyer JN 2014. Effects of 5′-fluoro-2-deoxyuridine on mitochondrial biology in Caenorhabditis elegans. Experimental Gerontology 56, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samikkannu T, Chen C-H, Yih L-H, Wang ASS, Lin S-Y, Chen T-C and Jan K-Y 2003. Reactive Oxygen Species Are Involved in Arsenic Trioxide Inhibition of Pyruvate Dehydrogenase Activity. Chemical Research in Toxicology 16, 409–414. [DOI] [PubMed] [Google Scholar]
- Sanders LH and Greenamyre JT 2013. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radical Biology and Medicine 62, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders LH, Howlett EH, McCoy J and Greenamyre JT 2014a. Mitochondrial DNA damage as a peripheral biomarker for mitochondrial toxin exposure in rats. Toxicol Sci 142, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B and Greenamyre JT 2014b. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiol Dis 70, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P and Marsden CD 1990. Mitochondrial Complex I Deficiency in Parkinson's Disease. Journal of Neurochemistry 54, 823–827. [DOI] [PubMed] [Google Scholar]
- Schouest K, Zitova A, Spillane C and Papkovsky DB 2009. Toxicological assessment of chemicals using Caenorhabditis elegans and optical oxygen respirometry. Environmental Toxicology and Chemistry 28, 791–799. [DOI] [PubMed] [Google Scholar]
- Schrier Vergano S, Rao M, McCormack S, Ostrovsky J, Clarke C, Preston J, Bennett MJ, Yudkoff M, Xiao R and Falk MJ 2014. In vivo metabolic flux profiling with stable isotopes discriminates sites and quantifies effects of mitochondrial dysfunction in C. elegans. Molecular Genetics and Metabolism 111, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A and Greenamyre JT 2003. Mechanism of Toxicity in Rotenone Models of Parkinson's Disease. The Journal of Neuroscience 23, 10756–10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LL, Ryde IT, Hartman JH, Romersi RF, Markovich Z and Meyer JN 2019. Strengths and limitations of morphological and behavioral analyses in detecting dopaminergic deficiency in Caenorhabditis elegans. Neurotoxicology 74, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter L, Li Y, Nakatsuka NJ, Calvo SE, Grabarek Z and Mootha VK 2014. CLYBL is a polymorphic human enzyme with malate synthase and β-methylmalate synthase activity. Hum Mol Genet 23, 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabatabaie T, Potts JD and Floyd RA 1996. Reactive Oxygen Species-Mediated Inactivation of Pyruvate Dehydrogenase. Archives of Biochemistry and Biophysics 336, 290–296. [DOI] [PubMed] [Google Scholar]
- Todt CE, Bailey DC, Pressley AS, Orfield SE, Denney RD, Snapp IB, Negga R, Bailey AC, Montgomery KM, Traynor WL and Fitsanakis VA 2016. Acute exposure to a Mn/Zn ethylene-bis-dithiocarbamate fungicide leads to mitochondrial dysfunction and increased reactive oxygen species production in Caenorhabditis elegans. NeuroToxicology 57, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ved R, Saha S, Westlund B, Perier C, Burnam L, Sluder A, Hoener M, Rodrigues CM, Alfonso A, Steer C, Liu L, Przedborski S and Wolozin B 2005. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem 280, 42655–42668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Mitani N, Ishii N and Miki K 2005. A mutation in a cuticle collagen causes hypersensitivity to the endocrine disrupting chemical, bisphenol A, in Caenorhabditis elegans. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 570, 71–80. [DOI] [PubMed] [Google Scholar]
- Wharton DC and Tzagoloff A 1967. Cytochrome oxidase from beef heart mitochondria Methods in Enzymology, Academic Press, pp. 245–250. [Google Scholar]
- Williams PL and Dusenbery DB 1988. Using the nematode Caenorhabditis elegans to predict mammalian acute lethality to metallic salts. Toxicology and industrial health 4, 469–478. [DOI] [PubMed] [Google Scholar]
- Worth AJ, Basu SS, Snyder NW, Mesaros C and Blair IA 2014. Inhibition of Neuronal Cell Mitochondrial Complex I with Rotenone Increases Lipid β-Oxidation, Supporting Acetyl-Coenzyme A Levels. Journal of Biological Chemistry 289, 26895–26903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J-Y, Kao H-J, Li S-C, Stevens R, Hillman S, Millington D and Chen Y-T 2004. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. The Journal of Clinical Investigation 113, 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Vu H, Liu L, Wang T-C and Schaefer WH 2011. Metabolic profiles show specific mitochondrial toxicities in vitro in myotube cells. Journal of Biomolecular NMR 49, 207–219. [DOI] [PubMed] [Google Scholar]
- Zheng S-Q, Ding A-J, Li G-P, Wu G-S and Luo H-R 2013. Drug Absorption Efficiency in Caenorhbditis elegans Delivered by Different Methods. PLOS ONE 8, e56877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Wang Z and Klaunig JE 2013. Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. Int J Biochem Mol Biol 4, 191–200. [PMC free article] [PubMed] [Google Scholar]
- Zuryn S, Kuang J, Tuck A and Ebert PR 2010. Mitochondrial dysfunction in Caenorhabditis elegans causes metabolic restructuring, but this is not linked to longevity. Mechanisms of Ageing and Development 131, 554–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.