

Abstract
Stroke elicits excessive immune activation in the injured brain tissue. This well-recognized neural inflammation in the brain is not just an intrinsic organ response but also a result of additional intricate interactions between infiltrating peripheral immune cells and the resident immune cells in the affected areas. Given that there is a finite number of immune cells in the organism at the time of stroke, the partitioned immune systems of the CNS and periphery must appropriately distribute the limited pool of immune cells between the two domains, mounting a necessary post-stroke inflammatory response by supplying a sufficient number of immune cells into the brain while maintaining peripheral immunity. Stroke pathophysiology has mainly been neurocentric in focus, but understanding the distinct roles of the CNS and peripheral immunity in their concerted action against ischemic insults is crucial. This review will discuss stroke-induced influences of the peripheral immune system on CNS injury/repair and of neural inflammation on peripheral immunity, and how comorbidity influences each.
Keywords: Ischemic stroke, Immunity, Inflammation, Comorbidity, Immunomodulation
1. INTRODUCTION
Ischemic stroke caused by the blockage of an artery that supplies blood to the brain accounts for more than 85% of strokes in United States. The resulting brain injury leads to severe disabilities and/or mortality. Although tremendous efforts have led to gene therapies and drugs that were found to be protective in pre-clinical studies, these treatments, except for tissue plasminogen activator (tPA) and thrombectomy in a limited population of patients, have not been successfully translated into clinical settings (http://www.strokecenter.org/trials/). The disappointing translational outcomes suggest an insufficient understanding of stroke pathophysiology, which hinges upon several issues including i) the complex stroke pathological mechanisms that involve excitotoxicity, inflammation, apoptosis, necrosis, and angiogenesis across different stages post-stroke, ii) limited validity of experimental approaches and design and insufficient statistical power, and iii) lack of inclusion of clinical comorbid conditions in experimental brain ischemic models.
Stroke induces sustained immune activation across post-stroke stages (Fury et al., 2020). A plethora of evidence shows that neuroinflammation is a major event involving injury development and repair processes. The stroke-induced neuroinflammatory cascades include the production of excessive cytokines/chemokines, reactive oxygen species (ROS), and nitric oxidative species (NOS), which result in the disruption of vascular integrity and cell death (Iadecola and Anrather, 2011). Despite its pathogenic nature, rapid and optimal neuroinflammation is a necessary response for subsequent injury repair and functional recovery (Chu et al., 2015; Gliem et al., 2015). The apparent controversy may derive from the time- and context-dependent role of inflammation. Moreover, the existing non-modifiable and modifiable risk factors for stroke further influence spatial and temporal neuroinflammation across different stages of stroke. To better understand stroke-induced changes in immune/inflammatory responses, this review evaluates recent findings on how peripheral and CNS immunity/inflammation are cross-regulated in cerebral ischemia in normal and comorbid conditions.
2. IMMUNITY IN STROKE: BRAIN vs. PERIPHERY
The immune system in the brain has been regarded a separate entity from the periphery. Due to the presence of the physical blood-brain barrier (BBB), deficiency of lymphatic drainage, and the absence of professional antigen presenting cells, the brain has been considered an immune-privileged organ. However, recent evidence reveals the presence of crosstalk between the brain and periphery even in normal physiologic conditions. Peripheral immune cells are found in intact brain parenchyma (Hickey et al., 1991; Posel et al., 2016; Song et al., 2016; Korin et al., 2017), and physiologic interactions between the immune system and the brain have been described (Schwartz and Kipnis, 2011; Norris and Kipnis, 2019). Given that the CNS drains cellular and soluble components through cerebrospinal fluid into the deep cervical lymph nodes, in which CNS-derived antigens induce immune responses (Kida et al., 1993; Aspelund et al., 2015; Louveau et al., 2015), the brain is an immune specialized organ. The neuroimmune interaction can be intensified in neurological conditions, such as stroke, in which the brain encounters damage-induced endogenous signals and peripheral antigens via compromised BBB integrity. Subsequent recruitment of immune cells from the periphery into the injured site leads full-blown neuroinflammation (Figure 1). The excessive neuroimmune reactions in the CNS, in turn, disturb the homeostasis of peripheral immunity, leading stroke-induced immune depression (Meisel et al., 2005). As the immune/inflammatory response at the time of stroke may affect injury development, mortality, and repair/recovery, understanding the compartmentalization and interaction between the CNS and peripheral immunity in stroke would facilitate the development of immune-based approaches to modulate the course of pathology.
Figure 1.
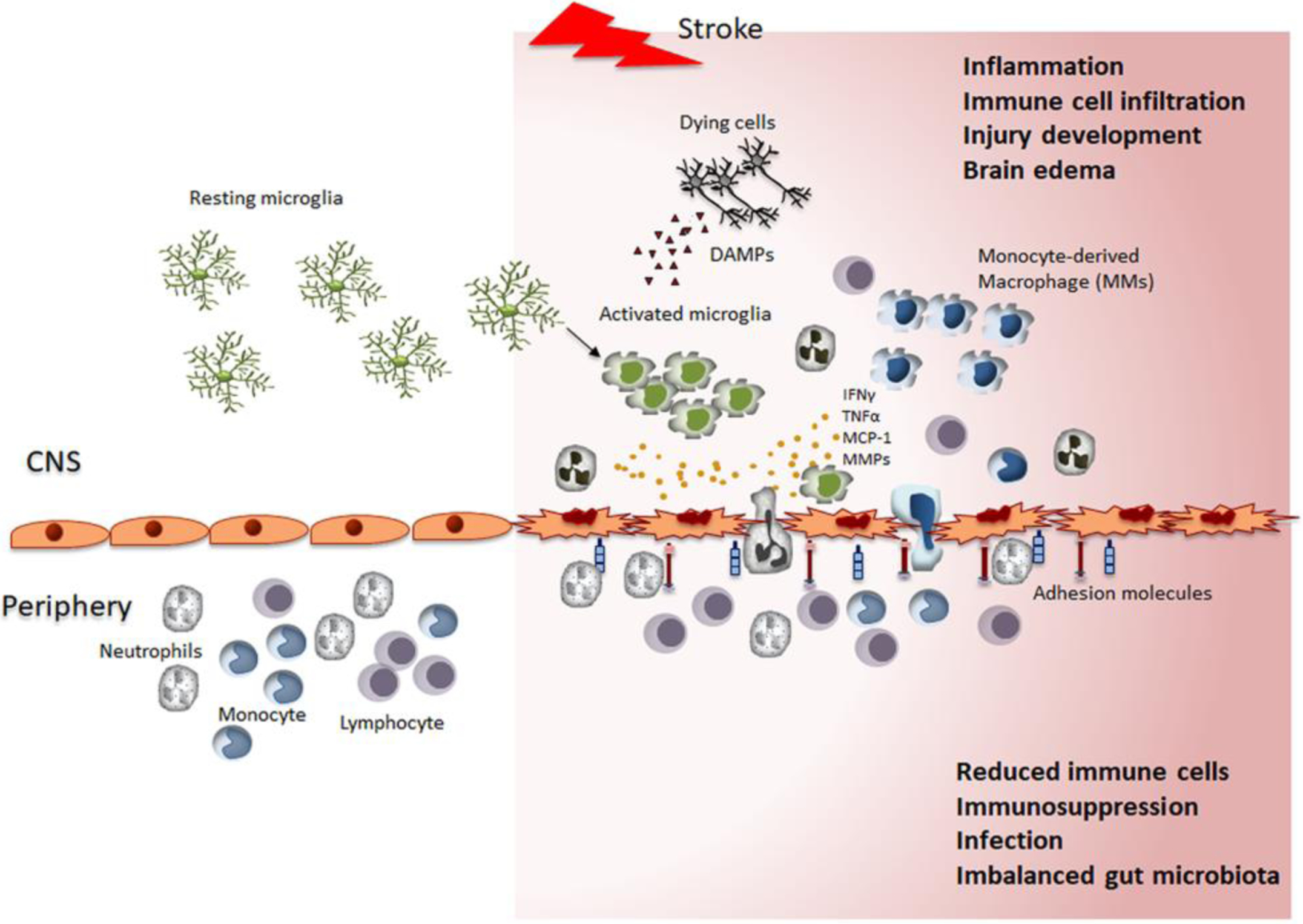
Interactions between the CNS and peripheral immune systems in response to stroke. Partitioned prior to stroke, the immune system of the CNS and periphery are connected through the disrupted BBB following stroke. Surveilling microglia encountering danger signals (e.g., DAMPs) in the affected area are activated and produce inflammatory mediators. Increased expression of adhesion molecules in vessels allows trans-endothelial migration of circulating immune cells. Infiltrated neutrophils, monocytes, and lymphocytes elicit neural inflammation. The deployment of peripheral immune cells to the injured brain and excessive neuroimmune reactions in the CNS profoundly disturb peripheral immunity and lead to immune depression, infection, and imbalanced gut microbiota. The stroke-induced alteration of immunity and the inflammatory response in CNS and periphery influence injury development and repair/remodeling processes.
2.1. Immune cell activation and infiltration in ischemic brain
Stroke-induced energy failure, excitotoxicity, and ion imbalance causes a spectrum of cell deaths including apoptosis and necrosis (Lipton, 1999). The so-called damage-associated molecular pattern (DAMPs) signals from dying cells, including modified/oxidized lipid species, cytoplasmic proteins, DNA, RNA, and extracellular matrix components (Matzinger, 2002), are recognized by pattern recognition receptors (PRRs), such as toll-like receptors and scavenger receptors on microglia, perivascular macrophages, and endothelial cells, which in turn elicit innate host immune responses (Marsh et al., 2009b). The stroke-induced immune cell activation and infiltration is temporally and spatially regulated, with the function of immune cells depending on the ischemic environment at different phases of stroke (Fumagalli et al., 2015). Activation of local microglia and astrocytes is the earliest event, occurring within hours of ischemic onset and is sustained several weeks after stroke (Gelderblom et al., 2009). These activated local immune cells and damaged neurons produce inflammatory substances that cause infiltration of circulating immune cells (Yilmaz and Granger, 2008; Zinnhardt et al., 2015). While their spatial and temporal pattern of trafficking during a stroke recovery phase has not been adequately defined, recent longitudinal transcriptome analyses identify the sustained innate and adaptive immune transcripts in ischemic brain and the infiltration of peripheral immune cells at acute, subacute, and recovery stages of stroke (Fury et al., 2020). It suggests long-lasting immune activation in the brain in chronic stroke.
2.2. Function of immune cells in stroke brain: Innate and adaptive immunity
The initial immune response to stroke occurs through innate immunity. In experimental brain ischemia, peripheral immune cells appear in the brain within 1 day and are sustained until 7 days post-stroke. Neutrophils are the first blood-borne immune cells that infiltrate the stroked brain, beginning at 30 min to a few hours, peaking at days 1–3, then steadily declining over time (Gelderblom et al., 2009; Jickling et al., 2015). Subsequently, monocytes/macrophages (the major infiltrating cells), dendritic cells, and natural killer cells appear in the injured brain, with a peak at 3–7 days after stroke (Stevens et al., 2002; Gelderblom et al., 2009; Kim et al., 2014b). This early innate immune activation is followed by activation of the adaptive immune system, while a relatively smaller number of T and B cells infiltrate into the post-ischemic brain (Stevens et al., 2002; Gelderblom et al., 2009). The roles of innate and adaptive immune activation have been explored in acute and sub-acute phases of stroke and are believed to be sustained during the chronic stages of stroke.
a. Neutrophils:
Neutrophils are the first blood-derived immune cells to be attracted to the ischemic brain. Neutrophils produce ROS, proteases, including MMP-9, neutrophil extracellular traps, which induce clot formation, and pro-inflammatory cytokines/chemokines, which are pivotal in BBB breakdown, thrombosis, and pro-inflammation in ischemic stroke (Gidday et al., 2005; Darbousset et al., 2012; Jickling et al., 2015). Despite the well-recognized association of neutrophils with infarct development in human and experimental animal models (Buck et al., 2008; Kumar et al., 2013; Garcia-Bonilla et al., 2014), results show conflicting roles of neutrophils on ischemic outcomes. A study reported that late neutrophil-derived MMP-9 inhibition worsens stroke outcome by reducing vascular remodeling, while early MMP-9 inhibition improves outcomes (Christoffersson et al., 2012). Additional studies reported anti-inflammatory effects of neutrophils by degradation of proinflammatory DAMPs (Cauwe et al., 2009), and by release of growth factors (Taichman et al., 1997), which may be related to better recovery after stroke (Emerich et al., 2002; Jickling et al., 2015).
Functional heterogeneity is associated with neutrophil polarization. Depending on extracellular stimuli, neutrophils switch from the neurotoxic N1 phenotype towards the neuroprotective N2 phenotype (Puellmann et al., 2006; Fridlender et al., 2009). Cuartero et al. have demonstrated that nuclear peroxisome proliferator-activated receptor (PPAR)-γ prompts neutrophil reprogramming to N2 phenotype and resolution of inflammation in the permanent middle cerebral artery occlusion (MCAO) in mice (Cuartero et al., 2013). The observed functional heterogeneity and cellular phenotypes of neutrophils suggest that manipulating neutrophil polarization can be a therapeutic strategy for stroke.
b. Microglia and monocyte-derived macrophages:
Among immune cells, microglia and infiltrating monocyte-derived macrophages (MDMs) play a major role in neuroinflammation in ischemic stroke. These are the major cell types producing cytotoxic substances (Colton and Gilbert, 1987; Clausen et al., 2008). Accordingly, the detrimental role of microglia/macrophages on neuronal death has been reported in vitro (Boje and Arora, 1992; Arantes et al., 2000) and ischemic injury in vivo (Hoehn et al., 2005; Liu et al., 2007). Evidence for the neuroprotective roles for microglia/macrophages in stroke is also conflicting. The depletion or inactivation of microglia/macrophages reduced neurogenesis and exacerbated neuroinflammation and stroke-induced injury (Faustino et al., 2011; Szalay et al., 2016). Microglia and MDMs release reactive oxygen and nitrogen species, pro-inflammatory cytokines (e.g., TNF, IL-1, IL-6), and MMPs that are known to be cytotoxic for injury development. On the other hand, these cells are important immune cells that also secrete anti-inflammatory factors including IL-10, TGF-b, and arginase I, which are critical for the resolution of inflammation and tissue remodeling (Jayaraj et al., 2019). These paradoxical observations of mononuclear phagocyte activity support the view that the immune-associated responses in stroke are context-dependent and the ischemic milieu dictates the functions of these cells in different post-stroke stages (Fumagalli et al., 2015).
Both microglia and MDMs express common antigens and have overlapping functions, including eliciting neural inflammation and inflammation resolution. Enhanced phagocytosis on viable neurons or oligodendrocyte precursor cells is associated with stroke-induced brain injury (Neher et al., 2013; Hayakawa et al., 2016). However, the removal of immunogenic intracellular contents and apoptotic cells prevents the propagation of cytotoxicity (Elliott and Ravichandran, 2010). Clean-up of cellular debris is an essential event for the inflammation resolution process and stroke recovery (Neumann et al., 2009; Ting et al., 2020). In addition, the uptake of infiltrated neutrophils by resident phagocytes in the brain seems to be beneficial in ischemic injury and functional recovery (Neumann et al., 2015; Otxoa-de-Amezaga et al., 2019). The dual functions in mononuclear phagocytes apparently derive from their phenotype polarization. In the stroked hemisphere, these cells are polarized either as classically activated pro-inflammatory M1or alternatively activated anti-inflammatory M2 subsets. The M2 phenotype is further divided into M2a, M2b, and M2c subtypes (Murray et al., 2014; Kim and Cho, 2016). In addition to these subsets, Mox (Kadl et al., 2010) and M3 phenotypes (Walker and Lue, 2015) have been also described, though without extensive studies. Generally, the M1 phenotype is usually considered to contribute to exacerbated ischemic injury and worsened neurological outcomes by releasing cytotoxic substances and impairing post-stroke neurogenesis and axonal regeneration (David and Kroner, 2011; Hu et al., 2012). By releasing growth factors, enhancing inflammatory resolution, and clearing apoptotic debris, the M2 phenotype is involved in wound healing and recovery after stroke (Mosser and Edwards, 2008; David and Kroner, 2011). Given that phagocytosis is an important function for pathogen clearance and repair/remodeling process after injury, there have been many attempts to distinguish microglia from MDMs and their specific functions. While this challenging issue has not been resolved, a recent study by Grassivaro et al. provided important insight into whether making such a distinction has biological meaning. Specifically, the study showed convergence between microglia and macrophage phenotypes in CNS tissue during neuroinflammation, demonstrating bidirectional changes in marker expression between microglia and infiltrating MDMs in the inflamed tissue (Grassivaro et al., 2020). This is likely due to the influence of the CNS milieu on the behavior of infiltrating immune cells (Fumagalli et al., 2015). In addition, Greenhalgh et al. reported that infiltrating MDMs influence resident microglia by regulating microglial phagocytic function (Greenhalgh et al., 2018). Given that microglia and MDMs interact to act as a functional unit rather than different cellular entities, these studies collectively cast doubt on whether it is possible to distinguish the individual cell type in the injured brain and to designate functional significance in stroke pathophysiology. The challenge of determining the specific role of each microglia/macrophage subset in the context of the stroke injury milieu still remains to be overcome.
c. T Lymphocytes:
Unlike innate immune cells, the number of infiltrating lymphocytes into the injured brain is relatively small (Gelderblom et al., 2009). Natural Killer (NK) cells are a type of cytotoxic lymphocytes that play a role in innate immunity. Without priming or prior activation steps, NK cells quickly respond to pathologic insults (Vivier et al., 2008). NK cells exacerbate brain infarction by enhancing cytotoxicity and inflammation (Gan et al., 2014). It has been also suggested that IFN-γ released by NK cells contributes to ischemic injury by recruiting macrophages or dendritic cells (Shi et al., 2011). In contrast, depletion of NK cells did not demonstrate benefits in MCAO models (Mracsko et al., 2014b). Moreover, IFN-γ can improve post-stroke survival by reducing bacterial infections while not affecting lesion size (Liu et al., 2017).
Studies have shown the contribution of T cells to neuroinflammation and secondary stroke progression (Iadecola and Anrather, 2011; Gill and Veltkamp, 2016). Transgenic mice lacking lymphocytes consistently demonstrated smaller infarct (Yilmaz et al., 2006; Subramanian et al., 2009; Kleinschnitz et al., 2010) while restoration of lymphocyte by adoptive transfer of splenocytes reversed the protective effect (Yilmaz et al., 2006). The depletion of the CD8+, CD4+, and γδ T cells subsets using antibodies also attenuated stroke lesion progression (Shichita et al., 2009; Liesz et al., 2011; Gelderblom et al., 2012). There have been contradicting reports on antigen dependency of T cell activation in stroke. A study has shown that T cells contribute to acute ischemic injury without adaptive immune mechanism such as antigen recognition or co-stimulatory pathways (Kleinschnitz et al., 2010). However, other studies have reported that T cell clonal expansion occurs in the brain and periphery within the first week after stroke (Liesz et al., 2013), and antigen-dependent activation of CD8+ cytotoxic T cells is associated with stroke lesion development (Mracsko et al., 2014b). Another T-cell subset, regulatory Tcells (Treg), has been identified as a protective effector in stroke. The mechanisms that are associated with the protective effect of Treg include anti-inflammatory cytokines, IL-10 (Liesz et al., 2009; Na et al., 2015), inhibition of MMP-9 activity, and maintaining BBB integrity (Li et al., 2014). Treg cells also play a role in limiting neuroinflammation and function of pro-inflammatory T cells subpopulations during the acute phase of stroke (Liesz and Kleinschnitz, 2016). Conversely, there is an evidence demonstrating the detrimental role of Treg cells in ischemic stroke. Selective depletion of Treg cells significantly reduced injury size and improved neurological function in stroke, and Treg cell-induced microvascular dysfunction was linked to the exacerbation (Kleinschnitz et al., 2013).
d. B Lymphocytes:
While most research in stroke immunology has focused on innate immune cells or T cells, local antibody production was observed in cerebrospinal fluid of stroke patients, suggesting the presence of B lymphocytes in the ischemic brain (Pruss et al., 2012). The role of B cells in stroke has been addressed in the mice lacking mature B cells (μMT knock-out (μMT−/−) mice with a mutation in the μ chain of the B cell receptor) (Kitamura et al., 1991). Significantly larger ischemic injury and higher mortality after MCAO was observed in these mice, which was rescued by adoptive transfer of B cells in these mice, showing a beneficial role of B lymphocytes in stroke. The protective effect of B cells was mediated by IL-10 (Ren et al., 2011; Chen et al., 2012). The exacerbated ischemic outcome in μMT/− mice were linked to an increased number of neutrophils, T cells, and microglia/macrophages in the ischemic brain, while IL-10-producing B cells decreased the neutrophil infiltration (Ren et al., 2011). In a separate study, an adoptive transfer of B cells reduced infarct size independent of other immune populations in recipient mice after transient MCAO, and the depletion of B cells delayed motor recovery, impaired spatial memory, and increased anxiety in post-stroke mice (Ortega et al., 2020). The study demonstrated that diapedesis of B cells throughout the brain, including remote areas, is involved in functional recovery (Ortega et al., 2020). These observations suggest that the beneficial effect of B cells on acute ischemic injury is mediated by inhibition of cytotoxic immune cells. A contrasting view, of a detrimental role of B cells in stroke, was also reported. Preclinical data from Doyle and colleagues showed sustained infiltration of B lymphocytes into the ischemic hemisphere for at least 12 weeks (Doyle et al., 2015). Interestingly, the delayed sustained B cell presence in the brain during the chronic phase of stroke was associated with cognitive decline, suggesting it as a predictor of dementia after stroke (Pendlebury and Rothwell, 2009; Levine et al., 2015). Additional supporting preclinical evidence that links B cells to stroke-induced cognitive decline is from the observation that B cell-deficient mice displayed attenuated cognitive decline after stroke (Doyle et al., 2015), and that cognitive deficits are linked to B cell-produced antibodies in mice (Becker et al., 2016). In patients, the number of B cells in the brain was significantly higher in the group with post-stroke dementia compared to the stroke group without dementia (Doyle et al., 2015). Alternatively, self-reactive antibodies have been shown to play a role in opsonization of degenerating myelin debris that facilitates phagocytosis of the myelin debris by phagocytes and promotes nerve regeneration (Vargas et al., 2010). Despite evidence supporting the injurious role of B cells involving cytokine- and/or antibody-dependent cell-mediated cytotoxicity, complement-mediated mechanisms, and possible direct induction of apoptosis, the evidence for the protective function of B cells suggests a context- and post-stroke stage-dependent role of B cells in the adaptive immune response.
2.3. Stroke-induced immune deficiency in the periphery
Stroke-induced neural inflammation does not spare peripheral immunity from disturbances. An increased of inflammatory cells and cytokines has been observed in the periphery during the acute phase of stroke, which was rapidly followed by immunosuppression (Offner et al., 2006b; Chapman et al., 2009). The immunosuppression is considered a compensatory mechanism against brain damage (Meisel et al., 2005; Urra et al., 2009) and is associated with impaired respiratory burst, compromising the defense functions of granulocytes and monocytes towards bacteria (Seki et al., 2010; Ruhnau et al., 2014). Experimental stroke studies have shown the contraction of lymphatic organs, such as the spleen and thymus. The spleen was identified as an immediate reservoir of monocytes that deploys them upon injury (Swirski et al., 2009). Accordingly, preclinical studies have reported rapid contraction of the spleen and deployment of monocytes into circulation upon stroke (Offner et al., 2006b; Kim et al., 2014b). The immediate immune cell supply by the spleen is considered vital to meet the immune challenge in response to stroke, because monocyte production from bone marrow takes several days after initial injury (Kim et al., 2014b). The majority of immune cells deployed from lymphatic organs in circulation infiltrate the injured brain to elicit neural inflammation.
Post-stroke immune depression involves the hypothalamic–pituitary-adrenal (HPA) axis and autonomic nervous system. Stroke-induced cytokines (i.e., IL-1β, TNFα, and IL-6) stimulate the hypothalamic paraventricular nucleus (PVN) to synthesize corticotropin-releasing factor (CRF). The secreted CRF induces adrenocorticotropin hormone (ACTH), which facilitates the secretion of glucocorticoids (GCs) (cortisol in human, corticosterone in rodents) from the adrenal cortex (Chamorro et al., 2007). Glucocorticoids downregulate pro-inflammatory mediators such as IL-1β, IL-11, IL-12, TNF-α, chemokines (IL-8), IFN-γ, prostaglandins, and nitric oxide, but upregulate anti-inflammatory mediators such as IL-4, IL-10, and TGFβ (Wilckens and De Rijk, 1997; Barrat et al., 2002). A synthetic glucocorticoid, dexamethasone, has shown strong pro-apoptotic and anti-proliferative effects in T lymphocytes (Tuosto et al., 1994). These properties of glucocorticoids are linked to the lymphocytopenia and altered pro-inflammatory/anti-inflammatory mediators following stroke (Offner et al., 2006a; Mracsko et al., 2014a). Elevated CRF in PVC also activates the sympathetic nervous system and leads to secretion of catecholamines in the bone marrow, thymus, spleen, and lymph nodes (Chamorro et al., 2007). In traumatic brain injury models, sympathetic activation triggers the secretion of IL-10 and deactivates monocytes (Woiciechowsky et al., 1998). Also, persistently increased catecholamine levels reduced circulating lymphocytes and immune organ atrophy after stroke (Prass et al., 2003). The anti-inflammatory effect of the GCs and catecholamines are considered as a mechanism for post-stroke immune depression.
The resulting immunodeficiency in the periphery renders increased susceptibility to secondary infections. (Shim and Wong, 2016). Preclinical studies have demonstrated that stroke-induced immunodeficiency leads to increased incidences of bacterial infections, which was reversed by adoptive transfer of T and NK cells from wild-type mice, or administration of IFN-γ (Prass et al., 2003). In addition, stroke causes higher susceptibility to aspiration pneumonia, which was attributed to sympathetic hyperactivity (Prass et al., 2006). Higher incidence of pneumonia and urinary tract infections negatively impact stroke outcome and mortality (Aslanyan et al., 2004). The reciprocal immune responses between the brain and periphery, respective excessive neural inflammation in the CNS, and immunodepression in the peripheral organs raises concerns on non-selective anti-inflammatory strategies to treat stroke patients. Thus, therapeutic strategies to boost peripheral immunity may include an approach to heighten endogenous protective mechanisms such as application of remote ischemic conditioning. A recent study provided evidence that post-stroke limb conditioning changes the composition of the circulating monocytes toward a pro-inflammatory status and this change has been associated with functional recovery (Yang et al., 2019b). Since the conditioning was applied in the limb away from the protected organ (brain), the study suggests that this immune-mediated peripheral mechanism underlies conditioning-induced benefits in stroke outcomes and underscored the importance of circulating CCR2+ pro-inflammatory monocytes for these benefits. Such an approach may serve as a therapeutic strategy to enhance peripheral immunity without compromising the necessary immune response in injured brain tissue.
2.4. Gut microbiota in stroke immunity
During evolution, several bacterial populations termed microbiota colonized different parts of the mammalian body and established a commensal alliance. Among the sites of microbiota colonization, the gastrointestinal tract has the most complex microbiota (Costello et al., 2009). Microbiota in the gut is involved in digestion, vitamin synthesis, and nutrition storage and metabolism (Lakshminarayanan et al., 2014). Gut microbiota is also linked to immune function. Gram-negative bacteria are necessary to induce the genesis of B cells in intestinal lymphoid organs such as Peyer’s patches and mesenteric lymph nodes (Bouskra et al., 2008). Germ-free mice colonized with Bacteroides fragilis corrected T cell deficiencies and TH1/TH2 imbalances and elicited appropriate cytokine production of T cells (Mazmanian et al., 2005). The gut microbiota inhibit growth of harmful microbes and their translocation into the different parts of body prevents inflammation (Belkaid and Naik, 2013; Yoo and Mazmanian, 2017).
The gut microbiota has a bidirectional interaction with CNS via neural, humoral, and immunological signaling, and the importance of their interaction has been reported under pathologic conditions (Carabotti et al., 2015; Winek et al., 2016a). In stroke, interactions between the gut and injured brain seems to be mediated by DAMPs and cytokines and migration of inflammatory cells (Round and Mazmanian, 2009; Ivanov and Honda, 2012; Maynard et al., 2012). Gastrointestinal complications accompanied by increased intestinal permeability resulting in poor stroke outcomes have been observed in stroke patients (Camara-Lemarroy et al., 2014). The leaky gut is associated with gut dysbiosis and bacterial translocation into circulation and the lymphatic system, causing systemic infection (Schulte-Herbruggen et al., 2009; Hagiwara et al., 2014). Experimental stroke studies have shown that lymphocyte numbers were significantly reduced in Peyer’s patches after stroke (Schulte-Herbruggen et al., 2009). Among the cells, γδ T cells in the gut migrate to the ischemic site and contribute to inflammation by secreting pro-inflammatory cytokines such as IL-17 (Shichita et al., 2009; Benakis et al., 2016), while anti-inflammatory Treg cells migrate from the gut into leptomeninges of stroked brain to exert neuroprotection (Benakis et al., 2016). In later phases of stroke, the Treg cells from lymph nodes migrate into gut and suppress differentiation of γδ T cells (Liesz et al., 2015; Benakis et al., 2016). Despite available reports that characterize interactions of the brain-gut axis following stroke in a normal condition, understandings on the brain-gut interaction where the body’s immune system has been modified by antibiotic treatment and in stroke patients with existing comorbidities are limited.
3. COMORBIDITY-MODIFIED IMMUNITY AND THE IMPACT IN STROKE
Several risk factors, including aging, hypertension, diabetes, and hyperlipidemia are associated with increased incidence of cardio- and cerebrovascular diseases including stroke (Pinto et al., 2004). These risk factors modify peripheral immunity and have been associated with chronic low-grade systemic inflammation characterized by higher levels of pro-inflammatory markers in the periphery. Despite recognized pro-inflammatory status, studies have consistently reported impaired/inappropriate immune responses in these comorbid conditions. Stroke pathology includes a host of pro-death cascades such as excitotoxicity, oxidative stress, edema, and lipid oxidation (Iadecola and Anrather, 2011). Activation of these pathogenic processes often converged into inflammatory responses, leading to cellular demise, and they are intensified in comorbid conditions and negatively affect stroke outcomes (Hu et al., 2017; Tun et al., 2017; Lin et al., 2019). This review will focus on comorbidity-modified changes in peripheral immunity prior to stroke. We will also address how the inclusion of these risk factors affects comorbidity-modified neuroimmune interactions in stroke on CNS disease progression and outcomes.
3.1. Aging
Aging is a non-modifiable risk factor for stroke. Inclusion of aged animals in preclinical studies, however, resulted in equivocal outcomes in stroke. While some studies demonstrated increased brain injury size and behavioral deficits (Popa-Wagner et al., 2007; DiNapoli et al., 2008; Doyle et al., 2010; Manwani et al., 2013), some others showed decreased infarct size and edema in aged animals (Liu et al., 2010a; Liu et al., 2012; Ritzel et al., 2018). The reason for this controversy is not clear, but the stroke-induced mortality rate seems consistently higher in aged mice regardless of injury size (Wang et al., 2003; Crapser et al., 2016; Ritzel et al., 2018).
Aging compromises and dysregulates inflammatory/immune reactions in the CNS injury. Particularly, both clinical and preclinical studies have shown that the condition diminished phagocytic activities of mononuclear phagocytes (Floden and Combs, 2011; Njie et al., 2012), reduced chemotaxis in neutrophils (Wenisch et al., 2000; Brubaker et al., 2013), and decreased cytotoxic activity in natural killer cells and macrophages (Tasat et al., 2003; Beli et al., 2011). There is also an impaired pro-inflammatory response in the macrophages/dendritic cells and microglia (Liang et al., 2009; Nyugen et al., 2010; Panda et al., 2010). Aging also substantially modifies adaptive immunity. The numbers of naïve T and B cells are eventually reduced by differentiation into memory cells (Wertheimer et al., 2014; Thompson et al., 2017). The function of lymphocytes is also altered in aging. For example, B cells in aged subjects exhibit defective transcription factors and enzymes related to class switching and somatic hypermutation, leading to a decreased antibody response and antibody-mediated protection (Frasca et al., 2004; Frasca et al., 2016). In addition, CD4+ or CD8+ T cells isolated from aged mice displayed impaired signaling in antigen recognition, cell propagation and differentiation, and cytokine production (Haynes et al., 1997; Tsukamoto et al., 2010; Garcia and Miller, 2011; Smithey et al., 2011).
The age-related deregulated inflammatory response is linked with gut microbiota. Elderly people have a different gut microbiota profile compared to younger healthy adults (O’Toole and Jeffery, 2015). Aging reduces the diversity of gut microbiota, with increased populations of harmful opportunist bacteria and reduced commensal bacteria (Hopkins and Macfarlane, 2002; Rea et al., 2012; O’Toole and Jeffery, 2015; Odamaki et al., 2016). The changes are related to the impaired immune tolerance and increased inflammation in the gut (Kumar et al., 2007; Pamer, 2007) and may be associated with increased systemic pro-inflammatory cytokines in aged subjects (Biagi et al., 2010). Age-altered gut microbiota influences host nutrition and metabolism and affect aging-related pathophysiologic phenomena including metabolic and autoimmune diseases, weight loss, cognitive decline, and sarcopenia (Rampelli et al., 2013; Ursell et al., 2014; Rios-Covian et al., 2016).
Taken together, the aging-induced chronic pro-inflammatory status potentially leads to blunted inflammatory responses (Sieber et al., 2011), dysregulated immune cell functions (Ritzel et al., 2018), and altered gut microbiota towards harmful populations (Spychala et al., 2018), which would alter the clearance of peripheral infections and the progression of injury development after stroke. With further studies to define mechanisms of the negative impact of aging in stroke, strategies aimed at rejuvenating immune cells and gut microbiota are suggested to improve stroke outcomes in the aging brain.
3.2. Hypertension
Hypertension, the second most prevalent risk factor for stroke, is correlated with worse stroke outcomes and higher mortality (Willmot et al., 2004; O’Donnell et al., 2010). The reported mechanisms underlying the condition are impaired autoregulatory responses of cerebral blood flow, vascular dysfunction, and enhanced oxidative stress (Iadecola and Davisson, 2008).
Chronic systemic inflammation in hypertension seems to be linked to the exacerbated stroke outcomes. Peripheral immunity is largely skewed towards pro-inflammatory status in hypertension. Angiotensin II is a major mediator of hypertension (Kvakan et al., 2009). A clinical study showed that plasma IL-6 levels were positively correlated with blood pressure (Kim et al., 2008b), and that angiotensin II-induced hypertension was completely abrogated in IL-6−/− mice (Brands et al., 2010). Hypertension increases the number of circulating leukocytes, and their adhesion is also increased due to overexpression of junctional adhesion molecules in endothelial cells (DeSouza et al., 1997). The identity and function of immune cells in hypertension have not been fully defined; however, involvement of both innate and adaptive immune cells has been suggested. Available but limited literature, however, indicates that hypertension significantly reduced microglial activation, which was associated with larger infarct (De Geyter et al., 2012), suggesting a beneficial role of injury-induced activation of microglia. Hypertension spontaneously activates circulating monocytes and increases reactive oxygen species production in immune cells (Dorffel et al., 1999; Kim and Vaziri, 2005). Proinflammatory M1 macrophages were the predominant cells in the aortas following 7 days treatment of angiotensin-II (Kossmann et al., 2013), followed by increased M2 macrophages by prolonged infusion of angiotensin-II for 14 to 28 days (Moore et al., 2015). Stroke increased periphery-derived CD45+ myeloid immune cells with larger stroke-induced brain injury in a hypertensive condition (Moller et al., 2015). Hypertension enhanced the expression of adhesion molecules in leukocytes that may contribute to the increased myeloid immune cell accumulation and inflammation in stroke. Angiotensin II promotes the proliferation of splenic lymphocytes and increases cytokine production through angiotensin I receptors on immune cells (Bataller et al., 2003; Ganta et al., 2005). The effect of hypertension in the cells of the CNS includes the activation of astrocytes and microglia and elevated expression of adhesion molecules in endothelial cells, causing leukocyte accumulation (Waki et al., 2008; Kaiser et al., 2014). Both M1 and M2 markers were upregulated in microglia, but not in monocytes in hypertensive mice, which may be explained by the neurogenic regulation of hypertension (Shen et al., 2015).
There is evidence to suggest the involvement of gut microbiota in hypertension. The gut microbial number and diversity are altered in hypertension. Kang et al. have listed the alteration of potentially beneficial or harmful gut bacteria in hypertension in their review (Kang and Cai, 2018). While bacteria that considered to be beneficial (e.g, Bacteroides, Faecalibacterium, Oscillibacter, Roseburia, Bifidobacterium, Coprococcus, and Butyrivibrio) are enriched in healthy controls, harmful bacteria such as Prevotella, Klebsiella, Porphyromonas, and Actinomyces are elevated in the hypertensive group (Kang and Cai, 2018). The gut microbiota can influence blood pressure by regulating hormones such as serotonin, dopamine, and norepinephrine and kidney function to excrete sodium load (Gomez-Guzman et al., 2015; Durgan et al., 2016). Chronic pro-inflammation in hypertension can be also the result of gut microbial action. Prevotella copri is enhanced in individuals with hypertension (Li et al., 2017) and contributes to inflammation by releasing superoxide reductase and phosphoadenosine phosphosulphate reductase (Scher et al., 2013; Li et al., 2017). Immune responses triggered by endotoxin from Prevotella and Klebsiella are also related to the chronic low-grade inflammation in hypertension (Cybulsky et al., 1988; Kohn and Kung, 1995). This evidence suggests that the alteration of gut microbiota can be used to normalize the pathology associated with hypertension. This is supported by studies showing that fecal transplants from normotensive to hypertensive rats, and vice versa, result in alteration of plasma acetate and heptanoate, sodium excretion, and blood pressure (Mell et al., 2015; Adnan et al., 2017). In addition, antibiotic administration reversed the altered bacteria ratio and blood pressure in hypertensive rats (Yang et al., 2015). Understanding how the immune response and gut-immune interaction is altered in hypertensive subjects and its effect on stroke outcomes remains to be addressed.
3.3. Hyperlipidemia
High levels of plasma cholesterol are associated with increased incidences of vascular diseases and worsened stroke outcome. Genetics and cholesterol rich diets affect plasma cholesterol levels (Griffin and Lichtenstein, 2013; van Dongen et al., 2013; Mente et al., 2017). Mice with genetic deficiency of apolipoprotein E (ApoE KO) and/or low-density lipoprotein receptor (LDL−/−) are used for modeling experimental hyperlipidemic conditions. ApoE KO mice fed a high fat diet for 8 weeks displayed 7–8 times higher blood cholesterol levels compared to C57 mice with normal diet (Kim et al., 2008a). There is also the interaction between cholesterol levels in circulation and microbiota richness and diversity (Cotillard et al., 2013; Dao et al., 2016). For instance, the gut microbiota regulates plasma cholesterol levels and atherosclerotic lesion development in germ-free ApoE KO mice (Kasahara et al., 2017; Lindskog Jonsson et al., 2018). Similarly, the depletion of gut microbiota by antibiotics increased plasma cholesterol levels and profoundly altered cholesterol metabolism. (Le Roy et al., 2019), The study also showed the colonization of microbiota from hypercholesterolemic mice induced hyperlipidemia, suggesting crosstalk between intestinal microbiota and circulating lipids. Among gut bacteria, Erysipelotrichaceae, Alistipes, Barnesiella, and Turicimonas have been reported to be positively correlated to plasma cholesterol (Fu et al., 2015; Le Roy et al., 2019). There are several bacterial taxa (Roseburia intestinalis, Faecalibacterium prausnitzii, and Anaerostipes, Eubacterium) that produce beneficial metabolites such as butyrate and lower plasma cholesterol levels and atherosclerosis development (Kasahara et al., 2018), through bile acid metabolism, or through entrapment of cholesterol (Liong and Shah, 2005). Thus, manipulating gut microbiota would be beneficial to improve lipid profiles, reduce peripheral inflammation, and attenuate stroke incidence.
Similar to the other comorbidities, hyperlipidemia is also associated with the chronic inflammation in the periphery. There was increased circulating cytokines and chemokines such as IL-6, TNF-α, MCP-1, and CCL5 (Olefsky and Glass, 2010). In addition to increasing humoral factors, hyperlipidemia increases production of hematopoietic progenitor cells in bone marrow, leading to monocytosis and neutrophilia (Murphy et al., 2011; Seijkens et al., 2014). Ly6Chigh pro-inflammatory monocytosis was observed in hyperlipemic mice (Swirski et al., 2007). The pro-inflammatory monocyte-driven macrophages excessively uptake oxidized/modified lipoproteins. These foamy macrophages produce excessive pro-inflammatory cytokines and are associated with the chronic inflammation seen in hyperlipidemia (Choi et al., 2011; Weber et al., 2011; Koltsova et al., 2012). Besides inflammation, hyperlipidemia also compromises tissue repair/remodeling process by impaired pathogen clearance (Angeli et al., 2004; Roufaiel et al., 2016). Hyperlipidemia also affects the function of lymphocytes. oxLDL uptaken antigen presenting cells stimulate T cells, leading the cells to be differentiated into pro-inflammatory T effector cells, rather than immunosuppressive Treg population (Maganto-Garcia et al., 2011; Newton and Benedict, 2014; Herold et al., 2017). B cell-deficient hyperlipidemic mice showed decreased atherosclerotic lesions (Caligiuri et al., 2002; Major et al., 2002). The study is in contrast to the report that showed B cell-produced antibodies against oxLDL have been proposed as the protective mechanism (Lewis et al., 2009).
Hyperlipidemic brains already exhibit increased microglial phagocytosis, microgliosis, and higher expression of vascular adhesion molecules, even prior to stroke. Larger numbers of activated myeloid phagocytes, T cells, and granulocytes were observed in the choroid plexus of the hyperlipidemic mice (Drake et al., 2011). The chronic proinflammatory environment in the periphery and brain is associated with the negative influence of hyperlipidemia in stroke injury. Previous studies have reported that plasma cholesterols levels were positively correlated with larger infarcts and edema formation (Kim et al., 2008a; ElAli et al., 2011). It has been also shown that higher low-density lipoprotein level is associated with long-term mortality after stroke (Xing et al., 2016). The hyperlipidemia-exacerbated stroke injury was associated with increased inflammatory response in the ischemic hemisphere via CD36, a multifunctional class B scavenger receptor (Kim et al., 2008a). By exchanging bone marrow cells between CD36-expressing and CD36-deficient mice, a subsequent study revealed that CD36 expressed in monocyte-derived macrophages plays a critical role in hyperlipidemia-exacerbated stroke injury and edema (Kim et al., 2012). Clinical evidence also shows the negative impact of hyperlipidemia in neurological outcomes. Patients with hyperlipidemia showed worse neurological outcomes, and administration of cholesterol-lowering drugs ameliorated neurological outcomes of stroke patients (Amarenco et al., 2006; Restrepo et al., 2009). Though these studies showed a detrimental effect of hyperlipidemia in stroke outcome, others showed an association between hyperlipidemia and a lower risk of short- and long-term mortality after stroke (Jimenez-Conde et al., 2010; Shigematsu et al., 2015; Yeramaneni et al., 2017). Moreover, hyperlipidemic stroke patients tend to have reduced white matter hyperintensity volume that predicts infarct progression (Arsava et al., 2009; Jimenez-Conde et al., 2010). Caveats of analyses of stroke outcome in hyperlipidemic conditions include potential treatment effect of cholesterol lowering drug in patients, since the drugs could exert pleotropic effects on microvascular integrity, inflammation, and oxidative stress (Liao, 2002; Zhao et al., 2014).
3.4. Diabetes
Diabetes has a high prevalence in stroke patients. Hallmarks of the conditions include hyperglycemia and insulin resistance, and chronic low-grade systemic inflammation with altered innate and adaptive immunity. Among immune cells, monocytes/macrophages are known to play a key role in inducing diabetes. Circulating monocytes isolated from patients or experimental diabetic animals have predominantly pro-inflammatory properties (Devaraj et al., 2006; Bradshaw et al., 2009; Hazra et al., 2013). The pro-inflammatory monocyte may differentiate into M1 macrophages in adipose tissue and contribute to inducing insulin resistance by releasing proinflammatory cytokines such as TNF-α, IL-6, and MCP-1 (Lumeng et al., 2007). Impaired insulin sensitivity in the liver and muscle disrupts peroxisome proliferator-activated receptor-γ (PPARγ) in macrophages (Odegaard et al., 2007), subsequently inhibiting M2 macrophage activation. Accumulating evidence exhibits the imbalance of T cell differentiation and function in a diabetic context. In patients with type 2 diabetes, CD4+ T cells tended to polarize into proinflammatory Th1 and Th17 cells in peripheral blood and adipose tissue. In contrast, CD4+ T cells differentiating into anti-inflammatory Th2 or Treg cells were decreased in the diabetic condition (Cortez-Espinosa et al., 2015; Nekoua et al., 2016; Lobo et al., 2018; Yuan et al., 2018). Although there are no significant differences in B cells in peripheral blood of diabetic or healthy subjects (Verboven et al., 2018), B cells play a central role in insulin resistance through the production of IgG antibodies and the activation of T cells and macrophages (Winer et al., 2011; Verboven et al., 2018). This is supported by a study demonstrating that mice deficient in mature B cells showed lower fasting glucose levels and improved glucose tolerance induced by high-fat diet intervention (Simar et al., 2014). B cells also affect the proliferation of Th17 and the production of proinflammatory cytokines in type 2 diabetic patients (Cao et al., 2016).
As in hyperlipidemic and hypertension, there is evidence that gut microbiota is also altered in diabetic conditions (Turnbaugh et al., 2006; Murphy et al., 2010). The imbalanced intestinal microbiota in diabetic conditions favor colonization of immune trigger population such as Enterobacteriaceae or Bacteroides, but decreased beneficial anaerobic bacteria (Davis-Richardson et al., 2014; Soyucen et al., 2014). It is unclear whether the alteration of gut microbiota is the cause or consequence of the metabolic condition. The colonization of germ-free mice with microbiota from obese mice leads to an increase in total body fat compared to the mice colonized with microbiota from lean mice (Turnbaugh et al., 2006), supporting a causal role of microbiota in obesity, and subsequently, the induction of insulin resistance. Furthermore, some microbial toxins have been reported to directly impair pancreatic β-cell function (Myers et al., 2003). LPS of the microbiota has been thought to be a trigger of the inflammatory response by binding with TLR4 and activating NF-κB in immune cells (Neal et al., 2006; Poggi et al., 2007). Thus, gut microbiota is likely a facilitating factor in the onset of diabetes by modulation of the immune response and contribution to chronic inflammation in this metabolically compromised condition.
Literature indicates the influence of a diabetes-induced shift towards pro-inflammatory status at the time of stroke and in the alteration of immune/inflammatory responses following stroke. While diabetes is a predictor for poor ischemic outcomes, including increased infarct volume and swelling and impaired neurological outcomes (Arboix et al., 2005; Ritter et al., 2011; Tureyen et al., 2011; Kim et al., 2014a), the means by which the altered peripheral immune/inflammatory status influences the progression of injury in diabetic stroke are not well defined. Potential mechanisms for diabetes-induced exacerbation of stroke outcome include increased numbers of macrophages/neutrophils, pro-inflammatory gene expression in the post-stroke brain (Tureyen et al., 2011), endothelial-neutrophil interaction and elevated ICAM expression (Ritter et al., 2011; Tureyen et al., 2011), and hemorrhagic transformation with increased MMP9 activity (Mishiro et al., 2014). In contrast, blunted/delayed immune response is also implicated with the worsened stroke outcomes. Post-stroke inflammatory responses and scar formation are delayed in diabetic db/db mice (Kumari et al., 2007). We have reported that peritoneal macrophages from diabetic mice show a blunted inflammatory response upon LPS stimulation and have reduced inflammatory cytokines/chemokines in the early post-stroke brain (Kim et al., 2014a). The blunted inflammatory response was also reported in diabetic macrophages of db/db mice, with decreased microglial activation and proinflammatory cytokines in the stroked brain (Kumari et al., 2007). As the inflammatory response is a necessary step to repair, the mounting of an effective, rapid, and orchestrated inflammatory response at the acute stage should be critical for subsequent tissue repair and remodeling (Barton, 2008). Since the impaired inflammation is partly accounted for by the exacerbated injury in diabetic stroke, the effect of diabetes-induced immune alteration and its influence in stroke-induced inflammatory responses needs to be further addressed.
4. IMMUNE-BASED STRATEGIES FOR STROKE THERAPY
The immune system undoubtedly influences stroke outcome and recovery/repair function. The distinction between the CNS and peripheral immune system at the time of stroke is lost, as the BBB is disrupted following stroke. The resulting bidirectional interactions between the two immune systems provides a unique opportunity to modulate the course of pathology and repair process. Several strategies are suggested and implemented to achieve benefit in acute and recovery stages of stroke.
4.1. Targeting inflammation
Anti-inflammatory strategies have mainly focused on reducing inflammation in the acute phase of stroke. NF-κB has been an important target in reducing acute stroke injury, as it is involved in the pathways of inflammation and apoptosis (Howell and Bidwell, 2020). Systemic injection of NF-κB inhibitors attenuated infarct size and neurological outcomes in rodent models of ischemia (Nurmi et al., 2004; Ali et al., 2020). Cytokines and their receptors have also been targeted to reduce acute injury. The IL-1 receptor antagonist (IL-1Ra) has been shown to reduce infarct volume in several preclinical studies (Banwell et al., 2009), with equivocal results in clinical trials (Emsley et al., 2005; Smith et al., 2018). Targeting microglial activation by minocycline and polarizing microglia to M2 phenotype by IL-33 or IL-4 improved stroke outcomes (Liu et al., 2016a; Yang et al., 2017). Attenuating neuroinflammation by targeting neutrophils (Jickling et al., 2015) or lymphocytes (Ducruet and Connolly, 2011) reduced the acute stroke injury. Inhibition of leukocyte adhesion is beneficial in reducing experimental stroke injury (Yilmaz and Granger, 2008), but did not show clincal efficacy (Enlimomab Acute Stroke Trial, 2001). Despite reports on anti-inflammatory approaches that effectively reduced excessive inflammation, leading to attenuated acute infarct size in preclinical studies, repeated failures in translating the strategy suggests potential benefit in eliciting neural inflammation for subsequent repair process. In addition, unintended suppression of peripheral immunity may cause secondary infection during the critical post-stroke period. Further studies that determine the effect of anti-inflammatory strategies beyond the acute phase and repair/remodeling processes in normal and comorbid conditions are needed.
4.2. Ischemic conditioning
Conditioning refers a phenomenon that the application of sub-lethal noxious stimuli induce endogenous protective mechanisms. Conditionoing-induced protection in the brain can be achieved by a variety of agents and stressors such as ischemia/hypoxia, inflammatory mediators, metabolic blockers, anesthetics, cortical spreading depression, and seizures (Dirnagl et al., 2009). The nature of conditioning-induced protection across organs allows its application areas remote to the organs requiring protection. Remote ischemic limb conditioning (RLC) is as non-invasive and feasible in clinical application. In the context of stroke, RLC has shown favorable effects in infarct reduction after cerebral ischemia (Yang et al., 2019a; Weir et al., 2020).
RLC-induced beneficial effects are associated with altering inflammatory pathways and immune cell composition. RLC modulates the NF-κB pathway and cytokine expression in lipopolysaccharide (LPS)-induced inflammation or lung injury models (Konstantinov et al., 2004; Kim et al., 2014c; Zhou et al., 2017). RLC induces lymphopenia in periphery in naïve or ischemic mice (Chen et al., 2018; Liu et al., 2019) and reduced neutrophil adhesion, phagocytic activity, and apoptosis in healthy subjects (Shimizu et al., 2010). In cerebral ischemia, RLC abolished stroke-induced release of splenic immune cells (T cell, B cell, and NK cell) leading reduced infiltration of those cells into the stroked brain (Chen et al., 2018). In addition, RLC increased non-inflammatory resident monocytes in experimental ischemic stroke (Liu et al., 2016b) and polarized macrophage to anti-inflammatory phenotype in hemorrhagic stroke, leading to increased hematoma clearance (Vaibhav et al., 2018).
Besides the overall benefits to health, exercise can be a form of conditioning stimuli. Exercise changes composition of immune cells in circulation (Tipton, 2014). Exercise regulates the number of lymphocytes and monocytes in the blood (Gleeson and Bishop, 2005; Lancaster and Febbraio, 2014) suggesting that an immune mechanism may be shared between exercise and RLC.
Exposure to low-dose lipopolysaccharide (LPS) before cerebral ischemia has been neuroprotective in stroke models. While activation of TLR4 is the potent underlying mechanism by the systemic administration of LPS (Ahmed et al., 2000; Vartanian et al., 2011), reprogramming of the immune system seems to be related to LPS-induced ischemic tolerance (Smith et al., 2002) leading to reduced postischemic proinflammatory gene expression, endothelial and microglial activation, as well as leukocyte infiltration (Bauer et al., 2000; Lin et al., 2009; Marsh et al., 2009a; Vartanian et al., 2011). On the other hand, Garcia-Bonilla et al. showed that accumulated pro-inflammatory Ly6Chi monocytes are associated with the LPS preconditioning-induced neuroprotection in experimental ischemic brain through suppression of meningeal inflammation (Garcia-Bonilla et al., 2018). In addition, RLC induced functional recovery in chronic stroke, which was associated with an early monocyte shift to pro-inflammatory Ly6Chi monocytes (Yang et al., 2019b), suggesting the critical role of RLC-induced inflammatory response on ischemic tolerance. Conditioning-induced changes of peripheral immune cells, therefore, may serve as a potential immune-based strategy to treat stroke.
4.3. Stem cell therapy
Stem cell therapy is now widely applied for clinical trials for human diseases including stroke (Krause et al., 2019). Various types of stem cells such as embryonic stem cells, neural stem/precursor cells (NSPC), mesenchymal stem cells (MSC), and adult stem cells from bone marrow (BM) or umbilical cord blood have been examined in stroke studies. Regardless of cell types and administration routes (via direct application to the CNS or periphery), transplant of the stem cells have shown beneficial effect on stroke outcomes in many pre-clinical studies (Boshuizen and Steinberg, 2018). Immunomodulation is one of the major mechanisms that the stem cells can exert their therapeutic effect on ischemic stroke. Both intravenous and intracerebral administration of MSC or NSPC decreased pro-inflammatory cytokines, but increased anti-inflammatory cytokines in ischemic brain (Bacigaluppi et al., 2009; Liu et al., 2009). In contrast, intravenous administration of BM-MSC increased TNF-α and IL-1β in the striatum and circulation in the early phase (2d post-stroke), but decreased in later (7d post-stroke) after ischemic stroke (Li et al., 2018). The early pro-inflammatory reaction may be induced by insufficiently activated MSCs after injection. Previous studies have shown that the activation of MSC requires a threshold concentration of cytokines to become immunosuppressive and the inactivated MSCs may be pro-inflammatory (Li et al., 2001; Li et al., 2012). These suggest that an optimal inflammatory environment is critical in order stem cells to exert a beneficial role. The effect of stem cell therapy on immune cell trafficking is also controversial. Administration of MSCs or NSPCs during acute and subacute phases of stroke decreased numbers of Iba1+ microglia/macrophages in ischemic brain (Bacigaluppi et al., 2009; Sheikh et al., 2011; Tsai et al., 2014). The report was contradicted by findings of increased microglia/macrophages by BM-MSC or NSPC treatment (Capone et al., 2007; Daadi et al., 2010; Li et al., 2018). These differing results could be derived from the influence of stem cells in immune cell polarization. Studies have shown that MSC induced anti-inflammatory M2 phenotype of microglia/macrophages in vitro (Kim and Hematti, 2009; Yan et al., 2013; Hegyi et al., 2014) and ischemic stroke in vivo (Ohtaki et al., 2008). This stem cell-induced anti-inflammatory status is associated with increased insulin-like growth factor-1, VEGF, BDNF, and TGF-β (Taguchi et al., 2004; Jiang et al., 2005; Li et al., 2018) and promotes neurogenesis and dendritic sprouting (Shen et al., 2006; Daadi et al., 2010; Liu et al., 2010b). A recent study showed superiority of interferon (IFN)-γ activated MSC compared to naïve MSC in inducing the benefits in ischemic stroke (Tobin et al., 2020).
4.4. Antibiotics
Peripheral infections including pneumonia and urinary tract infections are frequently observed in stroke patients, which contribute to mortality and unfavorable functional outcomes after stroke (Aslanyan et al., 2004). Antibiotics have been considered as treatments for post-stroke infection. Studies showed that antibiotics (e.g., enrofloxacin, minocycline, and ceftriaxone) reduced the incidence of infections, and improved mortality and neurological function in experimental brain ischemia (Yong et al., 2004; Lipski et al., 2007; Hetze et al., 2013) and in patients (Harms et al., 2008; Schwarz et al., 2008; Amiri-Nikpour et al., 2015). Antibiotics also modulate immune responses that may be linked to the beneficial effect in ischemic stroke. Minocycline reduced inflammation and microglial activation in an experimental stroke model (Yong et al., 2004). Ceftriaxone regulated expression of inflammatory cytokines in non-ischemic rat (Rao et al., 2014). However, there are preclinical reports that argue against the benefits of antibiotics in stroke. Antibiotic treatment exacerbated ischemic injury and functional outcomes in rodent models (Zierath et al., 2015; Winek et al., 2016b; Dong et al., 2017), and clinical trials also showed the insignificant impact of antibiotics in peripheral infection and stroke outcomes (Kohler et al., 2013; Kalra et al., 2015; Westendorp et al., 2015; Ulm et al., 2017). Therefore, a detailed understanding of the mechanism by which antibiotics modulate stroke-induced immune responses in the periphery and CNS is warranted in order to guide clinical usage of antibiotics in ischemic stroke.
4.5. Probiotic therapy
Several probiotics, Lactobacillus, Bifidobacterium, and Enterococcus are beneficial to human health (Ishibashi and Yamazaki, 2001). Immunoregulation is one of the mechanisms by which probiotics exert beneficial effects. In LPS-stimulated macrophages, Enterococcus and Lactobacillus reduce inflammation by reciprocal down-regulation of TNFa and upregulation of IL-10 (Divyashri et al., 2015). Oral administration of Enterococcus increased circulating IgG and IgA and the proportion of mature B cells in dogs (Benyacoub et al., 2003). Enterococcus administration increased concentration of plasma IgG in mice and the IL-4, IL-6, and IFN-γ levels in splenocytes (Sun et al., 2010). Literature also indicate that probiotics can influence CNS function and injury progression. Lactobacillus improved animal behavioral and cognitive function and reduced anxiety by regulation of brain-derived neurotrophic factor (BDNF) and neurotransmitters (serotonin, noradrenaline, and dopamine) in the brain (Bercik et al., 2011; Luo et al., 2014; Liang et al., 2015). Tan et al. have shown that daily oral intake of probiotics improved immunological imbalance and neurological function in patients with traumatic brain injury (Tan et al., 2011). Pretreatment of Clostridium butyricum reduced brain injury via suppression of apoptosis and oxidative stress in global ischemia (Sun et al., 2016). A mixture of Lactobacillus acidophilus, Lactobacillus rhamnosus, Lactobacillus bulgaricus, Bifidobacterium breve, Bifidobacterium longum, and Streptococcus thermophilus also attenuated hippocampal injury and cognitive impairements (Rahmati et al., 2019). A combination of Bifidobacterium breve, Lactobacillus casei, Lactobacillus bulgaricus, and Lactobacillus acidophilus reduced malondialdehyde and TNF-α, and provided neuroprotection against focal ischemia (Akhoundzadeh et al., 2018). The beneficial role of probiotics through immunomodulation thus suggests its potential as an immune-based therapy for stroke patients.
5. Perspective/Conclusion
The immune system is tightly controlled, and over- or insufficient activation may result in excessive inflammation or infection, respectively. In healthy individuals, immunity is in balance, but in pathological conditions, this balance is lost. Our understanding of immunity in stroke is heavily associated with overt inflammatory responses in the injured brain. Accordingly, numerous attempts have been made to reduce it. What has not been addressed is the relevance of neural inflammation in the disturbance of peripheral immunity. The distinction between the immune systems of the CNS and periphery prior to stroke is lost following stroke, and the consequence is immune interdependence between the periphery and CNS. The infiltration of peripheral immune cells to the injured brain could be a necessary response to overcome the ischemic insult. Nevertheless, the neural inflammation elicited in the injured brain inevitably disturbs peripheral immunity. The neurocentric view of stroke pathology, focusing solely on neuroinflammation, often comes at the expense of immune-depression and infection in the rest of body. It is also noteworthy that modifiable stroke risk factors, such as insulin resistance, hypertension, and hyperlipidemia, profoundly alter peripheral immune system prior to stroke. This comorbidity-modified peripheral immunity has a major impact on the progression of subsequent brain injuries and repair processes. Considering the fact that peripheral immunity influences CNS injury progression, promising development of the view is the use of peripheral immunity itself as a point of manipulation to limit or alter the course of the resulting pathology (Figure 2). While anti-inflammatory strategies are largely applied to reduce neuroinflammation, their effects on peripheral immunity should be considered. Other strategies that modulate peripheral immunity also include remote ischemic conditioning, in which cross tolerance among organs can be achieved by altering immune cell composition, as well as stem cell therapy following stroke. Prophylactic antibiotic and probiotic treatment are likely additional immune modulatory strategies to attenuate peripheral infection and modulate intestinal microbiota, which ultimately influence the process of stroke pathophysiology. In combination with thrombolysis/thrombectomy, the immunomodulatory approaches may induce a maximum efficacy to minimize ischemic injury and immunodeficiency, thereby preventing systemic infection and leading to enhanced post-stroke survival and long-term recovery.
Figure 2.
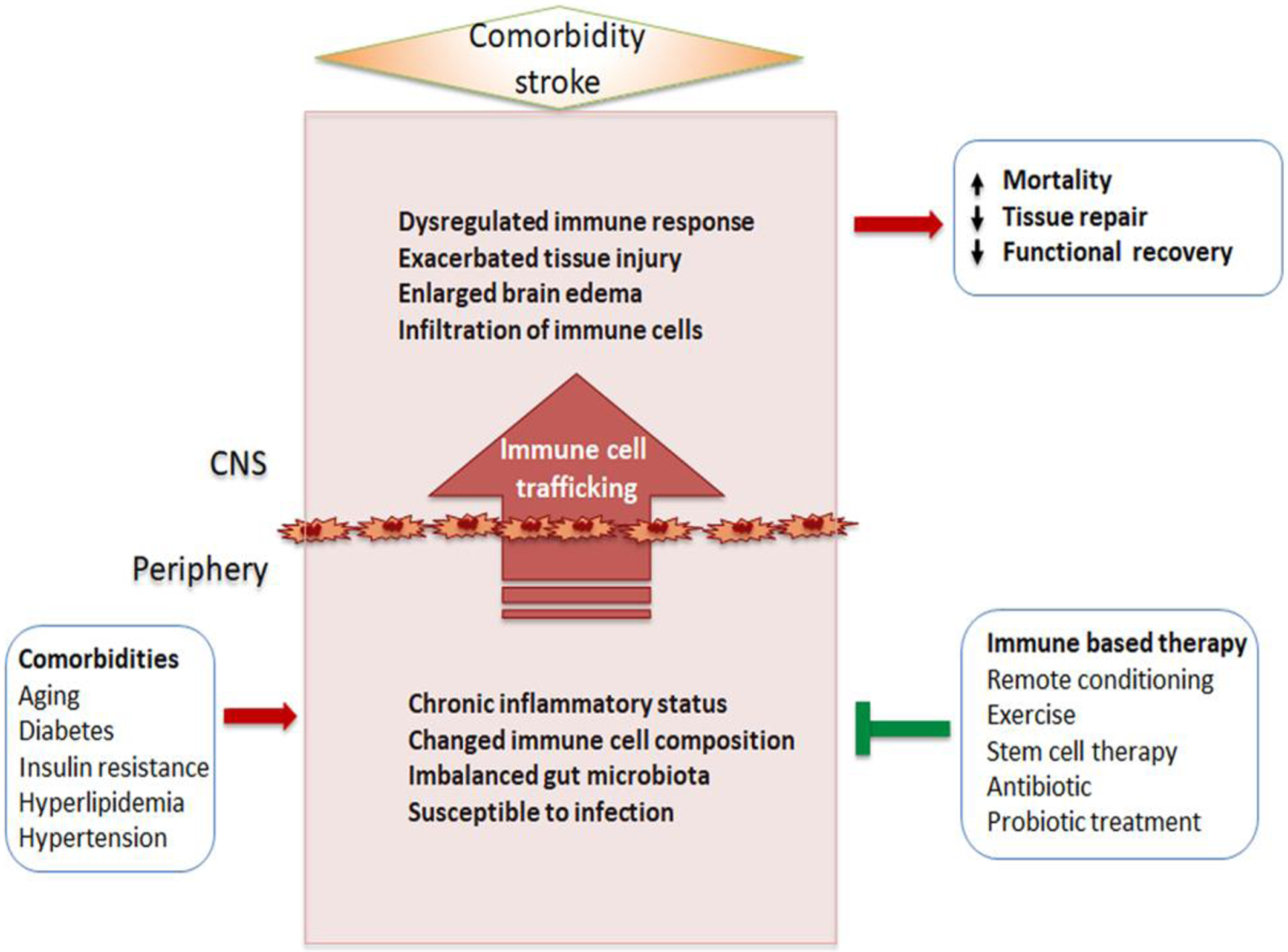
The impact of comorbidities in immune/inflammatory reaction in ischemic stroke. Comorbidities shared in cardio- and cerebrovascular diseases are often characterized by chronic low-grade inflammatory status in the periphery. The conditions alter immune cell composition and phenotypes. These comorbidity-primed immune cells enter into the CNS after stroke lead to impaired and inappropriate immune responses in the CNS and exacerbated stroke outcomes. Several immunomodulating strategies aim at the peripheral immune system are suggested to improve the acute injury and long-term recovery in the comorbid stroke.
Acknowledgements
This work was supported by NIH Grants NS095359 and NS111568 (SC). We thank Faariah Shakil for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adnan S et al. 2017. Alterations in the gut microbiota can elicit hypertension in rats. Physiol Genomics 49, 96–104. doi: 10.1152/physiolgenomics.00081.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH et al. 2000. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke 31, 193–199. doi: 10.1161/01.str.31.1.193. [DOI] [PubMed] [Google Scholar]
- Akhoundzadeh K et al. 2018. Effects of the Oral Ingestion of Probiotics on Brain Damage in a Transient Model of Focal Cerebral Ischemia in Mice. Iran J Med Sci 43, 32–40. [PMC free article] [PubMed] [Google Scholar]
- Ali A et al. 2020. NF-kappaB Inhibitors Attenuate MCAO Induced Neurodegeneration and Oxidative Stress-A Reprofiling Approach. Front Mol Neurosci 13, 33. doi: 10.3389/fnmol.2020.00033. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Amarenco P et al. 2006. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 355, 549–559. doi: 10.1056/NEJMoa061894. [DOI] [PubMed] [Google Scholar]
- Amiri-Nikpour MR et al. 2015. An open-label evaluator-blinded clinical study of minocycline neuroprotection in ischemic stroke: gender-dependent effect. Acta Neurol Scand 131, 45–50. doi: 10.1111/ane.12296. [DOI] [PubMed] [Google Scholar]
- Angeli V et al. 2004. Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity 21, 561–574. doi: 10.1016/j.immuni.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Arantes RM et al. 2000. Early damage of sympathetic neurons after co-culture with macrophages: a model of neuronal injury in vitro. Neuroreport 11, 177–181. doi: 10.1097/00001756-200001170-00035. [DOI] [PubMed] [Google Scholar]
- Arboix A et al. 2005. Cerebral infarction in diabetes: clinical pattern, stroke subtypes, and predictors of in-hospital mortality. BMC Neurol 5, 9. doi: 10.1186/1471-2377-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsava EM et al. 2009. Severity of leukoaraiosis correlates with clinical outcome after ischemic stroke. Neurology 72, 1403–1410. doi: 10.1212/WNL.0b013e3181a18823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanyan S et al. 2004. Pneumonia and urinary tract infection after acute ischaemic stroke: a tertiary analysis of the GAIN International trial. Eur J Neurol 11, 49–53. doi: 10.1046/j.1468-1331.2003.00749.x. [DOI] [PubMed] [Google Scholar]
- Aspelund A et al. 2015. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212, 991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacigaluppi M et al. 2009. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain 132, 2239–2251. doi: 10.1093/brain/awp174. [DOI] [PubMed] [Google Scholar]
- Banwell V et al. 2009. Systematic review and stratified meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke. J Stroke Cerebrovasc Dis 18, 269–276. doi: 10.1016/j.jstrokecerebrovasdis.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Barrat FJ et al. 2002. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med 195, 603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM 2008. A calculated response: control of inflammation by the innate immune system. J Clin Invest 118, 413–420. doi: 10.1172/JCI34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R et al. 2003. Prolonged infusion of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am J Physiol Gastrointest Liver Physiol 285, G642–651. doi: 10.1152/ajpgi.00037.2003. [DOI] [PubMed] [Google Scholar]
- Bauer P et al. 2000. Endothelial expression of selectins during endotoxin preconditioning. Am J Physiol Regul Integr Comp Physiol 279, R2015–2021. doi: 10.1152/ajpregu.2000.279.6.R2015. [DOI] [PubMed] [Google Scholar]
- Becker KJ et al. 2016. Antibodies to myelin basic protein are associated with cognitive decline after stroke. J Neuroimmunol 295–296, 9–11. doi: 10.1016/j.jneuroim.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beli E et al. 2011. Natural killer cell function is altered during the primary response of aged mice to influenza infection. Mech Ageing Dev 132, 503–510. doi: 10.1016/j.mad.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Naik S 2013. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol 14, 646–653. doi: 10.1038/ni.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C et al. 2016. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med 22, 516–523. doi: 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyacoub J et al. 2003. Supplementation of food with Enterococcus faecium (SF68) stimulates immune functions in young dogs. J Nutr 133, 1158–1162. doi: 10.1093/jn/133.4.1158. [DOI] [PubMed] [Google Scholar]
- Bercik P et al. 2011. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 141, 599–609, 609 e591–593. doi: 10.1053/j.gastro.2011.04.052. [DOI] [PubMed] [Google Scholar]
- Biagi E et al. 2010. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS One 5, e10667. doi: 10.1371/journal.pone.0010667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boje KM, Arora PK 1992. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res 587, 250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Boshuizen MCS, Steinberg GK 2018. Stem Cell-Based Immunomodulation After Stroke: Effects on Brain Repair Processes. Stroke 49, 1563–1570. doi: 10.1161/STROKEAHA.117.020465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskra D et al. 2008. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- Bradshaw EM et al. 2009. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol 183, 4432–4439. doi: 10.4049/jimmunol.0900576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands MW et al. 2010. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56, 879–884. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker AL et al. 2013. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol 190, 1746–1757. doi: 10.4049/jimmunol.1201213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck BH et al. 2008. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 39, 355–360. doi: 10.1161/STROKEAHA.107.490128. [DOI] [PubMed] [Google Scholar]
- Caligiuri G et al. 2002. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest 109, 745–753. doi: 10.1172/JCI7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara-Lemarroy CR et al. 2014. Gastrointestinal complications after ischemic stroke. J Neurol Sci 346, 20–25. doi: 10.1016/j.jns.2014.08.027. [DOI] [PubMed] [Google Scholar]
- Cao YL et al. 2016. Th1/Th2 cytokine expression in diabetic retinopathy. Genet Mol Res 15. doi: 10.4238/gmr.15037311. [DOI] [PubMed] [Google Scholar]
- Capone C et al. 2007. Neurosphere-derived cells exert a neuroprotective action by changing the ischemic microenvironment. PLoS One 2, e373. doi: 10.1371/journal.pone.0000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabotti M et al. 2015. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 28, 203–209. [PMC free article] [PubMed] [Google Scholar]
- Cauwe B et al. 2009. Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr Biol (Camb) 1, 404–426. doi: 10.1039/b904701h. [DOI] [PubMed] [Google Scholar]
- Chamorro A et al. 2007. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke 38, 1097–1103. doi: 10.1161/01.STR.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- Chapman KZ et al. 2009. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow Metab 29, 1764–1768. doi: 10.1038/jcbfm.2009.113. [DOI] [PubMed] [Google Scholar]
- Chen C et al. 2018. Splenic responses play an important role in remote ischemic preconditioning-mediated neuroprotection against stroke. J Neuroinflammation 15, 167. doi: 10.1186/s12974-018-1190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y et al. 2012. Intrastriatal B-cell administration limits infarct size after stroke in B-cell deficient mice. Metab Brain Dis 27, 487–493. doi: 10.1007/s11011-012-9317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH et al. 2011. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 35, 819–831. doi: 10.1016/j.immuni.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Christoffersson G et al. 2012. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 120, 4653–4662. doi: 10.1182/blood-2012-04-421040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HX et al. 2015. Evidence That Ly6C(hi) Monocytes are Protective in Acute Ischemic Stroke by Promoting M2 Macrophage Polarization. Stroke 46, 1929–1937. doi: 10.1161/STROKEAHA.115.009426. [DOI] [PubMed] [Google Scholar]
- Clausen BH et al. 2008. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation 5, 46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Gilbert DL 1987. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett 223, 284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- Cortez-Espinosa N et al. 2015. CD39 expression on Treg and Th17 cells is associated with metabolic factors in patients with type 2 diabetes. Hum Immunol 76, 622–630. doi: 10.1016/j.humimm.2015.09.007. [DOI] [PubMed] [Google Scholar]
- Costello EK et al. 2009. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotillard A et al. 2013. Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588. doi: 10.1038/nature12480. [DOI] [PubMed] [Google Scholar]
- Crapser J et al. 2016. Ischemic stroke induces gut permeability and enhances bacterial translocation leading to sepsis in aged mice. Aging (Albany NY) 8, 1049–1063. doi: 10.18632/aging.100952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuartero MI et al. 2013. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 44, 3498–3508. doi: 10.1161/STROKEAHA.113.002470. [DOI] [PubMed] [Google Scholar]
- Cybulsky MI et al. 1988. Acute inflammation and microthrombosis induced by endotoxin, interleukin-1, and tumor necrosis factor and their implication in gram-negative infection. Lab Invest 58, 365–378. [PubMed] [Google Scholar]
- Daadi MM et al. 2010. Human neural stem cell grafts modify microglial response and enhance axonal sprouting in neonatal hypoxic-ischemic brain injury. Stroke 41, 516–523. doi: 10.1161/STROKEAHA.109.573691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao MC et al. 2016. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65, 426–436. doi: 10.1136/gutjnl-2014-308778. [DOI] [PubMed] [Google Scholar]
- Darbousset R et al. 2012. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 120, 2133–2143. doi: 10.1182/blood-2012-06-437772. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A 2011. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 12, 388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Davis-Richardson AG et al. 2014. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol 5, 678. doi: 10.3389/fmicb.2014.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Geyter D et al. 2012. Spontaneously hypertensive rats display reduced microglial activation in response to ischemic stroke and lipopolysaccharide. J Neuroinflammation 9, 114. doi: 10.1186/1742-2094-9-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSouza CA et al. 1997. Elevated levels of circulating cell adhesion molecules in uncomplicated essential hypertension. Am J Hypertens 10, 1335–1341. doi: 10.1016/s0895-7061(97)00268-9. [DOI] [PubMed] [Google Scholar]
- Devaraj S et al. 2006. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes 55, 774–779. doi: 10.2337/diabetes.55.03.06.db05-1417. [DOI] [PubMed] [Google Scholar]
- DiNapoli VA et al. 2008. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging 29, 753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U et al. 2009. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 8, 398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divyashri G et al. 2015. Probiotic attributes, antioxidant, anti-inflammatory and neuromodulatory effects of Enterococcus faecium CFR 3003: in vitro and in vivo evidence. J Med Microbiol 64, 1527–1540. doi: 10.1099/jmm.0.000184. [DOI] [PubMed] [Google Scholar]
- Dong XH et al. 2017. Chronic Exposure to Subtherapeutic Antibiotics Aggravates Ischemic Stroke Outcome in Mice. EBioMedicine 24, 116–126. doi: 10.1016/j.ebiom.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorffel Y et al. 1999. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension 34, 113–117. doi: 10.1161/01.hyp.34.1.113. [DOI] [PubMed] [Google Scholar]
- Doyle KP et al. 2010. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation 7, 62. doi: 10.1186/1742-2094-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle KP et al. 2015. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci 35, 2133–2145. doi: 10.1523/JNEUROSCI.4098-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake C et al. 2011. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav Immun 25, 1113–1122. doi: 10.1016/j.bbi.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducruet AF, Connolly ES Jr. 2011. Targeting lymphocytes in ischemic stroke. World Neurosurg 76, 212–214. doi: 10.1016/j.wneu.2011.06.060. [DOI] [PubMed] [Google Scholar]
- Durgan DJ et al. 2016. Role of the Gut Microbiome in Obstructive Sleep Apnea-Induced Hypertension. Hypertension 67, 469–474. doi: 10.1161/HYPERTENSIONAHA.115.06672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElAli A et al. 2011. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke 42, 3238–3244. doi: 10.1161/STROKEAHA.111.615559. [DOI] [PubMed] [Google Scholar]
- Elliott MR, Ravichandran KS 2010. Clearance of apoptotic cells: implications in health and disease. J Cell Biol 189, 1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerich DF et al. 2002. The role of leukocytes following cerebral ischemia: pathogenic variable or bystander reaction to emerging infarct? Exp Neurol 173, 168–181. doi: 10.1006/exnr.2001.7835. [DOI] [PubMed] [Google Scholar]
- Emsley HC et al. 2005. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry 76, 1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enlimomab Acute Stroke Trial I. 2001. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 57, 1428–1434. doi: 10.1212/wnl.57.8.1428. [DOI] [PubMed] [Google Scholar]
- Faustino JV et al. 2011. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci 31, 12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floden AM, Combs CK 2011. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J Alzheimers Dis 25, 279–293. doi: 10.3233/JAD-2011-101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca D et al. 2004. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J Immunol 172, 2155–2162. doi: 10.4049/jimmunol.172.4.2155. [DOI] [PubMed] [Google Scholar]
- Frasca D et al. 2016. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine 34, 2834–2840. doi: 10.1016/j.vaccine.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlender ZG et al. 2009. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J et al. 2015. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ Res 117, 817–824. doi: 10.1161/CIRCRESAHA.115.306807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S et al. 2015. The ischemic environment drives microglia and macrophage function. Front Neurol 6, 81. doi: 10.3389/fneur.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fury W et al. 2020. Sustained increases in immune gene transcripts and immune cell trafficking during recovery after experimental brain ischemia. Stroke. doi: 10.1161/STROKEAHA.120.029440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y et al. 2014. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A 111, 2704–2709. doi: 10.1073/pnas.1315943111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganta CK et al. 2005. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol 289, H1683–1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- Garcia-Bonilla L et al. 2014. Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J Immunol 193, 2531–2537. doi: 10.4049/jimmunol.1400918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bonilla L et al. 2018. Endogenous Protection from Ischemic Brain Injury by Preconditioned Monocytes. J Neurosci 38, 6722–6736. doi: 10.1523/JNEUROSCI.0324-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia GG, Miller RA 2011. Age-related defects in the cytoskeleton signaling pathways of CD4 T cells. Ageing Res Rev 10, 26–34. doi: 10.1016/j.arr.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M et al. 2009. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Gelderblom M et al. 2012. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 120, 3793–3802. doi: 10.1182/blood-2012-02-412726. [DOI] [PubMed] [Google Scholar]
- Gidday JM et al. 2005. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 289, H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gill D, Veltkamp R 2016. Dynamics of T cell responses after stroke. Curr Opin Pharmacol 26, 26–32. doi: 10.1016/j.coph.2015.09.009. [DOI] [PubMed] [Google Scholar]
- Gleeson M, Bishop NC 2005. The T cell and NK cell immune response to exercise. Ann Transplant 10, 43–48. [PubMed] [Google Scholar]
- Gliem M et al. 2015. Hyperglycemia and PPARgamma Antagonistically Influence Macrophage Polarization and Infarct Healing After Ischemic Stroke. Stroke 46, 2935–2942. doi: 10.1161/STROKEAHA.115.010557. [DOI] [PubMed] [Google Scholar]
- Gomez-Guzman M et al. 2015. Antihypertensive effects of probiotics Lactobacillus strains in spontaneously hypertensive rats. Mol Nutr Food Res 59, 2326–2336. doi: 10.1002/mnfr.201500290. [DOI] [PubMed] [Google Scholar]
- Grassivaro F et al. 2020. Convergence between Microglia and Peripheral Macrophages Phenotype during Development and Neuroinflammation. J Neurosci 40, 784–795. doi: 10.1523/JNEUROSCI.1523-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh AD et al. 2018. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol 16, e2005264. doi: 10.1371/journal.pbio.2005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JD, Lichtenstein AH 2013. Dietary Cholesterol and Plasma Lipoprotein Profiles: Randomized-Controlled Trials. Curr Nutr Rep 2, 274–282. doi: 10.1007/s13668-013-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara S et al. 2014. Desulfovibrio desulfuricans bacteremia in a patient hospitalized with acute cerebral infarction: case report and review. J Infect Chemother 20, 274–277. doi: 10.1016/j.jiac.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Harms H et al. 2008. Preventive antibacterial therapy in acute ischemic stroke: a randomized controlled trial. PLoS One 3, e2158. doi: 10.1371/journal.pone.0002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K et al. 2016. CD200 restrains macrophage attack on oligodendrocyte precursors via toll-like receptor 4 downregulation. J Cereb Blood Flow Metab 36, 781–793. doi: 10.1177/0271678X15606148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes L et al. 1997. Age-related changes in CD4 T cells of T cell receptor transgenic mice. Mech Ageing Dev 93, 95–105. doi: 10.1016/s0047-6374(96)01826-x. [DOI] [PubMed] [Google Scholar]
- Hazra S et al. 2013. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia 56, 644–653. doi: 10.1007/s00125-012-2781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi B et al. 2014. Regulation of mouse microglia activation and effector functions by bone marrow-derived mesenchymal stem cells. Stem Cells Dev 23, 2600–2612. doi: 10.1089/scd.2014.0088. [DOI] [PubMed] [Google Scholar]
- Herold M et al. 2017. Liver X receptor activation promotes differentiation of regulatory T cells. PLoS One 12, e0184985. doi: 10.1371/journal.pone.0184985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetze S et al. 2013. Superiority of preventive antibiotic treatment compared with standard treatment of poststroke pneumonia in experimental stroke: a bed to bench approach. J Cereb Blood Flow Metab 33, 846–854. doi: 10.1038/jcbfm.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF et al. 1991. T-lymphocyte entry into the central nervous system. J Neurosci Res 28, 254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Hoehn BD et al. 2005. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke 36, 2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- Hopkins MJ, Macfarlane GT 2002. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J Med Microbiol 51, 448–454. doi: 10.1099/0022-1317-51-5-448. [DOI] [PubMed] [Google Scholar]
- Howell JA, Bidwell GL 3rd 2020. Targeting the NF-kappaB pathway for therapy of ischemic stroke. Ther Deliv 11, 113–123. doi: 10.4155/tde-2019-0075. [DOI] [PubMed] [Google Scholar]
- Hu X et al. 2017. Cerebral Vascular Disease and Neurovascular Injury in Ischemic Stroke. Circ Res 120, 449–471. doi: 10.1161/CIRCRESAHA.116.308427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X et al. 2012. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Davisson RL 2008. Hypertension and cerebrovascular dysfunction. Cell Metab 7, 476–484. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J 2011. The immunology of stroke: from mechanisms to translation. Nat Med 17, 796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi N, Yamazaki S 2001. Probiotics and safety. Am J Clin Nutr 73, 465S–470S. doi: 10.1093/ajcn/73.2.465s. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Honda K 2012. Intestinal commensal microbes as immune modulators. Cell Host Microbe 12, 496–508. doi: 10.1016/j.chom.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraj RL et al. 2019. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation 16, 142. doi: 10.1186/s12974-019-1516-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q et al. 2005. Investigation of neural progenitor cell induced angiogenesis after embolic stroke in rat using MRI. Neuroimage 28, 698–707. doi: 10.1016/j.neuroimage.2005.06.063. [DOI] [PubMed] [Google Scholar]
- Jickling GC et al. 2015. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 35, 888–901. doi: 10.1038/jcbfm.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Conde J et al. 2010. Hyperlipidemia and reduced white matter hyperintensity volume in patients with ischemic stroke. Stroke 41, 437–442. doi: 10.1161/STROKEAHA.109.563502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadl A et al. 2010. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res 107, 737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser D et al. 2014. Spontaneous white matter damage, cognitive decline and neuroinflammation in middle-aged hypertensive rats: an animal model of early-stage cerebral small vessel disease. Acta Neuropathol Commun 2, 169. doi: 10.1186/s40478-014-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra L et al. 2015. Prophylactic antibiotics after acute stroke for reducing pneumonia in patients with dysphagia (STROKE-INF): a prospective, cluster-randomised, open-label, masked endpoint, controlled clinical trial. Lancet 386, 1835–1844. doi: 10.1016/S0140-6736(15)00126-9. [DOI] [PubMed] [Google Scholar]
- Kang Y, Cai Y 2018. Gut microbiota and hypertension: From pathogenesis to new therapeutic strategies. Clin Res Hepatol Gastroenterol 42, 110–117. doi: 10.1016/j.clinre.2017.09.006. [DOI] [PubMed] [Google Scholar]
- Kasahara K et al. 2018. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol 3, 1461–1471. doi: 10.1038/s41564-018-0272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K et al. 2017. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J Lipid Res 58, 519–528. doi: 10.1194/jlr.M072165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S et al. 1993. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol 19, 480–488. doi: 10.1111/j.1365-2990.1993.tb00476.x. [DOI] [PubMed] [Google Scholar]
- Kim CH, Vaziri ND 2005. Hypertension promotes integrin expression and reactive oxygen species generation by circulating leukocytes. Kidney Int 67, 1462–1470. doi: 10.1111/j.1523-1755.2005.00223.x. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho S 2016. Microglia and Monocyte-Derived Macrophages in Stroke. Neurotherapeutics 13, 702–718. doi: 10.1007/s13311-016-0463-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E et al. 2014a. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation 11, 83. doi: 10.1186/1742-2094-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E et al. 2014b. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab 34, 1411–1419. doi: 10.1038/jcbfm.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E et al. 2008a. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia-induced exacerbation in ischemic brain injury. J Neurosci 28, 4661–4670. doi: 10.1523/JNEUROSCI.0982-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E et al. 2012. CD36 in the periphery and brain synergizes in stroke injury in hyperlipidemia. Ann Neurol 71, 753–764. doi: 10.1002/ana.23569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hematti P 2009. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol 37, 1445–1453. doi: 10.1016/j.exphem.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KI et al. 2008b. Association between blood pressure variability and inflammatory marker in hypertensive patients. Circ J 72, 293–298. doi: 10.1253/circj.72.293. [DOI] [PubMed] [Google Scholar]
- Kim YH et al. 2014c. Effect of remote ischemic post-conditioning on systemic inflammatory response and survival rate in lipopolysaccharide-induced systemic inflammation model. J Inflamm (Lond) 11, 16. doi: 10.1186/1476-9255-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura D et al. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350, 423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C et al. 2010. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 115, 3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C et al. 2013. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood 121, 679–691. doi: 10.1182/blood-2012-04-426734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler E et al. 2013. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta-analysis. Stroke 44, 2493–2499. doi: 10.1161/STROKEAHA.113.000780. [DOI] [PubMed] [Google Scholar]
- Kohn FR, Kung AH 1995. Role of endotoxin in acute inflammation induced by gram-negative bacteria: specific inhibition of lipopolysaccharide-mediated responses with an amino-terminal fragment of bactericidal/permeability-increasing protein. Infect Immun 63, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltsova EK et al. 2012. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest 122, 3114–3126. doi: 10.1172/JCI61758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinov IE et al. 2004. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics 19, 143–150. doi: 10.1152/physiolgenomics.00046.2004. [DOI] [PubMed] [Google Scholar]
- Korin B et al. 2017. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci 20, 1300–1309. doi: 10.1038/nn.4610. [DOI] [PubMed] [Google Scholar]
- Kossmann S et al. 2013. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol 33, 1313–1319. doi: 10.1161/ATVBAHA.113.301437. [DOI] [PubMed] [Google Scholar]
- Krause M et al. 2019. Cell-Based Therapies for Stroke: Are We There Yet? Front Neurol 10, 656. doi: 10.3389/fneur.2019.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A et al. 2007. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J 26, 4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AD et al. 2013. Leukocytosis in patients with neurologic deterioration after acute ischemic stroke is associated with poor outcomes. J Stroke Cerebrovasc Dis 22, e111–117. doi: 10.1016/j.jstrokecerebrovasdis.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R et al. 2007. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab 27, 710–718. doi: 10.1038/sj.jcbfm.9600382. [DOI] [PubMed] [Google Scholar]
- Kvakan H et al. 2009. Role of the immune system in hypertensive target organ damage. Trends Cardiovasc Med 19, 242–246. doi: 10.1016/j.tcm.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Lakshminarayanan B et al. 2014. Compositional dynamics of the human intestinal microbiota with aging: implications for health. J Nutr Health Aging 18, 773–786. doi:10.1007/s12603-014-0513-510.1007/s12603-014-0513-510.1007/s12603-014-0549-610.1007/s12603-014-0549-6 . [DOI] [PubMed] [Google Scholar]
- Lancaster GI, Febbraio MA 2014. The immunomodulating role of exercise in metabolic disease. Trends Immunol 35, 262–269. doi: 10.1016/j.it.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Le Roy T et al. 2019. The intestinal microbiota regulates host cholesterol homeostasis. BMC Biol 17, 94. doi: 10.1186/s12915-019-0715-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine DA et al. 2015. Trajectory of Cognitive Decline After Incident Stroke. JAMA 314, 41–51. doi: 10.1001/jama.2015.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MJ et al. 2009. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 120, 417–426. doi: 10.1161/CIRCULATIONAHA.109.868158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL et al. 2001. IL-17 and IFN-gamma mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol 116, 5–14. doi: 10.1016/s0165-5728(01)00264-8. [DOI] [PubMed] [Google Scholar]
- Li J et al. 2017. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 5, 14. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P et al. 2014. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke 45, 857–864. doi: 10.1161/STROKEAHA.113.004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W et al. 2012. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ 19, 1505–1513. doi: 10.1038/cdd.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X et al. 2018. Intravenously Delivered Allogeneic Mesenchymal Stem Cells Bidirectionally Regulate Inflammation and Induce Neurotrophic Effects in Distal Middle Cerebral Artery Occlusion Rats Within the First 7 Days After Stroke. Cell Physiol Biochem 46, 1951–1970. doi: 10.1159/000489384. [DOI] [PubMed] [Google Scholar]
- Liang S et al. 2009. Age-related alterations in innate immune receptor expression and ability of macrophages to respond to pathogen challenge in vitro. Mech Ageing Dev 130, 538–546. doi: 10.1016/j.mad.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S et al. 2015. Administration of Lactobacillus helveticus NS8 improves behavioral, cognitive, and biochemical aberrations caused by chronic restraint stress. Neuroscience 310, 561–577. doi: 10.1016/j.neuroscience.2015.09.033. [DOI] [PubMed] [Google Scholar]
- Liao JK 2002. Beyond lipid lowering: the role of statins in vascular protection. Int J Cardiol 86, 5–18. doi: 10.1016/s0167-5273(02)00195-x. [DOI] [PubMed] [Google Scholar]
- Liesz A, Kleinschnitz C 2016. Regulatory T Cells in Post-stroke Immune Homeostasis. Transl Stroke Res 7, 313–321. doi: 10.1007/s12975-016-0465-7. [DOI] [PubMed] [Google Scholar]
- Liesz A et al. 2013. Spectratype analysis of clonal T cell expansion in murine experimental stroke. J Neuroimmunol 257, 46–52. doi: 10.1016/j.jneuroim.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Liesz A et al. 2015. Functional role of regulatory lymphocytes in stroke: facts and controversies. Stroke 46, 1422–1430. doi: 10.1161/STROKEAHA.114.008608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A et al. 2009. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15, 192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liesz A et al. 2011. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 134, 704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Lin D et al. 2019. The Role of Oxidative Stress in Common Risk Factors and Mechanisms of Cardio-Cerebrovascular Ischemia and Depression. Oxid Med Cell Longev 2019, 2491927. doi: 10.1155/2019/2491927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HY et al. 2009. Lipopolysaccharide preconditioning reduces neuroinflammation against hypoxic ischemia and provides long-term outcome of neuroprotection in neonatal rat. Pediatr Res 66, 254–259. doi: 10.1203/PDR.0b013e3181b0d336. [DOI] [PubMed] [Google Scholar]
- Lindskog Jonsson A et al. 2018. Impact of Gut Microbiota and Diet on the Development of Atherosclerosis in Apoe(−/−) Mice. Arterioscler Thromb Vasc Biol 38, 2318–2326. doi: 10.1161/ATVBAHA.118.311233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liong MT, Shah NP 2005. Acid and bile tolerance and cholesterol removal ability of lactobacilli strains. J Dairy Sci 88, 55–66. doi: 10.3168/jds.S0022-0302(05)72662-X. [DOI] [PubMed] [Google Scholar]
- Lipski J et al. 2007. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience 146, 617–629. doi: 10.1016/j.neuroscience.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Lipton P 1999. Ischemic cell death in brain neurons. Physiol Rev 79, 1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu C et al. 2019. The changes of systemic immune responses during the neuroprotection induced by remote ischemic postconditioning against focal cerebral ischemia in mice. Neurol Res 41, 26–36. doi: 10.1080/01616412.2018.1523037. [DOI] [PubMed] [Google Scholar]
- Liu F et al. 2010a. Expression of Na-K-Cl cotransporter and edema formation are age dependent after ischemic stroke. Exp Neurol 224, 356–361. doi: 10.1016/j.expneurol.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F et al. 2012. Age-related changes in AMP-activated protein kinase after stroke. Age (Dordr) 34, 157–168. doi: 10.1007/s11357-011-9214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N et al. 2009. Expression of IL-10 and TNF-alpha in rats with cerebral infarction after transplantation with mesenchymal stem cells. Cell Mol Immunol 6, 207–213. doi: 10.1038/cmi.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q et al. 2017. Brain Ischemia Suppresses Immunity in the Periphery and Brain via Different Neurogenic Innervations. Immunity 46, 474–487. doi: 10.1016/j.immuni.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Liu X et al. 2016a. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke 47, 498–504. doi: 10.1161/STROKEAHA.115.012079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z et al. 2007. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 38, 146–152. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- Liu Z et al. 2010b. Bone marrow stromal cells enhance inter- and intracortical axonal connections after ischemic stroke in adult rats. J Cereb Blood Flow Metab 30, 1288–1295. doi: 10.1038/jcbfm.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZJ et al. 2016b. Remote Ischemic Preconditioning-Mediated Neuroprotection against Stroke is Associated with Significant Alterations in Peripheral Immune Responses. CNS Neurosci Ther 22, 43–52. doi: 10.1111/cns.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo TF et al. 2018. Impaired Treg and NK cells profile in overweight women with gestational diabetes mellitus. Am J Reprod Immunol 79. doi: 10.1111/aji.12810. [DOI] [PubMed] [Google Scholar]
- Louveau A et al. 2015. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN et al. 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117, 175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J et al. 2014. Ingestion of Lactobacillus strain reduces anxiety and improves cognitive function in the hyperammonemia rat. Sci China Life Sci 57, 327–335. doi: 10.1007/s11427-014-4615-4. [DOI] [PubMed] [Google Scholar]
- Maganto-Garcia E et al. 2011. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 124, 185–195. doi: 10.1161/CIRCULATIONAHA.110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major AS et al. 2002. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol 22, 1892–1898. doi: 10.1161/01.atv.0000039169.47943.ee. [DOI] [PubMed] [Google Scholar]
- Manwani B et al. 2013. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp Neurol 249, 120–131. doi: 10.1016/j.expneurol.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh B et al. 2009a. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci 29, 9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ et al. 2009b. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 158, 1007–1020. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P 2002. The danger model: a renewed sense of self. Science 296, 301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Maynard CL et al. 2012. Reciprocal interactions of the intestinal microbiota and immune system. Nature 489, 231–241. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK et al. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Meisel C et al. 2005. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci 6, 775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Mell B et al. 2015. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol Genomics 47, 187–197. doi: 10.1152/physiolgenomics.00136.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mente A et al. 2017. Association of dietary nutrients with blood lipids and blood pressure in 18 countries: a cross-sectional analysis from the PURE study. Lancet Diabetes Endocrinol 5, 774–787. doi: 10.1016/S2213-8587(17)30283-8. [DOI] [PubMed] [Google Scholar]
- Mishiro K et al. 2014. Diabetes mellitus aggravates hemorrhagic transformation after ischemic stroke via mitochondrial defects leading to endothelial apoptosis. PLoS One 9, e103818. doi: 10.1371/journal.pone.0103818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller K et al. 2015. Arterial Hypertension Aggravates Innate Immune Responses after Experimental Stroke. Front Cell Neurosci 9, 461. doi: 10.3389/fncel.2015.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP et al. 2015. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol 309, H906–917. doi: 10.1152/ajpheart.00821.2014. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP 2008. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8, 958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mracsko E et al. 2014a. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun 41, 200–209. doi: 10.1016/j.bbi.2014.05.015. [DOI] [PubMed] [Google Scholar]
- Mracsko E et al. 2014b. Antigen dependently activated cluster of differentiation 8-positive T cells cause perforin-mediated neurotoxicity in experimental stroke. J Neurosci 34, 16784–16795. doi: 10.1523/JNEUROSCI.1867-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ et al. 2011. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest 121, 4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EF et al. 2010. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59, 1635–1642. doi: 10.1136/gut.2010.215665. [DOI] [PubMed] [Google Scholar]
- Murray PJ et al. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MA et al. 2003. Dietary microbial toxins and type 1 diabetes. Ann N Y Acad Sci 1005, 418–422. doi: 10.1196/annals.1288.071. [DOI] [PubMed] [Google Scholar]
- Na SY et al. 2015. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke 46, 212–220. doi: 10.1161/STROKEAHA.114.007756. [DOI] [PubMed] [Google Scholar]
- Neal MD et al. 2006. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol 176, 3070–3079. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- Neher JJ et al. 2013. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 110, E4098–4107. doi: 10.1073/pnas.1308679110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekoua MP et al. 2016. Modulation of immune cells and Th1/Th2 cytokines in insulin-treated type 2 diabetes mellitus. Afr Health Sci 16, 712–724. doi: 10.4314/ahs.v16i3.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H et al. 2009. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J et al. 2015. Very-late-antigen-4 (VLA-4)-mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropathol 129, 259–277. doi: 10.1007/s00401-014-1355-2. [DOI] [PubMed] [Google Scholar]
- Newton AH, Benedict SH 2014. Low density lipoprotein promotes human naive T cell differentiation to Th1 cells. Hum Immunol 75, 621–628. doi: 10.1016/j.humimm.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Njie EG et al. 2012. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging 33, 195 e191–112. doi: 10.1016/j.neurobiolaging.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris GT, Kipnis J 2019. Immune cells and CNS physiology: Microglia and beyond. J Exp Med 216, 60–70. doi: 10.1084/jem.20180199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi A et al. 2004. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 35, 987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- Nyugen J et al. 2010. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol 30, 806–813. doi: 10.1007/s10875-010-9448-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell MJ et al. 2010. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 376, 112–123. doi: 10.1016/S0140-6736(10)60834-3. [DOI] [PubMed] [Google Scholar]
- O’Toole PW, Jeffery IB 2015. Gut microbiota and aging. Science 350, 1214–1215. doi: 10.1126/science.aac8469. [DOI] [PubMed] [Google Scholar]
- Odamaki T et al. 2016. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol 16, 90. doi: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI et al. 2007. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H et al. 2006a. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab 26, 654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H et al. 2006b. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol 176, 6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Ohtaki H et al. 2008. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc Natl Acad Sci U S A 105, 14638–14643. doi: 10.1073/pnas.0803670105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK 2010. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72, 219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Ortega SB et al. 2020. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc Natl Acad Sci U S A 117, 4983–4993. doi: 10.1073/pnas.1913292117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otxoa-de-Amezaga A et al. 2019. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol 137, 321–341. doi: 10.1007/s00401-018-1954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamer EG 2007. Immune responses to commensal and environmental microbes. Nat Immunol 8, 1173–1178. doi: 10.1038/ni1526. [DOI] [PubMed] [Google Scholar]
- Panda A et al. 2010. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol 184, 2518–2527. doi: 10.4049/jimmunol.0901022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendlebury ST, Rothwell PM 2009. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol 8, 1006–1018. doi: 10.1016/S1474-4422(09)70236-4. [DOI] [PubMed] [Google Scholar]
- Pinto A et al. 2004. Cerebrovascular risk factors and clinical classification of strokes. Semin Vasc Med 4, 287–303. doi: 10.1055/s-2004-861497. [DOI] [PubMed] [Google Scholar]
- Poggi M et al. 2007. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50, 1267–1276. doi: 10.1007/s00125-007-0654-8. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A et al. 2007. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol 113, 277–293. doi: 10.1007/s00401-006-0164-7. [DOI] [PubMed] [Google Scholar]
- Posel C et al. 2016. Isolation and Flow Cytometric Analysis of Immune Cells from the Ischemic Mouse Brain. J Vis Exp 53658. doi: 10.3791/53658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prass K et al. 2006. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke 37, 2607–2612. doi: 10.1161/01.STR.0000240409.68739.2b. [DOI] [PubMed] [Google Scholar]
- Prass K et al. 2003. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med 198, 725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruss H et al. 2012. Evidence of intrathecal immunoglobulin synthesis in stroke: a cohort study. Arch Neurol 69, 714–717. doi: 10.1001/archneurol.2011.3252. [DOI] [PubMed] [Google Scholar]
- Puellmann K et al. 2006. A variable immunoreceptor in a subpopulation of human neutrophils. Proc Natl Acad Sci U S A 103, 14441–14446. doi: 10.1073/pnas.0603406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmati H et al. 2019. Probiotic supplementation attenuates hippocampus injury and spatial learning and memory impairments in a cerebral hypoperfusion mouse model. Mol Biol Rep 46, 4985–4995. doi: 10.1007/s11033-019-04949-7. [DOI] [PubMed] [Google Scholar]
- Rampelli S et al. 2013. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging (Albany NY) 5, 902–912. doi: 10.18632/aging.100623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PS et al. 2014. Effects of ceftriaxone on systemic and central expression of anti- and pro-inflammatory cytokines in alcohol-preferring (P) rats exposed to ethanol. Alcohol Alcohol 49, 689. doi: 10.1093/alcalc/agu071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea MC et al. 2012. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol 50, 867–875. doi: 10.1128/JCM.05176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X et al. 2011. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 31, 8556–8563. doi: 10.1523/JNEUROSCI.1623-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo L et al. 2009. Impact of hyperlipidemia and statins on ischemic stroke outcomes after intra-arterial fibrinolysis and percutaneous mechanical embolectomy. Cerebrovasc Dis 28, 384–390. doi: 10.1159/000235625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios-Covian D et al. 2016. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front Microbiol 7, 185. doi: 10.3389/fmicb.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter L et al. 2011. Exaggerated neutrophil-mediated reperfusion injury after ischemic stroke in a rodent model of type 2 diabetes. Microcirculation 18, 552–561. doi: 10.1111/j.1549-8719.2011.00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM et al. 2018. Aging alters the immunological response to ischemic stroke. Acta Neuropathol 136, 89–110. doi: 10.1007/s00401-018-1859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roufaiel M et al. 2016. CCL19-CCR7-dependent reverse transendothelial migration of myeloid cells clears Chlamydia muridarum from the arterial intima. Nat Immunol 17, 1263–1272. doi: 10.1038/ni.3564. [DOI] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK 2009. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9, 313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhnau J et al. 2014. Stroke alters respiratory burst in neutrophils and monocytes. Stroke 45, 794–800. doi: 10.1161/STROKEAHA.113.003342. [DOI] [PubMed] [Google Scholar]
- Scher JU et al. 2013. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Herbruggen O et al. 2009. Differential affection of intestinal immune cell populations after cerebral ischemia in mice. Neuroimmunomodulation 16, 213–218. doi: 10.1159/000205514. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J 2011. A conceptual revolution in the relationships between the brain and immunity. Brain Behav Immun 25, 817–819. doi: 10.1016/j.bbi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz S et al. 2008. Effects of prophylactic antibiotic therapy with mezlocillin plus sulbactam on the incidence and height of fever after severe acute ischemic stroke: the Mannheim infection in stroke study (MISS). Stroke 39, 1220–1227. doi: 10.1161/STROKEAHA.107.499533. [DOI] [PubMed] [Google Scholar]
- Seijkens T et al. 2014. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J 28, 2202–2213. doi: 10.1096/fj.13-243105. [DOI] [PubMed] [Google Scholar]
- Seki Y et al. 2010. Suppressed neutrophil respiratory burst in patients with haemorrhagic stroke. J Clin Neurosci 17, 187–190. doi: 10.1016/j.jocn.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Sheikh AM et al. 2011. Mesenchymal stem cell transplantation modulates neuroinflammation in focal cerebral ischemia: contribution of fractalkine and IL-5. Neurobiol Dis 41, 717–724. doi: 10.1016/j.nbd.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Shen LH et al. 2006. Intracarotid transplantation of bone marrow stromal cells increases axon-myelin remodeling after stroke. Neuroscience 137, 393–399. doi: 10.1016/j.neuroscience.2005.08.092. [DOI] [PubMed] [Google Scholar]
- Shen XZ et al. 2015. Microglia participate in neurogenic regulation of hypertension. Hypertension 66, 309–316. doi: 10.1161/HYPERTENSIONAHA.115.05333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi FD et al. 2011. Organ-specific features of natural killer cells. Nat Rev Immunol 11, 658–671. doi: 10.1038/nri3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T et al. 2009. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med 15, 946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Shigematsu K et al. 2015. Influences of hyperlipidemia history on stroke outcome; a retrospective cohort study based on the Kyoto Stroke Registry. BMC Neurol 15, 44. doi: 10.1186/s12883-015-0297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim R, Wong CH 2016. Ischemia, Immunosuppression and Infection--Tackling the Predicaments of Post-Stroke Complications. Int J Mol Sci 17. doi: 10.3390/ijms17010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M et al. 2010. Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J Surg Res 158, 155–161. doi: 10.1016/j.jss.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Sieber MW et al. 2011. Attenuated inflammatory response in aged mice brains following stroke. PLoS One 6, e26288. doi: 10.1371/journal.pone.0026288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simar D et al. 2014. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism 63, 1188–1197. doi: 10.1016/j.metabol.2014.05.014. [DOI] [PubMed] [Google Scholar]
- Smith CJ et al. 2018. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke 49, 1210–1216. doi: 10.1161/STROKEAHA.118.020750. [DOI] [PubMed] [Google Scholar]
- Smith RM et al. 2002. Innate immunity and cardiac preconditioning: a putative intrinsic cardioprotective program. Cardiovasc Res 55, 474–482. doi: 10.1016/s0008-6363(02)00288-2. [DOI] [PubMed] [Google Scholar]
- Smithey MJ et al. 2011. Increased apoptosis, curtailed expansion and incomplete differentiation of CD8+ T cells combine to decrease clearance of L. monocytogenes in old mice. Eur J Immunol 41, 1352–1364. doi: 10.1002/eji.201041141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C et al. 2016. Expansion of brain T cells in homeostatic conditions in lymphopenic Rag2(−/−) mice. Brain Behav Immun 57, 161–172. doi: 10.1016/j.bbi.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyucen E et al. 2014. Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatr Int 56, 336–343. doi: 10.1111/ped.12243. [DOI] [PubMed] [Google Scholar]
- Spychala MS et al. 2018. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann Neurol 84, 23–36. doi: 10.1002/ana.25250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL et al. 2002. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res 932, 110–119. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- Subramanian S et al. 2009. Recombinant T cell receptor ligand treats experimental stroke. Stroke 40, 2539–2545. doi: 10.1161/STROKEAHA.108.543991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J et al. 2016. Clostridium butyricum pretreatment attenuates cerebral ischemia/reperfusion injury in mice via anti-oxidation and anti-apoptosis. Neurosci Lett 613, 30–35. doi: 10.1016/j.neulet.2015.12.047. [DOI] [PubMed] [Google Scholar]
- Sun P et al. 2010. Effects of Enterococcus faecium (SF68) on immune function in mice. Food Chemistry 123, 63–68. doi:doi: 10.1016/j.foodchem.2010.03.128. [DOI] [Google Scholar]
- Swirski FK et al. 2007. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 117, 195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK et al. 2009. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay G et al. 2016. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7, 11499. doi: 10.1038/ncomms11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A et al. 2004. Administration of CD34+ cells after stroke enhances neurogenesis via angiogenesis in a mouse model. J Clin Invest 114, 330–338. doi: 10.1172/JCI20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman NS et al. 1997. Human neutrophils secrete vascular endothelial growth factor. J Leukoc Biol 62, 397–400. doi: 10.1002/jlb.62.3.397. [DOI] [PubMed] [Google Scholar]
- Tan M et al. 2011. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care 15, R290. doi: 10.1186/cc10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasat DR et al. 2003. Age-dependent change in reactive oxygen species and nitric oxide generation by rat alveolar macrophages. Aging Cell 2, 159–164. doi: 10.1046/j.1474-9728.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- Thompson HL et al. 2017. Functional and Homeostatic Impact of Age-Related Changes in Lymph Node Stroma. Front Immunol 8, 706. doi: 10.3389/fimmu.2017.00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting SM et al. 2020. Brain Cleanup as a Potential Target for Poststroke Recovery: The Role of RXR (Retinoic X Receptor) in Phagocytes. Stroke 51, 958–966. doi: 10.1161/STROKEAHA.119.027315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton CM 2014. The history of “Exercise Is Medicine” in ancient civilizations. Adv Physiol Educ 38, 109–117. doi: 10.1152/advan.00136.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin MK et al. 2020. Activated Mesenchymal Stem Cells Induce Recovery Following Stroke Via Regulation of Inflammation and Oligodendrogenesis. J Am Heart Assoc 9, e013583. doi: 10.1161/JAHA.119.013583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MJ et al. 2014. Recovery of neurological function of ischemic stroke by application of conditioned medium of bone marrow mesenchymal stem cells derived from normal and cerebral ischemia rats. J Biomed Sci 21, 5. doi: 10.1186/1423-0127-21-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H et al. 2010. Bim dictates naive CD4 T cell lifespan and the development of age-associated functional defects. J Immunol 185, 4535–4544. doi: 10.4049/jimmunol.1001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tun NN et al. 2017. Diabetes mellitus and stroke: A clinical update. World J Diabetes 8, 235–248. doi: 10.4239/wjd.v8.i6.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuosto L et al. 1994. Analysis of susceptibility of mature human T lymphocytes to dexamethasone-induced apoptosis. Eur J Immunol 24, 1061–1065. doi: 10.1002/eji.1830240508. [DOI] [PubMed] [Google Scholar]
- Tureyen K et al. 2011. Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J Neurochem 116, 499–507. doi: 10.1111/j.1471-4159.2010.07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ et al. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Ulm L et al. 2017. The Randomized Controlled STRAWINSKI Trial: Procalcitonin-Guided Antibiotic Therapy after Stroke. Front Neurol 8, 153. doi: 10.3389/fneur.2017.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra X et al. 2009. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 158, 1174–1183. doi: 10.1016/j.neuroscience.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Ursell LK et al. 2014. The intestinal metabolome: an intersection between microbiota and host. Gastroenterology 146, 1470–1476. doi: 10.1053/j.gastro.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaibhav K et al. 2018. Remote ischemic post-conditioning promotes hematoma resolution via AMPK-dependent immune regulation. J Exp Med 215, 2636–2654. doi: 10.1084/jem.20171905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen J et al. 2013. Heritability of metabolic syndrome traits in a large population-based sample. J Lipid Res 54, 2914–2923. doi: 10.1194/jlr.P041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME et al. 2010. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A 107, 11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian KB et al. 2011. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation 8, 140. doi: 10.1186/1742-2094-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verboven K et al. 2018. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci Rep 8, 4677. doi: 10.1038/s41598-018-22962-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E et al. 2008. Functions of natural killer cells. Nat Immunol 9, 503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- Waki H et al. 2008. Specific inflammatory condition in nucleus tractus solitarii of the SHR: novel insight for neurogenic hypertension? Auton Neurosci 142, 25–31. doi: 10.1016/j.autneu.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF 2015. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 7, 56. doi: 10.1186/s13195-015-0139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY et al. 2003. Effect of age in rats following middle cerebral artery occlusion. Gerontology 49, 27–32. doi: 10.1159/000066505. [DOI] [PubMed] [Google Scholar]
- Weber C et al. 2011. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest 121, 2898–2910. doi: 10.1172/JCI44925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir P et al. 2020. A meta-analysis of remote ischaemic conditioning in experimental stroke. J Cereb Blood Flow Metab 271678X20924077. doi: 10.1177/0271678X20924077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenisch C et al. 2000. Effect of age on human neutrophil function. J Leukoc Biol 67, 40–45. doi: 10.1002/jlb.67.1.40. [DOI] [PubMed] [Google Scholar]
- Wertheimer AM et al. 2014. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol 192, 2143–2155. doi: 10.4049/jimmunol.1301721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp WF et al. 2015. The Preventive Antibiotics in Stroke Study (PASS): a pragmatic randomised open-label masked endpoint clinical trial. Lancet 385, 1519–1526. doi: 10.1016/S0140-6736(14)62456-9. [DOI] [PubMed] [Google Scholar]
- Wilckens T, De Rijk R 1997. Glucocorticoids and immune function: unknown dimensions and new frontiers. Immunol Today 18, 418–424. doi: 10.1016/s0167-5699(97)01111-0. [DOI] [PubMed] [Google Scholar]
- Willmot M et al. 2004. High blood pressure in acute stroke and subsequent outcome: a systematic review. Hypertension 43, 18–24. doi: 10.1161/01.HYP.0000105052.65787.35. [DOI] [PubMed] [Google Scholar]
- Winek K et al. 2016a. Gut microbiota impact on stroke outcome: Fad or fact? J Cereb Blood Flow Metab 36, 891–898. doi: 10.1177/0271678X16636890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winek K et al. 2016b. Depletion of Cultivatable Gut Microbiota by Broad-Spectrum Antibiotic Pretreatment Worsens Outcome After Murine Stroke. Stroke 47, 1354–1363. doi: 10.1161/STROKEAHA.115.011800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA et al. 2011. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17, 610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woiciechowsky C et al. 1998. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nat Med 4, 808–813. doi: 10.1038/nm0798-808. [DOI] [PubMed] [Google Scholar]
- Xing Y et al. 2016. Low Density Lipoprotein Cholesterol and the Outcome of Acute Ischemic Stroke: Results of a Large Hospital-Based Study. Eur Neurol 76, 195–201. doi: 10.1159/000450604. [DOI] [PubMed] [Google Scholar]
- Yan K et al. 2013. Bone marrow-derived mesenchymal stem cells maintain the resting phenotype of microglia and inhibit microglial activation. PLoS One 8, e84116. doi: 10.1371/journal.pone.0084116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J et al. 2019a. Peripheral Mechanisms of Remote Ischemic Conditioning. Cond Med 2, 61–68. [PMC free article] [PubMed] [Google Scholar]
- Yang J et al. 2019b. Remote Postischemic Conditioning Promotes Stroke Recovery by Shifting Circulating Monocytes to CCR2(+) Proinflammatory Subset. J Neurosci 39, 7778–7789. doi: 10.1523/JNEUROSCI.2699-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T et al. 2015. Gut dysbiosis is linked to hypertension. Hypertension 65, 1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y et al. 2017. ST2/IL-33-Dependent Microglial Response Limits Acute Ischemic Brain Injury. J Neurosci 37, 4692–4704. doi: 10.1523/JNEUROSCI.3233-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeramaneni S et al. 2017. Hyperlipidemia is associated with lower risk of poststroke mortality independent of statin use: A population-based study. Int J Stroke 12, 152–160. doi: 10.1177/1747493016670175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN 2008. Cell adhesion molecules and ischemic stroke. Neurol Res 30, 783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G et al. 2006. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 113, 2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Yong VW et al. 2004. The promise of minocycline in neurology. Lancet Neurol 3, 744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- Yoo BB, Mazmanian SK 2017. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 46, 910–926. doi: 10.1016/j.immuni.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan N et al. 2018. Expression of CD4+CD25+Foxp3+ Regulatory T Cells, Interleukin 10 and Transforming Growth Factor beta in Newly Diagnosed Type 2 Diabetic Patients. Exp Clin Endocrinol Diabetes 126, 96–101. doi: 10.1055/s-0043-113454. [DOI] [PubMed] [Google Scholar]
- Zhao J et al. 2014. The many roles of statins in ischemic stroke. Curr Neuropharmacol 12, 564–574. doi: 10.2174/1570159X12666140923210929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X et al. 2017. Remote ischemic preconditioning attenuates cardiopulmonary bypass-induced lung injury. PLoS One 12, e0189501. doi: 10.1371/journal.pone.0189501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierath D et al. 2015. Effect of Antibiotic Class on Stroke Outcome. Stroke 46, 2287–2292. doi: 10.1161/STROKEAHA.115.008663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinnhardt B et al. 2015. Multimodal imaging reveals temporal and spatial microglia and matrix metalloproteinase activity after experimental stroke. J Cereb Blood Flow Metab 35, 1711–1721. doi: 10.1038/jcbfm.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]