

Abstract
Skeletal muscles can undergo atrophy and/or programmed cell death (PCD) during development or in response to a wide range of insults, including immobility, cachexia, and spinal cord injury. However, the protracted nature of atrophy and the presence of multiple cell types within the tissue complicate molecular analyses. One model that does not suffer from these limitations is the intersegmental muscle (ISM) of the tobacco hawkmoth Manduca sexta. Three days before the adult eclosion (emergence) at the end of metamorphosis, the ISMs initiate a nonpathological program of atrophy that results in a 40% loss of mass. The ISMs then generate the eclosion behavior and initiate a nonapoptotic PCD during the next 30 h. We have performed a comprehensive transcriptomics analysis of all mRNAs and microRNAs throughout ISM development to better understand the molecular mechanisms that mediate atrophy and death. Atrophy involves enhanced protein catabolism and reduced expression of the genes involved in respiration, adhesion, and the contractile apparatus. In contrast, PCD involves the induction of numerous proteases, DNA methylases, membrane transporters, ribosomes, and anaerobic metabolism. These changes in gene expression are largely repressed when insects are injected with the insect steroid hormone 20-hydroxyecdysone, which delays death. The expression of the death-associated proteins may be greatly enhanced by reductions in specific microRNAs that function to repress translation. This study not only provides fundamental new insights into basic developmental processes, it may also represent a powerful resource for identifying potential diagnostic markers and molecular targets for therapeutic intervention.
Keywords: autophagy, Manduca sexta, metamorphosis, proteasome, ubiquitin
INTRODUCTION
Skeletal muscle is the largest tissue in the human body by mass and serves as its primary reservoir of amino acids. It is highly plastic and displays dramatic increases in both mass and strength during development and in response to eccentric exercise or anabolic steroids [reviewed (25, 74)]. Conversely, it can undergo atrophy and lose both mass and strength in response to any of a number of insults that include inactivity, starvation, denervation, weightlessness, cancer cachexia, HIV infection, sepsis, glucocorticoids, or tenotomy.
Atrophy is typically a protracted process that occurs over the course of weeks to years. For example, under extreme conditions it takes 4–6 wk of muscle unloading in humans to observe a 20% loss of mass in the quadriceps femoris muscle (20). Many adults over age 50 yr experience sarcopenia and lose muscle mass at a rate of ∼1% per year. While this may not seem dramatic, it can result in an ∼40% loss of muscle mass by age 80 yr, which has significant impacts on health (23, 60).
While atrophy can be reversible, there are a number of muscle disorders that result in the actual death of the muscle fiber. Acute injury can lead to the rapid necrotic death of muscle fibers and proinflammatory responses, a process known as rhabdomyolysis (13). However, muscles can also die over a more protracted time course. For example, repeated rounds of mechanical damage and regeneration lead to a decrease in the total number of muscle fibers in Duchenne muscular dystrophy via necroptosis (68, 72). Muscle fiber death can also occur nonpathologically during aging. For example, between ages 5 and 91 yr, there is an ∼60% reduction in the number of muscle fibers in the urethral rhabdosphincter, which is highly correlated with age-dependent incontinence (91).
Animal models have been invaluable for understanding some of the molecular mechanisms that mediate skeletal muscle atrophy and death. However, there are a number of limitations inherent in this approach. The first is that skeletal muscle is a complex tissue that typically contains not only multiple muscle fiber types (e.g., fast versus slow), but also different cell types such as satellite cells, endothelial cells, fibroblasts, pericytes, and macrophages (97). Therefore, it is difficult to determine if an observed change in gene expression occurs in the muscle fiber itself or within one of the other resident cell populations. Another issue is that researchers typically employ an acute insult such as denervation to induce skeletal muscle atrophy, which may not reflect the native processes associated with more developmentally regulated processes.
As a complementary approach, we have examined the intersegmental muscles (ISMs) from the tobacco hawkmoth Manduca sexta. During metamorphosis, the ISMs undergo rapid and sequential programs of atrophy and programmed cell death (PCD) as normal components of development [reviewed in (76)]. The ISMs are organized as sheets of individual giant muscle fibers, where each syncytial cell is ∼5 mm long and up to 1 mm in diameter (55) (Supplemental Fig. S1; supplemental material available at: https://doi.org/10.6084/m9.figshare.12240878). The ISMs attach at the segmental boundaries within the abdomen and provide the major motive force for both larval crawling and for adult eclosion (emergence) at the end of metamorphosis. On day 15 of pupal-adult development, the ISMs initiate a program of atrophy that results in an ∼40% loss of muscle mass by the time the adult ecloses 3 days later at the end of day 18 (84) (Supplemental Fig. S1). Atrophy is triggered by a decline in the circulating levels of the insect molting hormone 20-hydroxyecdysone (20E), which acts as a key regulator of developmental timing at the end of metamorphosis (84). ISM atrophy occurs without overt changes in physiological properties of the muscle such as resting membrane potential or tetanic force per cross-sectional area (81). Like atrophy in mammalian models, ISM atrophy involves increased ubiquitin-dependent proteolysis (31, 80).
Late on day 18, the release of the peptide eclosion hormone (EH) acts on the nervous system to generate the eclosion motor program that helps the animal emerge from the overlying pupal cuticle (102). It also acts directly on the ISMs to trigger PCD, which takes place just in advance of lights out (∼5:30 PM in our colony) (55, 85). The muscles then die during the subsequent 30 h and are no longer detectable. ISM death represents a distinct developmental process rather than the continuation of the atrophy program (81, 84).
The timing of developmental changes during metamorphosis in Manduca is tightly controlled by circadian changes in the circulating levels of the insect molting hormone 20E (84). Judicious administration of exogenous 20E on day 15 blocks both atrophy and death, while steroid treatment on day 17 blocks death and prolongs atrophy (84). However, once either of these sequential programs has been initiated, it cannot be reversed or delayed by hormone treatment. ISM death is nonapoptotic and instead occurs via an autophagic cell death program (77, 82).
The goal of this study is to dissect the molecular pathways that regulate skeletal muscle atrophy and death during development. Our hypothesis is that while there is enhanced catabolism during both atrophy and death, the underlying molecular mechanisms that drive each of these programs are fundamentally different. Atrophy results from the coordinated loss of the contractile proteins while death involves the destruction of the contractile apparatus. Earlier studies provided the identity for a few genes that are differentially expressed during ISM development that were invaluable for providing basic insights into the underlying processes [reviewed in (22)]. However, to obtain a more complete understanding of these programs, we have performed a comprehensive and unbiased RNA-Seq analysis across ISM development to examine the expression of 20,795 genes and 418 microRNAs (miRNAs). Atrophy is associated with the repression of a number of genes, including those encoding the extracellular matrix and the contractile apparatus. Death involves the induction of a number of proteolytic pathways and a shift to anaerobic metabolism. This study provides valuable insights into these developmental processes as well as a resource for hypothesis generation of skeletal muscle atrophy and death in humans.
MATERIALS AND METHODS
Animals.
The tobacco hawkmoth M. sexta was reared and staged as described previously (84). The lateral ISMs were dissected free from adjacent tissue under ice-cold saline, flash-frozen on dry ice, and stored in liquid nitrogen until used for RNA isolation. Some animals were injected on day 17 of pupal-adult development with 25 μg of 20E (Sigma) in 10% isopropanol to delay ISM death (83), and then the ISMs removed before the normal time of eclosion on day 18.
RNA isolation, library construction, and sequencing.
The ISMs of three or four animals per stage [eight developmental stages: days 13, 14, 15, 16, 17, 18, and 1 h posteclosion (PE); plus 20E injection on day 17 (20E)] were homogenized with a Polytron (Kinematica, Bohemia, NY) with the mirVana RNA Isolation kit (Life Technologies) and total RNA was isolated. For each stage of development, three independent biological replicates were constructed with total RNA for each stage of development, analyzed with a Bioanalyzer (Agilent Technologies; Santa Clara, CA), and then subjected to 50SE RNA-Seq using an Illumina HiSeq 2000 (San Diego, CA) by Beijing Genomics Institute (Hong Kong).
For small RNA-Seq library construction, 50 μg of total RNA was fractionated via 15% urea polyacrylamide gel electrophoresis, and the 18–30 nt fraction extracted for library construction. 3′- and 5′-Adaptors were ligated to the small RNA, and the cDNA reverse transcribed and PCR amplified. The libraries were purified by polyacrylamide gel electrophoresis (PAGE) and subjected to 50-SE sequencing as above. In some cases, independent sets of libraries were constructed and sequenced by the University of Massachusetts Deep Sequencing Core Facility.
Quantitative PCR quantification.
Total RNA was isolated from the ISMs on 3 days of pupal-adult development (days 13, 16, and 18). In addition, one group of animals was injected with 25 μg of 20E on day 17 and analyzed late on day 18. mRNA from three independent biological replicates was converted to cDNA using the Superscript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA) and quantified via quantitative qPCR using SYBR Select Master Mix (Applied Biosystems by Life Technologies, Carlsbad, CA) with the following primers (Supplemental Table S1).
Relative mRNA levels were compared by the ∆∆Ct method, and Renilla luciferase expression was normalized to firefly luciferase. All experiments were performed with three biological replicates and two to four technical replicates.
Genome sequence and the annotation.
We downloaded the genomic sequence of M. sexta (Msex1.0) and the transcript and the protein sequences (revised-OGS-June2012) from the Manduca Base (https://i5k.nal.usda.gov/data/Arthropoda/mansex-(Manduca_sexta)/Current%20Genome%20Assembly/2.Official%20or%20Primary%20Gene%20Set/OGS2.0/ (39). Since the gene annotation was not available in the database, we identified the genes in M. sexta with homologs in Drosophila melanogaster. We downloaded proteome data sets from UniProt (release 2013_12) (103) and mRNA sequences from FlyBase (FB2013_06) (39) as the D. melanogaster comprehensive annotations. Using the sequences in the Manduca Base as queries, we ran BLASTP, BLASTX, and BLASTN with E-value < 10−4 (BLAST+ version 2.2.28) (2) against the D. melanogaster proteome and transcriptome databases. The best BLASTP hits in D. melanogaster were assigned as the gene annotations of M. sexta. For the genes with no BLASTP hits, we assigned the best BLASTX and BLASTN hits to the gene annotations. For the genes in M. sexta that do not have any homologs in D. melanogaster, we conducted the same BLAST search with nr (version 2013/12/17) and nt (version 2013/12/16) databases downloaded from the National Center for Biotechnology Information website. The statistics of the annotated genes in M. sexta are shown in Table 1.
Table 1.
| Genes without Isoforms | Genes with Isoforms | |
|---|---|---|
| Total input | 18,841 | 20,795 |
| Best BLAST hits | 15,477 (82.14%) |
16,965 (81.58%) |
| Human homologs | 10,887 (57.78%) |
11,903 (57.24%) |
| Mouse homologs | 10,773 (57.18%) |
11,797 (56.73%) |
| Fly homologs | 11,968 (63.52%) |
13,124 (63.11%) |
For miRNA annotation, we downloaded M. sexta miRNA (mse.gff3) annotation from miRBase v20 (41, 110). In addition, we predicted potential miRNAs with mapped small RNAs in all time points with mirDeep2 (24).
Gene expression and differentially expressed gene clusters.
For RNA-Seq libraries, we mapped all reads in the three biological replicates per stage (days 13, 14, 15, 16, 17, 18 and 20E) to the genome with TopHat2 (v2.0.10) (40) allowing two mismatches. Multiply mapped reads were apportioned by the number of times they mapped to the genome and the RPKM values for the genes were calculated.
To identify differentially expressed genes between a pair of time points during ISM development, we ran the DESeq2 (v1.5.5) (56) algorithm implemented in R using mapped read counts as the input. Transcripts with a false discovery rate (FDR) ≤ 0.01 and absolute fold-change (fc) ≥ 2 were considered to be differentially expressed.
To cluster differentially expressed genes according to their RPKM values, we performed K-mean clustering with the fpc package (v2.1-7) implemented in R (33, 34). We used correlation distance as the distance measurement and bootstrapped each parameter setting 100 times. We tested the different number of clusters, k, from 3 to 20 and selected k = 6 based on the cluster stability measurement (i.e., Jaccard index and silhouette coefficient) and biological knowledge (33, 34).
Transcription factor binding site estimation.
We extracted 1 kb upstream sequences from transcription start sites (TSSs) of all genes in M. sexta. We calculated statistically significantly enriched motifs of transcription factor binding sites (TFBSs) with Analysis of Motif Enrichment (AME version 4.9.1) (61, 62) against the extracted upstream sequences. We further analyzed the detected transcription factor (TF) hits that were differentially expressed (i.e., TFs that belong to any gene clusters) during the ISM developmental stages. To estimate the position of TFBSs, we ran Find Individual Motif Occurrences (FIMO version 4.9.1) (27) against the same set of the upstream sequences. An FDR 1 × 10−3 threshold was used to select statistically significant hits for the AME and FIMO analysis. For the motif models, we downloaded the motifs from the TRANScription FACtor (TRANSFAC) database release 12.1 (69 manually selected insect models) (62), JASPAR Core Insect (131 models) (61), and Fly Factor Survey (652 models for 326 genes) (111).
To investigate potential protein-protein interactions among the detected TFs, we obtained the experimental, genetic text-mining, and interolog interaction data from the STRING database (95). We searched the interactions among the detected TFs including one interaction away and drew networks with Cytoscape 3.1.1 (88).
miRNA expression and differentially expressed miRNAs.
For small RNA-Seq libraries, we computationally stripped off the 3′-adaptor sequences and mapped reads to the genome using Bowtie (v1.1.0) (45). We used only perfectly matched reads to the genome and apportioned multiply mapped reads. We computed parts per million (ppm) to estimate the abundance of the annotated and the predicted miRNAs. We calculated differentially expressed miRNAs with the same protocol used for the detection of differentially expressed genes. However, we used a different cut-off for miRNAs to define differentially expressed miRNAs (P value ≤ 0.05, fc ≥ ± 2, and ppm ≥ 10).
miRNA target prediction and enrichment analysis.
We first collected 3′-UTR (untranslated region) sequences of mRNAs from the Manduca Base. For the mRNAs that lack their 3′-UTR annotations, we predicted the 3′-UTR sequences using the assembled transcripts with Cufflinks (v2.2.1) (100). Briefly, we assembled mRNAs based on the mapped RNA-Seq reads in all stages with the options implemented in Cufflinks: -u–pre-mrna-fraction 0.2–min-frags-per-transfrag 25 –overlap-radius 50. Taking into account the prediction results, we made the miRNA target sequence list composed of annotated 3′-UTRs and predicted 3′-UTRs whose length are ≥ 10 bp. The annotated and predicted 3′-UTRs used in this study can be found in the supplemental data. Using the miRNA and the 3′-UTR sequences, we calculated miRNA target sites in the 3′-UTRs with TargetScan 6.0 (49).
To remove false-positive miRNA target genes, we calculated the Pearson correlation coefficient between expression levels of a miRNA and the target genes. We extracted the target genes that have a correlation coefficient ≤−0.6. We also computed statistically significantly enriched miRNAs in a specific gene cluster by hypergeometric test (FDR ≤ 0.01).
miRNA regulation of mRNA stability and translatability.
To evaluate the ability of miRNAs to regulate death-associated transcripts, we subcloned the miR-92b target region of the small cytoplasmic leucine rich repeat protein (SCLP, Msex013021) 3′-UTR into the pFila dual luciferase vector (7) using the primers 5′ GAACAATAATTCTAGTCTCTATGGACTAAGCCTGTGA 3′ and 5′ AAGCGGCCGCTCTAGGACACTTAACATAACATCCCAAACC 3′. As a positive siRNA control, we made a construct that contained a 22-nucleotide sequence that was completely complementary to miR-92b using primers 5′ CTAGATAGAATTCTAGTGCAGGCCGGGATTGGTGCAATTGGGCC 3′ and the synthesized antisense strand was 3′ TACTTAAGATCACGTCCGGCCCTAACCACGTTAAC 5′.
COS-1 cells were maintained in high-glucose Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) and 1× penicillin-streptomycin (Gibco, Grand Island, NY). Cells were transfected with pFila plasmid DNA using Lipofectamine 2000 (Thermo Fisher, Waltham, MA) with or without 1 µl of 20 µM miR-92b mimic (QIAGEN, Valencia, CA). All transfections were performed in triplicate. Cells were lysed, and luciferase activity was quantified using the Dual Luciferase Reporter Assay System (Promega, Madison, WI) on a Polarstar plate reader. The fold-change in firefly luciferase was normalized to the levels of the constitutively expressed Renilla luciferase.
Total RNA was isolated, converted to cDNA with Superscript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA) and quantified via qPCR using SYBR Select Master Mix (Applied Biosystems by Life Technologies, Carlsbad, CA) with the following primers: Renilla forward 5′ CATGGGATGAATGGCCTGATA 3′ and reverse primers 5′ CAACATGGTTTCCACGAAGAAG 3′ and firefly forward 5′ CATAGCTTACTGGGACGAAGAC 3′ and reverse 5′ CCACCTGATAGCCTTTGTACTT 3′ primers. Relative mRNA levels were compared by the ∆∆Ct method, and Renilla luciferase expression was normalized to firefly luciferase. All experiments were performed in duplicate with three technical replicates.
RESULTS
Availability of data and material.
All the sequencing libraries are accessible from GSE80830 in Gene Expression Omnibus.
Differential gene expression during ISM development.
To determine which genes are differentially expressed during ISM development, we generated 21 RNA-Seq libraries composed of three biological replicates from the muscles on each day of pupal-adult development ranging from day 13, when the muscles are homeostatic, to day 18, when they are committed to die (Supplemental Fig. S1). We also utilized ISM RNA from day 18 animals that had been injected on day 17 with 20E to delay the normal timing of cell death. Since the Manduca genome was not annotated at the time that we conducted our initial analyses, we performed our own annotation to allow us to identify the Manduca genes, as well as the orthologs from the fruit fly (D. melanogaster), the silk moth (Bombyx mori), and mouse (Mus musculus). We found that 85.8–87.3% reads were uniquely mapped to the Manduca genome (Supplemental Table S2).
Using the read counts obtained from mapping (85.8–87.3% uniquely mapped reads; Supplemental Table S2), we computed differentially expressed genes of the pairs of all possible stages (Supplemental Fig. S2). There were almost no differences in the levels of gene expression between days 13 and 14, a time when the muscles are homeostatic and not undergoing discernible changes in either physiology or development (Fig. 1). Beginning on day 15, when the muscles initiate atrophy, there are a number of significant changes in gene expression, with 125 genes induced and 235 repressed. This trend continued for the next 2 days with gene repression outstripping induction (day 16: 226 induced vs. 591 repressed; day 17: 877 induced vs. 989 repressed). On day 18, more than double the number of genes that were differentially expressed compared with day 17; there were more newly induced genes than repressed (2,578 vs. 2,104).
Fig. 1.
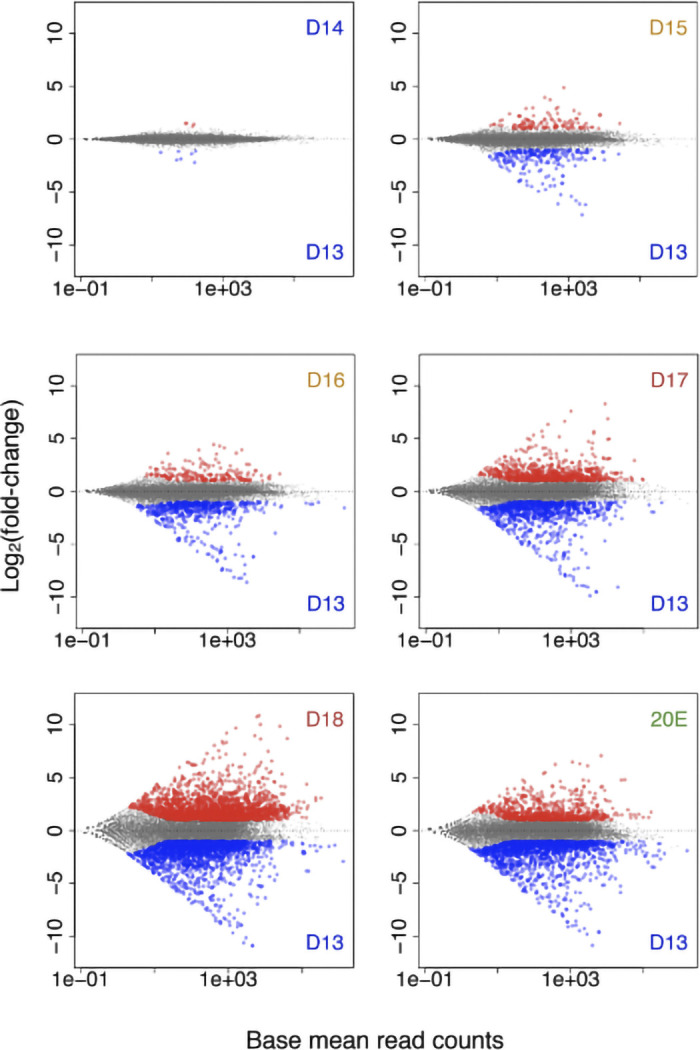
Differentially expressed genes during the intersegmental muscle (ISM) development. Scatter plots of differentially expressed genes between day 13 and the other time points during the ISM development as well as 20-hydroxyecdysone (20E)-treated animals (20E). Red dots, upregulated genes; blue dots, downregulated genes. The abundance of almost all transcripts was identical between days 13 and 14 when the muscles are homeostatic. Starting on day 15, when atrophy begins, almost twice as many genes were repressed than were induced. In contrast, when the ISMs became committed to die on day 18, more than twice as many genes were induced than were repressed. D, day; n = 3 independent libraries were analyzed for each developmental stage.
Twelve of the induced genes displayed more than a 256-fold monotonic increase (log2 fold-change = 8) relative to their expression on day 13. Two of these genes (Msex004867, Msex004866; FDR < 3.4 × 10−112) were homologs of the D. melanogaster takeout gene (CG11853), which encodes ligand binding proteins that include the juvenile hormone binding protein and is thought to be an output gene of the circadian clock (14). Two additional genes (Msex015696, Msex008658; FDR < 2.5 × 10−107) encode proteins that are homologous to astacin-like matrix metalloendopeptidase in D. melanogaster (CG6763). The remaining eight genes encoded a series of diverse proteins that include homologs of cuticular protein glycine-rich 29 precursor (Msex015422; FDR < 3.8 × 10−91), cysteine-rich secretory protein (Msex006668; FDR ≤ 3.1 × 10−204), inorganic phosphate cotransporter-like protein (Msex005594; FDR ≤ 1.1 × 10−48), lipase 4 (Lip4) (Msex010110; FDR ≤ 1.3 × 10−53), ejaculatory bulb protein III (PebIII) (Msex008696; FDR ≤ 9.7 × 10−47), an uncharacterized protein that has sequence similarity to protease inhibitor 4 from the moth Lonomia obliqua (Msex003322; FDR ≪ 1.0 × 10−300), and Acheron/LARP6, a Lupus antigen-related protein (Msex001882; FDR ≤ 1.5 × 10−199). Interestingly, Acheron (Msex001882) and the uncharacterized protein Msex003322 displayed remarkable levels of induction from day 13 to day 18: from 4.8 and 1.1 RPKM to 2339.5 and 1160.3 RPKM, respectively. The expression change of the other 10 induced genes were from 0.02–0.36 RPKM on day 13 to 57.4–531.0 RPKM on day 18. The dramatic upregulation of Acheron has been documented at both the RNA and protein levels via Northern and Western blotting (90, 104).
Published Northern blots confirm a number of the changes in gene expression that we observed, including actin, myosin heavy chain, small cytoplasmic leucine-rich repeat protein (SCLP), Acheron, polyubiquitin, proteasome subunits, and apolipoprotein III (37, 43, 78, 80, 93, 104). In addition, we selected nine differentially expressed genes for qPCR analysis using the constitutively expressed ubiquitin-fusion protein 80 (Ubf80; Msex008476) as a control: Acheron (Msex001882), RING Finger Protein 145 (Msex012524), heat shock protein 70B (Msex015761), vacuolar proton pump (Msex009238), ecdysone-induced protein 75 (Msex001561), nidogen (Msex009360), polyubiquitin (Msex002975), lipase 4 (Msex010110), and TPR repeat protein (Msex009658) (Fig. 2). RNA-Seq and qPCR measures of transcript abundance were comparable at every day of development examined and also in the 20E group. The absolute fold change between qPCR and RNA-Seq was typically within 1 PCR cycle of one another. Consequently, we concluded that the RNA-Seq data are fully informative about the patterns of gene expression in the ISMs.
Fig. 2.

Corroboration of RNA-Seq data with quantitative PCR (qPCR). The expression levels of the nine genes that were shown to be differentially expressed during atrophy and/or death with RNA-Seq (red) were assessed for equivalence with qPCR (blue). For each day of development, three biological replicates (n = 3) were obtained, and the data were analyzed as fold-change in expression relative to day 13. RNA-Seq and qPCR measures of transcript abundance were comparable at every day of development and also in the 20-hydroxyecdysone (20E) group; the absolute fold change between qPCR and RNA-Seq were typically within 1 PCR cycle of one another. Data are means ± SE. D, day.
The functional role of differentially expressed genes during ISM development.
We performed a Gene Ontology (GO) analysis on both up- and downregulated genes across development and in response to 20E (Fig. 3). In agreement with the patterns of differential gene expression shown in Fig. 1 and Supplemental Fig S2, there were no significant changes in gene expression during the period of homeostasis (days 14–15). However, with the initiation of atrophy (days 15–17), there was gene induction of transcripts encoding proteins associated with coagulation, histone H3-K4 methylation, acylglycerol transport, autophagy, fatty acid synthase activity, and vitamin binding. Concurrently, there was a repression of genes encoding components of chitin metabolism, adhesion, and ATP-dependent helicase activity. With the commitment to die on day 18, there was the dramatic induction of genes involved in protein catabolism, immune response, the proteasome, membrane transporters, fatty acid synthase, and DNA methylation. At the same stage, there was a reduction in ribosome gene expression, cuticle formation, cell adhesion, and nucleotide kinase activity. Injecting animals on day 17 with 20E delayed the normal eclosion of animals on day 18 (84, 85). While this blunted the anticipated induction of most of these pathways, it proved to be a potent inducer of immune system genes and a repressor of metabolism and energy utilization.
Fig. 3.

Gene Ontology (GO) enrichment analysis of differentially expressed genes during intersegmental muscle (ISM) development. A: GO terms enriched with the upregulated genes between day 13 and the other time points during the ISM development. The color shows the statistical significance measured in false discovery rate (FDR). B: GO terms enriched with the downregulated genes between day 13 and the other time points during the ISM development. The color shows the statistical significance measured in false discovery rate. n = 3 independent libraries were analyzed for each developmental stage. Atrophy (days 15–17) was associated with increased coagulation, histone H3-K4 methylation, acylglycerol transport, autophagy, fatty acid synthase activity, and vitamin binding and the repression of chitin metabolism, adhesion, and ATP-dependent helicase activity. The commitment to die on day 18 involved enhanced protein catabolism, immune response, the proteasome, membrane transporters, fatty acid synthase, and DNA methylation and the repression of ribosome production, cuticle formation, cell adhesion, and nucleotide kinase activity. 20-Hydroxyecdysone (20E) prevented most of the changes in death associated processes. D, day.
To obtain a more comprehensive understanding of the patterns of differentially expressed genes during atrophy and PCD, we performed K-mean clustering on gene expression quantification results, i.e., RPKM from day 13 to 18 to determine the number of possible gene clusters. Through bootstrapping, we tested the stability of between four and 20 different clusters and observed the best stability with six clusters (Jaccard coefficient: 0.92–0.86) (Fig. 4A). The clustered gene list and the gene expression RPKM values on each day of development are summarized in the Supplemental Table S2.
Fig. 4.

Expression and functional aspects of clustered genes. A: the patterns of differential gene expression across intersegmental muscle (ISM) development can be best explained with six distinct clusters. Black line, the center of the cluster; gray, represent 25% of the best-fitting genes. The y-axis denotes Log2 fold change (RPKM value), while the number of genes that fall within each cluster are denoted with the colored numbers within each panel. B–D: pathway enrichment among the six clusters (C1–C6). The columns represent the clusters, while the rows are the GO terms for cellular component (B), biological process (C), and molecular function (D). n = 3 independent libraries were analyzed for each developmental stage. D, day; FDR, false discovery rate.
Each cluster displayed a distinct pattern of gene expression during ISM development. We further performed GO analysis in each cluster to inspect the functional properties of the grouped genes they represent (Fig. 3). Cluster 1 (hereafter, C1) contained 538 genes whose expression continuously declined from day 13 to day 17 and then slightly increased on day 18. The GO terms mainly related to cell periphery and extracellular parts: “cell periphery” (GO:0071944), “extracellular matrix” (GO:0031012), “cuticle development” (GO:0042335), “biological adhesion” (GO:0022610), “structural molecule activity” (GO:0005198). Prominent in this cluster were the majority of the cuticle genes, which were coordinately and dramatically repressed during ISM development.
The 2,583 genes in C2 displayed relatively stable expression during the early phase of atrophy and then initiated a precipitous decline starting on day 16. Many of the genes in this cluster were represented by GO terms related to cuticle and extracellular matrix. In addition, there were the GO terms related to cellular respiration and metabolism: “cellular respiration” (GO:0045333), “respiratory chain” (GO:0070469), “oxidoreductase complex” (GO:1990204). Many of the major contractile protein genes are also represented in this cluster, including troponins T (Msex004726) and C (Msex006421), myosin heavy chain (Msex009968), and tropomyosin (Msex013366). We found that all of the downregulated genes shown in Table 2 belong to C2.
Table 2.
| Manduca Base ID | D13 | D18 | Log2(fold-change) | FDR | Gene Annotation | Organism |
|---|---|---|---|---|---|---|
| Msex013528-RA | 14,803.84 | 880.05 | −3.83 | 4.49E-45 | cuticular protein 49Ae (Cpr49Ae) | D. melanogaster |
| Msex013529-RA | 1,324.51 | 79.89 | −3.81 | 8.69E-46 | cuticular protein 67Fb (Cpr67Fb) | D. melanogaster |
| Msex005195-RA | 2,564.95 | 0.33 | −3.46 | 1.16E-05 | cuticle protein LCP65Ad (Lcp65Ad) | D. melanogaster |
| Msex009968-RA | 10,139.21 | 1,204.35 | −2.91 | 1.63E-40 | myosin heavy chain (Mhc) | D. melanogaster |
| Msex004528-RA | 1,527.35 | 265.01 | −2.16 | 7.02E-06 | CG44085 | D. melanogaster |
| Msex004529-RA | 2,400.22 | 418.15 | −2.15 | 1.08E-05 | N.A. | N.A. |
| Msex000706-RA | 3,264.96 | 712.19 | −2.06 | 2.43E-23 | myosin alkali light chain 1 (Mic1) | D. melanogaster |
| Msex001727-RA | 2,983.65 | 722.77 | −1.93 | 3.02E-40 | paramyosin (Prrn) | D. melanogaster |
| Msex011357-RA | 1,521.87 | 463.52 | −1.54 | 6.69E-07 | sallimus (sis) | D. melanogaster |
| Msex008530-RA | 27,229.8 | 8,788.53 | −1.49 | 4.95E-11 | actin 578 (Act578) | D. melanogaster |
| Msex002562-RA | 1,185.56 | 402.85 | −1.44 | 2.62E-17 | CG9986 | D. melanogaster |
| Msex015806-RA | 2,383.71 | 811.96 | −1.41 | 5.40E-09 | muscle LIM protein at 84B (Mlp84B) | D. melanogaster |
| Msex013227-RA | 1,233.87 | 418.34 | −1.39 | 6.02E-05 | sallimus (sis) | D. melanogaster |
| Msex017750-RA | 1,136.22 | 417.85 | −1.33 | 1.46E-09 | uncharacterized protein LOC100865369 | Apis florea |
| Msex013367-RA | 1,066.72 | 408.14 | −1.28 | 4.55E-21 | tropomyosin 1 (Tm1) | D. melanogaster |
| Msex015807-RA | 1,985.6 | 748.69 | −1.27 | 5.28E-07 | CG7484 | D. melanogaster |
| Msex013366-RA | 2,346.98 | 942.97 | −1.19 | 1.57E-07 | tropomyosin 2 (Tm2) | D. melanogaster |
| Msex013228-RA | 2,121.73 | 833.67 | −1.15 | 8.18E-03 | N.A. | N.A. |
| Msex013228-R8 | 2,771.25 | 1,147.37 | −1.08 | 9.92E-03 | N.A. | N.A. |
| Msex016926-RA | 1,213.93 | 516.96 | −1.05 | 8.40E-03 | N.A. | N.A. |
| Msex010227-RA | 1,425.23 | 660.61 | −1.01 | 7.50E-24 | 60S ribosomal protein LP0 (RpLP0) | D. melanogaster |
| Msex017592-RA | 1,249.95 | 579.2 | −1.00 | 1.78E-08 | 40S ribosomal protein S2 (RpS2) | D. melanogaster |
D, day; FDR, false discovery rate; N.A., not available.
The 667 genes in C3 displayed enhanced expression up to day 16, leveled off, and then declined dramatically in advance of death on day 18. The GO terms detected in this cluster were also related to respiratory pathways, for example “NADH dehydrogenase activity” (GO:0003954) and “generation of precursor metabolites and energy” (GO:0006091). Interestingly, we observed a dramatic biphasic expression pattern for ribosome biogenesis in C3, with large increases in expression during the early phase of atrophy followed by a dramatic decline in advance of cell death.
The expression of 582 genes in C4 increased continuously from days 13 to 17 and then declined on day 18. GO terms for this cluster include “transcriptionally active chromatin” (GO:0035327) and “positive regulation of histone H3-K4 methylation” (GO:0051571). Those GO terms were distinct terms in C4, although the GO terms related to metabolite transporter activities and catabolic activities were detected in both C4 and C5.
The 2,511 genes in C5 displayed a modest increase in expression during early development and then a dramatic rise on day 18. This cluster contains almost all of the genes involved in ubiquitin-proteasome-dependent protein degradation, such as polyubiquitin (Msex002996) and a large number of proteasome subunits (Table 3). All the upregulated genes shown in Table 1 were clustered into C5 with the exception of the novel gene Msex004609 (homolog of uncharacterized protein with no discernible domains, LOC101746963 in B. mori), which resides in C4.
Table 3.
| Manduca Base ID | D13 | D18 | Log2(fold·change) | FDR | Gene Annotation | Organism |
|---|---|---|---|---|---|---|
| Msex003322-RA | 1.09 | 1,160.33 | 9. 86 | 1.00E-300 | uncharacterized protein LOC101743112 | B. mori |
| Msex001882·RA | 4.75 | 2,339.50 | 8. 69 | 1.48E-199 | La-related protein (CG17386) | D. melanogaster |
| Msex008699-RA | 13.50 | 1,525.58 | 6. 57 | 1.18E-93 | ejaculatory bulb-specific protein 3 (PebIII) | D. melanogaster |
| Msex005579-RA | 282.34 | 5,868.86 | 4.38 | 2.09E-86 | CG34325 | D. melanogaster |
| Msex007599-RA | 76.46 | 1,290.55 | 4.14 | 4.66E-240 | glutamine synthetase 2 (Gs2) | D. melanogaster |
| Msex001006-RA | 151.28 | 1,494.01 | 3.12 | 5.85E-14 | CG45050 | D. melanogaster |
| Msex002996-RA | 774.12 | 7,048.24 | 3.10 | 1.55E-19 | N.A. | N.A. |
| Msex002996-RB | 1,513.91 | 13,773.77 | 3.10 | 1.91E-19 | ubiquitin-5E (Ubi-p5E) | D. melanogaster |
| Msex002997-RA | 2,827.45 | 25,383.64 | 3. 07 | 1.67E-17 | ubiquitin-63E (Ubi-p63E) | D. melanogaster |
| Msex000653-RA | 288.48 | 2,240.48 | 2. 90 | 4.42E-20 | hypothetical protein KGM_01767 | Danaus plexipus |
| Msex005540-RA | 344.60 | 2,383.63 | 2.85 | 1.77E-78 | thin (tn) | D. melanogaster |
| Msex014957-RA | 465.83 | 3,058.80 | 2.74 | 1.99E-32 | small cyloplasmic leucine rich repeat protein (Sclp) | D. melanogaster |
| Msex013021-RA | 874.32 | 5,637.41 | 2. 69 | 5.57E-24 | small cyloplasmic leucine rich repeat protein (Sclp) | D. melanogaster |
| Msex011306-RA | 190.43 | 1,072.99 | 2.43 | 1.89E-12 | laminin subunit beta-1 (LanB1) | D. melanogaster |
| Msex002976-RA | 331.63 | 1,625.77 | 2.34 | 8.34E-24 | ubiquitin conjugating enzyme 87F (Ubc87F) | D. melanogaster |
| Msex013343-RA | 343.39 | 1,627.28 | 2.33 | 3.56E-87 | neurochondrin | D. melanogaster |
| Msex011554-RA | 434.65 | 2,107.87 | 2. 30 | 3.64E-16 | heat shock protein 83 (Hsp83) | D. melanogaster |
| Msex012988-RA | 377.72 | 1,533.41 | 2.10 | 1.88E-51 | 26S proteasome non-ATPase regulatory subunit 1 (Rpn2) | D. melanogaster |
| Msex017264-RA | 469.41 | 1,922.63 | 2.07 | 1.51E-13 | muscle-specific protein 20 (Mp20) | D. melanogaster |
| Msex000788-RA | 449.82 | 1,815.78 | 2.06 | 1.17E-17 | CG43897 | D. melanogaster |
| Msex009386-RA | 368.65 | 1,370.78 | 1.98 | 1.06E-95 | 26S proteasome regulatory complex subunit p39A (Rpn9) | D. melanogaster |
| Msex015814-RA | 318.08 | 1,137.06 | 1.92 | 1.92E-53 | 26S protease regulatory subunit 4 (Pros26.4) | D. melanogaster |
| Msex004605-RA | 372.80 | 1,245.84 | 1.81 | 4.42E-19 | proteasome subunit beta type-4 (Prosbeta7) | D. melanogaster |
| Msex007686-RA | 411.06 | 1,317.94 | 1.77 | 4.53E-40 | 26S proteasome regulatory complex subunit p97 (Rpn1) | D. melanogaster |
| probable 26S proteasome non-ATPase regulatory subunit | ||||||
| Msex007558-RA | 463.23 | 1,425.76 | 1.71 | 2.95E-61 | 3 (Rpn3) | D. melanogaster |
| Msex004609-RA | 405.89 | 1,151.27 | 1.57 | 2.50E-10 | uncharacterized protein LOC101746963 | B. mori |
| Msex005351-RA | 425.53 | 1,166.59 | 1.52 | 4.78E-13 | proteasome maturation protein (Pomp) | D. melanogaster |
| Msex014545-RB | 419.37 | 1,131.53 | 1.52 | 1.43E-44 | uncharacterized protein LOC101746228 | B. mori |
| Msex012266-RA | 590.45 | 1,527.44 | 1.45 | 6.76E-19 | CG17737 | D. melanogaster |
| Msex010506-RA | 463.51 | 1,092.21 | 1.32 | 1.60E-11 | DNA repair protein Rad23 (Rad23) | D. melanogaster |
| Msex000119-RA | 2,144.14 | 4,967.49 | 1.30 | 7.11E-37 | N.A. | N.A. |
| Msex000118-RA | 1,812.05 | 4,071.05 | 1.26 | 1.30E-36 | 26S proteasome non-ATPase regulatory subunit 11 (Rpn6) | D. melanogaster |
| Msex017071-RA | 3,311.99 | 7,317.55 | 1.22 | 6.77E-09 | ADP,ATP carrier protein (sesB) | D. melanogaster |
| Msex009904-RA | 575.70 | 1,163.96 | 1.06 | 6.14E-04 | CG34417 | D. melanogaster |
| Msex012133-RA | 549.29 | 1,079.71 | 1.04 | 2.76E-05 | ferritin 1 heavy chain homolog (Fer1HCH) | D. melanogaster |
D, day; FDR, false discovery rate; N.A., not available.
The 679 genes in C6 were expressed at relatively low levels throughout atrophy and then increased dramatically on day 18. The GO terms in C6 relate to immune system pathways, e.g., “immune system process” (GO:0002376) and “antibacterial humoral response” (GO:0019731), and hormone receptor pathways, e.g., “receptor activity” (GO:0004872) and “steroid hormone receptor activity” (GO:0003707). The GO terms detected in C6 are very distinct from the GO terms detected in the other clusters; the genes in this cluster represent many of the presumptive death-associated transcripts.
Transcriptional regulation during the ISM development.
We computed statistically significantly enriched TFBSs within the 1 kb region 5′ to the TSSs of the genes that are differentially expressed in the ISMs. The TFBSs of C2 (42), C3 (5), C4 (2), C5 (42), and C6 (1) TFs were considered for further analysis (Supplemental Table S3). We could not identify any TFBSs that were statistically significant in the genes associated with C1. With the hypothesis that TFs that regulate genes in a specific cluster may also display developmental regulation themselves, we also checked differentially expressed TFs in each cluster. While promoters and enhancers can reside at great distances and be located either 3′ or 5′ to the gene of interest, the current annotation of the Manduca genome precludes performing a more detailed survey. Nevertheless, this analysis allows us to begin creating models of developmentally regulated gene expression in the ISMs.
For genes in C6, we found the enrichment of the binding sites for Enhancer of split mgamma protein (HLHmγ; Msex003078), a direct nuclear target of the Notch signaling pathway (1). The TFBSs of Hairy (h; Msex000196) and Stripe (sr; Msex006516) were enriched in the genes in C4. We found the enrichment of five TFBSs, GFI-1B/CG7368 (Msex006263), Cubitus interruptus (ci; Msex002239), Hunchback (hb; Msex004840), Klumpfuss (klu; Msex003545 and Msex009479), and jim (Msex007169 and Msex011615) in the genes in C3. However, the expression levels of all the TFs were almost negligible (0.02–5.98 RPKM) during the ISM development, except for Stripe (14.77–3.65 RPKM).
Using data from the literature, we created maps of putative protein-protein interactions among the detected TFs (Fig. 5). Despite the fact that the expression levels for some of these TFs were low, we nevertheless found that they formed three subnetworks of protein-protein interactions. Interestingly, in the largest subnetwork detected, Polycomb group protein (Pleiohomeotic, Pho; Msex006348) was the hub between the module of the TFs related to hedgehog pathway and the modules composed of the TFs of ecdysone and circadian rhythm pathways (Fig. 5A). Pho binding sites were overrepresented in the genes in C2 (2,388/2,583 genes; FDR = 5.41 × 10−6) and C5 (2,291/2,511 genes; FDR = 8.26 × 10−9). Pho itself was clustered into C5 and displayed a gradual increase until day 17 and then a dramatic induction from 4.46 to 22.07 RPKM on day 18.
Fig. 5.

Protein-protein interaction subnetwork of transcription factors (TFs) detected in the transcription factor binding site (TFBS) enrichment analysis. Each node represents each TF detected in the TFBS enrichment analysis. Each edge indicates a protein-protein interaction between TFs. The colors of the nodes show the gene clusters that the TFs belong to. The other TFs interacting with the detected TFs are also shown. A: TFBSs enriched in gene clusters and the expression of the TFs. B: TFBSs uniquely enriched in cluster 2 (C2) and the expression of the TFs. C: TFBSs uniquely enriched in C5 and the expression of the TFs. D: TFBSs uniquely enriched in repressed genes by 20-hydroxyecdysone (20E) and the expression of the TFs. Black arrows indicate upregulation or downregulation by 20E. Enhancer of split mgamma protein, a target of Notch signaling, was enriched for genes in cluster 6 (C6). The TFBSs of Hairy/h and Stripe/sr were enriched in the genes in C4. Five TFBSs, GFI-1B, Cubitus interruptus/ci), Hunchback/hb, Klumpfuss/klu, and jim, were enriched for genes in C3. Two TF subnetworks were detected for C2 genes: homeobox genes (Abd-B and Homothorax/hth) and ecdysone receptor signaling (Ultraspiracle/usp).
To further investigate the transcriptional regulation of the genes in C2 and C5, we focused on the TFBSs that are uniquely enriched in those clusters. Out of 42 enriched TFBSs in the two clusters, 12 and 13 of those were uniquely enriched in C2 and C5, respectively. For the TFBSs enriched in the genes of C2, two TF subnetworks were detected: 1) homeobox genes such as Abd-B (Msex000748) and Homothorax (hth; Msex016683, Msex016684, and Msex017338) and 2) ecdysone receptor signaling like Ultraspiracle (usp; Msex014339) (Fig. 5B). The subnetworks of the TFs regulating C5 also contain ecdysone signaling pathway components such as the Ecdysone-induced protein 74EF (Eip74EF; Msex009996) and the Ftz transcription factor 1 (ftz-f; Msex005610) (Fig. 5C). The binding sites of the TFs related to circadian rhythms were also enriched in C5 genes such as Clock (clk; Msex010427) and Cycle (cyc; Msex013484) genes (Fig. 5C). As with the target cluster of clk/cyc, these genes were also clustered in C5 and showed a twofold increase on day 18. Interestingly, the antagonistic factors to the genes such as Timeless (Msex000831) and Period (Msex001717) were clustered in the repressed gene cluster, C2. Furthermore, we found another circadian clock regulator, Cry (Msex007463) was induced during development.
We observed that ecdysone signaling pathway components were highly expressed during ISM development (Fig. 5D). The most upstream component of the pathway is a nuclear receptor complex consisting of ecdysone receptor (EcR; Msex001634) and Usp, followed by the activation of Eip74E, BR-C (Msex007362), Eip93F (Msex015245 and Msex017016) (98). Although the enrichment of the EcR TFBSs was not detected, the expression of EcR was induced from 6.95 RPKM on day 13 to 20.94 RPKM on day 17 (Fig. 6). Eip74FE, BR-C, and Eip97F were all clustered into C5 and also displayed a ∼3-fold increase on day 18 compared with day 13. H3K27me3 demethylase Utx (Msex004649), which regulates key apoptosis and autophagy genes controlled by the ecdysone signaling pathway (16, 87), was also induced on day 17 (26.31–51.95 RPKM). Furthermore, we also observed enhanced expression of ftz-f1 on day 18 (3.60–34.84 RPKM), a TF required for cell death in Drosophila (112). Although the downstream apoptosis initiator reaper (rpr; Msex002141) and caspase gene dronc (Msex014106) were not statistically significantly upregulated (1.19–2.51 RPKM), another caspase gene drice (Msex006408) was induced twofold, from 10.97 RPKM on day 13 to 22.77 RPKM on day 18.
Fig. 6.

Expression of ecdysone receptor signaling components during development. RNA-Seq expression during development is presented in RPKM. n = 3 independent libraries were analyzed for each developmental stage. An additional group of n = 3 animals were injected on day 17 with 20-hydroxyecdysone (20E) and then analyzed on day 18. Onset of atrophy on day 15 is accompanied by a downward trend in E75 and a transient elevation of 74EF. Commitment to die was accompanied by a further decline in E75 and elevations in both ECR and 74EF. Data are means ± SE. D, day.
Impact of 20E on gene expression and transcriptional regulation.
A decline in the circulating levels of 20E on day 15 serves to trigger atrophy, while a further decline on day 17 commits the ISMs to die following adult eclosion late on day 18 (84). Injecting day 17 animals with 20E delays both adult eclosion and ISM death. Consequently, to identify ecdysteroid-regulated genes we compared the gene expression in day 18 animals to comparably aged animals that had been injected on day 17 with 20E. Out of 20,795 genes, 1,463, and 2,145 were induced and repressed, respectively (Fig. 7).
Fig. 7.

Gene Ontology term enrichment analysis for gene regulation by 20-hydroxyecdysone (20E). Animals were injected on day 17 with 20E and analyzed on day 18 for differential gene expression. Induced genes are displayed on the left (red), while repressed genes are shown on the right (blue).
The genes induced by 20E were associated with GO terms involved in DNA metabolic processes such as “DNA-dependent DNA replication” (GO:0006261), “DNA conformation change” (GO:0071103), “helicase activity” (GO:0004386), “DNA binding” (GO:0003677) and “RNA binding” (GO:0003723) (Fig. 7). Along with the GO terms detected, we observed the upregulation of nucleosome assembly protein 1 (Nap1) (Msex000075) from 98.40 RPKM on day 18 to 381.53 RPKM following treatment with 20E (276.19 RPKM on day 13). In addition, the GO terms related to cell cycle and development were also observed, e.g., “positive regulation of cell cycle process” (GO:0090068), “ribosome biogenesis” (GO:0042254), “regulation of MAPK cascade” (GO:0043408) and “mitogen-activated protein kinase binding” (GO:0051019). In D. melanogaster, it is known that Lk6 kinase promotes cell growth and development by phosphorylating eukaryotic translation initiation factor 4E (eIF4E) (4). We found that the Lk6 kinase homolog in M. sexta (Msex013719) displayed a gradual decrease during the ISM development, from 156.66 to 160.15 RPKM on day 13 to 22.73–23.34 RPKM on day 18. However, the expression level of Lk6 in the 20E-treated ISMs returned to day 13 levels (169.29–175.20 RPKM). We also observed that eIF4E (Msex004105) was also induced, from 30.38 RPKM on day 18 to 139.13 RPKM in response to 20E. Interestingly “response to DNA damage stimulus” was also detected (GO:0006974). In agreement with this GO term, expression of DNA repair protein RAD51 (Msex010633) was induced from 41.68 RPKM on day 18 to 217.74 RPKM following 20E treatment (75.86 RPKM on day 13). Interestingly, we found the homolog of protease inhibitor-like protein in another moth genus, Antheraea mylitta (Msex008966), was induced sevenfold from 45.03 RPKM on day 18 to 330.45 RPKM in response to 20E.
Since we observed a dramatic and coordinate increase in the ubiquitin-proteasome components on day 18 (Table 2), it was not surprising that 20E treatment led to their repression, e.g., “proteasomal ubiquitin-dependent protein catabolic process” (GO:0043161); “proteasome regulatory particle” (GO:0005838), and “proteasome complex” (GO:0000502) (Fig. 7). Other proteases were likewise repressed, like “peptidase activity” (GO:0008233). The same was true for autophagy-related protein 101 (Atg101; Msex002714), which is consistent with the detected GO term “salivary gland cell autophagic cell death” (GO:0035071) enriched in the downregulated genes.
Using the promoter sequences for the induced and repressed genes, we calculated TFBS motif enrichment to investigate the transcriptional regulation by 20E. The 10 and 49 TFBS motifs were enriched in the promoters of the upregulated and downregulated genes by 20E respectively. We found the 10 TFBS motifs in the promoters of the induced genes by 20E overlapped with those of the repressed genes by 20E [Anterior open (aop), bric a brac 1 (bab1), ci, dendritic arbor reduction 1 (dar1), hb, jim, klu, longitudinals lacking (lola), Pho and sr; Supplemental Table S3]. Interestingly the protein-protein interaction among the TFs of enriched TFBSs in the repressed genes were very similar to the ones enriched in C5 (Fig. 5D). The largest TF subnetwork of enriched TFBSs in the 20E repressed genes were composed of ecdysone pathway and circadian rhythm pathway components. Many TFs upregulated during atrophy were repressed by 20E such as Clk, cry and Eip74EF. However, EcR was consistently upregulated by 20E treatment.
miRNA expression during the ISM development.
MiRNAs (miRs) are 22 nucleotide-long small interfering RNAs that can bind to target sequences within mRNAs and inhibit protein expression either via translational repression or transcript degradation (11, 17). One or more miRs can exert control over the expression of entire developmental pathways (3, 44).
To determine if small interfering RNAs might regulate mRNA stability or translatability during atrophy or death, we generated and sequenced small RNA libraries at each stage of development from day 13 to day 18, plus 1 h PE and 20E treated. Of the sequences obtained, between 71.65 and 80.66% reads were successfully mapped to the M. sexta genome (Supplemental Table S4). In the miRBase v20, we found 93 annotated miRNAs in M. sexta. In contrast, there are 487 annotated miRs in the genome of the silk moth B. mori (10). B. mori and M. sexta are in the same order, Lepidoptera, and are closely related phylogenetically (36, 42). Consequently, we hypothesized that there are likely many more miRNAs in the M. sexta genome than previously identified. Using mirDeep2 (24), we predicted putative miRNAs in the M. sexta genome and found an additional 325 novel miRNA candidates, which brings the total number in line with Bombyx: 418 miRNAs for Manduca and 487 for B. mori. [After we had conducted our analyses, two papers appeared that report additional miRNA sequences from Manduca (108, 109).]
We further sought to determine if the length of the reads mapped to the miRNAs in M. sexta distributes within miRNA length range (5, 30). As shown in Fig. 8, we confirmed a peak of the miRNA mapping reads at 22 nt length in all samples. [It should be noted that there is a second size peak at ∼27 nt, which we have determined to be piRNAs, a different class of small interfering RNAs (70)]. A detailed analysis of these data is being submitted elsewhere (Tsuji J, Thomson T, Brown CK, Theurkauf WE, Wang Z, and Schwartz LM, unpublished observations). We used the miRNA annotation in M. sexta for the downstream analysis. The miRNA annotation used in this study is summarized in Supplemental Table S5.
Fig. 8.

Abundance and length distribution of small RNAs during intersegmental muscle development. Authentic microRNAs (miRNAs) that map to the genome are in black, while the gray bars reflect sequences that were not annotated. The majority of the small RNAs are ∼22 nucleotides long and map as miRNAs. The smaller peak at 27 nt may reflect piRNAs, a distinct class of small interfering RNAs. 20E, 20-hydroxyecdysone; D, day; PE, posteclosion.
We calculated miR expression values with miR mapping reads (67.15–87.55% of total mapped reads). There were 111 miRs that were expressed at least 10 ppm at least at one time point. We detected 31 miRs that were differentially expressed between at least two time points in all possible comparisons throughout the ISM developmental stages and 20E, which represents ∼7.5% of all miR sequences (Supplemental Fig. S3).
Compared with the miR expression on day 13, we found 10 that were upregulated and seven that were repressed during the ISM development (Fig. 9A). For upregulated miRs, we found that mse-miR-2a, mse-miR-87, and mse-miR-965 were stably induced during atrophy but repressed with the commitment of the ISMs to die on day 18. mse-miR-190, mse-miR-745, and two predicted miRs (predicted-scaffold00132-1 and predicted-scaffold00018-5) were elevated from days 14 to 17 and then again PE, but not on day 18. In contrast, mse-miR-34 was induced both before and after the initiation of death on day 18. Treatment with 20E failed to repress its expression. For downregulated miRs, the expression of mse-miR-2765 and two predicted miRs (predicted-scaffold00553-1 and predicted-scaffold01801-1) fell precipitously after day 13 including in the 20E-treated individuals.
Fig. 9.

Developmentally regulated microRNAs (miRNAs, also miRs) and the target genes. A: pairwise scatter plots of differentially expressed miRNAs between all days from day 13 to day 18, plus posteclosion (PE) and 20-hydroxyecdysone (20E)-treated animals. x-Axis, normalized read counts; y-axis, log2 fold-change. Differentially expressed miRs are denoted by a symbol shown in the table inset. B: expression of miRNAs and their putative target genes. The two panels on the right show the expression of the developmentally repressed miRNAs and the targets; the two on the left show the expression of the incrementally induced miRNAs and the targets. C: Gene Ontology (GO) terms enriched in mse-miR-92b (top) and mse-miR-6096 (bottom). Black, medium gray, and light gray indicate the GO terms of cellular component, molecular function, and biological process.
Posttranscriptional regulation during the ISM development.
Along with analyzing the miRNA expression, we predicted their putative target genes. Out of 20,795 genes in M. sexta, 13,692 genes had annotated 3′-UTRs in the Manduca Base. For the rest of the genes that lack the 3′-UTR the annotation, we predicted the 1,164 putative 3′-UTRs using the assembled transcripts with Cufflinks (100). The median length of the annotated and predicted 3′-UTRs were 838 and 274 bp, respectively (the median length of total 3′-UTRs = 766.5 bp). Using TargetScan (49), we found 14,260 genes (68.57% of all genes) have at least one predicted miRNA target site in the 3′-UTRs, and 10,539 genes (50.68% of all genes) have multiple target sites of one miRNA species within the gene bodies.
For further analysis, we focused only on the 31 differentially expressed miRs (Supplemental Fig. S3) and the target genes that were anticorrelated against the miR expression (Pearson correlation coefficient ≤ −0.6). The differentially expressed miRs repressed 3–664 genes as the posttranscriptional targets (the miRs and the target genes are summarized in Supplemental Table S5). Out of the 31 differentially expressed miRs, the 16 miRs had specific GO terms enriched in the target genes.
The miRs that displayed a gradual decrease from day 13 to day 18 tended to have targets with the transcripts of upregulated genes in ecdysone signaling, the ubiquitin-proteasome pathway, and daytime circadian rhythms pathway (Fig. 9B, Supplemental Table S5). mse-miR-92b targets included Clk, Eip74EF, ftz-f1, and proteasome subunit proteins (Prosbeta7, Prosbeta2, Pros28.1, Rpt3). mse-miR-2767 targets EcR and ftz-f1 in the ecdysone pathway components. These miR target sites were statistically significantly enriched in the 3′-UTRs of the genes clustered into C5 (mse-miR-92b, FDR = 2.90 × 10−6; mse-miR-2767, FDR = 3.30 × 10−5). Interestingly the target genes of the miRs include the genes required for the autophagosome formation. We observed the GO term enriched in both targets, “Atg1p signaling complex” (GO:0034273) (Fig. 9C). In addition to Apoptosis-linked gene-2 (ALG-2) and Autophagy-related 13 and 16 genes (Atg13, Atg16), the SCLP mRNA, which is dramatically increased on day 18 also had a mse-miR-92b target site.
The miRNAs that displayed incremental induction during ISM development were predicted to function as suppressors of downregulated genes during development (Fig. 9B). mse-miR-34 and mse-miR-6096 target 238 genes and 577 genes clustered into C2 (the total target genes, 266 and 644 genes, respectively). The target sites of those miRNAs were statistically enriched in the 3′-UTRs of the genes clustered into C2 (mse-miR-34 FDR = 5.24 × 10−3, mse-miR-6096, FDR = 8.88 × 10−11). Although mse-miR-34 expression was not associated with any enriched GO terms, mse-miR-6096 was associated with enrichment of numerous metabolic GO terms as well as “mitogen-activated protein kinase binding” (GO:0051019) (Fig. 9C).
While the negative correlation between miRs and their potential mRNA targets is intriguing and suggestive of a potential regulatory mechanism for tuning protein expression during ISM development, we wanted to test this hypothesis more formally. We selected miR-92b, which declines continuously during ISM development, and one of its putative target transcripts, SCLP, which is transiently induced with atrophy and then more dramatically upregulated on day 18 (43). A 411nt portion of the SCLP 3′-UTR was cloned downstream from the luciferase gene from the sea pen Renilla within the pFila vector, which also expresses firefly luciferase as an internal normalization control (7). Mammalian COS-1 cells were transiently transfected with the constructs and then treated with different 22-nt double-stranded RNAs before performing dual luciferase assays. As a control, we treated cells with an miR that shared 100% complementarity with a target sequence within the test 3′-UTR, which functionally served as an siRNA. As anticipated, this treatment resulted in a substantial (∼80%) repression of expression (Fig. 10A). We then tested authentic miR-92b for its ability to alter luciferase activity and observed an ∼40% repression.
Fig. 10.

MicroRNA (miR)-92 represses small cytoplasmic leucine rich repeat protein (SCLP) expression but not transcript abundance. A: the normalized relative luciferase activity (Renilla/Firefly) of the SCLP constructs when treated with miR-92b. The relative luciferase activity was compared 24 h after cotransfection of COS-1 cells with the construct [SCLP-3′-untranslated region (UTR) or SCLP-siRNA] and 1 µl of 20 nM miR-92b mimic. The 100% complementarity between miR-92b against the SCLP-3′-UTR repressed luciferase expression and served as a positive control for silencing. The use of miR-92b that targets the SCLP 3′-UTR also repressed expression. B: miR-92b was cotransfected with either SCLP-siRNA, a vector with a 3′-UTR target site completely complementary to miR-92b, or SCLP-3′-UTR, a vector with a 3′-UTR target site amplified from the 3′-UTR of SCLP. After 24 h, the cells were lysed, and Renilla luciferase expression was measured via quantitative (q)PCR and compared by the ∆∆Ct- method. Blue = control and red = treated. The use of an siRNA that was 100% complementary to the 3′-UTR led to transcript degradation, while the use of miR-92b against the authentic SCLP 3′-UTR did not alter transcript abundance, suggesting a block in translation. The Student’s t test was used to test statistical significance for the pairwise comparisons.
The ability of miR-92b to repress expression could have occurred either by blocking translation or by facilitating transcript degradation. To determine which of these two processes might be involved, we repeated the transfections described above and performed qPCR analysis using primers directed against luciferase (Fig. 10B). The use of a miR that is 100% complementary to the target sequence in the luciferase mRNA (siRNA) served as a positive control and resulted in a 69.3% decline in mRNA abundance. In contrast, transfection with miR-92B did not alter luciferase mRNA abundance, suggesting that the block in luciferase expression in our experiments reflected transcriptional repression rather than transcript destruction.
DISCUSSION
The ISMs from Manduca afford a unique system for examining the molecular mechanisms that mediate naturally occurring skeletal muscle atrophy and death. In the current study we employed unbiased RNA-Seq-based methodology to examine simultaneously all of the differentially expressed mRNAs and microRNAs across development. We were able to verify the accuracy of our RNA-Seq data sets in several ways, including qPCR (Fig. 2), Northern blots, and Western blots (e.g., 31, 37, 43, 79, 80, 93, 104). In each case, the developmental changes observed via RNA-Seq were confirmed by the other independent methods.
ISM Atrophy.
Prior to day 15 of pupal-adult development, the ISMs are in a homeostatic state and are neither growing nor regressing. This is seen by examining muscle mass (Supplemental Fig. S1), cellular physiology (81), and by comparing the patterns of gene expression between days 13 and 14, where transcript abundance is almost identical (Fig. 1, Supplemental Fig. S2). In contrast, beginning on day 15 there are dramatic changes in muscle mass (Supplemental Fig. S1) and gene expression, with 125 induced and 235 repressed (Fig. 1, Supplemental Fig. S2). One key aspect of atrophy is a general shift toward catabolism. Genes associated with the extracellular matrix and the contractile apparatus, including actin and myosin heavy chain (cluster 2), are constitutively expressed and then decline precipitously (Fig. 4). Teleologically, it makes sense that the cells would not expend the energy required to make contractile proteins at a time when they are undergoing atrophy. It should be also noted that the coordinated loss of contractile protein mRNAs means that the contractile apparatus does not selectively lose specific essential proteins, meaning that the muscle retains its normal physiological properties even as it rapidly loses mass (81).
ISM atrophy is accompanied by enhanced expression of virtually all of the components of the ubiquitin-proteasome system (UPS) (cluster 5). This includes ubiquitin conjugases (E2s), ubiquitin ligases (E3s), and the enzyme protein NH2-terminal asparagine amidohydrolase (NTAN1), which serves as a mediator of ubiquitin-dependent protein degradation via the N-End rule (29). In addition, there is enhanced expression of more than 65 genes required for the assembly of the 26S proteasome (53). The coordinated induction of proteasome genes helps facilitate the stoichiometric assembly of the complex (65).
The expression of the majority ribosomal genes, including those encoding for RNA helicases and snoRNA binding, transiently increases at the beginning of atrophy and then decline precipitously (cluster 2). The helicases facilitate the recruitment and release of the snoRNAs that modify ribosomal RNA during ribosome biogenesis (59). The repression of these genes presumably plays a number of key roles in facilitating the rapid loss of protein that accompanies ISM atrophy.
Intermediary metabolism genes are also induced transiently during atrophy and then plummet (cluster 4). In particular, enzymes involved in glycogen degradation, such as 4-alpha-glucanotransferase and glycogen debrancher enzymes, are induced, which facilitates the production of glucose to support glycolysis. In agreement with this interpretation, several glycolytic enzymes are also induced, including fructose-bisphosphate aldolase and NADH dehydrogenase. Consequently, atrophy is associated with aerobic metabolism to generate the ATP required for protein degradation.
Atrophy is also associated with enhanced histone H3K4 methylation (cluster 4), a process that enhances transcriptional activation (105). Given the rapid and dramatic changes in gene expression that accompany ISM atrophy and death, it would be informative to map changes in histone methylation marks across development. Interestingly, despite the intense investigation of skeletal muscle atrophy, few studies have addressed methylation status (86).
Lastly, ISM atrophy is also associated with the repression of epithelial genes required for cuticle synthesis, including chitin (cluster 1). There are two likely reasons for this. The first is that the muscles attach to the epithelium at the intersegmental boundaries, and some of these cells were recovered when the ISMs were isolated for analysis since we didn’t want to damage the fibers during dissection. However, the main source of cuticle within the tissue is the tracheal system. Unlike mammals, which employ a closed circulatory system and a respiratory pigment to deliver oxygen to tissues, insects utilize a series of hollow cuticular tubes to facilitate gas exchange (6). Since the cuticular lining of the trachea is withdrawn through the old pupal cuticle during eclosion, there may be associated changes in its structure to make it more pliable.
ISM death.
On day 18, when the ISMs become committed to die, there is a dramatic shift in gene expression, with more genes induced than repressed. Unlike the changes that accompany atrophy where the amplitude of these changes is modest, the commitment to die involves some profound examples of gene induction. The expression of more than a dozen genes increases 250-fold or more in 24 h (cluster 6). For example, Acheron (also known as Larp6) mRNA is induced 1,000-fold on day 18 (90, 104). Acheron is a member of the Lupus Antigen family of RNA binding proteins (104) and functions as a survival protein that protects some terminally differentiated cells and cancers from cell death (89, 90). EH triggers ISM death by inducing the expression of cyclic GMP (83), which induces Acheron phosphorylation and degradation. This leads to the mitochondrial release and degradation of cytochrome c and cellular demise (90).
The loss of cytochrome c suggests that mitochondrial respiration collapses in the condemned ISMs. Indeed, there is an upregulation of genes associated with gluconeogenesis, which reflects a likely shift from aerobic to anaerobic metabolism. For example, the transcription factor Klf15, which regulates the hydrolysis of fat in skeletal muscle and drives gluconeogenesis and amino acid catabolism (32), is induced 13-fold during ISM atrophy in Manduca. The availability of ATP is likely critical for the efficient degradation of macromolecules since the ISMs are not phagocytosed when they die (38). In agreement with this, there is an induction of membrane transporters that presumably facilitate recycling of valuable macromolecules.
The two-stage induction of the UPS presumably serves distinct roles for the developing muscles. The transient increase with the onset of atrophy helps shift the cells from an anabolic state to a catabolic one, whereby muscle protein is progressively lost. The subsequent sharp increase on day 18 helps mediate the wholesale loss of muscle protein during death (31, 37). Biochemical studies have demonstrated that the percentage of ubiquitin that is covalently attached to cellular proteins increases from ∼50% before adult eclosion to almost 100% with the initiation of death (31). This dramatic shift reflects the aggressive destruction of cellular protein. In some tissues, the UPS system also plays regulatory roles in the control of cell death by selectively targeting key cell death pathway components like the antiapoptotic protein Death-associated inhibitor of apoptosis 1 (DIAP1) (19).
The two genes that are most dramatically induced with ISM death encode members of the astacin family of matrix metalloproteinases, which are upregulated ∼3,000-fold between days 16 and 18. Originally identified in the digestive gastric juices from crayfish, astacins are also found in the venom of the brown recluse spider Loxosceles and the tentacles of certain jellyfish (52, 101). Astacins have promiscuous substrate specificities that include skeletal muscle myosin heavy chain (96). One intriguing possibility is that these proteases initiate indiscriminate cleavage of ISM proteins, which in turn then generates fragments of misfolded proteins that are good substrates for the UPS.
Lastly, death is associated with the induction of immune system genes. The reason for this is unclear but innate immune system genes are also induced with muscle damage in Drosophila (28), although in Manduca, this change in gene expression precedes the initiation of muscle damage.
MiRNAs and muscle atrophy and death.
In addition to analyzing changes in transcript abundance during ISM development, we also examined the expression of small interfering RNAs. While our primary focus was on miRs, which are ∼22 nt long, we also noted a second peak of small RNAs that were ∼27 nt in length, which may represent PIWI-interacting RNA (piRNAs), a distinct class of small interfering RNAs (Fig. 8; Tsuji J, Thomson T, Brown CK, Theurkauf WE, Wang Z, and Schwartz LM, unpublished observations). piRNAs are largely confined to gonads where they function to repress transposon expression to protect the integrity of the germline genome (99). We found that piRNA expression is repressed when the ISMs become committed to die and that this is correlated with the derepression of certain DNA transposons. This may represent a novel mechanism for insuring genome damage in cells that do not die by apoptosis.
We found that ∼7.5% (31 unique miRs) were differentially expressed between at least two time points during ISM development. Single miRs can repress protein expression by either blocking translation or by facilitating transcript degradation (35). A single miRNA can modulate entire developmental pathways and exert dramatic effects on cellular physiology through the simultaneous regulation of multiple transcripts (44, 48). We functionally tested the hypothesis that miRs could influence atrophy- or death-associated gene expression by demonstrating that miR-92b could block the translation of SCLP mRNA (Fig. 10). Thus, the dramatic increase in SCLP protein and other death-associated proteins is likely a consequence of both an increase in transcription coupled with the derepression of translation (43).
Not surprisingly, each of the miRs investigated in this study has putative target sequences in a large number of mRNAs. However, we were struck by the observation that in most cases, these targets were all members of a single cluster. For example, more than 20% of the genes in cluster 5 (516 of the 2,511 genes) could be targeted by miR-92b. Thus, a change in the abundance of a single miR would likely modulate the expression of major pathways associated with atrophy and/or death. This observation may explain the disparity that we have noted between the abundance of mRNAs and their encoded proteins. For example, the abundance of mRNA for ubiquitin-proteasome pathway components increase transiently during atrophy and then again with the commitment to die, for a total increase of about fivefold (Figs. 2, 4). However, expression of proteasomal proteins increases at least two orders of magnitude on day 18 (37). While this presumably reflects a number of regulatory processes including protein stability, the loss of miRs is certainly well positioned to exert a major influence on the developmental regulation of protein abundance.
A number of the miRs identified in this study have been reported to influence atrophy and cell death in other organisms (63, 107). For example, miR-92 serves a survival role in a variety of different cell types including embryonic stem cells and rod cells within the retina, in part due to its ability to repress the expression of proapoptotic proteins like BIM (71, 94). This observation has clinical relevance to human muscle disorders since miR-92 expression is repressed in two types of human cardiac failure- idiopathic dilated cardiomyopathy and ischemic cardiomyopathy (92).
Control of ISM gene expression.
One surprising result of our analysis was that almost all of these changes in gene expression could be accounted for by only six discrete patterns. The coordinated control of gene expression during development by a common set of cis-acting regulatory sequences has been referred to as a “gene battery” (15). For example, ribosome biogenesis has been studied in detail in a variety of organisms and shown to be coordinately controlled by specific transcriptional machinery (e.g., 51). These data provide an opportunity to generate hypothetical transcriptional regulatory networks that control the expression patterns observed from our cluster analysis (Fig. 5). For example, the Polycomb complex serves as a central node in the control of gene expression in the ISMs. Polycomb regulatory sequences are found in 92.5% of the genes in cluster 5, which are dramatically induced on day 18. While Pho is typically associated with transcriptional repression (50), the closely related protein pleiohomeotic-like (Phol) can facilitate transcriptional activation (75). BLAST searches with both Drosophila Pho and Phol retrieved the same sequence in Manduca, suggesting that a distinct Phol may not exist in Manduca. One possibility is that Pho may serve multiple roles in transcriptional regulation, especially given its ability to recruit a wide range of transcription regulators to promoters (64).
In Manduca, ISM atrophy and death are regulated by the declining ecdysteroid levels (84). We injected day 17 animals with 20E to delay eclosion and examined the resulting patterns of gene expression (Fig. 3, Supplemental Table S4). 20E blocked the anticipated induction or repression of many pathway components and kept transcript abundance at day 17 levels, such as the induction of the UPS system and the anticipated induction of Acheron (Fig. 2). In addition to providing insight into the steroid control of cell death, these data provide a comprehensive list of 20E-regulated genes that may be involved in developmental processes in other tissues (Fig. 7).
Control of PCD in Manduca versus Drosophila.
Drosophila has been a widely used genetic model for cell death, most notably of larval muscles, salivary glands, and midgut cells following pupariation (69). The best studied example of muscle death focused on the dorsal exterior oblique muscle (DEOM) (112). Two transcription factors that are implicated in DEOM death are the orphan nuclear receptors, FTZ-F1 and HR39. These proteins share sequence similarity and bind to the same promoter regions but tend to be expressed reciprocally and to antagonize one another in cells that are programmed to die (112). In contrast to their pattern of expression in the DEOMs, we observed that HR39 and FTZ-F1 are coordinately induced in the ISMs on day 18, suggesting that they may play different roles in this tissue. This may reflect differences in the nature of the ecdysteroid control of development for the ISMs (rising vs. declining titers) or perhaps other regulatory differences in the process of cell death.
Two additional nuclear proteins have been shown to regulate muscle degeneration in Drosophila are Chromator, which drives muscle degeneration, and East, which represses it (106). However, neither East nor Chromator is differentially expressed in the ISMs on day 18, reducing the likelihood that they play significant roles in either the timing or nature of ISM cell death.
Muscle atrophy in Manduca and mammals.
One of the motivations for undertaking this study was to gain insights that might have relevance to skeletal muscle atrophy and cell death in mammals. There is a general model for mammalian muscle that suggests that a wide range of atrophic stimuli, as diverse as denervation and sepsis, leads to a decrease in PI3-kinase/AKT activity, which in turn activates FOXO transcription factors and the subsequent expression of several RING finger ubiquitin ligases, including Atrogen-1, MuRF1, and MUSA1 (9, 26, 67, 73). Knock-down of atrogen-1 and MuRF1 abrogates atrophy both in vitro and in vivo (9, 26). In addition to the UPS, the PI3K/AKT pathway also activates other proteolytic systems including lysosomal and autophagic pathways within the atrophic muscle (58). The effects of FOXO on atrophy are antagonized by the serine/threonine-protein kinase mTOR, which has been shown to be a negative regulator of skeletal muscle atrophy in both insects and mammals (67).
While these same upstream regulatory components are conserved in insects and regulate muscle growth and atrophy in the fruit fly Drosophila (66), they do not appear to be major regulators of ISM atrophy at the end of metamorphosis in Manduca. At the RNA level, there are no changes in the expression of FOXO, mTOR, or mTor pathway components such as Ragulator complex protein LAMTOR5 or the Regulatory-associated protein of mTOR (data not shown). Moreover, despite the fact that the UPS system is induced with ISM atrophy and death, there is no change in the expression of the Manduca Atrogen-1 ortholog during development. (A MuRF1 homolog has yet to be identified in Manduca.) These data suggest that the upstream regulatory pathways for ISM atrophy are distinct from those employed by mammalian muscle. This may not be so surprising since ISM atrophy is so tightly controlled by the titer of 20E rather than environmental stimuli like nutritional status or neuronal activity.
That said, it does appear that many of the downstream effectors of atrophy are conserved between Manduca and mammals. In each system, there are increases in the UPS and autophagic and lysosomal systems (8, 46, 47, 54) and decreases in both ribosome biogenesis (12, 57) and the expression of contractile protein genes (18, 54).
Summary
In summary, the data presented here provide a range of new insights into the molecular pathways that are differentially regulated during skeletal muscle atrophy and death. There was a dramatic reduction in the expression genes associated with the contractile apparatus, respiration, and adhesion and a concurrent induction of ribosome biogenesis and several proteolytic systems. These and other proteases were induced with death, in some cases by more than three orders of magnitude, such as the astacin family of matrix metalloproteinases. In addition to control of the level of transcription, it appears that a decline in the levels of specific miRNAs can greatly exacerbate expression at the protein level. In at least one case, this appears to be via derepression of translation. In addition, these data sets offer a unique resource for hypothesis generation, and perhaps a systems biology analysis to examine gene networks during development that complement other studies of skeletal muscle atrophy in mammals (21).
GRANTS
This work was supported by a Life Sciences Moment Fund grant from the University of Massachusetts Center for Clinical and Translational Science, the Eugene M. and Ronnie Isenberg Professorship Endowment to L. M. Schwartz, and National Institute of Child Health and Human Development Grant P01HD078253 to W. E. Theurkauf and Z. Weng.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.M.S., W.E.T., and Z.W. conceived and designed research; L.M.S., T.T., E.C., C.K.B., and J.O. performed experiments; J.T., L.M.S., T.T., C.B., and X.D. analyzed data; L.M.S., J.T., T.T., C.B., C.K.B., W.E.T., and Z.W. interpreted results of experiments; J.T., C.B., and L.M.S. prepared figures; J.T., and L.M.S. drafted manuscript; J.T., L.M.S., and T.T. edited and revised manuscript; L.M.S., T.T., E.C., C.K.B., J.O., C.B., X.D., W.E.T., and Z.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Wendy Smith for the Manduca, Yanzhen Bi for providing the pFila vector, and Stephen Richards for help with Manduca Base. We also thank a number of colleagues for helpful discussions, including: Drs. Leonid Pobezinsky, Craig Woodard, Alexander Gerson, and Michele Markstein.
Current addresses: J. Tsuji, Genomics Platform, Broad Institute of MIT and Harvard, Cambridge, MA; J. Oppenheimer and E. Chan, University of Massachusetts Medical School, Worcester, MA; X. Dong, Genomics and Bioinformatics Hub, Brigham and Women’s Hospital, Boston, MA.
REFERENCES
- 1.Almeida MS, Bray SJ. Regulation of post-embryonic neuroblasts by Drosophila Grainyhead. Mech Dev 122: 1282–1293, 2005. doi: 10.1016/j.mod.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 215: 403–410, 1990. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Ambros V. MicroRNAs and developmental timing. Curr Opin Genet Dev 21: 511–517, 2011. doi: 10.1016/j.gde.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arquier N, Bourouis M, Colombani J, Léopold P. Drosophila Lk6 kinase controls phosphorylation of eukaryotic translation initiation factor 4E and promotes normal growth and development. Curr Biol 15: 19–23, 2005. doi: 10.1016/j.cub.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297, 2004. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Best BT. Single-cell branching morphogenesis in the Drosophila trachea. Dev Biol 451: 5–15, 2019. doi: 10.1016/j.ydbio.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Bi Y, Zheng X, Shao C, Pan W, Jiang L, Ouyang H. Construction and application of a built-in dual luciferase reporter for microRNA functional analysis. Electron J Biotechnol 14: 2, 2011. doi: 10.2225/vol14-issue2-fulltext-9. [DOI] [Google Scholar]
- 8.Bialek P, Morris C, Parkington J, St Andre M, Owens J, Yaworsky P, Seeherman H, Jelinsky SA. Distinct protein degradation profiles are induced by different disuse models of skeletal muscle atrophy. Physiol Genomics 43: 1075–1086, 2011. doi: 10.1152/physiolgenomics.00247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 10.Cai Y, Yu X, Zhou Q, Yu C, Hu H, Liu J, Lin H, Yang J, Zhang B, Cui P, Hu S, Yu J. Novel microRNAs in silkworm (Bombyx mori). Funct Integr Genomics 10: 405–415, 2010. doi: 10.1007/s10142-010-0162-7. [DOI] [PubMed] [Google Scholar]
- 11.Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14: 100–112, 2013. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaillou T, Kirby TJ, McCarthy JJ. Ribosome biogenesis: emerging evidence for a central role in the regulation of skeletal muscle mass. J Cell Physiol 229: 1584–1594, 2014. doi: 10.1002/jcp.24604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chavez LO, Leon M, Einav S, Varon J. Beyond muscle destruction: a systematic review of rhabdomyolysis for clinical practice. Crit Care 20: 135, 2016. doi: 10.1186/s13054-016-1314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claridge-Chang A, Wijnen H, Naef F, Boothroyd C, Rajewsky N, Young MW. Circadian regulation of gene expression systems in the Drosophila head. Neuron 32: 657–671, 2001. doi: 10.1016/S0896-6273(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 15.Davidson EH. Genomic Regulatory Systems: Development and Evolution. San Diego: Academic Press, 2001. [Google Scholar]
- 16.Denton D, Aung-Htut MT, Lorensuhewa N, Nicolson S, Zhu W, Mills K, Cakouros D, Bergmann A, Kumar S. UTX coordinates steroid hormone-mediated autophagy and cell death. Nat Commun 4: 2916, 2013. doi: 10.1038/ncomms3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dexheimer PJ, Cochella L. MicroRNAs: From Mechanism to Organism. Front Cell Dev Biol 8: 409, 2020. doi: 10.3389/fcell.2020.00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhahbi JM, Atamna H, Boffelli D, Martin DI, Spindler SR. mRNA-Seq reveals complex patterns of gene regulation and expression in the mouse skeletal muscle transcriptome associated with calorie restriction. Physiol Genomics 44: 331–344, 2012. doi: 10.1152/physiolgenomics.00129.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, Deas E, Meier P. Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat Cell Biol 5: 467–473, 2003. doi: 10.1038/ncb984. [DOI] [PubMed] [Google Scholar]
- 20.Dudley GA, Hather BM, Buchanan P. Skeletal muscle responses to unloading with special reference to man. J Fla Med Assoc 79: 525–529, 1992. [PubMed] [Google Scholar]
- 21.Ehmsen JT, Kawaguchi R, Mi R, Coppola G, Höke A. Longitudinal RNA-Seq analysis of acute and chronic neurogenic skeletal muscle atrophy. Sci Data 6: 179, 2019. doi: 10.1038/s41597-019-0185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fahrbach SE, Nambu JR, Schwartz LM. Programmed cell death in insects In: Insect Molecular Biology and Biochemistry, edited by Gilbert LI. ScienceDirect: p. 419–449, 2012. [Google Scholar]
- 23.Fielding RA, Vellas B, Evans WJ, Bhasin S, Morley JE, Newman AB, Abellan van Kan G, Andrieu S, Bauer J, Breuille D, Cederholm T, Chandler J, De Meynard C, Donini L, Harris T, Kannt A, Keime Guibert F, Onder G, Papanicolaou D, Rolland Y, Rooks D, Sieber C, Souhami E, Verlaan S, Zamboni M; International Working Group on Sarcopenia . Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J Am Med Dir Assoc 12: 249–256, 2011. doi: 10.1016/j.jamda.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52, 2012. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao Y, Arfat Y, Wang H, Goswami N. Muscle atrophy induced by mechanical unloading: mechanisms and potential countermeasures. Front Physiol 9: 235, 2018. doi: 10.3389/fphys.2018.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445, 2001. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018, 2011. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green N, Walker J, Bontrager A, Zych M, Geisbrecht ER. A tissue communication network coordinating innate immune response during muscle stress. J Cell Sci 131: jcs217943, 2018. doi: 10.1242/jcs.217943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grigoryev S, Stewart AE, Kwon YT, Arfin SM, Bradshaw RA, Jenkins NA, Copeland NG, Varshavsky A. A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J Biol Chem 271: 28521–28532, 1996. doi: 10.1074/jbc.271.45.28521. [DOI] [PubMed] [Google Scholar]
- 30.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524, 2014. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 31.Haas AL, Baboshina O, Williams B, Schwartz LM. Coordinated induction of the ubiquitin conjugation pathway accompanies the developmentally programmed death of insect skeletal muscle. J Biol Chem 270: 9407–9412, 1995. doi: 10.1074/jbc.270.16.9407. [DOI] [PubMed] [Google Scholar]
- 32.Haldar SM, Jeyaraj D, Anand P, Zhu H, Lu Y, Prosdocimo DA, Eapen B, Kawanami D, Okutsu M, Brotto L, Fujioka H, Kerner J, Rosca MG, McGuinness OP, Snow RJ, Russell AP, Gerber AN, Bai X, Yan Z, Nosek TM, Brotto M, Hoppel CL, Jain MK. Kruppel-like factor 15 regulates skeletal muscle lipid flux and exercise adaptation. Proc Natl Acad Sci USA 109: 6739–6744, 2012. doi: 10.1073/pnas.1121060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hennig C. Dissolution point and isolation robustness: Robustness criteria for general cluster analysis methods. J Multivariate Anal 99: 1154–1176, 2008. doi: 10.1016/j.jmva.2007.07.002. [DOI] [Google Scholar]
- 34.Hennig C. Cluster-wise assessment of cluster stability. Comput Stat Data Anal 52: 258–271, 2007. doi: 10.1016/j.csda.2006.11.025. [DOI] [Google Scholar]
- 35.Iwakawa HO, Tomari Y. The Functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol 25: 651–665, 2015. doi: 10.1016/j.tcb.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int J Biol Sci 5: 351–365, 2009. doi: 10.7150/ijbs.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones ME, Haire MF, Kloetzel PM, Mykles DL, Schwartz LM. Changes in the structure and function of the multicatalytic proteinase (proteasome) during programmed cell death in the intersegmental muscles of the hawkmoth, Manduca sexta. Dev Biol 169: 436–447, 1995. doi: 10.1006/dbio.1995.1159. [DOI] [PubMed] [Google Scholar]
- 38.Jones ME, Schwartz LM. Not all muscles meet the same fate when they die. Cell Biol Int 25: 539–545, 2001. doi: 10.1006/cbir.2000.0669. [DOI] [PubMed] [Google Scholar]
- 39.Kanost MR, Arrese EL, Cao X, Chen YR, Chellapilla S, Goldsmith MR, Grosse-Wilde E, Heckel DG, Herndon N, Jiang H, Papanicolaou A, Qu J, Soulages JL, Vogel H, Walters J, Waterhouse RM, Ahn SJ, Almeida FC, An C, Aqrawi P, Bretschneider A, Bryant WB, Bucks S, Chao H, Chevignon G, Christen JM, Clarke DF, Dittmer NT, Ferguson LCF, Garavelou S, Gordon KHJ, Gunaratna RT, Han Y, Hauser F, He Y, Heidel-Fischer H, Hirsh A, Hu Y, Jiang H, Kalra D, Klinner C, König C, Kovar C, Kroll AR, Kuwar SS, Lee SL, Lehman R, Li K, Li Z, Liang H, Lovelace S, Lu Z, Mansfield JH, McCulloch KJ, Mathew T, Morton B, Muzny DM, Neunemann D, Ongeri F, Pauchet Y, Pu LL, Pyrousis I, Rao XJ, Redding A, Roesel C, Sanchez-Gracia A, Schaack S, Shukla A, Tetreau G, Wang Y, Xiong GH, Traut W, Walsh TK, Worley KC, Wu D, Wu W, Wu YQ, Zhang X, Zou Z, Zucker H, Briscoe AD, Burmester T, Clem RJ, Feyereisen R, Grimmelikhuijzen CJP, Hamodrakas SJ, Hansson BS, Huguet E, Jermiin LS, Lan Q, Lehman HK, Lorenzen M, Merzendorfer H, Michalopoulos I, Morton DB, Muthukrishnan S, Oakeshott JG, Palmer W, Park Y, Passarelli AL, Rozas J, Schwartz LM, Smith W, Southgate A, Vilcinskas A, Vogt R, Wang P, Werren J, Yu XQ, Zhou JJ, Brown SJ, Scherer SE, Richards S, Blissard GW. Multifaceted biological insights from a draft genome sequence of the tobacco hornworm moth, Manduca sexta. Insect Biochem Mol Biol 76: 118–147, 2016. doi: 10.1016/j.ibmb.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42, D1: D68–D73, 2014. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kristensen NP, Skalski AW. Phylogeny and paleontology in Lepidoptera: Moths and butterflies, 1 Evolution, systematics, and biogeography In: Handbook of Zoology (Kristensen NP, editor). Berlin, New York: De Gruyter, 1999, vol IV, Part 35, p. 7–25. [Google Scholar]
- 43.Kuelzer F, Kuah P, Bishoff ST, Cheng L, Nambu JR, Schwartz LM. Cloning and analysis of small cytoplasmic leucine-rich repeat protein (SCLP), a novel, phylogenetically-conserved protein that is dramatically up-regulated during the programmed death of moth skeletal muscle. J Neurobiol 41: 482–494, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 44.Lai X, Wolkenhauer O, Vera J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res 44: 6019–6035, 2016. doi: 10.1093/nar/gkw550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25, 2009. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51, 2004. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 47.Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, Goldberg AL. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest 104: 1411–1420, 1999. doi: 10.1172/JCI7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science 294: 862–864, 2001. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 49.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20, 2005. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 50.Lewis EB. A gene complex controlling segmentation in Drosophila. Nature 276: 565–570, 1978. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Zheng Y, Hu H, Li X. Integrative analyses shed new light on human ribosomal protein gene regulation. Sci Rep 6: 28619, 2016. doi: 10.1038/srep28619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu G, Zhou Y, Liu D, Wang Q, Ruan Z, He Q, Zhang L. Global transcriptome analysis of the tentacle of the jellyfish cyanea capillata using deep sequencing and expressed sequence tags: insight into the toxin- and degenerative disease-related transcripts. PLoS One 10: e0142680, 2015. doi: 10.1371/journal.pone.0142680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, Ciechanover A. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res 26: 869–885, 2016. doi: 10.1038/cr.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Llano-Diez M, Fury W, Okamoto H, Bai Y, Gromada J, Larsson L. RNA-sequencing reveals altered skeletal muscle contraction, E3 ligases, autophagy, apoptosis, and chaperone expression in patients with critical illness myopathy. Skelet Muscle 9: 9, 2019. doi: 10.1186/s13395-019-0194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lockshin RA, Williams CM. Programmed Cell Death–I. Cytology of degeneration in the intersegmental muscles of the Pernyi silkmoth. J Insect Physiol 11: 123–133, 1965. doi: 10.1016/0022-1910(65)90099-5. [DOI] [PubMed] [Google Scholar]
- 56.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550–558, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Machida M, Takeda K, Yokono H, Ikemune S, Taniguchi Y, Kiyosawa H, Takemasa T. Reduction of ribosome biogenesis with activation of the mTOR pathway in denervated atrophic muscle. J Cell Physiol 227: 1569–1576, 2012. doi: 10.1002/jcp.22871. [DOI] [PubMed] [Google Scholar]
- 58.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471, 2007. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 59.Martin R, Straub AU, Doebele C, Bohnsack MT. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol 10: 4–18, 2013. doi: 10.4161/rna.21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marty E, Liu Y, Samuel A, Or O, Lane J. A review of sarcopenia: Enhancing awareness of an increasingly prevalent disease. Bone 105: 276–286, 2017. doi: 10.1016/j.bone.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 61.Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, Lim J, Shyr C, Tan G, Zhou M, Lenhard B, Sandelin A, Wasserman WW. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42 Database issue: D142–D147, 2014. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34 Database issue: D108–D110, 2006. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCormick R, Goljanek-Whysall K. MicroRNA Dysregulation in aging and pathologies of the skeletal muscle. Int Rev Cell Mol Biol 334: 265–308, 2017. doi: 10.1016/bs.ircmb.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 64.Meier K, Brehm A. Chromatin regulation: how complex does it get? Epigenetics 9: 1485–1495, 2014. doi: 10.4161/15592294.2014.971580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Krüger E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J Biol Chem 278: 21517–21525, 2003. doi: 10.1074/jbc.M301032200. [DOI] [PubMed] [Google Scholar]
- 66.Mensah LB, Davison C, Fan SJ, Morris JF, Goberdhan DC, Wilson C. Fine-tuning of PI3K/AKT signalling by the tumour suppressor PTEN is required for maintenance of flight muscle function and mitochondrial integrity in ageing adult Drosophila melanogaster. PLoS One 10: e0143818, 2015. doi: 10.1371/journal.pone.0143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA, Sandri M. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 6: 6670, 2015. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morgan JE, Prola A, Mariot V, Pini V, Meng J, Hourde C, Dumonceaux J, Conti F, Relaix F, Authier FJ, Tiret L, Muntoni F, Bencze M. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat Commun 9: 3655–3659, 2018. [Erratum in Nat Commun 9: 4107, 2018] doi: 10.1038/s41467-018-06057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nicolson S, Denton D, Kumar S. Ecdysone-mediated programmed cell death in Drosophila. Int J Dev Biol 59: 23–32, 2015. doi: 10.1387/ijdb.150055sk. [DOI] [PubMed] [Google Scholar]
- 70.Ozata DM, Gainetdinov I, Zoch A, O’Carroll D, Zamore PD. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20: 89–108, 2019. doi: 10.1038/s41576-018-0073-3. [DOI] [PubMed] [Google Scholar]
- 71.Pernaute B, Spruce T, Smith KM, Sánchez-Nieto JM, Manzanares M, Cobb B, Rodríguez TA. MicroRNAs control the apoptotic threshold in primed pluripotent stem cells through regulation of BIM. Genes Dev 28: 1873–1878, 2014. doi: 10.1101/gad.245621.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 201: 499–510, 2013. doi: 10.1083/jcb.201212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004. doi: 10.1016/S0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280: 4294–4314, 2013. doi: 10.1111/febs.12253. [DOI] [PubMed] [Google Scholar]
- 75.Schuettengruber B, Ganapathi M, Leblanc B, Portoso M, Jaschek R, Tolhuis B, van Lohuizen M, Tanay A, Cavalli G. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol 7: e13, 2009. doi: 10.1371/journal.pbio.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwartz LM. Skeletal muscles do not undergo apoptosis during either atrophy or programmed cell death-Revisiting the myonuclear domain hypothesis. Front Physiol 9: 1887, 2019. doi: 10.3389/fphys.2018.01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schwartz LM, Brown C, McLaughlin K, Smith W, Bigelow C. The myonuclear domain is not maintained in skeletal muscle during either atrophy or programmed cell death. Am J Physiol Cell Physiol 311: C607–C615, 2016. doi: 10.1152/ajpcell.00176.2016. [DOI] [PubMed] [Google Scholar]
- 78.Schwartz LM, Jones ME, Kosz L, Kuah K. Selective repression of actin and myosin heavy chain expression during the programmed death of insect skeletal muscle. Dev Biol 158: 448–455, 1993. doi: 10.1006/dbio.1993.1202. [DOI] [PubMed] [Google Scholar]
- 79.Schwartz LM, Kosz L, Kay BK. Gene activation is required for developmentally programmed cell death. Proc Natl Acad Sci USA 87: 6594–6598, 1990. doi: 10.1073/pnas.87.17.6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwartz LM, Myer A, Kosz L, Engelstein M, Maier C. Activation of polyubiquitin gene expression during developmentally programmed cell death. Neuron 5: 411–419, 1990. doi: 10.1016/0896-6273(90)90080-Y. [DOI] [PubMed] [Google Scholar]
- 81.Schwartz LM, Ruff RL. Changes in contractile properties of skeletal muscle during developmentally programmed atrophy and death. Am J Physiol Cell Physiol 282: C1270–C1277, 2002. doi: 10.1152/ajpcell.01275.2000. [DOI] [PubMed] [Google Scholar]
- 82.Schwartz LM, Smith SW, Jones ME, Osborne BA. Do all programmed cell deaths occur via apoptosis? Proc Natl Acad Sci USA 90: 980–984, 1993. doi: 10.1073/pnas.90.3.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwartz LM, Truman JW. Cyclic GMP may serve as a second messenger in peptide-induced muscle degeneration in an insect. Proc Natl Acad Sci USA 81: 6718–6722, 1984. doi: 10.1073/pnas.81.21.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schwartz LM, Truman JW. Hormonal control of rates of metamorphic development in the tobacco hornworm Manduca sexta. Dev Biol 99: 103–114, 1983. doi: 10.1016/0012-1606(83)90257-9. [DOI] [PubMed] [Google Scholar]
- 85.Schwartz LM, Truman JW. Peptide and steroid regulation of muscle degeneration in an insect. Science 215: 1420–1421, 1982. doi: 10.1126/science.6278594. [DOI] [PubMed] [Google Scholar]
- 86.Seaborne RA, Strauss J, Cocks M, Shepherd S, O’Brien TD, Someren KAV, Bell PG, Murgatroyd C, Morton JP, Stewart CE, Mein CA, Sharples AP. Methylome of human skeletal muscle after acute & chronic resistance exercise training, detraining & retraining. Sci Data 5: 180213, 2018. doi: 10.1038/sdata.2018.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seenundun S, Rampalli S, Liu QC, Aziz A, Palii C, Hong S, Blais A, Brand M, Ge K, Dilworth FJ. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J 29: 1401–1411, 2010. doi: 10.1038/emboj.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504, 2003. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shao R, Scully SJ Jr, Yan W, Bentley B, Mueller J, Brown C, Bigelow C, Schwartz LM. The novel lupus antigen related protein acheron enhances the development of human breast cancer. Int J Cancer 130: 544–554, 2012. doi: 10.1002/ijc.26015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sheel A, Shao R, Brown C, Johnson J, Hamilton A, Sun D, Oppenheimer J, Smith W, Visconti PE, Markstein M, Bigelow C, Schwartz LM. Acheron/LARP6 is a survival protein that protects skeletal muscle from programmed cell death during development. Front Cell Dev Biol 8: 622, 2020. doi: 10.3389/fcell.2020.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strasser H, Tiefenthaler M, Steinlechner M, Eder I, Bartsch G, Konwalinka G. Age dependent apoptosis and loss of rhabdosphincter cells. J Urol 164: 1781–1785, 2000. doi: 10.1016/S0022-5347(05)67106-6. [DOI] [PubMed] [Google Scholar]
- 92.Sucharov C, Bristow MR, Port JD. miRNA expression in the failing human heart: functional correlates. J Mol Cell Cardiol 45: 185–192, 2008. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun D, Ziegler R, Milligan CE, Fahrbach S, Schwartz LM. Apolipophorin III is dramatically up-regulated during the programmed death of insect skeletal muscle and neurons. J Neurobiol 26: 119–129, 1995. doi: 10.1002/neu.480260110. [DOI] [PubMed] [Google Scholar]
- 94.Sundermeier TR, Zhang N, Vinberg F, Mustafi D, Kohno H, Golczak M, Bai X, Maeda A, Kefalov VJ, Palczewski K. DICER1 is essential for survival of postmitotic rod photoreceptor cells in mice. FASEB J 28: 3780–3791, 2014. doi: 10.1096/fj.14-254292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39, Database: D561–D568, 2011. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tamori J, Kanzawa N, Tajima T, Tamiya T, Tsuchiya T. Purification and characterization of a novel isoform of myosinase from spear squid liver. J Biochem 126: 969–974, 1999. doi: 10.1093/oxfordjournals.jbchem.a022542. [DOI] [PubMed] [Google Scholar]
- 97.Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest 120: 11–19, 2010. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thummel CS. Molecular mechanisms of developmental timing in C. elegans and Drosophila. Dev Cell 1: 453–465, 2001. doi: 10.1016/S1534-5807(01)00060-0. [DOI] [PubMed] [Google Scholar]
- 99.Tóth KF, Pezic D, Stuwe E, Webster A. The piRNA pathway guards the germline genome against transposable elements. Adv Exp Med Biol 886: 51–77, 2016. doi: 10.1007/978-94-017-7417-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515, 2010. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Trevisan-Silva D, Gremski LH, Chaim OM, da Silveira RB, Meissner GO, Mangili OC, Barbaro KC, Gremski W, Veiga SS, Senff-Ribeiro A. Astacin-like metalloproteases are a gene family of toxins present in the venom of different species of the brown spider (genus Loxosceles). Biochimie 92: 21–32, 2010. doi: 10.1016/j.biochi.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 102.Truman JW, Riddiford LM. Neuroendocrine control of ecdysis in silkmoths. Science 167: 1624–1626, 1970. doi: 10.1126/science.167.3925.1624. [DOI] [PubMed] [Google Scholar]
- 103.UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res 43, D1: D204–D212, 2015. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Valavanis C, Wang Z, Sun D, Vaine M, Schwartz LM. Acheron, a novel member of the Lupus Antigen family, is induced during the programmed cell death of skeletal muscles in the moth Manduca sexta. Gene 393: 101–109, 2007. doi: 10.1016/j.gene.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16: 178–189, 2015. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 106.Wasser M, Bte Osman Z, Chia W. EAST and Chromator control the destruction and remodeling of muscles during Drosophila metamorphosis. Dev Biol 307: 380–393, 2007. doi: 10.1016/j.ydbio.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 107.Zhang S, Chen N. Regulatory Role of MicroRNAs in muscle atrophy during exercise intervention. Int J Mol Sci 19: 405, 2018. doi: 10.3390/ijms19020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang X, Zheng Y, Cao X, Ren R, Yu XQ, Jiang H. Identification and profiling of Manduca sexta microRNAs and their possible roles in regulating specific transcripts in fat body, hemocytes, and midgut. Insect Biochem Mol Biol 62: 11–22, 2015. doi: 10.1016/j.ibmb.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang X, Zheng Y, Jagadeeswaran G, Ren R, Sunkar R, Jiang H. Identification of conserved and novel microRNAs in Manduca sexta and their possible roles in the expression regulation of immunity-related genes. Insect Biochem Mol Biol 47: 12–22, 2014. doi: 10.1016/j.ibmb.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang X, Zheng Y, Jagadeeswaran G, Ren R, Sunkar R, Jiang H. Identification and developmental profiling of conserved and novel microRNAs in Manduca sexta. Insect Biochem Mol Biol 42: 381–395, 2012. doi: 10.1016/j.ibmb.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, Sinha S, Wolfe SA, Brodsky MH. FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res 39, Database: D111–D117, 2011. doi: 10.1093/nar/gkq858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zirin J, Cheng D, Dhanyasi N, Cho J, Dura JM, Vijayraghavan K, Perrimon N. Ecdysone signaling at metamorphosis triggers apoptosis of Drosophila abdominal muscles. Dev Biol 383: 275–284, 2013. doi: 10.1016/j.ydbio.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]