

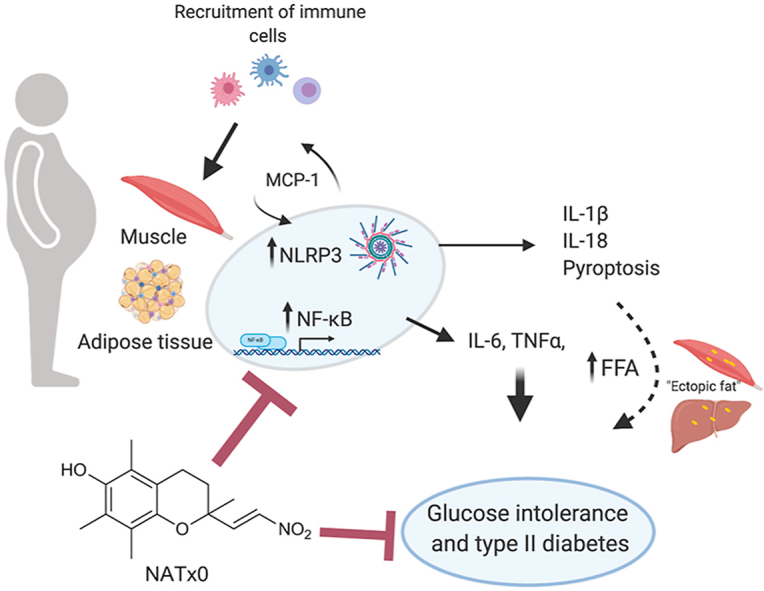
Abstract
Chronic metabolic diseases, like obesity, type II diabetes and atherosclerosis often involve a low-grade and sterile systemic inflammatory state, in which activation of the pro-inflammatory transcription factor NF-kB and the NLRP3 inflammasome play a major role. It is well established that genetic inhibition of the NLRP3 inflammasome ameliorates acute and chronic inflammation. Indeed, accumulating experimental evidences in murine models and also in humans suggest that inhibition of the NLRP3 inflammasome might be a suitable approach to tackle the deleterious effects of chronic metabolic diseases. In this work, we explored our previously synthesized nitroalkene-Trolox™ derivative named NATx0, as a non-conventional anti-inflammatory strategy to treat chronic inflammatory diseases, such as obesity-induced glucose intolerance. We found that NATx0 inhibited NF-kB nuclear translocation and pro-inflammatory gene expression in macrophages in vitro. In addition, treatment with NATx0 prevented NLRP3 inflammasome activation after LPS/ATP stimulation in macrophages in vitro. When tested acutely in vivo, NATx0 inhibited neutrophil recruitment in zebrafish larvae, and also diminished IL-1β production after LPS challenge in mice. Finally, when NATx0 was administered chronically to diet-induced obese mice, it decreased muscle tissue inflammation and glucose intolerance, leading to improved glucose homeostasis. In conclusion, we propose that this novel nitroalkene-Trolox derivative is a suitable tool to tackle acute and chronic inflammation in vitro and in vivo mainly due to inhibition of NF-kB/NLRP3 activation.
Keywords: Inflammation related diseases, Inflammasomes, Nitroalkenes, Diet induced obesity, Glucose intolerance
Graphical abstract
Highlights
-
•
Low-grade chronic inflammation plays a crucial role in non-communicable diseases, such as cancer, obesity and type II diabetes.
-
•
NLRP3 inflammasome inhibition could ameliorate glucose intolerance and systemic inflammation.
-
•
We designed a novel nitroalkene-Trolox™ derivative as an anti-inflammatory candidate to treat chronic diseases.
-
•
NATx0 inhibits the NLRP3 inflammasome, ameliorating tissue inflammation and glucose intolerance during diet-induced obesity.
1. Introduction
Inflammatory responses occurring on chronic non-communicable diseases, NCD's, such as cardiovascular diseases, cancer, obesity, type II diabetes and insulin resistance, are a common feature of all NCD's [1,2]. There is an important amount of evidence linking inflammation with obesity and its metabolic sequelae [[3], [4], [5], [6], [7], [8], [9], [10], [11]] and involves activation of the pro-inflammatory transcription factor NF-kB and the inflammasomes [[12], [13], [14], [15], [16], [17], [18], [19]], especially NRLP3. Therefore, it is conceivable that aiming to attack chronic low-grade inflammation could allow treating these diseases that are collectively responsible for almost 37% of all deaths worldwide [20].
During inflammatory processes several oxidized/electrophilic lipid mediators are formed that overall, may exert anti-inflammatory actions and participate in the resolution of inflammation [21]. Inflammation favors lipid peroxidation chain reactions and these reactive intermediates may be the substrate for lipid nitration, reacting with .NO or its derivatives (nitrite, nitrate, peroxynitrite and nitrogen dioxide) [[22], [23], [24]]. Oxidative stress and inflammation lead to the formation of nitrated unsaturated fatty acids, which are endogenous nitroalkenes (a nitro group attach to a C that is forming a double bond) that exert potent anti-inflammatory actions by different cell mechanisms [[25], [26], [27], [28], [29], [30], [31]]. The presence of the nitro group makes these nitrated unsaturated fatty acids strong electrophiles that creates covalent bonding with nucleophiles in proteins (thiols and histidine residues) and low molecular weight thiols (e.g. GSH) via Michael addition reactions [32]. The downstream signaling actions of electrophilic fatty acid derivatives are predominantly ascribed to post-translational Michael addition with critical nucleophilic amino acids located in a subproteome of electrophile-sensitive protein [29,32]. Nitrated-unsaturated fatty acids modulate four main signaling cascades via reversible covalent Michael addition reactions in key regulatory or receptors proteins, altering downstream signaling events: activation of PPARγ [31]; Nrf-2/Keap1 [33,34]; and the Heat Shock Response [34]; and inhibition of NF-kB [35,36].
Inflammasomes, including the NLRP3 inflammasome (nucleotide-binding domain and leucine-rich repeat containing family, pyrin domain containing 3), are multiprotein cytosolic signaling complexes that can be activated by pathogen or danger signals through a class of cytosolic receptors termed Nod-like receptor proteins (NLR's) [[37], [38], [39], [40]]. The first consequence of the NLRP3 inflammasome activation is the formation of the mature hetero-tetramer of caspase 1 by dimerization and auto-proteolysis, which processes the pro-forms of the immature cytokines IL-1β and IL-18 into its mature forms [40]. The second consequence of inflammasome activation is the induction of pyroptosis, a form of cell death characterized by being pro-inflammatory as it provokes pore formation at the membrane of the host cell favoring the release of the generated cytokines among other molecules [41,42].
NLRP3 receptor is potentially activated through indirect interactions with several endogenous stress molecules, which includes oligomers amyloid polypeptide, glucose, ceramide, oxidized low density lipoprotein, ATP and cholesterol crystals, as well as bacterial, viral and fungal pathogens [43,44]. Activation of NLRP3 inflammasome requires two signals. The first is an NF-kB -activating stimulus that induces the expression of the pro-forms of interleukins 1β and 18 and of the NLRP3 receptor itself. The second signal produces the assembly of the inflammasome and is provided by different signals such as ceramide, ATP and cholesterol crystals. The presence of this second signal finally produces the mature forms of the cytokines which are released from the cell [43,44]. These pro-inflammatory cytokines are implicated in disrupting insulin signaling. Indeed, mice that are knock out (KO) for NLRP3 are protected against diet-induced obesity, insulin resistance and systemic inflammation [45]. Moreover, it has been shown that the blockade of IL-1β using a recombinant human IL-1 receptor antagonist (Anakinra) in clinical trials ameliorates type II diabetes and systemic inflammation [46]. Finally, a previously described nitroalkene compound was found to inhibit NLRP3 ATPase activity [51].
Recently, our group described a novel pharmacological strategy for the treatment of atherosclerosis, based on the design and development of a nitroalkene-α-tocopherol derivative (NATOH) [47]. Mice treated with NATOH showed down-regulation of NF-kB-dependent pro-inflammatory markers (including IL-1β and adhesion molecules) and amelioration of atherosclerosis. However, as NATOH is transported mainly by lipoproteins, it was delivered to aortic lesions with little or no biological activity on other tissues. We decided then to modify NATOH structure to make it more hydrophilic but keeping the nitroalkene properties. For that, we designed and synthesized a water-soluble molecule, derived from Trolox™ (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), but lacking the isoprenoid tail [48], as shown in Fig. 1A. The resulting molecule (2,5,7,8-tetramethyl-2-[(E)-2-nitrovinyl]chroman-6-ol), was named NATx0. As we previously described [48], NATx0 reacted with similar second order constants to nitro-fatty acids when challenged with a model nucleophile and reacted significantly faster with GAPDH, an enzyme containing an acidic thiol in its active site. In this work, we explored the anti-inflammatory properties of NATx0, in particular its capability to inhibit NF-kB-dependent signaling in vitro and in vivo, including NLRP3 modulation. Finally, we studied NATx0 effects in a high fat diet (HFD)-fed mouse model of obesity-induced glucose intolerance.
Fig. 1.

NATx0 inhibits acute inflammatory response in zebrafish larvae. A, α- Tocopherol Vitamin E, its hydrosoluble form (Trolox™) and Trolox-derived nitroalkene NATx0 chemical structures are shown. B, NATx0 inhibits neutrophil recruitment in an acute inflammatory assay in zebrafish larvae. Briefly, 3 dpf zebrafish neutrophil-specific larvae Tg (mpx:GFP) were pretreated with NATx0, Trolox or Ibuprofen for 2 h and wounded in tail fins by transection with a scapel. After, larvae were incubated again in the presence of the compounds during 4 h and then recruited neutrophils were imaged and quantified. Experimental diagram and representative pictures obtained corresponding to DMSO, NATx0 0.2 μM and NATx0 0.5 μM are shown. Graph represent the quantification of the neutrophils recruited to wounded fins inside a defined ROI. Bar graphs shown are the mean ± S.D. (error bars) of at least 17 larvae in each condition. Data represents two independent experiments. Statistical analysis: One-way ANOVA with Bonferroni's multiple comparison test, * = 0.0487; DMSO vs Tx 2 μM ns = 0.992; DMSO vs Tx 4 μM ns = 0.844; **** <0.0001; **** <0.0001.
We found that NATx0 inhibits inflammation both in vitro and in vivo, specifically affecting NF-kB and NLRP3-dependent cytokine production. Mechanistically, we found that NATx0 inhibits p65/ReLA nuclear translocation, similar to other nitroalkenes. Interestingly, we found that NATx0 inhibits NLRP3-dependent IL-1β secretion by interfering with ASC oligomerization and inflammasome complex formation. Finally, we found that treatment of diet-induced obese mice with NATx0 decreased inflammasome activation in tissue, ameliorating glucose intolerance. We conclude that scaffold selection is important to potentiate nitroalkene salutatory effects in vivo, opening a novel possibility for pharmacological interventions aimed to treat chronic metabolic conditions with inflammatory background, like glucose intolerance and type II diabetes during obesity.
2. Methods
2.1. Materials
2.1.1. Cell culture
THP-1 cells (ATCC TIB-202; 2015) were grown in RPMI media with 10% FBS (Gibco) and differentiated to macrophages with Phorbol 12-myristate 13-acetate (PMA) 200 nM (Sigma Aldrich; P8139) for 48 h, at 37 °C in 5% CO2, and then were used for the different experiments. RAW 264.7 cells (ATCC TIB-71) were maintained in complete media containing DMEM with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, at 37 °C in 5% CO2. All treatments with NATx0 or Tx were done in serum-free RPMI or DMEM in order to avoid possible nitroalkene interaction with serum proteins. NATx0 and Tx were always dissolved in DMSO (except for the obesity induced in mice experiment: see Animals and Experimental Design) and the final doses were accomplished using the corresponding media. DMSO final concentration was always below 0.1%.
2.1.2. Cell toxicity studies
Differentiated THP-1 and RAW 264.7 cells were treated with increasing doses of NATx0 (0, 2.5, 5, 10, 17.5, 25, 50 and 100 μM) for 24 h at 37 °C in serum-free media. A standard MTT assay was performed by measuring the mitochondrial-dependent reduction of MTT added to cells at a 0.5 mg/mL final concentration. Formazan crystals were dissolved in isopropanol and absorbance was read at 570 nm using a microplate spectrophotometer. IC50 were calculated using GraphPad Prism 6.0 and refer to NATx0 dose at which cell viability is reduced by half. 100% viability corresponds to DMSO control.
2.1.3. Quantitative real-time PCR
The effect of NATx0 over NF-kB -dependent gene expression was studied in THP-1 cells by qPCR. Differentiated THP-1 cells were incubated with NATx0 (5 and 10 μM) or vehicle for 2 h in serum-free media. Then, cells were treated with LPS (1 μg/mL) for 3 h. To evaluate HO-1 and GCLM induction, NATx0 (5 and 10 μM) or vehicle were incubated for 5 h in serum-free media. Supernatant were removed and mRNA was isolated with TRIzol reagent (Invitrogen). Total RNA was retrotranscribed to cDNA using SuperScript™ II Reverse Transcriptase (Invitrogen) using a Piko 24 Thermal Cycler (Thermo Scientific). Gene expression analysis was calculated using the ΔΔCt method with β-Actin as the house keeping gene. SYBR Green (Roche) was used as DNA binding dye and the RT-PCR was done in an Eco Illumina thermocycler. Primers sequences used were: β- Actin forward: 5′-CATGTACGTTGCTATCCAGGC-3'; β- Actin reverse: 5′CTCCTTAATGTCACCCACGAT-3'; IL-6 forward: 5′-AGTGAGGAACAAGCCAGAGC-3'; IL-6 reverse: 5′-ATTTGTGGTTGGGTCAGGGG-3'; TNFα forward: 5′-GCCTCTTCTCCTTCCTGATCG-3'; TNFα reverse: 5′-TCGAGAAGATGATCTGACTGCC-3'; HO-1 forward: 5′-AAGACTGCGTTCCTGCTCAA-3'; HO-1 reverse: 5′-GGGGGCAGAATCTTGCACTT-3'; GCLM forward: 5′-AGACGGGGAACCTGCTGAA-3'; GCLM reverse: 5′-TCATGAAGCTCCTCGCTGTC-3'.
2.1.4. Cytokine detection by ELISA
For the analysis of the pro-inflammatory cytokines, RAW 264.7 cells were seeded in 12-or 24-well plates the night before each treatment in complete DMEM. The next day, cells were incubated with NATx0 (5 and 10 μM) or vehicle for 2 h in serum-free media and then treated with LPS (1 μg/mL) for 16 h. Supernatant was collected and the production of IL-6 and MCP-1 cytokines was analyzed by ELISA (BD 555240 and BD 555260, respectively, BD OptEIA).
2.1.5. Immunocytochemistry for NF-kB
Differentiated THP-1 cells were incubated with NATx0 for 2 h (10 μM) or vehicle. Cells were later activated with LPS from Escherichia coli serotype 0111:B4 (1 μg/mL; Sigma Aldrich) for 30 min and then fixed with 4% paraformaldehyde for 30 min at room temperature, permeabilized with 2% BSA - Triton 0.3% in PBS for 30 min, and blocked with 3% BSA in PBS. NF-kB labeling was performed overnight at 4 °C using rabbit anti-p65 antibody (Cell Signaling) and then Alexa Fluor 488-conjugated secondary antibody against rabbit was used. Nucleus were stained using DAPI. Images were acquired using an Olympus IX81 epifluorescence microscope equipped with an Orca-Hamamatsu CCD camera. Images were processed using Image J 1.51 h (Wayne Rasband, National Institutes of Health USA).
2.1.6. NLRP3 inflammasome activation assays
THP-1 monocytes were differentiated to macrophages in 12 or 24-well plates. 48 h later, the medium was removed and replaced with serum-free medium containing LPS (250 ng/mL from Escherichia coli serotype 0111:B4, + control) or LPS plus NATx0 (0.5–20 μM) or DMSO (1:1000) (“first signal protocol”) for 3 h at 37 °C. A negative control was added with only medium. Cells were then stimulated with the inflammasome activator adenosine 5′-triphosphate disodium salt hydrate ATP (5 mM) or ATP plus NATx0 (0.5–20 μM) or DMSO (1:1000) (“second signal protocol”) for 45 min. Cells were analyzed for viability by MTT as described above. Supernatants were removed and analyzed for IL-1β concentration using ELISA kits (BD 557953BD, BD OptEIA™), according to the manufacturer's instructions. LDH release was measured using Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific).
2.1.7. Western blotting
THP-1 monocytes were differentiated to macrophages in 6-well plates and treated for inflammasome activation, as detailed before. In these experiments, second signal protocol was always used (compounds added in concomitant with ATP). After treatments, cells were washed with cold PBS and cell lysates were prepared in lysis buffer (20 mM Tris pH 8, 100 mM NaCl, 1 mM EDTA, 0.5% NP40 with 1 M NaF, 1 M Nicotinamide, 1 M β-glycerophosphate pH 7.9 and protease inhibitor cocktail, Roche). Protein quantification was achieved using the Bradford method. Proteins released to culture media (sup.) were precipitated with cold acetone using a volume four times that of the protein sample, incubating 1 h at −20 °C. After centrifuging 10 min at 13.000 g pellets were repeatedly washed with cold acetone. Protein samples were prepared in Laemmli sample buffer 4x, resolved on 12% SDS-PAGE gels and transferred onto nitrocellulose membranes using a semi-dry transfer system. Membranes were blocked in 5% dried milk in TBS-T for 2 h at room temperature. After that, membranes were incubated with primary antibody diluted in 2% BSA in TBS-T overnight at 4 °C. Finally, membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody diluted in 2% BSA in TBS-T for 1 h and developed using chemiluminescent substrates (Thermo). Primary antibodies used were: β-Actin (Abcam ab8224), caspase-1 (Santa Cruz sc-515), IL-1β (Santa Cruz sc-7884), ASC (Santa Cruz sc-514414), p-Akt (Cell Signaling T308), Akt (Cell Signaling 9272), GAPDH (Cell Signaling 14C10). Secondary HRP-conjugated antibodies used were anti-mouse IgG and anti-rabbit IgG from Sigma.
2.1.8. ASC oligomers detection
Inflammasome activation procedure was done as described for western blotting. For the cross-linking assay, 16% formaldehyde was added in RMPI to plates and left 10 min at room temperature under slow agitation. 1.25 M Glycine was added to inhibit non-reactive formaldehyde and after, cells were washed twice with cold PBS. Cells were scraped in cold PBS and centrifuge at 5.000 rpm for 10 min. Pellets were dissolved in RIPA buffer and mixed for 1 h in cold chamber. Protein quantification was done by Bradford method and samples were prepared in Laemmli sample buffer for western blotting without heating the samples. For the detection of ASC specks by fluorescence microscopy, cells were fixed with 4% paraformaldehyde after treatments, permeabilized with PBS- BSA 2% - Triton 0.3%, and blocked with PBS containing 3% BSA. Cells were stained with anti-ASC antibody and Alexa Fluor 633-conjugated secondary antibody. DAPI was used to stain nuclei. Cell images were taken with Olympus IX81 epifluorescence microscope.
2.1.9. Animals and Experimental Design
All mice and zebrafish used in this study were maintained at the Institut Pasteur de Montevideo Animal facility (UATE). The experimental protocol was approved by the Institutional Animal Care and Use Committee of the Institut Pasteur de Montevideo (CEUA, Protocol numbers 014-14 and 019–15). Studies were performed according to the methods approved in the protocol.
For the LPS challenging experiment in mice, seven C57BL/6 mice were injected intraperitoneally (i.p) with 20 mg/kg NATx0 or vehicle (DMSO) 1 h before i.p. injection of 10 mg/kg LPS Escherichia coli 055:B5 (Sigma-Aldrich) or PBS. After 2 h mice were killed and levels of IL-1β in serum and peritoneum was measured by ELISA (BD 559603, BD OptEIA).
For the inflammation assay in zebrafish, fish were raised and maintained according to standard protocols. The neutrophil-specific zebrafish line Tg (mpx:GFP)i114, referred to as Tg(mpx:GFP), was used for the acute inflammation assays as previously described [48]. Larvae were pre-incubated for 2 h with the compounds at the doses indicated in Fig. 1B. Larvae tail fins were transected with a sterile scalpel at the region indicated in Fig. 1B. After, they were incubated in the presence of the compounds for 4 h and fixed in 4% paraformaldehyde overnight. To assess neutrophil number, whole mount immunohistochemistry was performed using rabbit polyclonal anti-GFP (Invitrogen A-11122). Images were captured in an inverted fluorescence microscope (Olympus) at 10x magnification and neutrophils in the region posterior to the circulatory loop were counted.
For the diet-induced obesity in mice experiment, male C57BL/6 mice (starting at 24 weeks of age) were randomly assigned in two groups and were treated by gavage with NATx0 (150 mg/kg) or vehicle for 4 weeks once per day 5 days a week (Monday through Friday) under normal rodent chow diet. Vehicle consisted in a 1% carboxymethylcelullose solution with 1% Tween 80 (MC/Tween 1%) in high purity water. The compound needed to be sonicated in the vehicle until a proper suspension was obtained. Compound preparation was performed once a week according to the average weights of the group. After, animals were fed with HFD (42% fat and 0.25% cholesterol) and treated by gavage with NATx0 (150 mg/kg) or vehicle over 11 weeks once per day Monday through Friday. Basal glycaemia was controlled at the beginning of the experiment and before starting administration. Glucose tolerance tests (GTT) were performed by ip injection of 1.5 g/kg body weight of glucose solution to 12 h-fasted mice. Plasma glucose concentrations were measured in blood from the tail using a hand-held glucometer (Accu-Chek, Roche) before and after 15, 30, 60, and 120 min of the injection. Area under the curve (AUC) was calculated by using the trapezoid rule. For blood and organ extraction mice were injected with 0.5 U/kg body weight of insulin 20 min prior they were anesthetized with ketamine/xylacine solution. Plasma was separated by centrifugation, aliquoted and stored at −80 °C. Tissues of interest were frozen in liquid nitrogen and stored at −80 °C. Protein extraction was performed from muscle. In all experiments, groups consisted of 4–5 mice per group.
2.1.10. Statistical analysis
Statistical analysis was performed using Ordinary one-way ANOVA followed by the Bonferroni post hoc or unpaired Student's t-test, in which a value of p < 0.05 was considered statistically significant. All data is presented as mean ± SD. Calculations were done using GraphPad Prism 6.0.
3. Results
3.1. NATx0 inhibits acute inflammatory response in zebrafish larvae
In order to characterize the potential anti-inflammatory properties of a novel α-tocopherol nitroalkene derivative NATx0 (Fig. 1A), we first evaluated its effect using a widely validated zebrafish inflammation model. We used a transgenic zebrafish line that expresses GFP under the neutrophil-specific myeloperoxidase promoter [49]. Zebrafish larvae are transparent, which allows the visualization of fluorescence proteins in cellular processes in vivo, including neutrophil movement, which could be identifiable from 48 h after fertilization. In this widely validated system, transection of the tail of the larvae induces an inflammatory focus which is resolved over a similar time course to mammalian systems [48,50]. Three days post fertilization (3 dpf) larvae were pretreated with NATx0, Trolox or Ibuprofen for 2 h and after wound was performed in tail fins (Fig. 1B). Wounded larvae were incubated again in the presence of the compounds during 4 h and then recruited neutrophils were imaged and quantified in the region of interest (ROI) (Fig. 1B). NATx0 pre-treatment in larvae inhibited the recruitment of neutrophils to the site of injury, improving the effect of the well-known anti-inflammatory Ibuprofen used at even higher dose. Even more, NATx0 backbone (Trolox) had no effect over neutrophil recruitment used at higher doses than NATx0 (2 and 4 μM compared with 0.2 and 0.5 μM of NATx0, see Fig. 1B).
3.2. NATx0 inhibits NF-kB-dependent signaling and activates anti-oxidant responses through Nrf2/keap1 system
In order to determine the mechanism of action of NATx0 in preventing inflammation, we measured inflammatory response in macrophages in vitro. First, we evaluated cellular in vitro toxicity of NATx0 in macrophages. In order to avoid cell-dependent effects, we used murine and human macrophages for the in vitro experiments. RAW 264.7 and THP-1 macrophages were treated with increasing concentrations of NATx0 for 24 h resulting in a calculated IC50 for each cell line of 28.1 and 18.3 μM, respectively (Fig. 2A). Next, we assayed the effect of NATx0 on NF-kB translocation to the nucleus in differentiated THP-1 macrophages, using anti-p65 subunit antibody and Alexa-488 secondary antibody (Fig. 2B). In the control condition, p65 subunit is cytoplasmic (represented in green and pointed by arrowheads) and when cells are treated with LPS, p65 is found almost completely in the nucleus (DMSO plus LPS condition). We found that NATx0 partially inhibits the translocation of p65 to the nucleus when macrophages are treated with LPS in the presence of the compound (Fig. 2B). Moreover, gene expression of IL-6 and TNFα downstream targets of NF-kB activation and nuclear translocation were inhibited by NATx0 in macrophages after an LPS stimulus (Fig. 2C), which was paralleled by decreased cytokines released to the culture media (Fig. 2D). This inhibition was not observed when Trolox was incubated in the presence of LPS at the same concentrations (see Fig. 2C and D). We saw a slight and significant decrease in IL-6 secretion with Trolox (Fig. 2D). However the inhibition with NATx0 was significantly more profound (Fig. 2D). Even more, incubation with NATx0 activated Keap1-Nrf2 pathway. HO-1 and GCLM gene-overexpression was observed in macrophages incubated with NATx0 at 5 and 10 μM and also HO-1 and GCLM protein were over-expressed (Fig. 2E and F, respectively), compared with the control condition, showing that NATx0 also regulates the canonical nitroalkene regulated pathways already known [33,34]. Again, Trolox was not able to induce Keap1-Nrf2 pathway at the same doses as NATx0 (see Fig. 2E).
Fig. 2.

NATx0 inhibits NF-kB-dependent signaling and activates anti-oxidant responses through Nrf2/keap1 system. A, Differentiated THP-1 and Raw 264.7 macrophages were incubated with increasing concentrations of NATx0 (0, 2.5, 5.0, 10, 25, 50 and 100 μM) for 24 h. Then a standard MTT assay was performed. DMSO control was added resulting in 100% viability. IC50 refers to NATx0 dose at which cell viability is reduced by half. B, Differentiated THP-1 cells were incubated with NATx0 for 2 h (10 μM) or vehicle and activated with 1 μg/mL LPS for 30 min and then fixed with 4% PFA. Rabbit anti-p65 antibody and Alexa Fluor 488-conjugated secondary antibody were used for labeling NF-kB-p65 subunit (green). Nucleus were stained using DAPI (blue). Arrowheads point nuclear or cytoplasmic localization of p65/RelA subunit. Graph shows the percentage of nuclear signal quantification after stimulation with PBS (control) or LPS after pre-incubation with DMSO (control +) or NATx0 10 μM. One-way ANOVA with Bonferroni's multiple comparison test: **** <0.0001; *** = 0.0002. C, Differentiated THP-1 cells were incubated with 5 and 10 μM of NATx0 or Tx for 2 h and treated with LPS (1 μg/mL) for 3 h. IL-6 and TNFα gene expression was studied by qPCR. One-way ANOVA with Bonferroni's multiple comparison test, IL-6: LPS vs Tx 5 μM ns = 0.9796; LPS vs Tx 10 μM ns >0.999; LPS vs NATx0 5 μM * = 0.0186; LPS vs NATx0 10 μM *** = 0.0002. TNFα: LPS vs Tx 5 μM ns = 0.9454; LPS vs Tx 10 μM ns = 0.998; LPS vs NATx0 5 μM ns = 0.1522; LPS vs NATx0 10 μM *** = 0.0005. D, Raw 264.7 macrophages were incubated with NATx0 or Tx (5 and 10 μM) or vehicle for 2 h and then treated with LPS (1 μg/mL) for 16 h. IL-6 and MCP-1 cytokines released to supernatant were analyzed by ELISA. Control refers to untreated cells. One-way ANOVA with Bonferroni's multiple comparison test, IL-6: LPS vs Tx 5 μM ** = 0.0032; LPS vs Tx 10 μM **** <0.0001; LPS vs NATx0 5 μM **** <0.0001; LPS vs NATx0 10 μM **** <0.0001. MCP-1: LPS vs Tx 5 μM ns >0.999; LPS vs Tx 10 μM ** = 0.0076; LPS vs NATx0 5 μM ns = 0.1002; LPS vs NATx0 10 μM **** <0.0001; Tx 5 μM vs NATx0 5 μM ** = 0.0052; Tx 10 μM vs NATx0 10 μM **** <0.0001. E, HO-1 and GCLM gene expression after 5 h incubation with NATx0 and Tx (5 and 10 μM) was studied by qPCR. One-way ANOVA with Bonferroni's multiple comparison test, HO-1: Control vs Tx 5 and 10 μM ns >0.999; Control vs NATx0 5 μM * = 0.010; Control vs NATx0 10 μM ns = 0.1979; Tx 5 μM vs NATx0 5 μM <0.0001; Tx 10 μM vs NATx0 10 μM <0.0001. F, HO-1 and GCLM protein expression by western blot after 5 h incubation with NATx0 5 and 10 μM.Data are representative of two or three independent experiments. Bar graphs shown are the mean ± S.D. (error bars). . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. NATx0 modulates NLRP3 inflammasome in vitro
The NLRP3 inflammasome has been proposed to play a major role in the regulation of chronic inflammatory diseases in humans [[14], [15], [16]]. It is well established that its chronic activation, along with other inflammatory processes, generates a low-grade systemic chronic inflammation that instigates the development of type II diabetes, atherosclerosis, cancer, dementia and other pathologies [15,16]. NLRP3 protein can recognize, directly or indirectly, several danger signals through its LRR domain leading to its oligomerization via its NOD domain. After, oligomerized NLRP3 can recruit the adaptor protein ASC via PYD-PYD domain interactions that also provokes ASC to oligomerize and recruit caspase-1 via CARD-CARD domain, to generate the final NLRP3 inflammasome complex. Finally, caspase-1 is activated and able to cleave proIL-1β and proIL-18 to produce its mature forms for secretion. The liberation of the mature cytokines from the cell is thought to be through pores formed by gasdermin D in the cell membrane, also a consequence of inflammasome assembly and caspase-1 activation [[40], [41], [42], [43], [44]]. In this work, we assayed the capability of NATx0 to inhibit inflammasome activation both during the priming step (“first signal”) as well as during the complex assembly step (“second signal”). In our in vitro experiments, THP-1 macrophages were incubated with first signal (3 h-treatment LPS 250 ng/mL). Later on, the second signal consisted in ATP incubation for 45 min. First, we tested cell viability after NLRP3 activation and NATx0 treatment (Fig. 3A). When NATx0 treatment was performed during the first signal (together with LPS) a protective effect of NATx0 was observed at 10 μM. This protection was not evidenced when NATx0 was applied together with second signal, however no cell toxicity was observed under those conditions (see Fig. 3A, graph corresponding to NATx0 in concomitant with first signal and second signal). Moreover, a dose-dependent inhibitory effect of NATx0 on IL-1β release was observed (Fig. 3B), when applied either during the first signal or with the second signal, with an IC50 of 8.8 μM and 6.1 μM, respectively. Since NATx0 inhibits nuclear NF-kB translocation (Fig. 2), and considering that the IL-1β transcriptional step is dependent of NF-kB, the inhibition effect of NATx0 observed during the first signal was expected. However, to our surprise, we found an even greater inhibition of IL-1β release when NATx0 was applied during the second signal, were NF-kB-dependent transcription was already established. Moreover, mature 17 KDa form of IL-1β was detected in the culture media of LPS/ATP treated macrophages (Fig. 3C, DMSO + LPS/ATP). When the compound was applied concomitant with the second signal at 10 μM (Fig. 3C), mature IL-1β was almost undetectable and proIL-1β was detected as expected. In a previous work [51], another nitroalkene compound (3,4-methylenedioxy-β-nitrostyrene) prevented ASC-speck formation by binding directly to NLRP3 and inhibiting its ATPase activity. Due to the homology of the nitrostyrene molecule with NATx0, we thought to investigate if NATx0 was preventing ASC oligomerization in LPS/ATP treated macrophages (Fig. 3D and E). The oligomerization of ASC was detected using fluorescence microscopy evaluating the formation of ASC “specks” or by immunoblotting of oligomers after chemical crosslinking. NATx0 diminished ASC dimer formation (48 KDa) at 10 μM incubated together with the second signal and did not affect ASC monomer which was expressed constitutively in untreated cells (control) or treated with LPS/ATP only (DMSO + LPS/ATP). ASC “speck” inhibition was also observed in LPS/ATP stimulated macrophages treated with NATx0 by fluorescence microscopy (Fig. 3E). These results implicate that NATx0 inhibition is upstream to ASC oligomerization and/or by inhibiting ASC itself. LDH release inhibition was also observed when stimulated macrophages were treated with NATx0 at 5 μM and 10 μM at the same time than the second signal (Fig. 3F), implicating that inflammasome formation is inhibited and therefore pyroptosis is blocked.
Fig. 3.

NATx0 modulates NLRP3 inflammasome in differentiated THP-1 cells. A, % Cell viability calculated by MTT after NLRP3 activation, when NATx0 was applied in concomitant with LPS (“first signal protocol”) for 3 h and after with ATP for 45 min; or when NATx0 was applied together with ATP for 45 min (“second signal protocol”) after 3 h LPS stimulus. One-way ANOVA with Bonferroni's multiple comparison test: **** <0.0001. B, IL-1β formation is inhibited by NATx0 in a dose-dependent manner when it is applied together with first signal or second signal. One-way ANOVA with Bonferroni's multiple comparison test: *** = 0.001 0 vs. 10 μM NATx0; *** = 0.0002 0 vs. 20 μM NATx0 (left graph); *** = 0.0024; **** <0.0001 (right graph). C, Western blot analysis of proIL-1β, pro-caspase 1 (p45) and active IL-1β after inflammasome activation in the presence of 10 μM NATx0 or DMSO (vehicle) when applied in concomitant with “second signal”. D, Western blot analysis of ASC oligomers formation in crosslinked cells after inflammasome activation in the presence of 10 μM NATx0 or DMSO (vehicle) when applied in concomitant with “second signal”. ASC monomers were detected at 24 kDa, dimers at 48 kDa and oligomers over 65 kDa (dotted arrows). E, ASC “speck” detection by fluorescence microscopy after inflammasome activation in the presence of 10 μM NATx0 or DMSO when applied in concomitant with “second signal”. Cells were fixed, permeabilized and stained for ASC (green). Specks are denoted in white arrows. Percentage of ASC speck formation over ASC- positive cells is shown. One-way ANOVA with Bonferroni's multiple comparison test: **** <0.0001; ** = 0.0087. F, LDH (lactate dehydrogenase) release after inflammasome activation in the presence of 5 and 10 μM NATx0 or DMSO when applied in concomitant with “second signal”. One-way ANOVA with Bonferroni's multiple comparison test: *** = 0.0002; ** = 0.0098. Control in all cases refers to untreated cells.
3.4. NATx0 inhibits acute NLRP3 activation and IL-1β production in vivo
We next decided to further investigate NATx0 capability of inhibiting NLRP3 inflammasome and IL-1β production in vivo. For that, we assayed serum and peritoneum levels of IL-1β in C57BL/6 J after an i.p. injection of LPS and measured IL-1β release, a surrogate of in vivo NLRP3 activation [52]. Mice were pretreated with NATx0 or vehicle for 1 h before a 2-h LPS treatment. Plasma and peritoneum levels of IL-1β in NATx0-pretreated animals were greatly diminished (Fig. 4) as compared with animals that were pretreated with vehicle (control).
Fig. 4.

NATx0 inhibits acute NLRP3 activation and IL-1β production in vivo. C57BL/6 mice were injected intraperitoneally (i.p) with 20 mg/kg NATx0 or vehicle (DMSO) 1 h before i.p. injection of 10 mg/kg LPS Escherichia coli or PBS. After 2 h mice were killed and levels of IL-1β in serum and peritoneum was measured by ELISA. One-way ANOVA with Bonferroni's multiple comparison test: *** <0.0001; **** <0.0001; ** = 0.0004 (n = 7 per group).
3.5. NATx0 reduces glucose intolerance in a mouse model of diet induced obesity
Finally, the effect of NATx0 was assayed on a diet-induced obesity mouse model that generates glucose intolerance in C57BL6/J mice [[53], [54], [55]]. The purpose was to inhibit the pro-inflammatory signaling cascades involved in the onset of inflammation in metabolic tissues that pathogenically links obesity to glucose intolerance and therefore improve this parameter in the mouse. C57BL6/J mice were pretreated with NATx0 (150 mg/kg gavage, five days a week) during 4 weeks fed with normal diet. Pre -treatment was done in order to turn on the molecular pathways in which the nitroalkene is involved, for example the activation of Nrf2/keap1 antioxidant system and other protective mechanisms known for nitroalkenes, shown elsewhere. These mechanisms could include activation of PPARγ and heat shock responses [31,34]. After, mice were fed with a high fat diet (HFD) for another 11 weeks plus NATx0 (150 mg/kg) or vehicle (gavage administration). Mice gained similar weight and ate similar amount of food independently of whether they received NATx0 or vehicle during the experiment (Fig. 5A and B). However, when challenged with a GTT, NATx0 treated mice showed improved glucose tolerance compared with vehicle-treated mice (Fig. 5C).
Fig. 5.

NATx0 reduces glucose intolerance in a mouse model of diet-induced obesity. C57BL/6 mice were treated orally with NATx0 (150 mg/kg) or vehicle for 4 weeks under normal rodent chow diet. After, animals were fed with high fat diet (HFD) and treated orally with NATx0 (150 mg/kg) or vehicle for another 11 weeks. A, Graph shows animal weight evolution during the course of the experiment. At week 4, mice started to ate HFD. B, At week 12, consumed water, diuresis, feces and food intake parameters were measured in metabolic cages within 24 h in NATx0 treated or vehicle treated mice. Two-tailed unpaired t-test: ns > 0.25. C, Glucose tolerance test (GTT) was performed by i.p injection of 1.5 g/kg body weight of glucose solution to fasted mice and plasma glucose was measured in blood before and after 15, 30, 60, and 120 min of the injection. Two-tailed unpaired t-test: ** = 0.0031. Inset shows calculated area under the curve (AUC) for NATx0 or vehicle treated mice glucose response. Two-tailed unpaired t-test: * = 0.0319. D, At the end of the experiment, mice were injected with 0.5 U/kg body weight of insulin and sacrificed to obtain muscle. Western blot analysis was performed to analyze p-Akt, Akt and IL-1β protein expression in muscle. Two-tailed unpaired t-test: vehicle vs. NATx0 * = 0.0147 for p-Akt/Akt and * = 0.0131 for pro-IL-1β/IL-1β.
Furthermore, after an injection of insulin, animals were sacrificed and increase in the Akt phosphorylation level was observed at muscle in the NATx0 treated animals as compared with the vehicle group (Fig. 5D). This result reinforces the positive effect of the compound ameliorating insulin sensitivity, as mice treated with NATx0 exhibited improved Akt response under this experimental setup. We were also able to probe that active IL-1β was reduced in the NATx0 treated group in muscle as compared to the vehicle treated animals (Fig. 5D). This validates our hypothesis that NATx0 can reduce the chronic inflammation generated during obesity and therefore diminish its metabolic consequences.
4. Discussion
We have previously demonstrated that a rationally designed nitroalkene derived from a natural and safe drug delivery system (e.g. α-tocopherol or Vitamin E), was a novel and thriving pharmacological strategy for the treatment of atherosclerosis [47]. This strategy, which was named NATOH (5-nitroethenyl-γ-tocopherol) and was specifically designed to be carried by the VLDL/LDL system to atheroma lesions by homologizing the α-tocopherol structure, probed to work as a non-conventional anti-inflammatory compound in vitro and in vivo. Moreover, NATOH was able to decrease the atherosclerotic plaque formation and to inhibit the expression of relevant cytokines and adhesion molecules liberated at the lesions site in an atherosclerosis mouse model (ApoE knock out mice) [47]. In this work, we show that a novel and hydro soluble derivative of NATOH, named NATx0, previously described by our group [48], has promissory beneficial effects over inflammatory processes occurring during obesity, glucose intolerance and type II diabetes.
Our results show that NATx0 effects are mainly anti-inflammatory and cytoprotective, as it inhibits NF-kB, NLRP3 inflammasome and induce the Keap1-Nrf2 pathway. Nuclear factor-κB controls cell survival, proliferation and different pro-inflammatory mediators, including cytokines, chemokines, and adhesion molecules and is one of the principal mediators in obesity-induced insulin resistance [12]. Nitro-linoleic acid and nitro-oleic acid inhibit NF-kB-dependent cytokine expression via alkylation of p65 subunit of NF-kB [35]. More recently, Villacorta et al. demonstrated that nitrated fatty acids also inhibit NF-kB signaling by disrupting the Toll Like Receptor 4 (TLR4) [36]. NATx0 inhibited the translocation of p65/RelA to the nucleus and inhibited NF-kB dependent-gene expression and cytokines release to culture media in macrophages in vitro. Also, NATx0 induced the expression of hemoxigenase-1 and the glutamate-cysteine ligase modifier subunit (GCLM), both phase II enzymes controlled by the keap1-Nrf2 pathway, a major regulator of cytoprotective responses caused by reactive oxygen and nitrogen species (ROS, RNS) and electrophiles.
NATx0 anti-inflammatory effects were also assayed in vivo using a widely validated zebrafish inflammation model. In that system, neutrophil response to injury can be visualized in genetically manipulated zebrafish larvae that expresses GFP under the neutrophil-specific myeloperoxidase promoter. The mpo:GFP expression can be observed under the microscope in vivo, in anesthetized animals, because zebrafish larvae are transparent and neutrophils are already apparent 48 h after fertilization [49,50]. Neutrophil recruitment to the site of injury was dose-dependently diminished by NATx0 as compared to the vehicle group or even when compared with a group treated with Ibuprofen (positive control). This could mean that the damage response was resolved faster in the presence of NATx0 which is essential to avoid irreparable tissue damage mediated by toxic granule release of persisting neutrophils. The effect over neutrophil survival and pro-inflammatory activation is mainly regulated by NF-kB [56].
Emerging evidence unveiled that NLRP3 inflammasome, an intracellular supramolecular complex that regulate caspase-1 activation and the subsequent processing of pro-IL-1β and pro-IL-18, triggers inflammatory responses and instigates obesity-induced inflammation and insulin resistance [17]. Interestingly, NATx0 inhibited the NLRP3 inflammasome and IL-1β production both in vitro and in vivo, by possibly inhibiting NF-kB translocation to the nucleus, but also during the activation step by inhibiting up-stream ASC oligomerization or ASC itself. In fact, this effect may be involved at least partially on its anti-inflammatory effects in vivo. Other mechanisms could be as well involved in the observed NLRP3 inhibition by NATx0, including the modulation of xanthine oxidoreductase which has been previously reported to regulate NLRP3 inflammasome and to be potently inhibited by other nitroalkene [60].
Actually, activation of NLRP3 inflammasome has been shown to be a major pathogenic pathway in obesity-induced insulin resistance [17]. From a clinical perspective, a lot of expectation is centered in the inhibition of this pathway to treat type II diabetes, Coronary Artery Disease (CAD) and Cardiovascular Diseases (CVD) [46,[57], [58], [59]].
Obesity is associated with self-directed tissue inflammation where local or systemic factors, other than infectious agents activate the cells of the innate immune system. Spiegelman and colleagues were pioneers in the identification of the first links between inflammation, obesity and insulin resistance [[61], [62], [63]], showing that the adipose tissue of obese animals express tumor necrosis factor alpha (TNF-α), a pro-inflammatory cytokine that was found to promote insulin resistance via serine phosphorylation of insulin receptor substrate 1 (IRS1) [62]. By increasing bioavailability and electrophilicity of NATOH, we designed NATx0. The results obtained are promissory for the treatment of obesity-glucose intolerance in mice. NATx0 treated animals showed protection against glucose intolerance despite gaining similar weight than control mice. This was accompanied by increased Akt response in skeletal muscle after insulin exposure in both groups, confirming the in vivo results. However, we acknowledge that additional supportive evidence is necessaire to determine insulin resistance levels in the diet-induced obesity mice, such as euglycemic clamp or insulin levels determinations. These approaches will be incorporated for future studies with NATx0. Active IL-1β was also found diminished in skeletal muscle implying that the effects observed in vivo are at least partially due to inflammasome inhibition.
Overall, this work describes a potential new strategy to tackle inflammatory related-conditions. Using different biologically relevant molecular scaffolds to carry the nitroalkene group to different target tissues/organs may contribute to open novel pharmacological strategies aimed to ameliorate chronic inflammation and metabolic diseases. Accumulating evidence shows that acute and chronic inflammation are not necessarily regulated similarly by pharmacological interventions. As such, providing evidence that an anti-inflammatory novel molecule has an impact on such an important metabolic disease as obesity and glucose intolerance may open new venues for therapeutic interventions.
Author contributions
CE, CB, and GVL designed the study. RD, GG, and JRD performed in vitro experiments with cells. RD, JRD and AK performed experiments with zebrafish larvae. RD, MB and JRD did experiments with mice. RD, CB, GVL and CE analyzed data. RD, CB, GVL and CE prepared the manuscript. All authors contributed to the discussion of the results, edited and approved the final version of the manuscript.
Funding
This work was supported by Institut Pasteur de Montevideo – FOCEM Mercosur (COF 03/11), Agencia Nacional de Investigación e Innovación (ANII), Programa de Desarrollo de las Ciencias Básicas (PEDECIBA), Sistema Nacional de Investigadores (SNI), EOLO PHARMA S.A and affiliates.
Declaration of competing interest
GVL, CE and CB hold shares in EOLO PHARMA S.A. and affiliates.
Acknowledgments
We want to thank the staff from the Transgenic and Experimental Animal Unit and the zebrafish platform unit from Institut Pasteur de Montevideo.
Contributor Information
Carlos Batthyány, Email: batthyany@pasteur.edu.uy.
Gloria V. López, Email: vlopez@fq.edu.uy.
Carlos Escande, Email: escande@pasteur.edu.uy.
References
- 1.Phillips C.M., Chen L.W., Heude B., Bernard J.Y., Harvey N.C., Duijts L., Mensink-Bout S.M., Polanska K., Mancano G., Suderman M., Shivappa N., Hébert J.R. Dietary inflammatory index and non-communicable disease risk: a narrative review. Nutrients. 2019;11(8):1873. doi: 10.3390/nu11081873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 3.Heymsfield S.B., Wadden T.A. Mechanisms, pathophysiology, and management of obesity. N. Engl. J. Med. 2017;376:254–266. doi: 10.1056/NEJMra1514009. [DOI] [PubMed] [Google Scholar]
- 4.Chawla A., Nguyen K.D., Goh Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leibel R.L. Molecular physiology of weight regulation in mice and humans. Int. J. Obes. 2008;32(Suppl 7):S98–S108. doi: 10.1038/ijo.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrante A.W., Jr. Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J. Intern. Med. 2007;262:408–414. doi: 10.1111/j.1365-2796.2007.01852.x. [DOI] [PubMed] [Google Scholar]
- 7.Lumeng C.N., Saltiel A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoelson S.E., Lee J., Goldfine A.B. Inflammation and insulin resistance. J. Clin. Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shulman G.I. Cellular mechanisms of insulin resistance. J. Clin. Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahn B.B., Flier J.S. Obesity and insulin resistance. J. Clin. Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker R.G., Hayden M.S., Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metabol. 2011;5(1):11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlsen H., Haugen F., Zadelaar S., Kleemann R., Kooistra T., Drevon C.A., Blomhoff R. Diet-induced obesity increases NF-kappaB signaling in reporter mice. Genes Nutr. 2009;4(3):215–222. doi: 10.1007/s12263-009-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Targeting the nlrp3 inflammasome in chronic inflammatory diseases: current perspectives. J. Inflamm. Res. 2015;8:15–27. doi: 10.2147/JIR.S51250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo H., Callaway J.B., Ting J., Infammasomes P. Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strowig T., Henao-Mejia J., Elinav E., Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 17.Vandanmagsar B., Youm Y.H., Ravussin A., Galgani J.E., Stadler K., Mynatt R.L., Ravussin E., Stephens J.M., Dixit V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahechu P., Zozaya G., Martí P., Hernández-Lizoáin J.L., Baixauli J., Unamuno X., Frühbeck G., Catalán V. NLRP3 inflammasome: a possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front. Immunol. 2018;9:2918. doi: 10.3389/fimmu.2018.02918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rheinheimer J., de Souza B.M., Cardoso N.S., Bauer A.C., Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. 2017;74:1–9. doi: 10.1016/j.metabol.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Cardiovascular Diseases (CVDs) 2017. http://www.who.int/mediacentre/factsheets/fs317/en/ [Google Scholar]
- 21.Groeger A.L., Cipollina C., Cole M.P., Woodcock S.R., Bonacci G., Rudolph T.K., Rudolph V., Freeman B.A., Schopfer F.J. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat. Chem. Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker P.R., Schopfer F.J., O'Donnell V.B., Freeman B.A. Convergence of nitric oxide and lipid signaling: anti-inflammatory nitro-fatty acids. Free Radical Biol. Med. 2009;46:989–1003. doi: 10.1016/j.freeradbiomed.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonacci G., Baker P.R., Salvatore S.R., Shores D., Khoo N.K., Koenitzer J.R., Vitturi D.A., Woodcock S.R., Golin-Bisello F., Cole M.P., Watkins S., St Croix C., Batthyany C.I., Freeman B.A., Schopfer F.J. Conjugated linoleic acid is a preferential substrate for fatty acid nitration. J. Biol. Chem. 2012;287:44071–44082. doi: 10.1074/jbc.M112.401356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khoo N.K., Freeman B.A. Electrophilic nitro-fatty acids: anti-inflammatory mediators in the vascular compartment. Curr. Opin. Pharmacol. 2010;10:179–184. doi: 10.1016/j.coph.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker P.R., Lin Y., Schopfer F.J., Woodcock S.R., Groeger A.L., Batthyany C., Sweeney S., Long M.H., Iles K.E., Baker L.M., Branchaud B.P., Chen Y.E., Freeman B.A. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker P.R., Schopfer F.J., Sweeney S., Freeman B.A. Red cell membrane and plasma linoleic acid nitration products: synthesis, clinical identification, and quantitation. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freeman B.A., Baker P.R., Schopfer F.J., Woodcock S.R., Napolitano A., d'Ischia M. Nitro-fatty acid formation and signaling. J. Biol. Chem. 2008;283:15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schopfer F.J., Baker P.R., Giles G., Chumley P., Batthyany C., Crawford J., Patel R.P., Hogg N., Branchaud B.P., Lancaster J.R., Jr., Freeman B.A. Fatty acid transduction of nitric oxide signaling. Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J. Biol. Chem. 2005;280:19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 29.Schopfer F.J., Batthyany C., Baker P.R., Bonacci G., Cole M.P., Rudolph V., Groeger A.L., Rudolph T.K., Nadtochiy S., Brookes P.S., Freeman B.A. Detection and quantification of protein adduction by electrophilic fatty acids: mitochondrial generation of fatty acid nitroalkene derivatives. Free Radical Biol. Med. 2009;46:1250–1259. doi: 10.1016/j.freeradbiomed.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schopfer F.J., Cipollina C., Freeman B.A. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schopfer F.J., Lin Y., Baker P.R., Cui T., Garcia-Barrio M., Zhang J., Chen K., Chen Y.E., Freeman B.A. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc. Natl. Acad. Sci. U. S. A. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batthyany C., Schopfer F.J., Baker P.R., Duran R., Baker L.M., Huang Y., Cervenansky C., Branchaud B.P., Freeman B.A. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kansanen E., Bonacci G., Schopfer F.J., Kuosmanen S.M., Tong K.I., Leinonen H., Woodcock S.R., Yamamoto M., Carlberg C., Yla-Herttuala S., Freeman B.A., Levonen A.L. Electrophilic nitro-fatty acids activate nrf2 by a keap1 cysteine 151-independent mechanism. J. Biol. Chem. 2011;286:14019–14027. doi: 10.1074/jbc.M110.190710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kansanen E., Jyrkkanen H.K., Volger O.L., Leinonen H., Kivela A.M., Hakkinen S.K., Woodcock S.R., Schopfer F.J., Horrevoets A.J., Yla-Herttuala S., Freeman B.A., Levonen A.L. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009;284:33233–33241. doi: 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui T., Schopfer F.J., Zhang J., Chen K., Ichikawa T., Baker P.R., Batthyany C., Chacko B.K., Feng X., Patel R.P., Agarwal A., Freeman B.A., Chen Y.E. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J. Biol. Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villacorta L., Chang L., Salvatore S.R., Ichikawa T., Zhang J., Petrovic-Djergovic D., Jia L., Carlsen H., Schopfer F.J., Freeman B.A., Chen Y.E. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting lps-dependent tlr4 signalling in lipid rafts. Cardiovasc. Res. 2013;98:116–124. doi: 10.1093/cvr/cvt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Man S.M., Kanneganti T.D. Regulation of inflammasome activation. Immunol. Rev. 2015;265(1):6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinon F., Mayor A., Tschopp J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2019;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 39.Franchi L., Muñoz-Planillo R., Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroder K., Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 41.Bergsbaken T., Fink S.L., Cookson B.T. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H., Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;7(7610):153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin Y., Pastrana J.L., Li X., Huang X., Mallilankaraman K., Choi E.T., Madesh M., Wang H., Yang X.F. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front. Biosci. 2013;18:638–649. doi: 10.2741/4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo H., Callaway J., Ting J. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vandanmagsar B., Youm Y., Ravussin A. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larsen C.M., Faulenbach M., Vaag A., Volund A., Ehses J.A., Seifert B., Mandrup-Poulsen T., Donath M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez‐Duarte J., Galliussi G., Dapueto R., Rossello J., Malacrida L., Kamaid A., Schopfer F.J., Escande C., López G.V., Batthyány C. A novel nitroalkene‐α‐tocopherol analogue inhibits inflammation and ameliorates atherosclerosis in Apo E knockout mice. Br. J. Pharmacol. 2019;176:757–772. doi: 10.1111/bph.14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodriguez-Duarte J., Dapueto R., Galliussi G., Turell L., Kamaid A., Khoo N., Schopfer F.J., Freeman B.A., Escande C., Batthyány C., Ferrer-Sueta G., López G.V. Electrophilic nitroalkene-tocopherol derivatives: synthesis, physicochemical characterization and evaluation of anti-inflammatory signaling responses. Sci. Rep. 2018;8:12784. doi: 10.1038/s41598-018-31218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Renshaw S.A., Loynes C.A., Trushell D., Elworthy S., Ingham P.W., Whyte M. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108(13):3976–3978. doi: 10.1182/blood-2006-05-024075. [DOI] [PubMed] [Google Scholar]
- 50.Robertson A.L., Holmes G.R., Bojarczuk A.N., Burgon J., Loynes C.A., Chimen M., Sawtell A.K., Hamza B., Willson J., Walmsley S.R., Anderson S.R., Coles M.C., Farrow S.N., Solari R., Jones S., Prince L.R., Irimia D., Rainger G.E., Kadirkamanathan V., Whyte M., Renshaw S.A. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci. Transl. Med. 2014;6:225ra29. doi: 10.1126/scitranslmed.3007672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He Y., Varadarajan S., Muñoz-Planillo R., Burberry A., Nakamura Y., Núñez G. 3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 2014;10(2):1142–1150. doi: 10.1074/jbc.M113.515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coll R., Robertson A., Chae J. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Escande C., Chini C.C., Nin V., Dykhouse K.M., Novak C.M., Levine J., van Deursen J., Gores G.J., Chen J., Lou Z., Chini E.N. Deleted in breast cancer–1 regulates SIRT1 activity and contributes to high-fat diet–induced liver steatosis in mice. J. Clin. Invest. 2010;120(2):545–558. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Escande C., Nin V., Price N.L., Capellini V., Gomes A.P., Barbosa M.T., O'Neil L., White T.A., Sinclair D.A., Chini E.N. Flavonoid apigenin is an inhibitor of the NAD+ase CD38. Diabetes. 2013;62(4):1084–1093. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Escande C., Nin V., Pirtskhalava T., Chini C.C., Tchkonia T., Kirkland J.L., Chini E.N. Deleted in breast cancer 1 limits adipose tissue fat accumulation and plays a key role in the development of metabolic syndrome phenotype. Diabetes. 2014;63:1–11. doi: 10.2337/db14-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mussbacher M., Salzmann M., Brostjan C., Hoesel B., Schoergenhofer C., Datler H., Hohensinner P., Basílio J., Petzelbauer P., Assinger A., Schmid J.A. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front. Immunol. 2019;10:85. doi: 10.3389/fimmu.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S.D., Kastelein J.J.P., Cornel J.H., Pais P., Pella D., Genest J., Cifkova R., Lorenzatti A., Forster T., Kobalava Z., Vida-Simiti L., Flather M., Shimokawa H., Ogawa H., Dellborg M., Rossi P.R.F., Troquay R.P.T., Libby P., Glynn R.J., Group C.T. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 58.Ridker P.M., Luscher T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014;35:1782–1791. doi: 10.1093/eurheartj/ehu203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ridker P.M., Thuren T., Zalewski A., Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the canakinumab anti-inflammatory thrombosis outcomes study (cantos) Am. Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 60.Ives A., Nomura J., Martinon F., Roger T., LeRoy D., Miner J., Simon G., Busso N., So A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015;6:6555. doi: 10.1038/ncomms7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hotamisligil G.S., Johnson R.S., Distel R.J., Ellis R., Papaioannou V.E., Spiegelman B.M. Uncoupling of obesity from insulin resistance through a targeted mutation in ap2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 62.Hotamisligil G.S., Peraldi P., Budavari A., Ellis R., White M.F., Spiegelman B.M. Irs-1-mediated inhibition of insulin receptor tyrosine kinase activity in tnf-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 63.Hotamisligil G.S., Shargill N.S., Spiegelman B.M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]