

Key Points
B7-H6 was affinity matured by yeast surface display.
NK cell engagers harboring optimized B7-H6 have improved cytotoxic capacities.
NK cell engagers based on improved B7-H6 display a differentiated cytokine profile.
Visual Abstract
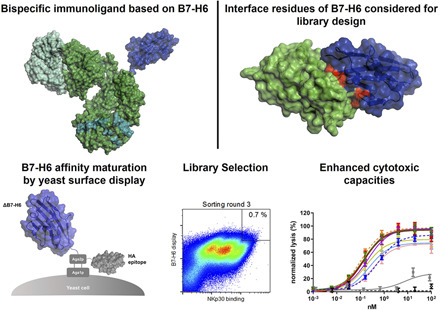
Abstract
Activating NK cell receptors represent promising target structures to elicit potent antitumor immune responses. In this study, novel immunoligands were generated that bridge the activating NK cell receptor NKp30 on NK cells with epidermal growth factor receptor (EGFR) on tumor cells in a bispecific IgG-like format based on affinity-optimized versions of B7-H6 and the Fab arm derived from cetuximab. To enhance NKp30 binding, the solitary N-terminal IgV domain of B7-H6 (ΔB7-H6) was affinity matured by an evolutionary library approach combined with yeast surface display. Biochemical and functional characterization of 36 of these novel ΔB7-H6–derived NK cell engagers revealed an up to 45-fold–enhanced affinity for NKp30 and significantly improved NK cell–mediated, EGFR-dependent killing of tumor cells compared with the NK cell engager based on the wild-type ΔB7-H6 domain. In this regard, potencies (EC50 killing) of the best immunoligands were substantially improved by up to 87-fold. Moreover, release of IFN-γ and TNF-α was significantly increased. Importantly, equipment of the ΔB7-H6–based NK cell engagers with a human IgG1 Fc part competent in Fc receptor binding resulted in an almost 10-fold superior killing of EGFR-overexpressing tumor cells compared with molecules either triggering FcγRIIIa or NKp30. Additionally, INF-γ and TNF-α release was increased compared with molecules solely triggering FcγRIIIa, including the clinically approved Ab cetuximab. Thus, incorporating affinity-matured ligands for NK cell–activating receptors might represent an effective strategy for the generation of potent novel therapeutic agents with unique effector functions in cancer immunotherapy.
Introduction
Natural killer cells are innate lymphocytes that recognize discontinuity and danger in multiple tissue compartments by integrating positive and negative signals (1). The negative signals are generally mediated by the interaction between self–MHC class I on tissues and either killer Ig-like receptor (KIR) family members or NK group 2A (NKG2A) (2). Positive signals are transduced via the interaction of an array of NK activation receptors, including the natural cytotoxicity receptors (NCRs; NKp30, NKp46, NKp44), NKG2D, and DNAM-1, as well as costimulatory molecules, including 4-1BB and their ligands (3–5). For the NCRs and NKG2D, many of the ligands are “danger signals” that are upregulated on stressed and diseased tissues, including virally infected cells and tumor cells (6–8). Another mechanism by which NK cells are activated is the bridging of the low-affinity–activating FcγRIIIa (CD16a) on NK cells with cells opsonized with IgG Abs or bispecific Abs (9, 10). Unlike the NCRs and NKG2D, signaling through FcγRIIIa is often more robust in resting NK cells but is modulated by multiple variables, including functionally distinct polymorphic variants of FcγRIIIa as well as competition for binding with circulating IgG (11–15). Ultimately, the balance of activation and inhibitory signal determines whether an NK cell will become activated. As such, NK cells have an endogenous capacity to differentiate between healthy and diseased tissues (16, 17).
Recently, several early clinical trials employing the adoptive transfer of wild-type or genetically modified (e.g., CAR) NK cells, either alone or in combination with Abs as a therapeutic modality for cancer, have been initiated with encouraging early results for hematological malignancies (18–21). Although adoptive cell therapy with ex vivo–activated NK cells represents a promising approach (22), the logistic complexity has also driven the development of NK cell–directed Ab-based approaches to cancer immunotherapy, such as bispecific or trifunctional entities that form a bridge between an activation receptor on NK cells and a tumor-associated Ag on the tumor cell, referred to as NK cell engager or immunoligand (11, 23, 24). Bispecific Abs targeting a tumor-associated Ag (e.g., CD20) and NKp46 (25), NKG2D (26) and NKp30 (27, 28), either via an Ab moiety or a recombinant form of the ectodomain of a ligand (e.g., ULBP2) (29), have demonstrated potent target-dependent cytotoxicity and cytokine release in vitro.
NKp30 is an activation receptor expressed on the majority of NK cells (30). Its cell-bound ligand, B7-H6, is upregulated on tumor cells and absent on most normal cells (7). The other less well-characterized ligand is HLA-B–associated transcript 3 (BAT3)/Bcl2–associated athanogene 6 (BAG6), which is expressed in the nucleus and can be transported to the plasma membrane or released in exosomes (31, 32). Importantly, decreased NKp30 expression has been correlated with reduced survival in acute myeloid leukemia (33), and a lower number of NK cells expressing NKp30 were found in patients with gastric or breast cancer, compared with healthy donors (34, 35). Together, these data suggest that the NKp30 receptor axis may play an important role in tumor surveillance of different tumor entities. Therefore, potent strategies modulating the NKp30 axis may represent promising approaches to promote antitumor NK cell responses.
In this work, we designed human EGFR × NKp30 NK cell engagers (i.e., immunoligands) to trigger NKp30-mediated tumor cell elimination using bispecific Ab-like fusion proteins containing a humanized Fab variant derived from cetuximab and a panel of affinity-optimized variants of the N-terminal Ig-like V-type domain of B7-H6 (ΔB7-H6) engineered by yeast surface display. The bispecific NK cell engagers harboring affinity-matured variants facilitated substantially increased NK cell–mediated killing of tumor cells of up to 87-fold compared with molecules harboring the wild-type ΔB7-H6 domain. Importantly, target-dependent cytokine release of TNF-α and IFN-γ was also fundamentally elevated. Intriguingly, this proinflammatory cytokine release was also significantly increased compared with therapeutic Ab cetuximab. Furthermore, we demonstrated that expression of ΔB7-H6–based immunoligands in an effector-competent human IgG1 backbone further enhanced killing of EGFR-overexpressing tumor cells by one order of magnitude compared with molecules either triggering FcγRIIIa or NKp30 alone. Taken together, our data suggest that an EGFR × NKp30 NK cell engager based on affinity-engineered variants of B7-H6 can elicit potent NK cell–mediated lysis with a differentiated profile of released cytokines that could be envisioned to result in targeted inflammation of tumors.
Materials and Methods
Yeast surface display and affinity maturation of ΔB7-H6 variants
Saccharomyces cerevisiae strain EBY100 [MATa URA3-52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1 GAL (pIU211:URA3)] (Thermo Fisher Scientific) was used for yeast surface display. Initially, cells were cultivated in yeast extract peptone dextrose medium composed of 20 g/l peptone, 20 g/l dextrose, and 10 g/l yeast extract supplemented with 10 ml/l penicillin–streptomycin (Life Technologies). Homologous recombination in yeast (gap repair cloning) was used for generation of the ΔB7-H6 library. Eight residues of the N-terminal IgV domain of B7-H6 at the binding interface of B7-H6 and NKp30 (Protein Data Bank [pdb]: 4ZSO) were randomized via TRIM technology at GeneArt (Thermo Fisher Scientific). As described previously, sequences were cloned in a pYD-derived backbone as destination vector (pDest) in frame with Aga2p C-terminally carrying an HA epitope to enable yeast cell surface presentation and detection of full-length molecules (36). Cells harboring library plasmids were cultivated at 30°C and 120 rpm in minimal SD-base medium with dropout mix composed of all essential amino acids (except those from tryptophan, to maintain selection pressure according to the manufacturer’s instructions) (Clontech Laboratories), supplemented with 5.4 g/l Na2HPO4 and 8.6 g/l NaH2PO4 × H2O. ΔB7-H6 expression for library sorting was achieved by culture of 1 × 107 cells/ml for 48 h at 20°C in SG medium with dropout mix, in which glucose was replaced by galactose (Clontech Laboratories) and 10% (w/v) polyethylene glycol 8000. NKp30 binding of yeast surface-expressed ΔB7-H6 was monitored by indirect immunofluorescence using His-tagged NKp30 (Abcam) in combination with Penta-His Alexa Fluor 647 Ab (diluted 1:20; QIAGEN). Simultaneously, ΔB7-H6 surface expression was detected by an anti–HA-PE Ab (diluted 1:20; Abcam). Detection and sorting of yeast candidates was done on an SH800S cell sorter (Sony) using a 70-μm sorting chip in three successive rounds. For the first sorting round, 1 × 108 yeast cells were incubated with 1 μM NKp30 followed by a second round with 100 nM NKp30 and a third round with 50 nM NKp30. Incubation was performed for 1 h on ice prior to washing with PBS and sorting. For the second and third sorting round, cells were incubated for 30 min in 3 and 30 ml of PBS, respectively, to increase sorting stringencies.
Expression and purification of NK cell engagers
Unique ΔB7-H6 sequences were fused to SEED AG chain and cloned into pTT5 (either in house or at GeneArt [Thermo Fisher Scientific]) to allow full-length bispecific SEED production by combination with humanized cetuximab (hu225) Fab on the SEED GA chain. Molecules were either produced in an effector-silenced backbone by introduction of amino acid exchanges L234A, L235A, P329G (SEED_PGLALA) or in an effector-competent IgG1 backbone (SEED). To this end, Expi293 cells were transiently transfected with respective expression vectors according to manufacturer’s instructions (Thermo Fisher Scientific). Ab containing supernatants were harvested and purified via MabSelect Ab purification chromatography resin (GE Healthcare). NK cell engagers were dialyzed overnight against PBS (pH 6.8) using Pur-A-Lyzer Maxi 3500 Dialysis Kit (Sigma-Aldrich). Protein concentrations were determined by UV-visible spectrophotometric measurement (Nanodrop ND-1000; Peqlab Biotechnologie), and thermal stabilities were evaluated by differential scanning fluorimetry on a Prometheus NT.48 (NanoTemper Technologies). To assess purity and aggregations, proteins were analyzed by analytical size-exclusion chromatography with a TSKgel SuperSW3000 column (4.6 × 300 mm; Tosoh Bioscience) in an Agilent HPLC system with a flow rate of 0.35 ml/min.
Biolayer interferometry
Kinetic measurements were performed on an Octet RED96 system (Pall Life Science; ForteBio) at 25°C and 1000 rpm. Immunoligands (5 μg/ml in PBS) were loaded on anti-human Fc biosensors for 5 min followed by 60 s rinsing of the sensor with kinetics buffer (PBS plus 0.1% Tween 20 plus 1% BSA). Afterwards, association to human NKp30 (Abcam) in various concentrations (15.6–1000 nM) was measured for 60 s followed by dissociation measurement for 180 s in kinetics buffer. An irrelevant Ag was measured as negative control in each experiment. Data were fitted and analyzed with ForteBio data analysis software 8.0 using a 1:1 binding model after Savitzky–Golay filtering.
Cell culture
EGFR-expressing epidermoid carcinoma cell line A431 and non–small cell lung carcinoma cell line A549 were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen and cultured in RPMI 1640 GlutaMAX-I or DMEM supplemented with 10% FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin (R10+ and D10+), respectively (all components from Thermo Fisher Scientific). Human Expi293 cells for production of immunoligands were cultivated in suspension with complete Expi293 Expression Medium (Thermo Fisher Scientific).
Tumor cell killings assays
Experiments were approved by the Ethics Committee of the Christian-Albrechts-University of Kiel (Kiel, Germany) and in accordance with Merck internal guidelines and with the Declaration of Helsinki. Preparation of PBMCs from healthy donors was performed as previously described after receiving written informed consent (37). NK cells were isolated by negative selection using an NK Cell Isolation Kit (Miltenyi Biotec) and maintained overnight at a density of 2 × 106 cells/ml in R10+ medium. Cytotoxicity was analyzed in standard 4-h 51Cr release assays performed in 96-well microtiter plates in a total volume of 200 μl, as described previously (37). Human PBMCs or purified NK cells were used as effector cells at E:T ratios of 80:1 and 10:1, respectively. NK cell engagers or cetuximab were applied at the concentrations indicated. For blocking experiments, cells were preincubated for 15 min with 50 μg/ml of either anti-NKp30 mouse IgG2A Ab (mouse IgG2A isotype control Ab was used as a control in these experiments; both were from R&D Systems) to block NKp30 binding on NK cells or oa_hu225-SEED-PGLALA to block EGFR binding on tumor cells before the respective NK cell engagers were added. Percentage lysis was calculated as follows from cpm: percentage lysis = (experimental cpm − basal cpm)/(maximal cpm − basal cpm) × 100 and normalized (0% = percentage lysis of oa_hu225-SEED-PGLALA control molecule; 100% = percentage lysis of cetuximab at saturating concentration) to allow for direct comparison of affinity-matured ΔB7-H6–derived molecules between individual donors. Inhibited lysis was calculated as follows: 100% − (percentage lysisblocked*100)/percentage lysis).
Cytokine release assay
For cytokine release assays, NK cells were isolated with an EasySep Human NK Cell Isolation Kit (STEMCELL Technologies) and incubated overnight in AIM V Medium containing 100 U/ml recombinant human IL-2 (R&D Systems). A total of 2,500 A431 cells were seeded in 384-well microtiter plates (Greiner Bio-One) and incubated for 3 h. Immunoligands were added to a final concentration of 85 nM followed by addition of NK cells at an E:T ratio of 5:1. After 24 h, supernatants were analyzed using human IFN-γ or TNF-α HTRF kits (Cisbio) according to manufacturer’s instructions. Plates were measured with PHERAstar FSX (BMG Labtech), and data were analyzed by MARS software (v.3.32; BMG Labtech), enabling a four-parameter logistic (4PL 1/y2) model fitting of the standard curve.
NK cell activation assay
A total of 20.000 A431 cells per well were seeded in 96-well V-bottom microtiter plates (Thermo Fisher Scientific) and incubated for 3 h prior addition of 100,000 NK cells per well (E:T ratio of 5:1), which were treated with 100 U/ml recombinant human IL-2 overnight. Immunoligands were added at a final concentration of 85 nM followed by 24-h incubation at 37°C. Cells were washed two times with PBS plus 1% BSA prior to incubation with LIVE/DEAD Fixable Near-IR Dead Cell Stain (Thermo Fisher Scientific), anti-human CD56-PE (Miltenyi Biotec), and anti-human CD69- allophycocyanin (Abcam) for 1 h on ice. After washing, cells were measured via flow cytometry with Intellicyt iQue Screener PLUS (Sartorius). For compensation of fluorochromes, Ab capturing analysis beads (OneComp eBeads compensation beads; Thermo Fisher Scientific) were employed according to the manufacturer’s instructions. The gating strategy is shown in Supplemental Fig. 1.
Data processing and statistical analysis
Graphical and statistical analyses were performed with GraphPad Prism 8 software. The p values were calculated employing repeated measures ANOVA and the Bonferroni or Tukey posttest, as recommended, or the Student t test when appropriate. The p values ≤0.05 were regarded as statistically significant.
Results
Extracellular IgV domain of B7-H6 is sufficient to engage NK cells for tumor cell lysis
In previous studies, it was shown that B7-H6 single chain variable fragment fusion proteins trigger NK cell–mediated lysis of tumor cells (28, 38). Because the NKp30 binding site is located in the IgV domain of B7-H6, we hypothesized that this isolated domain alone is sufficient to trigger NK cell activation and tumor cell killing. To address this question, the N-terminal Ig-like V-type domain (Asp25–Ala144) of the extracellular region of B7-H6 (ΔB7-H6) was used to design a novel EGFR-targeting NKp30 NK cell engager (Fig. 1A, 1B). To completely abolish Fc-mediated immune effector functions, the LALA-PG amino acid exchanges (L234A, L235A, P329G) were introduced (39). Kinetic studies of this ΔB7-H6_wt-SEED-PGLALA molecule revealed functional binding to NKp30 (Fig. 1C), in accordance with previously published work (40, 41). Most importantly, significant lysis of EGFR-expressing A431 tumor cells was triggered by this novel immunoligand (Fig. 1D), demonstrating that the isolated ΔB7-H6 domain is sufficient to promote NK cell–mediated lysis of target cells. However, the extent of tumor cell lysis with the monovalent EGFR-binding NK cell engager ΔB7-H6_wt-SEED-PGLALA (EC50 = 244.8 pM) was significantly reduced compared with a similarly designed one-armed (oa) EGFR-targeting molecule (oa_hu225-SEED) lacking the NKp30 binding domain but harboring an active IgG1 Fc region-triggering FcγRIIIa, one of the strongest known trigger molecules expressed on NK cells (EC50 = 27.8 pM).
FIGURE 1.

B7-H6 IgV domain is sufficient to elicit NK cell–mediated tumor cell lysis. (A) Structure of the extracellular domain of B7-H6 with N-terminal IgV domain colored in dark blue and the IgC part depicted in light blue. Model based on pdb entry 3pv7 and was generated with PyMOL v0.99. (B) Structural model (left) and scheme (right) of generated immunoligands for NK cell redirection consisting of ΔB7-H6 (dark blue) and the humanized Fab of cetuximab (hu225) in an effector-silenced IgG1 SEED backbone comprising amino acid exchanges L234A, L235A, P329G. The V region H chain and L chain are colored in green-cyan, Ab backbone in green, SEED GA and AG in CH3 domains indicated by deep teal coloring. Model based on pdb entries 5dk3 and 3pv7 and constructed using PyMOL v0.99. Scheme generated with BioRender (http://www.biorender.com). (C) Biolayer interferometry analysis of wild-type (wt) ΔB7-H6 immunoligand binding to NKp30. (D) Killing property of ΔB7H6_wt-SEED-PGLALA (solid square) was compared with oa hu225-SEED activating FcγRIIIa (solid diamond) and Fc-silenced oa_hu225-SEED-PGLALA lacking the B7-H6 IgV domain (open diamond) as a control molecule in standard 4-h 51Cr release experiments using freshly isolated human NK cells of healthy donors and A431 tumor cells in an E:T ratio of 10:1. Normalized percentage tumor cell lysis of three independent experiments with different donors is shown as mean ± SEM. ***p < 0.001, **p < 0.01 of oa_hu225-SEED versus ΔB7-H6-SEED-PGLALA.
Affinity maturation of the B7-H6 IgV domain by yeast display
To improve the cytotoxic activity of the novel ΔB7-H6–based NK cell engager, we designed a focused combinatorial mutant library of ΔB7-H6 for yeast surface display, using trinucleotide mutagenesis technology, with the aim of enhancing affinity of ΔB7-H6 for NKp30. ΔB7-H6 was displayed successfully and showed specific binding to recombinant NKp30 (Supplemental Fig. 2). Based on the cocrystal structure of B7-H6 and NKp30 eight residues of B7-H6 (Ser60, Gly62, Phe82, Gly83, Val125, Thr127, Leu129, and Lys130) at the contact interface with NKp30 were chosen for library design (Fig. 2A). Randomization of these residues was designed in a semirational way, such that only amino acids that were thought to be potentially beneficial for binding affinity were taken into account. As shown in Fig. 2B, the synthesized library matched fairly well with the initial design, showing only a little bias. The resulting library (comprising ∼1 × 107 unique clones) was sorted by FACS against recombinant NKp30 to isolate variants with significantly enhanced affinities (Fig. 2C). To this end, a two-dimensional labeling strategy was employed to simultaneously detect full-length ΔB7-H6 and binding to NKp30. Because of the low-affinity interaction of B7-H6 and NKp30 that was reported to be in the micromolar range (40), an NKp30 protein concentration of 1 μM was used in the first selection round and subsequently reduced to 100 and 50 nM, respectively (Fig. 2C). Library output after three rounds of selection was analyzed for NKp30 binding in direct comparison with the wild-type ΔB7-H6 and revealed improved NKp30 binding of the vast majority of affinity-matured ΔB7-H6 clones (Supplemental Fig. 2C, 2D). After sequencing of ∼200 clones from sorting rounds two and three, we were able to identify 47 unique clones (Supplemental Table I). Interestingly, none of those comprised mutations at positions Thr127 or Lys130. Consequently, it is tempting to speculate that these two residues are essential for B7-H6 binding to NKp30 or for the structural integrity of the molecule.
FIGURE 2.

Affinity maturation of ΔB7-H6 by yeast surface display. (A) Cocrystal structure of ΔB7-H6 (blue) with NKp30 extracellular domain (green). Residues of ΔB7-H6 at the interface with NKp30 and considered for focused randomization are colored in red. This illustration is based on pdb entry 4ZSO and was generated using PyMOL v0.99. (B) Designated and observed amino acid distribution for specified positions of ΔB7-H6 as determined by sequencing of 96 clones. X (ambiguous). (C) FACS selection for the enrichment of affinity-improved variants of ΔB7-H6 by yeast surface display. A two-dimensional staining strategy for correctly displayed library candidates simultaneously binding to NKp30 was employed. 1 μM, 100 nM, and 50 nM NKp30 were used for sorting rounds one, two, and three, respectively, to enrich high-affinity binders.
Generation and characterization of affinity-matured ΔB7-H6 NK cell engagers
All 47 unique affinity-matured ΔB7-H6 clones were reformatted as Fc-silenced SEEDbodies with monovalent EGFR binding by using the humanized Fab arm of cetuximab (hu225), as shown already for the wild-type ΔB7-H6 NK cell engager (Fig. 1B). As summarized in Supplemental Table I, besides two variants that showed either very low expression (S2#8) or no productivity at all (S2#9), expression yields of all variants after protein A chromatography were in the double- to triple-digit milligram-per-liter scale. This is generally acceptable for transient expression of bifunctional Ab-like fusion proteins. Moreover, melting temperatures of all variants were quite similar to the denaturation point of the SEEDbody comprising the ΔB7-H6 wild-type domain (Supplemental Table I), giving clear evidence that no substantial loss in stability was observed by introducing mutations into ΔB7-H6. Most importantly, the vast majority of the affinity-matured variants showed significantly enhanced affinities for NKp30 of up to 45.1-fold (S3#18) compared with the wild-type ΔB7-H6 NK cell engager (Supplemental Fig. 2E, Table I). In general, improved off-rates contributed substantially more to the overall affinity maturation than modulations of on-rates of the ΔB7-H6 SEED-PGLALA immunoligands. Interestingly, variant S3#13, which did not bind NKp30, was the only clone isolated that contained a mutation of Gly62 (Gly62Ile). This observation suggests that, in addition to Thr127 or Lys130, Gly62 may also be indispensable for ΔB7-H6 binding to NKp30. For subsequent functional evaluation of the affinity-matured ΔB7-H6–derived NK cell engagers, we focused on immunoligands showing at least 85% monomers (i.e., a maximum of 15% aggregates or low molecular weight species) as determined by size-exclusion chromatography. Accordingly, 36 affinity-matured ΔB7-H6 NK cell engagers were selected for functional experiments (Table I).
Table I. Biochemical characterization of immunoligands based on affinity-optimized ΔB7-H6 variants.
| Identifier | KD (M) | x-Fold KD Improvementa | Kon (1/Ms) | Koff (1/s) | x-Fold Koff Improvementa | Mutationsb |
|---|---|---|---|---|---|---|
| ΔB7-H6_wt | 4.09 × 10−7 | — | 4.12 × 105 | 1.68 × 10−1 | — | — |
| ΔB7-H6_S3#1 | 3.91 × 10−8 | 10.5 | 7.54 × 105 | 2.95 × 10−2 | 5.7 | S60T, F82W, L129Y |
| ΔB7-H6_S3#3 | 1.04 × 10−7 | 3.9 | 1.05 × 106 | 1.09 × 10−1 | 1.5 | S60H, V125F, L129Y |
| ΔB7-H6_S3#5 | 2.25 × 10−8 | 18.2 | 7.99 × 105 | 1.80 × 10−2 | 9.3 | S60V, F82W, L129Y |
| ΔB7-H6_S3#6 | 3.31 × 10−8 | 12.4 | 7.37 × 105 | 2.44 × 10−2 | 6.9 | S60H, F82W, L129Y |
| ΔB7-H6_S3#7 | 1.10 × 10−7 | 3.7 | 4.78 × 105 | 5.25 × 10−2 | 3.2 | S60H, F82Y, L129Y |
| ΔB7-H6_S3#8 | 8.07 × 10−8 | 5.1 | 5.64 × 105 | 4.55 × 10−2 | 3.7 | S60H, F82W, V125I |
| ΔB7-H6_S3#9 | 5.78 × 10−8 | 7.1 | 9.70 × 105 | 5.60 × 10−2 | 3.0 | S60H, L129Y |
| ΔB7-H6_S3#10 | 1.45 × 10−7 | 2.8 | 6.89 × 105 | 9.98 × 10−2 | 1.7 | F82Y, V125F, L129Y |
| ΔB7-H6_S3#11 | 3.91 × 10−8 | 10.5 | 1.11 × 106 | 4.32 × 10−2 | 3.9 | S60L, F82W, V125I, L129F |
| ΔB7-H6_S3#12 | 1.29 × 10−7 | 3.2 | 6.71 × 105 | 8.65 × 10−2 | 1.9 | S60E, L129Y |
| ΔB7-H6_S3#14 | 3.45 × 10−8 | 11.9 | 9.33 × 105 | 3.22 × 10−2 | 5.2 | S60T, F82Y, L129Y |
| ΔB7-H6_S3#15 | 1.24 × 10−8 | 33.0 | 8.17 × 105 | 1.01 × 10−2 | 16.6 | S60I, F82W, L129Y |
| ΔB7-H6_S3#16 | 2.87 × 10−8 | 14.3 | 1.01 × 106 | 2.89 × 10−2 | 5.8 | S60L, F82Y, L129Y |
| ΔB7-H6_S3#17 | 5.91 × 10−8 | 6.9 | 9.69 × 105 | 5.73 × 10−2 | 2.9 | F82Y, V125F |
| ΔB7-H6_S3#18 | 9.06 × 10−9 | 45.1 | 1.50 × 106 | 1.36 × 10−2 | 12.4 | S60Y, F82W, L129Y |
| ΔB7-H6_S3#19 | 1.46 × 10−7 | 2.8 | 6.73 × 105 | 9.81 × 10−2 | 1.7 | S60Q, F82W, L129Y |
| ΔB7-H6_S3#21 | 1.79 × 10−8 | 22.8 | 1.18 × 106 | 2.11 × 10−2 | 8.0 | S60L, F82W, L129Y |
| ΔB7-H6_S3#22 | 9.52 × 10−8 | 4.3 | 4.63 × 105 | 4.41 × 10−2 | 3.8 | S60E, F82W, L129Y |
| ΔB7-H6_S3#23 | 1.10 × 10−7 | 3.7 | 4.79 × 105 | 5.25 × 10−2 | 3.2 | S60E, F82Y, L129Y |
| ΔB7-H6_S3#24 | 1.49 × 10−8 | 27.5 | 7.02 × 105 | 1.04 × 10−2 | 16.1 | S60I, F82Y, L129Y |
| ΔB7-H6_S3#25 | 6.52 × 10−8 | 6.3 | 7.45 × 105 | 4.86 × 10−2 | 3.5 | S60W, F82Y, L129Y |
| ΔB7-H6_S3#26 | 1.13 × 10−7 | 3.6 | 4.09 × 105 | 4.64 × 10−2 | 3.6 | S60T, F82W, G83D, L129Y |
| ΔB7-H6_S3#27 | 8.30 × 10−8 | 4.9 | 6.69 × 105 | 5.56 × 10−2 | 3.0 | S60T, F82W, V125I, L129Y |
| ΔB7-H6_S3#28 | 4.43 × 10−7 | 0.9 | 3.06 × 105 | 1.36 × 10−1 | 1.2 | S60H, F82W |
| ΔB7-H6_S3#29 | 2.48 × 10−8 | 16.5 | 7.05 × 105 | 1.75 × 10−2 | 9.6 | S60V, F82W, V125I. L129Y |
| ΔB7-H6_S3#30 | 1.25 × 10−7 | 3.3 | 6.22 × 105 | 7.76 × 10−2 | 2.2 | S60E, F82W, V125I |
| ΔB7-H6_S3#31 | 4.36 × 10−8 | 9.4 | 6.16 × 105 | 2.69 × 10−2 | 6.3 | S60I, F82Y |
| ΔB7-H6_S2#1 | 2.05 × 10−7 | 2.0 | 3.80 × 105 | 7.80 × 10−2 | 2.2 | F82Y, L129Y |
| ΔB7-H6_S2#2 | 2.17 × 10−7 | 1.9 | 3.40 × 105 | 7.37 × 10−2 | 2.3 | F82W, L129Y |
| ΔB7-H6_S2#3 | 1.94 × 10−7 | 2.1 | 5.46 × 105 | 1.06 × 10−1 | 1.6 | S60T, F82W |
| ΔB7-H6_S2#7 | 1.91× 10−7 | 2.1 | 5.04 × 105 | 9.64 × 10−2 | 1.7 | F82Y, V125I, L129Y |
| ΔB7-H6_S2#10 | 1.87 × 10−7 | 2.2 | 5.97 × 105 | 1.11 × 10−1 | 1.5 | S60L, F82W |
| ΔB7-H6_S2#11 | 2.34 × 10−7 | 1.7 | 4.91 × 105 | 1.15 × 10−1 | 1.5 | S60T, F82W |
| ΔB7-H6_S2#13 | 2.05 × 10−7 | 2.0 | 4.10 × 105 | 8.42 × 10−2 | 2.0 | F82W, L129Y |
| ΔB7-H6_S2#14 | 2.17 × 10−7 | 1.9 | 5.70 × 105 | 1.24 × 10−1 | 1.4 | S60T, L129Y |
| ΔB7-H6_S2#15 | 3.50 × 10−7 | 1.2 | 4.01 × 105 | 1.40 × 10−1 | 1.2 | S60E, F82Y |
All molecules were expressed as Fc immune effector-silenced variants harboring the LALA-PG amino acid exchanges.
x-fold improvement values represent the improvement factor of respective overall affinities and off-rates against NKp30 compared with the ΔB7-H6 wild-type NK cell engager.
Mutations describes the incorporated amino acid exchanges of the variants compared with the ΔB7-H6 wild-type NK cell engager.
Affinity-matured ΔB7-H6 immunoligands elicit strongly enhanced NK cell–mediated cytotoxicity against EGFR-expressing tumor cells
As an initial screening, the 36 remaining ΔB7-H6, EGFR-targeting NK cell engagers were tested for their capacity to mediate tumor cell killing using PBMCs (containing 5–20% NK cells) from healthy donors as effector cells and the EGFR+ tumor cell line A431 as target cells in chromium release assays. To allow comparison and ranking of the 36 molecules, data sets derived from different effector cell donors, who usually show various donor-dependent maximal lytic activity, were normalized. An internal positive control was set to 100% and oa_hu225-SEED-PGLALA–mediated lysis to 0% because this molecule is functionally inactive and reflects the tumor cell lysis induced by the NK cells alone (Fig. 1D). In these experiments, shown in Supplemental Fig. 3, the Fc effector–silenced wild-type ΔB7-H6 NK cell engager ΔB7-H6_wt SEED-PGLALA triggered significant tumor cell lysis (EC50 = 1.1 nM). Of note, all 36 ΔB7-H6 affinity-matured SEED-PGLALA molecules mediated higher maximum tumor cell lysis and achieved enhanced potency of up to 187-fold (S3#25) compared with the wild-type ΔB7-H6 immunoligand (Supplemental Fig. 3). Among them, 11 constructs bound to NKp30 with affinity improvements of more than 10-fold (KD <40 nM) and five variants comprised enhanced affinities between 5-fold and 10-fold, whereas the majority of clones comprised optimized affinities of <5-fold (KD >120 nM; Supplemental Fig. 3, Table I). Six of the 11 NK cell engagers constructed with the high-affinity ΔB7-H6 clones (KD <40 nM) (i.e., S3#14, S3#15, S3#16, S3#18, S3#24, and S3#29) induced NK cell–mediated killing of tumor cells with EC50 values ranging from 10.6 to 71.1 pM, representing a 16-fold to 106-fold increase compared with the wild-type ΔB7-H6 NK cell engager (Supplemental Fig. 3). Of the NK cell engagers using ΔB7-H6 clones with optimized affinities for NKp30 between 5-fold and 10-fold, S3#25 and S3#31 were the most potent, displaying NKp30 affinity enhancements of 6.3-fold and 9.4-fold, respectively. These molecules induced NK cell–mediated killing of tumor cells with half-maximal killing activity of 6 pM (S3#25) and 25.1 pM (S3#31). From the variants comprising optimized NKp30 affinities of <5-fold, clone S3#27 (KD = 83 nM) elicited significant tumor cell lysis (EC50 = 27.6 pM) with 41-fold improved potency compared with wild-type ΔB7-H6, and it was also selected for further characterization.
Next, the potency of the nine most active candidates from the initial screening (i.e., S3#14, S3#15, S3#16, S3#18, S3#24, S3#25, S3#27, S3#29, and S3#31) was further compared via chromium release assays using A431 and A549 cells with PBMCs from three different donors (Fig. 3A). As observed for A431 cells, the nine candidates also mediated substantially improved maximum killing and significantly enhanced potency in tumor cell lysis compared with the NK cell engager harboring wild-type ΔB7-H6 against tumor cell line A549 expressing significantly lower EGFR levels compared with A431 cells (Fig. 3A). Although wild-type ΔB7-H6 NK cell engager ΔB7-H6_wt SEED-PGLALA showed significant but limited activity (EC50 A431 = 1.8 nM), the most active affinity-optimized NK cell engager, S3#15, demonstrated improved potency at low picomolar concentrations with PBMCs of three different donors (EC50 = 29.9 pM). The extent of lysis of A549 cells achieved by the most effective affinity-matured ΔB7-H6 NK cell engager, S3#24 (EC50 = 108.3 pM), is ∼3.6-times higher compared with A431 cells (Table II). This might most likely reflect the impact of the lower EGFR expression level on A549 target cells, a well-known parameter affecting effector cell killing. To further characterize the four most potent NK cell engagers, S3#15, S3#18, S3#24, and S3#25, we performed killings assays with purified, nonstimulated NK cells (Fig. 3B). Using A431 cells, the four ΔB7-H6 affinity-maturated NK cell engagers mediated tumor cell killing in the single-digit picomolar range, representing an increase in potency of up to 87-fold (S3#15) compared with the NK cell engager harboring the wild-type ΔB7-H6 domain (Table II). Even for A549 target cells, potencies of all four leading hits were still in the picomolar range. Notably, the efficacy of wild-type ΔB7-H6 NK cell engager was not substantially different from the negative control in NK cell Ab-dependent cell-mediated cytotoxicity with A549 cells (Fig. 3A, 3B). Finally, EGFR-specific and NKp30-mediated killing by the four leading NK cell engagers was further verified by either blocking EGFR on the cell surface of A431 cells or by preincubation of NK cells with an excess of an anti-human NKp30 Ab. As shown in Fig. 3C, lysis of A431 tumor cells was significantly inhibited for all four NK cell engagers under both conditions, demonstrating the specificity and prerequisite of bispecific engagement of these novel B7-H6 affinity-matured immunoligands (Fig. 3C).
FIGURE 3.

Cytotoxic activity of affinity-optimized ΔB7-H6–based NK cell engager. Standard 4-h 51Cr release assays were performed with A431 cells expressing high EGFR levels (left) and A549 cells expressing low EGFR levels (right) using human PBMCs at an E:T ratio of 80:1 (A) or purified NK cells at an E:T ratio of 10:1 (B) to analyze dose-dependent killing of the leading ΔB7-H6 SEED-PGLALA NK cell engager. The affinity-matured ΔB7-H6 variants were compared with wild-type ΔB7-H6 wt_hu225-SEED-PGLALA (gray) and a control molecule lacking ΔB7-H6 but still binding oa to EGFR via the humanized cetuximab Fab-fragment (oa_hu225-SEED-PGLALA; dotted black line). To allow comparison of the results, data of each experiment were normalized. Graphs show normalized mean ± SEM of three experiments performed with PBMCs or NK cell from different donors. (C) Specificity of A431 lysis mediated by the leading four NK cell engagers (applied at 0.6 nM) was verified by either blocking EGFR binding by preincubation of A431 cells with 50 μg/ml of the Fc-silenced oa_hu225-SEED-PGLALA (left graph) or NKp30 binding by preincubation of NK cells with 50 μg/ml anti-NKp30 Ab (right graph). Graphs show percentage of inhibited lysis as mean ± SEM of four individual experiments. ***p < 0.001, *p < 0.05 compared with NK cells only (left graph) or cetuximab (right graph).
Table II. Potencies of selected EGFR-targeting, affinity-matured ΔB7-H6 immunoligands.
| Molecule | A431 EC50 with PBMC (pM) | A549 EC50 with PBMC (pM) | A431 EC50 with NK Cells (pM) | A549 EC50 with NK cells (pM) |
|---|---|---|---|---|
| oa_hu225 | no lysis | no lysis | no lysis | no lysis |
| B7-H6_wt | 1839 | n.a. | 244.8 | n.a. |
| B7-H6_S3#14 | 150.1 | 556.5 | n.d. | n.d. |
| B7-H6_S3#15 | 29.9 | 133.2 | 2.80 | 31.31 |
| B7-H6_S3#16 | 87.3 | 205.8 | n.d. | n.d. |
| B7-H6_S3#18 | 51.1 | 132.0 | 4.31 | 42.47 |
| B7-H6_S3#24 | 39.7 | 108.3 | 3.55 | 35.19 |
| B7-H6_S3#25 | 37.8 | 134.9 | 4.25 | 34.62 |
| B7-H6_S3#27 | 60.2 | 355.6 | n.d. | n.d. |
| B7-H6_S3#29 | 33.6 | 172.3 | n.d. | n.d. |
| B7-H6_S3#31 | 34.8 | 223.8 | n.d. | n.d. |
Boldface numbers represent leading four immunoligands; italic numbers represent the best molecule in the respective category. All molecules were produced as Fc immune effector-silenced variants harboring the LALA-PG amino acid exchanges.
n.a., not applicable because of insignificant killing.
ΔB7-H6 affinity-maturated NK cell engagers exhibit a distinct NK cell activation profile
Next, we set out to scrutinize activation of NK cells in the presence of tumor cells overexpressing EGFR (A431) and the four leading immunoligands via analyzing CD69 upregulation as an early NK cell activation marker. As expected, negative control (i.e., oa_hu225-SEED-PGLALA) mediated neglectable NK cell activation, whereas the NK cell engager harboring wild-type ΔB7-H6 elicited CD69 expression on ∼17% of NK cells on average (Fig. 4A). Compared with the wild-type ΔB7-H6 immunoligand, the four most potent affinity-optimized ΔB7-H6 immunoligands activated nearly twice as many NK cells, ranging from 30.7 to 33.8% CD69+ NK cells (Fig. 4A). In addition, all four leading NK cell engagers promoted significantly increased NK cell production of proinflammatory cytokines IFN-γ and TNF-α in a target-dependent manner (Fig. 4B, 4C). In comparison with the wild-type ΔB7-H6 molecule, which induced release of 141 pg/ml IFN-γ and 37 pg/ml TNF-α, S3#15, S3#18, S3#24, and S3#25 induced release of 615–824 pg/ml IFN-γ and 152–214 pg/ml TNF-α on average (Fig. 4B, 4C). Thus, affinity-engineered NK cell engagers induced up to 5.8-fold increased levels of IFN-γ and TNF-α compared with the wild-type ΔB7-H6 immunoligand. These findings are in line with the substantially enhanced killing potencies and efficacies of the affinity-engineered ΔB7-H6 NK cell engagers observed before.
FIGURE 4.

NK cell activation, IFN-γ, and TNF-α release of the four leading ΔB7-H6 SEED-PGLALA immunoligands. Human purified NK cells were cocultured for 24 h with A431 cells at an E:T ratio of 5:1 prior to analysis of NK cell activation (A), IFN-γ (B), and TNF-α (C) release. Affinity-matured ΔB7-H6 NK cell engagers and oa_hu225-SEED-PGLALA were compared with wild-type ΔB7-H6_wt-SEED-PGLALA (all at 85 nM). Percentage of CD69+ NK cells was determined by double-staining of viable NK cells with CD56-PE and CD69-allophycocyanin using flow cytometric analyses. Human cytokine HTRF kits were used for the quantification of IFN-γ and TNF-α release of NK cells. Graphs show box and whisker plots as superimposition with dot plots of six (A) and eight (B and C) individual experiments, respectively. ***p < 0.001, **p < 0.01, *p < 0.05. n.s., not significant compared with ΔB7-H6_wt-SEED-PGLALA.
ΔB7-H6 affinity-maturated NK cell engagers elucidate improved killing by concomitant engagement of FcγRIIIa
Finally, we tested the NK cell engager ΔB7-H6 S3#18, which has the highest binding affinity for NKp30 (KD = 9 nM), in the background of an Fc region capable of FcγRIIIa binding (SEEDbody lacking the L234A, L235A, P329G amino acid exchanges) to analyze whether tumor cell lysis can be further enhanced by concomitant NKp30 and FcγRIIIa activation of NK cells. The oa_hu225-SEED molecule harboring an Fc domain capable of FcR binding but lacking a ΔB7-H6 domain was as potent as the affinity-matured Fc-silenced ΔB7-H6 S3#18 SEED-PGLALA molecule, which solely activates NKp30 (Fig. 5A). Intriguingly, incorporation of a functional Fc in the NK cell engager S3#18 (i.e., ΔB7-H6 S3#18-SEED) significantly improved half-maximal killing by 9.2-fold (Fig. 5A). Furthermore, as shown in Fig. 5B, half-maximal killing achieved with ΔB7-H6 S3#18-SEED was in the range or even slightly exceeded the cytotoxic activity of the clinically applied Ab cetuximab. These data give clear evidence that activating NK cells via NKp30 with a high-affinity–engineered B7-H6–derived NK cell engager is as effective as activating NK cells via FcγRIIIa and that the combination of NKp30 and FcγRIIIa activation of NK cells in one molecule can further potentiate the lysis of EGFR-expressing tumor cells.
FIGURE 5.

Synergistic cytotoxic activity by concomitant NKp30 and FcγRIIIa engagement of ΔB7-H6–based immunoligands with functional IgG1 Fc. The highest-affinity variant for NKp30 binding, S3#18, was tested in 4-h 51Cr release assays with A431 cells and purified NK cells at an E:T ratio of 10:1 to compare dose-dependent killing of (A) the Fc-silenced ΔB7-H6 S3#18-SEED-PGLALA (dark green), oa_hu225-SEED (gray) lacking ΔB7-H6, and ΔB7-H6 S3#18-SEED with a functional Fc (light green) or (B) cetuximab (black) and ΔB7-H6 S3#18 SEED, both carrying a functional Fc. Fc-silenced oa_hu225-SEED-PGLALA (dotted black line) lacking also ΔB7-H6 and thus NKp30 activation was used as control. Graph shows percentage of lysis as mean ± SEM of seven independent experiments. Cytokine release of IFN-γ (C) and TNF-α (D) as well as NK cell activation (E) for effector silenced and effector-competent ΔB7-H6 S3#18-SEED molecules as well as effector silenced and effector-competent oa_hu225-SEED and cetuximab (all at 85 nM). Cytokine release was analyzed after 24-h incubation of purified NK cells with A431 cells at an E:T ratio of 5:1 with human cytokine HTRF kits. Percentage of CD69+ NK cells was determined by double-staining of viable NK cells with CD56-PE and CD69-allophycocyanin using flow cytometry. Human cytokine HTRF kits were used for the quantification of IFN-γ and TNF-α release of NK cells. Profiles show box and whisker plots as superimpositions, with dot plots of up to eight independent experiments. ***p < 0.001, **p < 0.01, *p < 0.05. n.s., not significant compared with the ΔB7-H6 S3#18-SEED-PGLALA.
Finally, we compared the capacity to trigger IFN-γ and TNF-α by the molecule engaging both NKp30 and FcγRIIIa with constructs engaging either NKp30 or FcγRIIIa and with the clinically approved Ab cetuximab (Fig. 5C, 5D). Interestingly, the capacity to trigger cytokine release was maintained in the molecule engaging both activating receptors compared with the molecule solely engaging NKp30, with a trend toward higher TNF-α release induced by the molecule with an effector-competent IgG1 backbone. Of note, compared with the therapeutic Ab cetuximab, the release of IFN-γ and TNF-α was substantially elevated, irrespective of the used Fc. Moreover, activation of NK cells was comparable to cetuximab (Fig. 5E) Ultimately, our data demonstrate that the engineered NK cell engagers in this study are capable in triggering NK cell–mediated lysis with similar effectiveness as the clinically approved Ab cetuximab, but in addition, they trigger significant IFN-γ and TNF-α release to potentially modulate antitumor immune responses.
Discussion
To efficiently trigger antitumor NK cell responses using activating NK cell engagers, the solitary ΔB7-H6 was affinity maturated by a semirational design and screening approach. Engineered molecules with increased NKp30 binding activity resulted in significantly improved NK cell–mediated lysis of tumor cells and enhanced secretion of proinflammatory cytokines INF-γ and TNF-α. Coligation of NKp30 and FcγRIIIa on NK cells further enhanced their cytotoxic activity by almost 10-fold compared with molecules engaging either receptor alone. Furthermore, the most potent novel NK cell engager was similarly effective in triggering NK cell–mediated lysis of EGFR-expressing tumor cells as the clinically approved Ab cetuximab but additionally promoted considerably higher release of proinflammatory cytokines. These findings suggest that the novel NK cell–activating molecules may represent attractive agents to elicit strong NK cell–mediated antitumor responses.
NK cell–based therapeutic interventions represent promising approaches to improve cancer therapy by initiating a multifaceted immune response (1, 42). Because NK cell activation is tightly regulated by integrating signals of a large set of activating and inhibitory receptors (16, 17), potential therapeutic options have been proposed by either blocking inhibitory NK cell checkpoints, such as NKG2A (43), or by enhancing stimulatory signals, such as NKG2D, NKp30, NKp46, or the FcγRIIIa (23, 25, 26, 29). Because tumor cells evade NK cell recognition by down-modulation or shedding of the natural ligands of activating NK cell receptors, such as MICA/B or B7-H6 (44–48), restoring NK cell recognition by enhancing surface density of activating ligands may represent a promising approach for cancer therapy. In previous studies, this has been realized by fusing the natural ligands for activating NK cell receptors to single chain variable fragments or full Abs (27–29, 38). In this study, we designed (to our knowledge) novel IgG-based bispecific, multifunctional fusion proteins using the SEED technology (49) and an engineered minimal NKp30 binding domain derived from B7-H6. Because these molecules display IgG-like properties in terms of stability and harbor an IgG Fc domain allowing FcRn binding, such molecules may show favorable characteristics compared with previously described B7-H6–based immunoligands when applied in vivo (50). We were able to demonstrate that the IgV domain of B7-H6 is sufficient to trigger NK cell activation. Affinity maturation of the ΔB7-H6 domain by exchanging key residues at the B7-H6/NKp30 contact interface, using a combinatorial library approach, resulted in significantly enhanced tumor cell lysis by NK cells. These data clearly demonstrate that, similar to the Fc/FcγRIIIa system, which could be potently modulated by Fc-engineering, high-affinity receptor engagement of NKp30 results in fundamentally improved cytotoxic response. Also, interestingly, variants displaying only a moderate affinity improvement resulted in significantly increased lytic activity, which is in line with other bispecific effector cell engagers and engineered Fc domains, for which it has been demonstrated that maximal affinity enhancement may not necessarily result in improved cytolytic activity (37, 51–53). Because the observed cytolytic activity of our novel molecules does not directly correlate with binding affinity of the receptor-ligand interaction, the mechanisms of enhanced recognition might be more complex. From our experiments, it could not be excluded that engineering the ΔB7-H6 domain may, for example, impact protein flexibility, which in turn may have a strong impact on receptor clustering or activating synapse formation. Intriguingly, one of the best affinity-engineered ΔB7-H6 candidates triggered NK cell–mediated lysis of tumor cells via activation of NKp30 with similar effectiveness as molecules activating FcγRIIIa, one of the most potent known trigger molecules on NK cells that is, moreover, expressed at significantly higher levels compared with NKp30 [70,000 versus 1,000 specific Ag binding sites (27)]. Additionally, concomitant activation of FcγRIIIa and NKp30 by incorporating an effector-competent IgG1 Fc backbone resulted in an almost 10-fold improvement in lytic capacity. This is in line with previous findings that combining mAbs with B7-H6–based immunoligands demonstrate improved cytolytic activity (28). Moreover, Gauthier and coworkers (25) were able to demonstrate that simultaneous triggering of NKp46 and FcγRIIIa substantially enhances efficiencies of multifunctional Abs. The precise underlying mechanism is not fully understood because NKp30 and FcγRIIIa signal via the same adaptor chains [CD3ζ, common γ-chain (54)]. Beyond this, engagement of NKp30 via improved B7-H6–based engagers clearly differs from exclusive FcγRIIIa engagers in their capacity to trigger the production of proinflammatory cytokines. NKp30 engagers demonstrated up to 5-fold increased production of TNF-α and INF-γ compared with molecules solely triggering FcγRIIIa. This feature was also observed with molecules allowing concomitant NKp30 and FcγRIIIa engagement, indicating that FcγRIIIa triggering has no inhibitory effect in NKp30-mediated cytokine release. Whether this is due to distinct signaling events triggered by the two receptor systems or by activating distinct NK cell subsets must be further investigated.
Cao and colleagues (55) demonstrated that different therapeutic treatments, such as 5-FU, cisplatin, radiation, and cytokine therapy, with TNF-α resulted in the upregulation of B7-H6, rendering tumor cells more sensitive to NK cell–mediated killing in a NKp30-dependent fashion. The optimized ΔB7-H6–based agents may therefore significantly increase levels of surface-exposed B7-H6 by two different mechanisms. Although obviously “artificial” decoration of tumor cells using bispecific EGFR-directed ΔB7-H6 fusion proteins results in higher surface density of ΔB7-H6, the affinity-enhanced variants also significantly triggered TNF-α release by NK cells, which might in turn, as described by Cao and colleagues (55), lead to upregulation of endogenous B7-H6 on tumor cells in close vicinity. This might be especially interesting in situations in which the target Ag addressed by Ab-based approaches is inhomogeneously expressed because this mechanism might sensitize tumor cells sensitive to NK cell lysis, also in an Ag-independent way.
Besides TNF-α production, by triggering NKp30 the immunoligands described in this study also promoted INF-γ release in a strictly targeted fashion and to a significantly larger extent than molecules, such as cetuximab, exclusively activating NK cells via FcγRIIIa. Considering the pleiotropic effects of IFN-γ (56–59) on several different immune cell subsets present in the tumor microenvironment, this may have major consequences when applied in vivo, especially in the treatment of solid tumors. Although IFN-γ signaling has been demonstrated to promote tumor elimination by inhibiting suppressive immune cells, such as regulatory T cells (60), myeloid‐derived suppressor cells (61), and tumor‐associated macrophages, IFN-γ is also critical for NK, NKT, and T cell trafficking into tumors through chemokine induction (62, 63). In addition, IFN-γ activates APCs and cytotoxic T lymphocytes. Hence, the NK cell engagers described in this work might be able to turn cold tumors hot and may therefore also represent interesting candidates in combination with other immunomodulatory agents such as immune checkpoint inhibitors (64, 65).
In conclusion, the B7-H6–based NK cell engagers engineered in this study combine a unique set of effector functions to directly eliminate tumor cells by triggering NK cell cytotoxicity and eliciting a multifaceted immune reaction by promoting release of proinflammatory cytokines and therefore represent an interesting new class of immunomodulatory agents.
Supplementary Material
Acknowledgments
We thank Britta von Below and Anja Muskulus for excellent technical assistance. Moreover, we are grateful to Kerstin Hallstein, Laura Unmuth, Stephan Keller, Alexander Müller, Sigrid Auth, Pia Stroh, Marion Wetter, Thomas Rysiok, Janina Klemm, Stefan Becker, and Dirk Müller-Pompalla for experimental support. This work is dedicated to Prof. Stefan Hüttenhain on the occasion of his retirement.
This work was supported by research funding from German Cancer Aid (Mildred Scheel Professorship Program) to M.P.
The online version of this article contains supplemental material.
- ΔB7-H6
- N-terminal Ig-like V-type domain of B7-H6
- NCR
- natural cytotoxicity receptor
- oa
- one-armed
- pdb
- Protein Data Bank.
Disclosures
M.B., B.V., L.T., S.K., T.G., B.R., and S.Z. are employees of Merck Healthcare KGaA or EMD Serono. A patent related to this work is actually pending.
References
- 1.Chiossone L., Dumas P.-Y., Vienne M., Vivier E. 2018. Natural killer cells and other innate lymphoid cells in cancer. [Published erratum appears in 2018 Nat. Rev. Immunol. 18: 726.] Nat. Rev. Immunol. 18: 671–688. [DOI] [PubMed] [Google Scholar]
- 2.Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. 2008. Functions of natural killer cells. Nat. Immunol. 9: 503–510. [DOI] [PubMed] [Google Scholar]
- 3.Carlsten M., Järås M. 2019. Natural killer cells in myeloid malignancies: immune surveillance, NK cell dysfunction, and pharmacological opportunities to bolster the endogenous NK cells. Front. Immunol. 10: 2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrow A. D., Martin C. J., Colonna M. 2019. The natural cytotoxicity receptors in health and disease. Front. Immunol. 10: 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moretta L., Bottino C., Cantoni C., Mingari M. C., Moretta A. 2001. Human natural killer cell function and receptors. Curr. Opin. Pharmacol. 1: 387–391. [DOI] [PubMed] [Google Scholar]
- 6.Kruse P. H., Matta J., Ugolini S., Vivier E. 2014. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 92: 221–229. [DOI] [PubMed] [Google Scholar]
- 7.Brandt C. S., Baratin M., Yi E. C., Kennedy J., Gao Z., Fox B., Haldeman B., Ostrander C. D., Kaifu T., Chabannon C., et al. 2009. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 206: 1495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spear P., Wu M.-R., Sentman M.-L., Sentman C. L. 2013. NKG2D ligands as therapeutic targets. Cancer Immun. 13: 8. [PMC free article] [PubMed] [Google Scholar]
- 9.Seidel U. J. E., Schlegel P., Lang P. 2013. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 4: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schubert I., Kellner C., Stein C., Kügler M., Schwenkert M., Saul D., Stockmeyer B., Berens C., Oduncu F. S., Mackensen A., Fey G. H. 2012. A recombinant triplebody with specificity for CD19 and HLA-DR mediates preferential binding to antigen double-positive cells by dual-targeting. MAbs 4: 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koch J., Tesar M. 2017. Recombinant antibodies to arm cytotoxic lymphocytes in cancer immunotherapy. Transfus. Med. Hemother. 44: 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weng W.-K., Levy R. 2003. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 21: 3940–3947. [DOI] [PubMed] [Google Scholar]
- 13.Musolino A., Naldi N., Bortesi B., Pezzuolo D., Capelletti M., Missale G., Laccabue D., Zerbini A., Camisa R., Bisagni G., et al. 2008. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 26: 1789–1796. [DOI] [PubMed] [Google Scholar]
- 14.Bibeau F., Lopez-Crapez E., Di Fiore F., Thezenas S., Ychou M., Blanchard F., Lamy A., Penault-Llorca F., Frébourg T., Michel P., et al. 2009. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 27: 1122–1129. [DOI] [PubMed] [Google Scholar]
- 15.Preithner S., Elm S., Lippold S., Locher M., Wolf A., da Silva A. J., Baeuerle P. A., Prang N. S. 2006. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol. Immunol. 43: 1183–1193. [DOI] [PubMed] [Google Scholar]
- 16.Morvan M. G., Lanier L. L. 2016. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16: 7–19. [DOI] [PubMed] [Google Scholar]
- 17.Shimasaki N., Jain A., Campana D. 2020. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 19: 200–218. [DOI] [PubMed] [Google Scholar]
- 18.Rezvani K. 2019. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant. 54(Suppl. 2): 785–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu E., Marin D., Banerjee P., Macapinlac H. A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. 2020. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382: 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Björklund A. T., Carlsten M., Sohlberg E., Liu L. L., Clancy T., Karimi M., Cooley S., Miller J. S., Klimkowska M., Schaffer M., et al. 2018. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin. Cancer Res. 24: 1834–1844. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka J., Miller J. S. 2020. Recent progress in and challenges in cellular therapy using NK cells for hematological malignancies. Blood Rev. 44: 100678. [DOI] [PubMed] [Google Scholar]
- 22.Granzin M., Wagner J., Köhl U., Cerwenka A., Huppert V., Ullrich E. 2017. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front. Immunol. 8: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellwanger K., Reusch U., Fucek I., Wingert S., Ross T., Müller T., Schniegler-Mattox U., Haneke T., Rajkovic E., Koch J., et al. 2019. Redirected optimized cell killing (ROCK®): a highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. MAbs 11: 899–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Felices M., Lenvik T. R., Kodal B., Lenvik A. J., Hinderlie P., Bendzick L. E., Schirm D. K., Kaminski M. F., McElmurry R. T., Geller M. A., et al. 2020. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engager. Cancer Immunol. Res. 8: 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gauthier L., Morel A., Anceriz N., Rossi B., Blanchard-Alvarez A., Grondin G., Trichard S., Cesari C., Sapet M., Bosco F., et al. 2019. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell 177: 1701–1713.e16. [DOI] [PubMed] [Google Scholar]
- 26.Chan W. K., Kang S., Youssef Y., Glankler E. N., Barrett E. R., Carter A. M., Ahmed E. H., Prasad A., Chen L., Zhang J., et al. 2018. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol. Res. 6: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peipp M., Derer S., Lohse S., Staudinger M., Klausz K., Valerius T., Gramatzki M., Kellner C. 2015. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget 6: 32075–32088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kellner C., Günther A., Humpe A., Repp R., Klausz K., Derer S., Valerius T., Ritgen M., Brüggemann M., van de Winkel J. G., et al. 2015. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. OncoImmunology 5: e1058459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Strandmann E. P., Hansen H. P., Reiners K. S., Schnell R., Borchmann P., Merkert S., Simhadri V. R., Draube A., Reiser M., Purr I., et al. 2006. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 107: 1955–1962. [DOI] [PubMed] [Google Scholar]
- 30.Pende D., Parolini S., Pessino A., Sivori S., Augugliaro R., Morelli L., Marcenaro E., Accame L., Malaspina A., Biassoni R., et al. 1999. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 190: 1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pogge von Strandmann E., Simhadri V. R., von Tresckow B., Sasse S., Reiners K. S., Hansen H. P., Rothe A., Böll B., Simhadri V. L., Borchmann P., et al. 2007. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 27: 965–974. [DOI] [PubMed] [Google Scholar]
- 32.Simhadri V. R., Reiners K. S., Hansen H. P., Topolar D., Simhadri V. L., Nohroudi K., Kufer T. A., Engert A., Pogge von Strandmann E. 2008. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS One 3: e3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fauriat C., Just-Landi S., Mallet F., Arnoulet C., Sainty D., Olive D., Costello R. T. 2007. Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 109: 323–330. [DOI] [PubMed] [Google Scholar]
- 34.Han B., Mao F. Y., Zhao Y. L., Lv Y. P., Teng Y. S., Duan M., Chen W., Cheng P., Wang T. T., Liang Z. Y., et al. 2018. Altered NKp30, NKp46, NKG2D, and DNAM-1 expression on circulating NK cells is associated with tumor progression in human gastric cancer. J. Immunol. Res. 2018: 6248590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nieto-Velázquez N. G., Torres-Ramos Y. D., Muñoz-Sánchez J. L., Espinosa-Godoy L., Gómez-Cortés S., Moreno J., Moreno-Eutimio M. A. 2016. Altered expression of natural cytotoxicity receptors and NKG2D on peripheral blood NK cell subsets in breast cancer patients. Transl. Oncol. 9: 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roth L., Krah S., Klemm J., Günther R., Toleikis L., Busch M., Becker S., Zielonka S. 2020. Isolation of antigen-specific VHH single-domain antibodies by combining animal immunization with yeast surface display. Methods Mol. Biol. 2070: 173–189. [DOI] [PubMed] [Google Scholar]
- 37.Repp R., Kellner C., Muskulus A., Staudinger M., Nodehi S. M., Glorius P., Akramiene D., Dechant M., Fey G. H., van Berkel P. H. C., et al. 2011. Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J. Immunol. Methods 373: 67–78. [DOI] [PubMed] [Google Scholar]
- 38.Kellner C., Maurer T., Hallack D., Repp R., van de Winkel J. G. J., Parren P. W. H. I., Valerius T., Humpe A., Gramatzki M., Peipp M. 2012. Mimicking an induced self phenotype by coating lymphomas with the NKp30 ligand B7-H6 promotes NK cell cytotoxicity. J. Immunol. 189: 5037–5046. [DOI] [PubMed] [Google Scholar]
- 39.Schlothauer T., Herter S., Koller C. F., Grau-Richards S., Steinhart V., Spick C., Kubbies M., Klein C., Umaña P., Mössner E. 2016. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 29: 457–466. [DOI] [PubMed] [Google Scholar]
- 40.Li Y., Wang Q., Mariuzza R. A. 2011. Structure of the human activating natural cytotoxicity receptor NKp30 bound to its tumor cell ligand B7-H6. J. Exp. Med. 208: 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joyce M. G., Tran P., Zhuravleva M. A., Jaw J., Colonna M., Sun P. D. 2011. Crystal structure of human natural cytotoxicity receptor NKp30 and identification of its ligand binding site. Proc. Natl. Acad. Sci. USA 108: 6223–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huntington N. D., Cursons J., Rautela J. 2020. The cancer-natural killer cell immunity cycle. Nat. Rev. Cancer 20: 437–454. [DOI] [PubMed] [Google Scholar]
- 43.van Hall T., André P., Horowitz A., Ruan D. F., Borst L., Zerbib R., Narni-Mancinelli E., van der Burg S. H., Vivier E. 2019. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 7: 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reiners K. S., Topolar D., Henke A., Simhadri V. R., Kessler J., Sauer M., Bessler M., Hansen H. P., Tawadros S., Herling M., et al. 2013. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 121: 3658–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W., Guo H., Geng J., Zheng X., Wei H., Sun R., Tian Z. 2014. Tumor-released Galectin-3, a soluble inhibitory ligand of human NKp30, plays an important role in tumor escape from NK cell attack. J. Biol. Chem. 289: 33311–33319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlecker E., Fiegler N., Arnold A., Altevogt P., Rose-John S., Moldenhauer G., Sucker A., Paschen A., von Strandmann E. P., Textor S., Cerwenka A. 2014. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 74: 3429–3440. [DOI] [PubMed] [Google Scholar]
- 47.Chitadze G., Lettau M., Bhat J., Wesch D., Steinle A., Fürst D., Mytilineos J., Kalthoff H., Janssen O., Oberg H.-H., Kabelitz D. 2013. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 133: 1557–1566. [DOI] [PubMed] [Google Scholar]
- 48.Salih H. R., Rammensee H.-G., Steinle A. 2002. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 169: 4098–4102. [DOI] [PubMed] [Google Scholar]
- 49.Davis J. H., Aperlo C., Li Y., Kurosawa E., Lan Y., Lo K.-M., Huston J. S. 2010. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 23: 195–202. [DOI] [PubMed] [Google Scholar]
- 50.Pyzik M., Rath T., Lencer W. I., Baker K., Blumberg R. S. 2015. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J. Immunol. 194: 4595–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bortoletto N., Scotet E., Myamoto Y., D’Oro U., Lanzavecchia A. 2002. Optimizing anti-CD3 affinity for effective T cell targeting against tumor cells. Eur. J. Immunol. 32: 3102–3107. [DOI] [PubMed] [Google Scholar]
- 52.Staflin K., Zuch de Zafra C. L., Schutt L. K., Clark V., Zhong F., Hristopoulos M., Clark R., Li J., Mathieu M., Chen X., et al. 2020. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight 5: e133757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellerman D. 2019. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 154: 102–117. [DOI] [PubMed] [Google Scholar]
- 54.Vivier E., Raulet D. H., Moretta A., Caligiuri M. A., Zitvogel L., Lanier L. L., Yokoyama W. M., Ugolini S. 2011. Innate or adaptive immunity? The example of natural killer cells. Science 331: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao G., Wang J., Zheng X., Wei H., Tian Z., Sun R. 2015. Tumor therapeutics work as stress inducers to enhance tumor sensitivity to natural killer (NK) cell cytolysis by up-regulating NKp30 ligand B7-H6. J. Biol. Chem. 290: 29964–29973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burke J. D., Young H. A. 2019. IFN-γ: a cytokine at the right time, is in the right place. Semin. Immunol. 43: 101280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alspach E., Lussier D. M., Schreiber R. D. 2019. Interferon γ and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb. Perspect. Biol. 11: a028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ivashkiv L. B. 2018. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18: 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castro F., Cardoso A. P., Gonçalves R. M., Serre K., Oliveira M. J. 2018. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9: 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Overacre-Delgoffe A. E., Chikina M., Dadey R. E., Yano H., Brunazzi E. A., Shayan G., Horne W., Moskovitz J. M., Kolls J. K., Sander C., et al. 2017. Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell 169: 1130–1141.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Medina-Echeverz J., Haile L. A., Zhao F., Gamrekelashvili J., Ma C., Métais J.-Y., Dunbar C. E., Kapoor V., Manns M. P., Korangy F., Greten T. F. 2014. IFN-γ regulates survival and function of tumor-induced CD11b+ Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur. J. Immunol. 44: 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Groom J. R., Luster A. D. 2011. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Melero I., Rouzaut A., Motz G. T., Coukos G. 2014. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 4: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma P., Allison J. P. 2020. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 20: 75–76. [DOI] [PubMed] [Google Scholar]
- 65.Sharma P., Allison J. P. 2015. The future of immune checkpoint therapy. Science 348: 56–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.