

Abstract
Transmission Ratio Distortion (TRD), the uneven transmission of an allele from a parent to its offspring, can be caused by allelic differences affecting gametogenesis, fertilization or embryogenesis. However, TRD remains vaguely studied at a genomic scale. We sequenced the diploid and haploid genomes of three boars from leukocytes and spermatozoa at 50x to shed light into the genetic basis of spermatogenesis-caused Allelic Ratio Distortion (ARD). We first developed a Binomial model to identify ARD by simultaneously analysing all three males. This led to the identification of 55 ARD SNPs, most of which were animal-specific. We then evaluated ARD individually within each pig by a Fisher’s exact test and identified two shared genes (TOP3A and UNC5B) and four shared genomic regions harbouring distinct ARD SNPs in the three boars. The shared genomic regions contained candidate genes with functions related to spermatogenesis including AK7, ARID4B, BDKRB2, GSK3B, NID1, NSMCE1, PALB2, VRK1 and ZC3H13. Using the Fisher’s test, we also identified 378 genes containing variants with protein damaging potential in at least one boar, a high proportion of which, including FAM120B, TDRD15, JAM2 or AOX4 among others, are associated to spermatogenesis. Overall, our results show that sperm is subjected to ARD with variants associated to a wide variety of genes involved in different stages of spermatogenesis.
Keywords: transmission ratio distortion, allelic ratio distortion, sperm, whole genome sequencing, swine
1. Introduction
Allelic transmission ratio distortion (TRD) can be defined as the preferential transmission of one allele from a heterozygous parent to the offspring and consequently, the departure from the expected ratio of 0.5:0.5 under the Mendelian law of inheritance. Despite their potential implications for male fertility, both for human medicine and animal breeding, only few studies have explored TRD at a genomic level in mammals. Some of these studies are based on the genotypes of heterozygous parents and their offspring in mouse,1 pig2 and cattle,3 and have led to the identification of a few hundreds of loci displaying TRD. As TRD studies become more powerful with large families, animal models such as livestock with large pedigrees are better placed than humans to carry this research. In swine, Casellas et al.2 scanned the swine genome with 29,373 SNPs in 5 boars and their 352 offspring using a Bayesian Factor tool. The authors identified 84 SNPs that were heterozygous in at least one boar and displayed significant TRD. As TRD can be caused by defects compromising spermatogenesis, fertilizing ability or embryo development,4 TRD analysis could become an approach complementary to genome-wide association studies (GWAS) as it could help mapping genomic regions influencing reproductive performance that would, otherwise, remain undetected. However, the exploration of the potential impact on TRD caused by allelic ratio distortion (ARD) in the haploid sperm due to defects in spermatogenesis has not been explored thus far.
A Whole Genome Sequencing (WGS) approach to study TRD in sire to offspring designs is currently near to unfeasible due to the large number of animals that would need to be sequenced individually. The alternative of sequencing pools of gDNA from the offspring is neither a practical option because this would not allow controlling for the maternal allelic contribution. This limitation does not exist when studying ARD in sperm as the sequencing of one ejaculate allows calculating the allelic ratio in the population of haploid spermatozoa, and thus determine the existence of this ARD. In other words, each spermatozoon can be considered as a single individual carrying a haploid genome.
The aim of this study was to identify variants under ARD in the ejaculate of three boars from an artificial insemination stud. We have sequenced the genomes of these boars from leukocytes (diploid cells) and ejaculated spermatozoa (haploid cells) and used the number of reads carrying each allele at heterozygous sites as proxies of the allelic frequency to estimate ARD in sperm. We hypothesize that these SNPs displaying ARD are indicating the presence of loci influencing the efficiency of spermatogenesis and that these may have an impact on sire to offspring TRD.
2. Materials and methods
gDNA from blood from three boars of the Pietrain breed from different commercial boar studs were extracted with the Maxwell® RSC Whole Blood DNA Kit (Promega Biotech Ibérica SL, Alcobendas, Madrid, Spain) and treated with DNAse-free RNAse (Hoffmann-La Roche, Basel, Switzerland). Ejaculated sperm from the same animals was obtained by the hand glove method and purified as described by Gòdia et al.5 and gDNA was extracted as in Hammoud et al.6 Blood and sperm samples were collected by specialized professionals. The six WGS libraries were prepared with TruSeq DNA PCR-Free Kit (Illumina, Inc., San Diego, CA, USA) and sequenced to generate 150 bp paired end reads in an Illumina’s HiSeq X Ten System. The WGS fastq files were deposited in the NCBI Sequence Read Archive (SRA) under SRA experiment SRX7136525.
Raw sequencing reads were filtered to remove adaptors and low-quality reads with Trimmomatic v.0.36.7 Filtered reads were aligned to the porcine reference genome (Sscrofa11.1) with the Burrows-Wheeler Aligner (BWA) ‘mem’ v.0.7.128 and duplicate reads were removed using Picard v.2.18.7 (http://broadinstitute.github.io/picard/) MarkDuplicates. Variant calling was carried with GATK v.3.8.19 with base quality score recalibration. SNPs were discovered and filtered with standard hard filtering parameters along with a cluster filter (maximum of three variants in a cluster of 50 bp). Indels were discarded from further analysis. The resulting single nucleotide polymorphism (SNP) variants were then filtered for a minimum read depth of 20 and a maximum of two standard deviations from the average coverage. The predicted effect of the variants was assessed with SnpEff v.4.3T.10 Gene ontology analysis was carried with the Cytoscape v.3.6.0 plugin CluGO v.2.5.7.11P-values were Bonferroni-corrected (q-value).
2.1. Assessment of allelic ratio distortion in sperm
We used two statistical approaches to analyse ARD. In the first approach, we used a Binomial model adapted from Casellas et al.2 to evaluate ARD analysing the three boars simultaneously. Taking the ith SNP with two alleles (A and B) as example, the likelihood for A reads from WGS was defined as:
and p(B) = 1 – p(A). Note that αi was the sire-specific ARD parameter for the ith SNP, Hij was the proportion of A reads in the diploid genome of the jth boar and βi was a regression coefficient aiming to accommodate technological biases from WGS technology previously observed in the diploid genome. For each SNP, the Binomial model was solved by maximum likelihood. Statistical significance was tested by a standard likelihood ratio test.
It was applied to all the variants that were heterozygous in the blood samples for all three boars. Within each of the six sequenced samples, we used the number of reads carrying each allele to calculate the ratio based on the number of reads for a given allele divided by the total number of reads in that site. ARD was calculated in sperm (haploid) after correcting its allelic ratio by the ratio in white blood cells (diploid). The rationale behind this is that the ratio in blood should be 0.5 and any deviation from this value should be considered technical and therefore may also affect sperm (βi ≠ 0). Moreover, all the heterozygous variants with a ratio below 0.4 or above 0.6 in blood were considered to be prone to technical errors and were thus removed from the analysis. We also used the results from this model to compare this ARD with the TRD in swine2.
In the second approach, and in order to evaluate ARD independently within each animal, we first identified the heterozygous site in each pig in blood (again within the allele ratio 0.4–0.6) and then used the Fisher’s exact test to compare the allelic ratio between blood and sperm within each animal. Only variants in ARD in sperm above >0.6 or <0.4 were considered. To correct for multiple testing, a false discovery rate (FDR) method was employed. This was applied to:
identify coding variants in common genes affected in the three boars. This was based on the hypothesis that ARD variants may not be shared in the three boars but may affect common genes with similar functional consequences. SNPs located in coding regions were extracted with BEDTools intersect v.2.17.0.12 Coding regions were extracted from the Ensembl (v96) porcine annotation. The variant effect on protein sequence was predicted with SnpEff v.4.3T.10
identify ARD regions shared (less than 1 Mbp apart) in the three boars which could be indicative of a common affected regulatory element. ARD regions were determined by identifying these genomic segments containing at least three ARD SNPs with consecutive distances between SNPs below 1 Mbp within each pig. The ARD regional overlap between the three pigs was evaluated with BEDTools closest and intersect v.2.17.0.12
identify ARD variants with moderate or high functional potential in genes known to be related to spermatogenesis or sperm quality in each pig regardless of whether they are shared or not in these pigs. The hypothesis here was that a large number of different genes and biological pathways may lead to ARD and thus each pig might have its own set of functions altered which may not be necessarily shared in the three boars.
With the aim to assess whether our findings of ARD in sperm were the result of stochastic effects, we also employed the Fisher’s exact test, this time identifying first heterozygous sites in balanced allelic ratios (0.4–0.6) (considering sperm as the reference diploid genome) and then evaluating the allelic ratio to identify ARD in these SNPs in blood.
2.2. Variant validation by Sanger sequencing
We selected 10 variants for genotype validation using PCR coupled with Sanger sequencing (Supplementary Table S1). We focused on ARD variants mapping within genes with known function on sperm biology, spermatogenesis or meiosis. Amplification reactions ranged between 1.5 or 2.5 mM dNTPs, 0.3 µM of each primer, 1.5–2.5 mM MgCl2, 30 ng of genomic DNA and 0.75 Unit of Amplitaq Gold DNA Polymerase (Thermo Fisher Scientific, Barcelona, Spain). The final volume of the reactions was 15 µl. The thermal profile included a denaturation step at 95°C for 10 min, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 60°C for 1 min and extension at 72°C for 1 min, plus a final extension step at 72°C for 7 min. The specific conditions for each reaction are detailed in Supplementary Table S1. Amplicons with the expected size were purified with the ExoSAP‐IT PCR Clean‐up kit (Thermo Fisher Scientific, Barcelona, Spain) and sequenced with the BigDye Terminator Cycle Sequencing Kit v3.1 (Applied Biosystems, Foster City, CA, USA) and with the forward or reverse primers listed in Supplementary Table S1. Sequencing reactions were electrophoresed in an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA).
3. Results and discussion
3.1. WGS, mapping and variant calling
In average, 458 M PE reads were obtained per sample (Supplementary Table S2). Up to 99.5% of the reads mapped to the porcine genome (Sscrofa11.1). In average, 14.2% of reads were duplicates and were thus discarded for further analysis. A genome coverage between 46 and 55x was obtained per sample (Supplementary Table S2). The average number of SNPs per sample was 10 M and 6.3 M of these passed quality control filters. From these, in average, 2.8 M SNPs were heterozygous in the blood of each animal (Supplementary Table S2). The reference allele ratio of all the heterozygous SNPs displayed very similar distribution in both blood and sperm in the three boars (Supplementary Figure S1).
3.2. Analysis to detect ARD in the SNPs heterozygous in the three boars
Under the hypothesis that ARD variants could be common in a population, we applied a Binomial model in order to take statistical advantage of analysing the three boars simultaneously. This method allowed the identification of ARD in the polymorphic sites that were heterozygous in the three boars, regardless of whether this ARD was present in one or more pigs. A total of 302,384 SNPs were heterozygous in the three samples. Fifty-five SNPs displayed statistically significant ARD using this Binomial model (Fig. 1).
Figure 1.
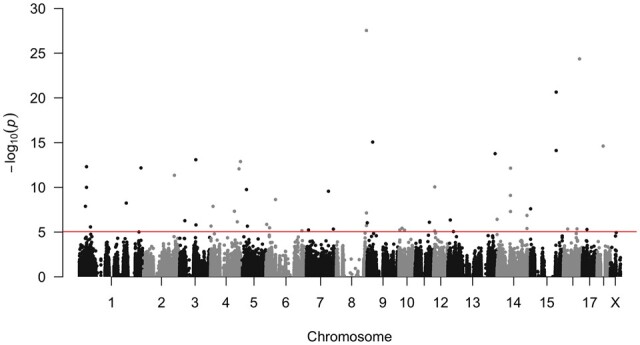
Manhattan plot of the allelic ratio distortion across the porcine chromosomes. The Binomial model identified 55 significant SNPs in allelic ratio distortion.
We then evaluated ARD independently within each animal by comparing the allelic events of blood and sperm using the Fisher’s exact test. Most of the 55 variants identified with the Binomial model presented ARD with the Fisher’s exact test in only one pig (Supplementary Table S3) and 17 did not display ARD in any animal with the Fisher’s exact test.
These results suggest that most of the 55 variants are not the ARD causal variant or that ARD is animal-specific. As a matter of fact, this approach included only those variants that were heterozygous in the blood of the three pigs, thereby discarding a large proportion of potential candidates. One intergenic SNP presented ARD in all the three pigs (SNP_ARD_26; Supplementary Table S3).
With the exclusion of 2 SNPs that located within unplaced scaffolds, 53 of the variants identified with the Binomial model, grouped into 44 regions containing 1 or more SNPs with consecutive SNP distances below 1 Mbp (Supplementary Figure S2). Thirty-seven, 5 and 2 regions contained 1, 2 and 3 SNPs, respectively (Supplementary Table S3). The previous work from Casellas et al.2 identified 84 SNPs in TRD. Of these, seven SNPs could not be liftover into the coordinates of the Sscrofa11.1 genome assembly or mapped into unplaced scaffolds. The remaining TRD SNPs were arranged by proximity in 63 regions (Supplementary Figure S2; Supplementary Table S3). Ten out of the 44 ARD regions, containing 12 ARD SNPs, were less than 2 Mbp apart from a TRD segment and 1 additional ARD region marked by 2 SNPs was just 2.08 Mbp from a TRD segment (Table 1; Supplementary Table S3). These results suggest a possible shared biological basis and also that a proportion of the TRD may be originated during spermatogenesis.
Table 1.
List of ARD regions in close proximity or overlapping to TRD segments
| ARD or TRD | rsID | Chr | Position | Distance between ARD and TRD regions | Closest gene distance between gene and ARD SNP |
|---|---|---|---|---|---|
| TRD | 4 | 111,484,345 | 1.38 Mbp | ||
| ARD | rs327579254 | 4 | 112,863,646 | None | |
| TRD | 7 | 95,211,457 | 52 kbp | ||
| ARD | rs342810440 | 7 | 95,263,998 | SIPA1L1 (90 kbp) | |
| TRD | 9 | 23,775,901 | 0.82 Mbp | ||
| ARD | rs342042877 | 9 | 24,599,014 | None | |
| TRD | 11 | 60,336,131 | 1.22 Mbp | ||
| ARD | rs339426473 | 11 | 61,564,228 | None | |
| ARD | novel | 13 | 200,024,203 | 1.86 Mbp | MORC3 (intronic) |
| TRD | 13 | 201,881,431 | |||
| ARD | rs325570178 | 14 | 56,797,388 | 1.14 Mbp | SLC35F3 (intronic) |
| ARD | rs340156423 | 14 | 57,037,751 | ||
| ARD | rs337352239 | 14 | 57,200,494 | KCNK1 (60 kbp) | |
| TRD | 14 | 58,345,290 | |||
| ARD | rs339246273 | 15 | 691,771 | 0.97 Mbp | NEB (intronic) |
| TRD | 15 | 1,657,293 | |||
| ARD | rs1111577152 | 15 | 109,256,963 | 2.08 Mbp | INO80D (54 kbp) |
| ARD | rs1113494508 | 15 | 109,256,979 | ||
| TRD | 15 | 111,342,012 | |||
| TRD | 16 | 16,901,264 | 1.05 Mbp | ||
| ARD | rs325913039 | 16 | 17,955,129 | GOLPH3 ortholog (51 kbp) | |
| ARD | rs694882285 | 17 | 19,909,268 | 0.50 Mbp | None |
| TRD | 17 | 20,406,228 | |||
| TRD | 18 | 23,809,561 | 1.36 Mbp | ||
| TRD | 18 | 24,134,625 | |||
| ARD | rs788330877 | 18 | 25,496,780 | FAM3C (70 kbp) |
In italics, the SNPs identified in the TRD study by Casellas et al. Chr: chromosome. The rsID variant is only provided for the ARD variants identified in our study.
These 14 ARD SNPs were less than 100 kbp away from nine coding genes (Table 1). Two of the ARD variants (rs1111577152 and rs1113494508), located 16 bp apart to each other, mapped 54 kbp downstream from INO80D (Table 1), a INO80 Complex Subunit member of the chromatin-remodelling complex expressed in developing spermatocytes, which plays a key role in DNA damage repair as it is essential for successful meiosis and spermatogenesis in mice.13 Other ARD variants mapped within introns of genes with no reported links with spermatogenesis (Table 1).
3.3. ARD coding variants in common genes
As spermatogenesis includes a set of complex processes including different stages such as proliferation and differentiation of spermatogonia, meiosis, spermiogenesis and sperm maturation, we hypothesized that ARD could be related to a wide variety of biological pathways. Under this assumption, we expected ARD variants to be rare, or at least not common, and thus not shared between the three pigs. In fact, Huang et al. already suggested that TRD variants tend to be rare because they are wiped out from the population as one allele is preferentially transmitted to the offspring over the other.4 We therefore sought to identify ARD variants independently in each pig using the Fisher’s exact test, which despite being different in the three pigs, would affect common genes or regulatory elements.
The three pigs presented coding variants in ARD in two genes involved in spermatogenesis: TOP3A and UNC5B (Table 2). TOP3A is a topoisomerase that plays a relevant role in meiotic recombination, as it has been found to promote the dissolution of double Holliday junctions.14,15UNC5B is an upstream effector of the Elmo1/Dock180 complex,16 which when disrupted in mice results in aberrant seminiferous epithelium, multinucleated giant cells, uncleared apoptotic germ cells and decreased sperm output.17 In our survey, TOP3A was affected by three ARD variants. A synonymous ARD SNP in TOP3A was shared by two boars and one of these boars also presented a missense ARD variant (Table 2). All the variants detected in TOP3A were novel whilst the variants in UNC5B were already annotated in dbSNP. However, as we do not know their allelic frequency in any population, these variants could be thus rare or uncommon. The fact that only two genes harboured ARD coding variants in the three pigs highlights the complexity and the multi-aetiological nature of ARD.
Table 2.
List of ARD variants affecting a common gene in the three boars
| Sample | Chr | Start | rsID | Closest gene | P-value | Ratio in blood | Ratio in sperm | snpEff | Read depth (blood/sperm) | Allele (Ref/Alt) |
|---|---|---|---|---|---|---|---|---|---|---|
| S2 | 12 | 60,452,676 | novel | TOP3A | 0.03 | 0.60 | 0.35 | synonymous | 47/40 | G/C |
| S1 | 12 | 60,465,223 | novel | TOP3A | 0.04 | 0.58 | 0.35 | missense | 48/48 | A/G |
| S1 | 12 | 60,466,709 | novel | TOP3A | 0.05 | 0.43 | 0.65 | synonymous | 37/46 | T/C |
| S3 | 12 | 60,466,709 | novel | TOP3A | 0.03 | 0.54 | 0.28 | synonymous | 41/43 | T/C |
| S3 | 14 | 74,186,268 | rs324649834 | UNC5B | 0.04 | 0.60 | 0.38 | synonymous | 40/55 | A/C |
| S2 | 14 | 74,199,368 | rs339908015 | UNC5B | 0.02 | 0.59 | 0.32 | synonymous | 41/38 | T/C |
| S1 | 14 | 74,204,519 | rs337527282 | UNC5B | 0.04 | 0.60 | 0.38 | synonymous | 62/39 | C/T |
The ratios were calculated based on the reference allele. Chr: Chromosome; S: sample.
3.4. Shared ARD regions in the three boars
We also considered the possibility that ARD variants could be rare and only present in one of the three pigs but affect common regulatory regions of relevance in spermatogenesis. We extracted the regions in each pig that contained at least three SNPs with consecutive SNP distance below 1 Mbp and then selected those that overlapped or were less than 1 Mbp apart in the three pigs. We identified four genomic regions in chromosomes 3, 7, 11 and 14 that contained a total 55 genes (Table 3; Supplementary Figure S3), several of which play a role at different stages of spermatogenesis (Table 3).
Table 3.
List of ARD regions in close vicinity or overlapping in the three samples
| Chr | S1 | S2 | S3 | Genes in the region |
|---|---|---|---|---|
| 3 |
19,346,924-21,139,827 & 22,281,850-24,847,672 (14) |
19,698,060-20,410,463 & 24,847,578-24,911,195 (7) |
20,623,640-21,407,898 (5) |
GTF3C1, NSMCE1, ERN2, PALB2, NDUFAB1 a , EARS2, GGA2, COG7, ENSSSCG00000031197, USP31, IGSF6, CDR2a, PDZD9, CRYM, ZP2 |
| 7 |
116,030,235-117,205,191 (4) |
117,052,038-117,122,913 (5) |
116,439,959-118,289,816 (7) |
RF00322, GLRX5, RF02192, RF02193, TCL1B, C14orf132, BDKRB2, BDKRB1, GSK3B a , AK7, PAPOLA a , VRK1 |
| 11 |
20,080,154-20,761,357 (5) |
20,283,123-20,824,460 (4) |
21,085,554-21,441,712 (3) |
HTR2A, ESD, RUBCNL, LCP1, ENSSSCG00000034648, CPB2, ZC3H13 |
| 14 |
55,926,799-57,746,832 (9) |
54,287,808-55,764,609 (6) |
54,474,132-57,200,642 (11) |
RF00001, RF00019, HEATR1, ERO1B, NID1, LYST, GNG4, RF00026, B3GALNT2, ARID4B, RF00425, TOMM20 a , RF00397, IRF2BP2, TARBP1, RF00026, PCNX2 |
Columns 2, 3 and 4 indicate the ARD genomic intervals for each sample (S1, S2 and S3, respectively). The number of ARD SNPs representing these intervals in each sample is indicated between brackets. Chr: chromosome; S: sample.
Gene name from orthologous genes.
Some of the detected genes (VRK1, GSK3B, NID1 PALB2, ZC3H13 or NSMCE1, among others) are related to early stages of spermatogenesis (spermatogonia proliferation and meiosis). Defects in VRK1 have not only been related to spermatogonia loss and infertility in male mice18 but also to meiosis in females.19GSK3B contributes to the induction of meiosis20 and NID1 is related to the distribution of meiotic crossover.21 Also related to recombination, PALB222 and ZC3H1323,24play a role in DNA repair during homologous recombination, whereas NSMCE1 is relevant for meiotic chromosome segregation.25,26
Other genes that arise from this study (i.e. AK7, BDKRB2, ZC3H13, ARID4B or HTR2A, among others) have been associated to spermiogenesis, the process in which spermatids mature into spermatozoa in the epithelium of the seminiferous tubules and to the acquisition of sperm motility in the epididymis. This is the case, for example, of AK7, which has been linked to spermatogenic failure and male infertility probably related to defects on the tail formation.27BDKRB2 regulates the AQP9 water channel in the murine epididymis28 and ion transport in the vas deferens of human and pig.29ZC3H13 is a member of the m6A methyltransferase complex, which is involved in the late maturation of spermatids by regulating the expression of key genes.30ARID4B is involved in Sertoli cell function and is linked to spermatogenic arrest at the stages of meiotic spermatocytes and post-meiotic haploid spermatids,31 whereas HTR2A has been associated to sperm count and motility, a property that is acquired by spermatozoa in the epididymis.32
3.5. ARD in genes related to spermatogenesis within each boar
Finally, we also considered the possibility that ARD may originate from a large number of genes and processes throughout the post-meiotic stages of spermatogenesis, and thus, ARD variants may affect non-shared genes. For each boar, we extracted the ARD variants with a predicted moderate or high damaging effect on protein sequence thereby potentially altering the protein function. We identified 408 (131, 129 and 148 for sample S1, S2 and S3, respectively) ARD variants with moderate or high protein damaging effect, none of them was shared between animals and they mapped to 378 genes (Supplementary Table S4). Six of these variants showed significant ARD after correction for multiple testing (FDR ≤ 0.05) and affected five genes (Supplementary Table S4). Four of these (ENSSSCG00000034083, ENSSSCG00000030031, ENSSSCG00000033287 and ENSSSCG00000039784) are novel genes with unknown function predicted by Ensembl Genebuild after mapping the transcripts and protein sequences from EMBL, GenBank, DDBJ, UniProtKB and RefSeq databases to the pig genome. The remaining gene, FAM120B, has been shown to play a role in adipogenesis as a transactivator of PPARG.33 Noteworthy, PPARG has been linked to sperm function and energy metabolism in pigs34 and humans35 and it has also been shown to be expressed in mouse late spermatids and primary Sertoli cells previously stimulated in vitro with lipopolysaccharide.36 Thus, it seems plausible that FAM120B modulates sperm maturation at the later stages of spermatogenesis through the regulation of PPARG.
The catalogue of 378 genes was enriched for biological functions related to replication fork processing (q-value: 4.4 × 10−2), damage DNA checkpoint (q-value: 4.8 × 10−2) and filament cytoskeleton organization (q-value: 2.2 × 10−2), which all are relevant processes involved in: (i) the maintenance of genome integrity during meiosis and (ii) the formation of the sperm. Of these variants, four had a predicted high impact on TDRD15, JAM2, PCDHGA9 and AOX4. The TDRD family is associated to piwi RNA biology which is essential to keep genome stability during spermatogenesis and TDRD15 has been shown to be upregulated in mature versus immature horse testes.37 Little is known however, about the PCDHGA9 protocadherin, but protocadherins have been linked to cell adhesion and in addition, PCDHGA9 is mainly expressed in human testes.38JAM2 has been directly linked to cell adhesion of Sertoli cells to form the blood–testis barrier and to spermatogenesis disruption.39 Finally, AOX4 has been found to be upregulated in germ cells compared with Sertoli cells during a synchronized first round of spermatogenesis.40 Of note, the alternative alleles of TDRD15 and AOX4, predicted to cause a premature stop codon on the protein sequence of these genes, were more abundant in the spermatozoa of samples S2 and S3, respectively. This phenomenon could be caused, at least, by two different scenarios. One possibility is that the reference allele, the one that is present in the reference genome assembly, is not necessarily the most frequent or the most beneficial allele in a population. Alternatively, a detrimental allele could have been hijacked by the allele from another SNP with a stronger influence on spermatogenesis in close linkage disequilibrium with the flagged SNP. The hijacking scenario is interesting but difficult to test as most likely, ARD alleles with high impact on the sequence of their host proteins would tend to be rare and thus a large population would have to be screened to identify enough animals with all the existing haplotypes and robustly measure linkage disequilibrium.
A careful inspection of the 378 genes associated to ARD variants yielded a large proportion of genes whose functions are relevant for spermatogenesis. The first group of genes were involved in the formation and repair of double-strand breaks during meiosis, which result in homologous recombination. This group included well-described genes such as BRCA2,41EME1,42GEN1,43HSF1,44MEI1,45,46RAD51B,47RAD9B, a paralog of RAD9A, which is involved in DNA double-strand break repair during meiosis in mice,48MSH249 and PMS2.50
The second group of genes associated to ARD variants included genes related to the formation and maturation of sperm. That is the case of HRB, a gene that is essential for acrosome formation with deficient mice showing meiosis and spermiogenesis defects leading to abnormal sperm and infertility.51,52HIPK4, which is associated to abnormal round-headed spermatozoa53 and CFAP100,54KIF24,55HAP154 and MARCH10,56 all linked to ciliogenesis. Other genes within this group were related to sperm maturation in the epididymis with impact on sperm motility, acrosome formation, mitochondria homeostasis or capacitation, such as KCNK17,57PLA2G3,58PPP3CC,59SLC26A8,60CCDC18961 and PINK1.62
Overall, these multiple functions described by the genes linked to ARD variants are a reflex of the complexity of spermatogenesis, suggesting that ARD can arise at any moment from meiosis to sperm maturation.
To the best of our knowledge, this pioneer study is the first to evaluate the potential forces of spermatogenesis that could drive TRD by evaluating ARD at the sperm level using WGS. One of the advantages of WGS over genotyping platforms is that it allows the interrogation of practically the whole genome and has thus the potential to identify the causal variants. Moreover, WGS allows querying ARD at the sperm level, which would be impossible with genotyping arrays.
To validate our results, we first confirmed by PCR followed by Sanger sequencing, 10 variants identified from the different analyses we carried and selected for mapping near or for altering the protein sequence of genes with known functions on spermatogenesis (Supplementary Table S1). Two amplicons did not amplify. For the other 8 ARD variants, we confirmed all the heterozygous genotypes (Supplementary Figure S4). Moreover, one of the amplicons that did not amplify corresponded to SNP_ARD_26, the ARD variant that appeared in the Binomial model and showed ARD in all the three pigs. Thus, we could not confirm the existence of the heterozygous state in any of the three samples. We then carried an experiment using the Fisher’s exact test to assess the allelic ratio in blood when considering only the variants that were heterozygous in sperm with allelic ratios between 0.4 and 0.6. In other words, we queried ARD in blood taking sperm as the diploid reference in which ARD should not happen. As expected, the number of heterozygous sites was very similar between blood and sperm across the three boars (Fig. 2A). However, the number of SNPs showing allelic ratio deviations (ARD < 0.4 or ARD > 0.6) at P ≤ 0.05 in blood versus the sperm reference, was one-third of the number of SNPs in ARD detected in sperm (Fig. 2B). Moreover, while the genes harbouring coding variants in ARD in sperm were enriched for pathways that are relevant for spermatogenesis and meiosis (replication fork processing, damage DNA checkpoint and filament cytoskeleton organization), the genes encompassing coding variants displaying ARD in blood were associated to pathways not related to sperm biology, such as microtubule anchoring (q-value: 0.01), maintenance of animal organ identity (q-value: 1.6 × 10−5), vitamin transport (q-value: 0.01) and cardiac muscle cell contraction (q-value: 0.02). This, together with the fact that we used stringent criteria to select the SNPs that would be subjected to the ARD study, suggests that our findings of ARD in sperm are real and have a biological basis.
Figure 2.

Comparison of the extent of ARD assessed in sperm and assessed in blood. (A) Number of heterozygous SNPs (allelic ratio 0.4–0.6) in each tissue. (B) Number of significant SNPs in ARD (allelic ratios <0.4 or >0.6; *P < 0.05) using the Fisher’s exact test.
In conclusion, our survey using WGS at 50x depth in three boars indicates the presence of ARD at the sperm level and shows that ARD can arise from multiple stages during spermatogenesis. Forthcoming studies to more deeply investigate ARD in sperm should include a larger number of boars and augmented sequencing depth. This combination would drastically increase the power to identify ARD variants and clarify the biological basis of spermatogenesis and its consequences on TRD. Moreover, the variants that we identified in this study should be tested in a larger sire : offspring pedigree to assess their allelic frequency and confirm the TRD effect. Additionally, if sufficiently frequent, these variants should be included in genetic association studies for sperm quality and male fertility to assess their potential implication on the male’s reproductive ability.
Supplementary data
Supplementary data are available at DNARES online.
Supplementary Material
Acknowledgements
We thank Sam Balasch (grup Gepork S.A.) for providing the blood and sperm samples. We thank Betlem Cabrera (CRAG) for her lab support.
Accession numbers
SRR10441782, SRR10441783, SRR10441781, SRR10441780, SRR10441779, SRR10441778.
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO) under grant AGL2013-44978-R and grant AGL2017-86946-R and by the CERCA Programme/Generalitat de Catalunya. AGL2017-86946-R was also funded by the Spanish State Research Agency (AEI) and the European Regional Development Fund (ERDF). We thank the Agency for Management of University and Research Grants (AGAUR) of the Generalitat de Catalunya (Grant Numbers 2014 SGR 1528 and 2017 SGR 01060). We also acknowledge the support of the Spanish Ministry of Economy and Competitivity for the Center of Excellence Severo Ochoa 2016–2019 (Grant Number SEV-2015-0533) grant awarded to the Centre for Research in Agricultural Genomics (CRAG). MG acknowledges a Ph.D. studentship from MINECO (Grant Number BES-2014-070560). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflict of interest
None declared.
References
- 1. Casellas J., Gularte R.J., Farber C.R., et al. 2012, Genome scans for transmission ratio distortion regions in mice, Genetics, 191, 247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casellas J., Manunza A., Mercader A., Quintanilla R., Amills M. 2014, A flexible Bayesian model for testing for transmission ratio distortion, Genetics, 198, 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Id-Lahoucine S., Cánovas A., Jaton C., et al. 2019, Implementation of Bayesian methods to identify SNP and haplotype regions with transmission ratio distortion across the whole genome: TRDscan v.1.0, J. Dairy Sci., 102, 3175–88. [DOI] [PubMed] [Google Scholar]
- 4. Huang L.O., Labbe A., Infante-Rivard C. 2013, Transmission ratio distortion: review of concept and implications for genetic association studies, Hum. Genet., 132, 245–63. [DOI] [PubMed] [Google Scholar]
- 5. Gòdia M., Mayer F.Q., Nafissi J., et al. 2018, A technical assessment of the porcine ejaculated spermatozoa for a sperm-specific RNA-seq analysis, Syst. Biol. Reprod. Med., 64, 291–303. [DOI] [PubMed] [Google Scholar]
- 6. Hammoud S.S., Nix D.A., Zhang H., Purwar J., Carrell D.T., Cairns B.R. 2009, Distinctive chromatin in human sperm packages genes for embryo development, Nature, 460, 473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bolger A.M., Lohse M., Usadel B. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li H. 2013, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, arXiv, 1303.3997.
- 9. DePristo M.A., Banks E., Poplin R., et al. 2011, A framework for variation discovery and genotyping using next-generation DNA sequencing data, Nat. Genet., 43, 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cingolani P., Platts A., Wang le L., et al. 2012, A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3, Fly (Austin ), 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bindea G., Mlecnik B., Hackl H., et al. 2009, ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks, Bioinformatics, 25, 1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quinlan A.R., Hall I.M. 2010, BEDTools: a flexible suite of utilities for comparing genomic features, Bioinformatics, 26, 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Serber D.W., Runge J.S., Menon D.U., Magnuson T. 2016, The mouse INO80 chromatin-remodeling complex is an essential meiotic factor for spermatogenesis, Biol. Reprod., 94, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dorn A., Rohrig S., Papp K., et al. 2018, The topoisomerase 3 alpha zinc-finger domain T1 of Arabidopsis thaliana is required for targeting the enzyme activity to Holliday junction-like DNA repair intermediates, PloS Genet., 14, e1007674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin C.A., Sarlos K., Logan C.V., et al. 2018, Mutations in TOP3A cause a bloom syndrome-like disorder, Am. J. Hum. Genet., 103, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaker K., Bartsch S., Patry C., et al. 2015, The bipartite Rac1 Guanine nucleotide exchange factor engulfment and cell motility 1/dedicator of cytokinesis 180 (elmo1/dock180) protects endothelial cells from apoptosis in blood vessel development, J. Biol. Chem., 290, 6408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elliott M.R., Zheng S., Park D., et al. 2010, Unexpected requirement for ELMO1 in clearance of apoptotic germ cells in vivo, Nature, 467, 333–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wiebe M.S., Nichols R.J., Molitor T.P., Lindgren J.K., Traktman P. 2010, Mice deficient in the serine/threonine protein kinase VRK1 are infertile due to a progressive loss of spermatogonia, Biol. Reprod., 82, 182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schober C.S., Aydiner F., Booth C.J., Seli E., Reinke V. 2011, The kinase VRK1 is required for normal meiotic progression in mammalian oogenesis, Mech. Dev., 128, 178–90. [DOI] [PubMed] [Google Scholar]
- 20. Guo T.B., Chan K.C., Hakovirta H., et al. 2003, Evidence for a role of glycogen synthase kinase-3 beta in rodent spermatogenesis, J. Androl., 24, 332–42. [DOI] [PubMed] [Google Scholar]
- 21. Jeffreys A.J., Neumann R. 2005, Factors influencing recombination frequency and distribution in a human meiotic crossover hotspot, Hum. Mol. Genet., 14, 2277–87. [DOI] [PubMed] [Google Scholar]
- 22. Yan Z.C., Fan D.D., Meng Q.J., et al. 2016, Transcription factor ZFP38 is essential for meiosis prophase I in male mice, Reproduction, 152, 431–7. [DOI] [PubMed] [Google Scholar]
- 23. Knuckles P., Lence T., Haussmann I.U., et al. 2018, Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d, Genes Dev., 32, 415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin Z., Hsu P.J., Xing X.D., et al. 2017, Mettl3-/Mettl14-mediated mRNA N-6-methyladenosine modulates murine spermatogenesis, Cell Res., 27, 1216–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taylor E.M., Copsey A.C., Hudson J.J.R., Vidot S., Lehmann A.R. 2008, Identification of the proteins, including MAGEG1, that make up the human SMC5-6 protein complex, Mol. Cell. Biol., 28, 1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hwang G., Verver D.E., Handel M.A., Hamer G., Jordan P.W. 2018, Depletion of SMC5/6 sensitizes male germ cells to DNA damage, Mol. Biol. Cell., 29, 3003–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lorès P., Coutton C., El Khouri E., et al. 2018, Homozygous missense mutation L673P in adenylate kinase 7 (AK7) leads to primary male infertility and multiple morphological anomalies of the flagella but not to primary ciliary dyskinesia, Hum. Mol. Genet., 27, 1196–211. [DOI] [PubMed] [Google Scholar]
- 28. Belleannée C., Da Silva N., Shum W.W., et al. 2009, Segmental expression of the bradykinin type 2 receptor in rat efferent ducts and epididymis and its role in the regulation of aquaporin 9, Biol. Reprod., 80, 134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pierucci-Alves F., Schultz B.D. 2008, Bradykinin-stimulated cyclooxygenase activity stimulates vas deferens epithelial anion secretion in vitro in swine and humans, Biol. Reprod., 79, 501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu R.C., Jiang M., Beaudet A.L., Wu M.Y. 2013, ARID4A and ARID4B regulate male fertility, a functional link to the AR and RB pathways, Proc. Natl. Acad. Sci. USA, 110, 4616–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cortés-Rodriguez M., Royo J.L., Reyes-Palomares A., Lendinez A.M., Ruiz-Galdon M., Reyes-Engel A. 2018, Sperm count and motility are quantitatively affected by functional polymorphisms of HTR2A, MAOA and SLC18A, Reprod. Biomed. Online, 36, 560–7. [DOI] [PubMed] [Google Scholar]
- 32. Li D.C., Kang Q.H., Wang D.M. 2007, Constitutive coactivator of peroxisome proliferator-activated receptor (PPAR gamma), a novel coactivator of PPAR gamma that promotes adipogenesis, Mol. Endocrinol., 21, 2320–33. [DOI] [PubMed] [Google Scholar]
- 33. Santoro M., Guido C., De Amicis F., et al. 2013, Sperm metabolism in pigs: a role for peroxisome proliferator-activated receptor gamma (PPAR gamma), J. Exp. Biol., 216, 1085–92. [DOI] [PubMed] [Google Scholar]
- 34. Aquila S., Bonofiglio D., Gentile M., et al. 2006, Peroxisome proliferator-activated receptor (PPAR)gamma is expressed by human spermatozoa: its potential role on the sperm physiology, J. Cell. Physiol., 209, 977–86. [DOI] [PubMed] [Google Scholar]
- 35. Wang G., Cheng S.T., Zhang S.S., Zhu Y., Xiao Y., Ju L.G. 2020, LPS impairs steroidogenesis and ROS metabolism and induces PPAR transcriptional activity to disturb estrogen/androgen receptor expression in testicular cells, Mol. Biol. Rep., 47, 1045–56. [DOI] [PubMed] [Google Scholar]
- 36. Li B., He X., Zhao Y., et al. 2019, Identification of piRNAs and piRNA clusters in the testes of the Mongolian horse, Sci. Rep., 9, 5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmidt T., Samaras P., Frejno M., et al. 2018, ProteomicsDB, Nucleic Acids Res., 46, D1271–D81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paul C., Robaire B. 2013, Impaired function of the blood-testis barrier during aging is preceded by a decline in cell adhesion proteins and GTPases, PloS One, 8, e84354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evans E., Hogarth C., Mitchell D., Griswold M. 2014, Riding the spermatogenic wave: profiling gene expression within neonatal germ and sertoli cells during a synchronized initial wave of spermatogenesis in mice, Biol. Reprod., 90, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharan S.K., Pyle A., Coppola V., et al. 2004, BRCA2 deficiency in mice leads to meiotic impairment and infertility, Development, 131, 131–42. [DOI] [PubMed] [Google Scholar]
- 41. Toledo M., Sun X., Brieno-Enriquez M.A., et al. 2019, A mutation in the endonuclease domain of mouse MLH3 reveals novel roles for MutL gamma during crossover formation in meiotic prophase I, PloS Genet., 15, e1008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matos J., Blanco M.G., Maslen S., Skehel J.M., West S.C. 2011, Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis, Cell, 147, 158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akerfelt M., Vihervaara A., Laiho A., et al. 2010, Heat shock transcription factor 1 localizes to sex chromatin during meiotic repression, J. Biol. Chem., 285, 34469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Libby B.J., De La Fuente R., O'Brien M.J., et al. 2002, The mouse meiotic mutation mei1 disrupts chromosome synapsis with sexually dimorphic consequences for meiotic progression, Dev. Biol., 242, 174–87. [DOI] [PubMed] [Google Scholar]
- 45. Ben Khelifa M., Ghieh F., Boudjenah R., et al. 2018, A MEI1 homozygous missense mutation associated with meiotic arrest in a consanguineous family, Hum. Reprod., 33, 1034–7. [DOI] [PubMed] [Google Scholar]
- 46. Da Ines O., Degroote F., Amiard S., Goubely C., Gallego M.E., White C.I. 2013, Effects of XRCC2 and RAD51B mutations on somatic and meiotic recombination in Arabidopsis thaliana, Plant J. ., 74, 959–70. [DOI] [PubMed] [Google Scholar]
- 47. Lyndaker A.M., Vasileva A., Wolgemuth D.J., Weiss R.S., Lieberman H.B. 2013, Clamping down on mammalian meiosis, Cell Cycle, 12, 3135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kolas N.K., Svetlanov A., Lenzi M.L., et al. 2005, Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I, J. Cell Biol., 171, 447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baker S.M., Bronner C.E., Zhang L., et al. 1995, Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis, Cell, 82, 309–19. [DOI] [PubMed] [Google Scholar]
- 50. Kang-Decker N., Mantchev G.T., Juneja S.C., McNiven M.A., van Deursen J.M. 2001, Lack of acrosome formation in Hrb-deficient mice, Science, 294, 1531–3. [DOI] [PubMed] [Google Scholar]
- 51. Juneja S.C., van Deursen J.M. 2005, A mouse model of familial oligoasthenoteratozoospermia, Hum. Reprod., 20, 881–93. [DOI] [PubMed] [Google Scholar]
- 52. Crapster J.A., Rack P.G., Hellmann Z.J., et al. 2020, HIPK4 is essential for murine spermiogenesis, Elife, 9, e50209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Firat-Karalar E.N., Sante J., Elliott S., Stearns T. 2014, Proteomic analysis of mammalian sperm cells identifies new components of the centrosome, J. Cell Sci., 127, 4128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kobayashi T., Tsang W.Y., Li J., Lane W., Dynlacht B.D. 2011, Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis, Cell, 145, 914–25. [DOI] [PubMed] [Google Scholar]
- 55. Iyengar P.V., Hirota T., Hirose S., Nakamura N. 2011, Membrane-associated RING-CH 10 (MARCH10 protein) is a microtubule-associated E3 ubiquitin ligase of the spermatid flagella, J. Biol. Chem., 286, 39082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dube E., Hermo L., Chan P.T., Cyr D.G. 2008, Alterations in gene expression in the caput epididymides of nonobstructive azoospermic men, Biol. Reprod., 78, 342–51. [DOI] [PubMed] [Google Scholar]
- 57. Sato H., Taketomi Y., Isogai Y., et al. 2010, Group III secreted phospholipase A2 regulates epididymal sperm maturation and fertility in mice, J. Clin. Invest., 120, 1400–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miyata H., Satouh Y., Mashiko D., et al. 2015, Sperm calcineurin inhibition prevents mouse fertility with implications for male contraceptive, Science, 350, 442–5. [DOI] [PubMed] [Google Scholar]
- 59. Rode B., Dirami T., Bakouh N., et al. 2012, The testis anion transporter TAT1 (SLC26A8) physically and functionally interacts with the cystic fibrosis transmembrane conductance regulator channel: a potential role during sperm capacitation, Hum. Mol. Genet., 21, 1287–98. [DOI] [PubMed] [Google Scholar]
- 60. Iso-Touru T., Wurmser C., Venhoranta H., et al. 2019, A splice donor variant in CCDC189 is associated with asthenospermia in Nordic Red dairy cattle, BMC Genomics, 20, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Horibe A., Eid N., Ito Y., Otsuki Y., Kondo Y. 2019, Ethanol-induced autophagy in sertoli cells is specifically marked at androgen-dependent stages of the spermatogenic cycle: potential mechanisms and implications, Int. J. Mol. Sci., 20, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Horibe A., Eid N., Ito Y., Otsuki Y., Kondo Y. 2019, Ethanol-induced autophagy in sertoli cells is specifically marked at androgen-dependent stages of the spermatogenic cycle: potential mechanisms and implications, Int. J. Mol. Sci., 20, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.