

Abstract
Motivation
The interaction between proteins and nucleic acids plays a crucial role in gene regulation and cell function. Determining the binding preferences of nucleic acid-binding proteins (NBPs), namely RNA-binding proteins (RBPs) and transcription factors (TFs), is the key to decipher the protein–nucleic acids interaction code. Today, available NBP binding data from in vivo or in vitro experiments are still limited, which leaves a large portion of NBPs uncovered. Unfortunately, existing computational methods that model the NBP binding preferences are mostly protein specific: they need the experimental data for a specific protein in interest, and thus only focus on experimentally characterized NBPs. The binding preferences of experimentally unexplored NBPs remain largely unknown.
Results
Here, we introduce ProbeRating, a nucleic acid recommender system that utilizes techniques from deep learning and word embeddings of natural language processing. ProbeRating is developed to predict binding profiles for unexplored or poorly studied NBPs by exploiting their homologs NBPs which currently have available binding data. Requiring only sequence information as input, ProbeRating adapts FastText from Facebook AI Research to extract biological features. It then builds a neural network-based recommender system. We evaluate the performance of ProbeRating on two different tasks: one for RBP and one for TF. As a result, ProbeRating outperforms previous methods on both tasks. The results show that ProbeRating can be a useful tool to study the binding mechanism for the many NBPs that lack direct experimental evidence. and implementation
Availability and implementation
The source code is freely available at <https://github.com/syang11/ProbeRating>.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Knowledge about the binding profiles of nucleic acid-binding proteins (NBPs) is a vital prerequisite to detecting potential NBP binding targets (i.e. RNAs and DNAs) in the cell, and understanding gene regulation and evolution (Dong et al., 2018; Tak Leung et al., 2019; Yang et al., 2011). Currently, the binding profiles are mainly determined computationally (Lambert et al., 2018; Pan et al., 2019) from high-throughput experimental data such as protein-binding microarray (PBM) (Berger et al., 2006; Weirauch et al., 2014) or chromatin immunoprecipitation (ChIP)-seq (Barski and Zhao, 2009; Park, 2009) for transcription factors (TFs), and RNAcompete assay (Ray et al., 2009, 2013) or crosslinking immunoprecipitation (CLIP) (Konig et al., 2012; Wang et al., 2015) for RNA-binding proteins (RBPs). Despite the continuous advances in these large-scale experimental technologies, binding data generated from them still only cover a limited portion of the known NBPs across different species due to the high cost. This is especially the case for the RBPs, which are less studied than the TFs. For example, the most extensive compendium (Ray et al., 2013) for RBPs (207 unique proteins) by far was generated by RNAcompete assays. The single largest RBP family in the compendium, the RNA recognition motif (RRM) family, has 171 entries. But, there are more than 5000 known and predicted RRM RBPs according to the CISBP-RNA database (Ray et al., 2013), mostly unexplored. Therefore, predicting the binding profiles for unexplored NBPs by leveraging the limited experimental data is needed. Especially, since most of the unexplored NBPs only have their sequences information available, predicting the binding profiles for NBPs directly from their sequences is critically desired.
There have been various researches seeking to determine an NBP’s binding preference throughout the years. The models focusing on sequence specificity have evolved from consensus motif or position weight matrix (PWM) in the early days (Bailey et al., 2009; Stormo, 2000), to classic machine learning models like SVM with k-mer features and so on (Ghandi et al., 2014; Orenstein et al., 2016; Pelossof et al., 2015), to the latest deep neural network realm (Alipanahi et al., 2015; Ghanbari and Ohler, 2019; Pan and Shen, 2018b; Zeng et al., 2016). Since RNA has secondary structures that DNA does not have, many approaches dealing with RBP-RNA interactions also incorporate RNA structure information to binding specificity (Gandhi et al., 2018; Hiller et al., 2006; Kazan et al., 2010). Despite the numerous efforts that have been devoted in this field, previous researches on binding preference modeling have several limitations.
First and foremost, the majority of the works are protein specific, i.e. they construct a binding preference model of a single protein, without considering the relationship among different proteins. Since the model is built on RNA or DNA targets of each protein independently, patterns learned from protein A typically cannot be efficiently transferred to protein B. One key reason for the previous methods being protein specific is that the available experimental data are highly limited and imbalanced: very few NBPs compared to the number of nucleic acids. For example, the RNAcompete assay typically contains >200 000 RNA probes; yet only a few hundred RBPs have been gauged so far (Ray et al., 2013). Other experimental data types are in similar situations. Therefore, for an experimentally unexplored NBP, it is still difficult to know its binding preference at this moment.
In addition, most existing methods predict the NBP binding preference as a highly reduced summarization of the actual binding data, like a consensus or PWM motif (Kazan et al., 2010; Stormo, 2000) conventionally, or a convolutional neural network filter (Alipanahi et al., 2015; Pan and Shen, 2018b) recently. These so called ‘binding specificity’ prediction approaches are very useful and have been intensively studied. However, using these reduced summarizations, one may not capture all the details of the binding data so that if the binding preference of protein A is transferred to protein B, information that is important to protein B but not A may be lost.
Lastly, a substantial body of research for nucleic acid–protein interactions have been focusing on structural data like RNA–protein/DNA–protein complex structures from the Protein Data Bank (PDB) (Berman, 2000); while some more recent studies focus on CLIP or ChIP-seq data which are high-throughput in vivo data. The former group includes a wide range of studies from identifying nucleic acids-binding amino acid residues in protein sequences (Jung et al., 2018; Peng and Kurgan, 2015; Walia et al., 2017; Yan et al., 2016; Zhang et al., 2019) to predicting DNA/RNA–protein interaction pairs (Bellucci et al., 2011; Suresh et al., 2015; Yi et al., 2018), and so on. These are related tasks to predicting an NBP’s binding preference. However, for a protein in interest, structural complexes in PDB often only cover limited individual interactions between fragments of nucleic acids and fragments of that protein, which are not diverse enough to determine the nucleic acids binding preference of the protein. In contrast, ChIP-seq and CLIP experiments generate high-throughput binding data, which contains a much larger number of diverse nucleic acid targets for a given protein. Computational methods focusing on ChIP or CLIP data essentially formulate nucleic acid–protein binding as a classification task and determine a protein’s binding preference model through binary labeled data (positive: bound, negative: unbound) (Li et al., 2017; Maticzka et al., 2014; Pan and Shen, 2018b). However, a subtle problem is that there are no defined unbound cases, i.e. the ChIP or CLIP experiments only report positive nucleic acids that are putatively bound by the given protein. Strategies like shuffling nucleotides in the positive sequences and so on are often used as a rough workaround to generate negative samples.
A few existing studies try to address the above limitations (Pelossof et al., 2015; Ray et al., 2013; Yang et al., 2018). One study (Ray et al., 2013) determines binding preference motifs as PWMs from numerically labeled RNAcompete data, and it proposes to infer the binding motif of an unexplored RBP to be the same as the PWM of that RBP’s well-studied nearest neighbor. It uses binding domain sequence similarity to define the nearest neighbor, and it predicts PWMs for thousands of unexplored RBPs in the CISBP-RNA database. Following this line, a later study (Yang et al., 2018) utilizes the co-evolution assumption between RNA and protein, and it combines K nearest neighbors’ PWMs to make the prediction. We refer to it as Co-Evo method. It reports better performance than the first study (Ray et al., 2013). Although the sequence similarity used by both studies is a simple metric to compare two proteins, and the PWMs are reduced summarizations of the real binding motifs, the two studies do provide feasible solutions for the unexplored NBPs problem. By far, to our knowledge, the best solution in terms of prediction quality is supplied by AffinityRegression (Pelossof et al., 2015). AffinityRegression utilizes a recommender system (Ricci et al., 2011) formulation, where NBPs are like users, and RNAs or DNAs are like products to be recommended. Given a family of TF or RBP domains and their binding profile data (PBM or RNAcompete assay), AffinityRegression learns protein family-level binding patterns from amino acid and nucleotide k-mer features, through bilinear regression and matrix factorization. For an unexplored NBP in the same family, AffinityRegression directly recommends its full binding profile, i.e. predicted binding affinity values of the NBP against each RNA or DNA probe in the assay, instead of a reduced summarization of the binding profile. However, although AffinityRegression has made significant progress, it assumes linearity in its regression model, which is unlikely held in practice. Its use of k-mer frequency features to represent the NBPs and nucleic acids is also disputable since the context information around potential binding sites would be lost and the feature dimension goes too quickly as k grows. For example, 3-mer amino acid features would be 8000 dimensions; while the number of proteins in the largest PBM or RNAcompete dataset is less than 300.
To close the gaps, here, we present a new method called ProbeRating to infer binding profiles for unexplored NBPs, using only sequence information. ProbeRating extends AffinityRegression by incorporating non-linearity to the model and improving features with more sophisticated representation techniques. ProbeRating does its job via a two-stage framework (Fig. 1). It first adapts the word embedding method FastText (Bojanowski et al., 2017) to extract distributed representations from the NBP sequences and the nucleic acid probes, respectively. It then takes the new representations as input features to train a supervised recommender system to recommend probes to unexplored NBPs. We implement the functionality of representation learning in a package called FastBioseq. For the recommender, we develop a novel neural network-based approach to incorporate the non-linearity. We show that ProbeRating significantly outperforms AffinityRegression for both RBP binding prediction and TF binding prediction tasks. Same as AffinityRegression, ProbeRating is also capable of recommending a full binding profile directly for a test NBP, instead of a reduced summarization.
Fig. 1.

Schematic diagram of our workflow. (a) Protein and RNA sequences are embedded as numerical feature vectors by FastBioseq. (b) The protein and RNA features are fed into ProbeRating’s recommender system to infer binding intensity profiles for unexplored proteins. Performance is evaluated to compare with the nearest-neighbor methods and AffinityRegression
2 Materials and methods
2.1 Input features
Instead of using k-mer features as AffinityRegression does, we adapt the FastText (Bojanowski et al., 2017) method which is an extension of the widely used Word2Vec (Mikolov et al., 2013) method in the field of natural language processing, and we extract features from protein and nucleic acid sequences. Word2Vec encodes words to numerical vectors, through training on a corpus of text documents for a classification task of predicting a center word based on its nearby words. It assumes the meaning of a word is characterized by its context, and it thus could capture semantics in the vectors. FastText extends Word2Vec by decomposing each word into subword n-grams and treats each n-gram (instead of each word) as a unit during the learning of representations. By doing this, FastText can deal with word that is not seen in the training corpus (out-of-vocabulary word) or get better embedding vectors for rare words, which cannot be done by Word2Vec. These properties make FastText very attractive for biological sequence embedding since: (i) mutations occur through the evolution, and (ii) the predefined biological ‘word’ (will explain later) may not precisely match the actual binding sites. Although the idea of using Word2Vec to encode proteins has been explored before and shown effective (Asgari and Mofrad, 2015; Pan and Shen, 2018a), to our knowledge, we are the first one that uses the more advanced FastText in binding preference prediction.
Here, we briefly introduce how our FastBioseq package utilizes FastText to generate protein and nucleic acid feature vectors. The details can be found in Supplementary Note. If we treat each k-mer of amino acids or nucleotides as a word, then a protein or DNA/RNA sequence can be decomposed as k sentences of non-overlapping k-mers, as shown in Figure 1a (taking RBP sequence and RNA sequence as an example). Different sentences correspond to different reading frames on the sequence, denoted with different colors in Figure 1a. For each sentence, FastBioseq uses the continuous bag-of-words (CBOW) algorithm (Bojanowski et al., 2017) of FastText to train on every k-mer’s contextual information, which is considered important for NBP binding sites recognition. After training, FastBioseq can embed the k-mers onto a vector space with user-defined dimensions. The resultant k-mer vectors are then combined to construct the sentence vectors which in turn produce the sequence vectors. In this way, FastBioseq solves both the high dimensionality and loss of context problems that the conventional k-mer frequency feature suffers from.
In natural language word embedding, a large corpus of sentences is typically required to provide enough coverage of different contexts for different words. This is also true for biological sequence embedding. So, we compiled several large corpora to train our FastBioseq embedding models, which will be described in Section 2.3. Moreover, we also implemented a Word2Vec version of sequence embedding as well as a Doc2Vec (Le and Mikolov, 2014) version which is another popular extension of Word2Vec to explicitly embed the entire document (i.e. entire biological sequence) to a vector instead of combining word vectors. These implementations can be easily used from our FastBioseq package.
2.2 The recommender model
Once we get the protein and nucleic acid features extracted from FastBioseq, we input them to our ProbeRating model, as Figure 1b shown. To explain the ProbeRating model, here we take RBPs as an example again. The model works the same for the TFs. Like several previous studies (Corrado et al., 2016; Pelossof et al., 2015), ProbeRating uses a recommender system setting to model the interaction of RBP features and RNA features. In this setting, given a list of users with their ratings over a list of products, it is known as the ‘cold start’ problem when the recommender is used to predict a new user’s ratings to the products. Here, we treat RBPs as users, RNA probes as products and their binding intensity scores as ratings.
2.2.1 The naïve model
A natural approach to address the cold start problem is to incorporate content information from the interacting objects themselves (i.e. content features of users-products, or RBPs–RNAs), based on matrix trifactorization. Formally, let be the RBP feature matrix containing RBPs each has features, be the RNA probe feature matrix containing probes with features, and be the binding intensity matrix corresponding to all the RBP–RNA pairs. The trifactorization approximates by , where is the weight matrix that explains the interaction between the RBP features and the RNA features. In our naïve model, we mimic this approach but formulate a feedforward neural network to add non-linearity. Namely,
| (1) |
where and are outputs from two subneural networks, one for RNA features and one for protein features. More details of this naïve model can be found in Supplementary Note.
However, for the binding profile prediction task, we are facing, a practical issue for this naïve model is that there are too few NBPs in the dataset to be learned from. As mentioned in earlier sections, the most extensive RBP compendium (Ray et al., 2013) available is generated by RNAcompete assays. In the compendium, the number of RBPs with binding profile data available is only <250 and is times smaller than the number of RNAs probes in the RNAcompete assay. TFs are in a similar situation. If we used conventional sequence features like k-mer frequencies, the model could easily overfit since the high dimensionality of the features. Even if we alleviated overfitting with regularizations, or with low-dimensional features extracted by FastBioseq, we could hardly learn a sophisticated parametric model given the small number of RBP instances. Our results showed that this naïve model did not perform well (Supplementary Note).
2.2.2 The final model
To address the issue in the naïve model, we adapt and extend a strategy used by AffinityRegression to convert the ‘binding intensity prediction’ problem to a ‘similarity prediction’ problem, solve it and then convert back. AffinityRegression solves the ‘similarity prediction’ problem with a regular bilinear regression model, and it does the conversion through a series of linear transformation and matrix factorization operations. We solve with the more expressive neural network model and use a more straightforward conversion approach, which can be interpreted as a non-parametric tweak to our naïve model.
To convert the RBP ‘binding intensity prediction’ problem to an RBP ‘similarity prediction’ problem, the original intensity matrix is transformed to , as shown in Figure 2. Each column of is a vector of normalized binding intensity scores of an RBP against all RNA probes. So becomes the cosine similarity matrix for all pairs of RBPs. We now predict the similarity value of RBPs and by from our neural network model:
| (2) |
where and are parameters. is a function to merge and , and it corresponds to a merge layer with no trainable parameters in the neural network. and are outputs from two shallow subneural networks:
| (3) |
| (4) |
where is a sigmoid function. is a row vector in the RBP feature matrix . is a row vector from the matrix . is used to match , and can be considered as a compression of . and are parameters to project protein features to a latent feature space with dimensions. Similarly, and are parameters for RNA features. All the above parameters, denoted by , are solved by the Adam optimizer to minimize the regularized sum of squared loss:
| (5) |
where is a regularization function (we use L2 norm), with coefficient . After we solve this ‘similarity prediction’ problem, we need to convert it back to the original problem. For a given testing RBP , we feed its FastBioseq feature vector to the neural network and output its predicted similarity values against each training RBPs. To reconstruct the binding intensity values from , we implement two simple yet effective options:
| (6) |
| (7) |
Fig. 2.

The architecture of the neural network model. Note that, the left multiplication with YT on the input RNA feature matrix and the output binding intensity matrix transforms the intensity prediction problem into a similarity prediction problem. With the similarity output from the model, we convert it back to the final intensity value by doing a non-parametric reconstruction
The first reconstruction treats the similarity values in as weights and does a weighted sum to obtain . The second reconstruction multiplies a Moore–Penrose pseudoinverse of to . It is inspired by AffinityRegression’s approach, and it comes directly from the above fact that . We found the first option generally performed better in our preliminary experiments and used it in this study. The reconstruction is non-parametric since the prediction needs the entire binding intensity matrix of the training data, i.e. . Intuitively, these approaches work because we first solve a much easier similarity prediction problem requiring much fewer data from the RBP–RBP pairs, and then leverage the original training data to reconstruct the prediction for the harder RBP–RNA binding intensity problem.
Furthermore, we explore different structures for the neural network. As shown in Figure 2, there could be deeper network architectures in the two subnets and after merging them by stacking more layers. Additionally, for the merge layer , we develop four different types of merging (please refer to Supplementary Note for details). For the results reported below, we use the shallow architecture and the merge layer with the fewest parameters.
2.3 Datasets
The primary datasets of our study include an RNAcompete dataset for the RBPs and a PBM dataset for the TFs. Both datasets are commonly used benchmark datasets from previous studies (Alipanahi et al., 2015; Gandhi et al., 2018; Koo et al., 2018; Orenstein et al., 2016; Yang et al., 2018), including AffinityRegression. The first dataset, called RRM162 (as shown in Table 1), is derived from the largest compendium of RBP binding assay (Ray et al., 2013) and the AffinityRegression paper. It contains 162 binding domains from the RNA Recognition Motif (RRM) family. We choose the RRM family since it is the largest family in the compendium and is also one of the most abundant RBP families in nature (Maris et al., 2005). Each RRM domains in RRM162 are measured against 241 357 RNA probes, which results in 162*241 357 binding intensity scores in total. Similarly, the second dataset, called Homeo215 (as shown in Table 1), is derived from the AffinityRegression paper, and it contains 215 Homeodomain sequences and their binding intensity Z-scores (Berger et al., 2008) against >30 000 DNA 8-mers. The reason we use 8-mer Z-scores instead of probe intensities in Homeo215 is described in Supplementary Note and later in Section 3.
Table 1.
Summary of datasets in this study
| Datasets | # proteins | # nucleic acids | Type |
|---|---|---|---|
| RRM162 | 162 | 241 357 | RNAcompete binding data |
| Homeo215 | 215 | 32 896 | PBM binding data |
| Uniprot400k | 428 109 | – | Diverse protein sequences |
| RRM3k | 3213 | – | RRM sequences |
| Homeo8k | 8302 | – | Homeo sequences |
In addition, to pretrain the FastBioseq protein embedding models as mentioned in Section 2.1, we compile three large corpora of protein sequences. The first dataset is Uniprot400k (Table 1), which contains >400 000 protein sequences from diverse families and species, downloaded from the Uniprot database (https://www.uniprot.org/). The second dataset, RRM3k (Table 1), contains >3000 RRM domain sequences extracted from the CISBP-RNA database (http://cisbp-rna.ccbr.utoronto.ca/). The third dataset, Homeo8k (Table 1), contains >8000 Homeodomain sequences obtained from the CISBP database (http://cisbp.ccbr.utoronto.ca/). Note that here, the protein embedding models are pretrained on these three datasets and are used later to extract protein features on a different dataset (RRM162 or Homeo215). This simple pretraining procedure involves the idea of transfer learning (Pan and Yang, 2010) that knowledge from one task is transferred to another task that usually has much fewer data.
The two primary datasets are used to train and test ProbeRating and the other methods to be compared with. We perform a series of preprocessing procedures to remove redundancy, normalize the intensity scores, etc., for each of the datasets (details in Supplementary Note). The three pretraining datasets are used to pretrain the FastBioseq embedding model so that the model can be used to convert protein sequences in the primary datasets to numerical vectors. We also remove the redundant proteins in each of the pretraining datasets, respectively, and we further remove the proteins overlapping with those in the primary datasets so that the pretraining datasets have no intersections with the two primary datasets. During evaluation, for each of ProbeRating and the other methods, we perform the training and testing as following: we first randomly divide the proteins in the dataset of RRM162 or Homeo215 into 10 folds and leave one fold out as an independent test set. For the remaining ninefolds, we do cross validation to use 90% of them as the training set to tune the model parameters and the rest 10% as the validation set to tune the model hyperparameters. By doing this, the test sets are completely independent from the training and validation process. The same procedure is applied to test on all proteins. We repeat the process 20 times with different random divisions of the dataset and report the average test performance. Same as the AffinitRegression paper, Spearman correlation coefficient (SCC) is used to assess the regression performance since the binding intensity scores are quantile normalized. The details of the redundancy removal, the experimental setup, the implementation, the hardware specification and the runtime summarization can be found in Supplementary Note.
3 Results
In this section, the capability of ProbeRating will be demonstrated by first showing that it is better than three baseline methods, and then showing it also outperforms the more sophisticated AffinityRegression.
3.1 ProbeRating outperformed nearest-neighbor baselines
Since we use a non-parametric reconstruction approach to leverage the training data to predict for unexplored NBPs, a natural sanity check is to compare ProbeRating with the simple but often effective nearest-neighbor approach. Given a testing NBP, if we find its nearest-neighbor by some similarity/distance metric, then the neighbor’s binding profile will be the prediction for this NBPs.
The most common similarity metric is the sequence similarity percent identity (PID), which can be obtained from the BLOSUM amino acid similarity matrix using sequence alignment algorithms. The BLOSUM nearest-neighbor approach has been used by previous studies (Ray et al., 2013; Yang et al., 2018) to infer the PWM binding motifs for unexplored NBPs and has also been compared with AffinityRegression. We evaluated the performance of ProbeRating against the BLOSUM nearest-neighbor baseline on both the RRM162 and the Homeo215 datasets. As shown in Table 2, ProbeRating achieved an average SCC of 0.864 across all RBPs on the RRM162 dataset and an average SCC 0.772 on the Homeo215 dataset. The SCCs were significantly better than BLOSUM baseline’s 0.771 and 0.676 with both P-values< based on the two-tailed Wilcoxon signed-rank test. Moreover, as we could see from Figure 3A and B, the blue dots are mostly above the x = y line in both plots, which indicates ProbeRating almost always outperformed BLOSUM baseline on the two datasets.
Table 2.
Summary of the performance of different methods
| Method | Performancea |
|
|---|---|---|
| RRM162 | Homeo215 | |
| BLOSUM nearest-neighbor baseline | 0.771 | 0.676 |
| k-mer nearest-neighbor baseline | 0.795 | 0.671 |
| FastBioseq nearest-neighbor baseline | 0.804 | 0.647 |
| AffinityRegression | 0.823 | 0.739 |
| AffinityRegression with FastBioseq feature | 0.827 | 0.747 |
| Co-Evo | 0.211 | 0.410 |
| ProbeRating | 0.864 | 0.772 |
Spearman correlation averaged over all tested proteins.
Fig. 3.
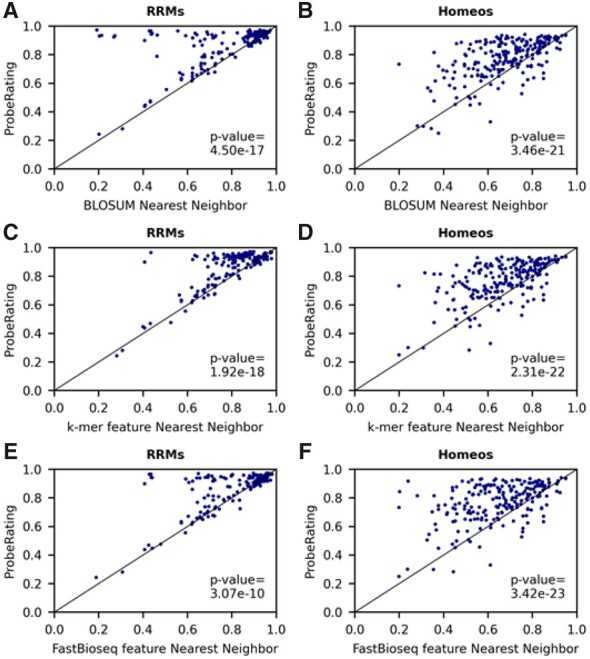
Performance of ProbeRating compared to three nearest-neighbor baselines. (A, B) ProbeRating against the BLOSUM nearest-neighbor baseline. The results for RRM162 are shown on the left in (A) and Homeo215 on the right in (B). The x-axis indicates the SCC between the BLOSUM baseline predicted intensity and the true intensity values for each protein. The y-axis indicates the SCC between the ProbeRating predicted intensity and the true intensity values. Each blue dot in the scatter plot represents the performance of a protein. The straight black line represents x = y. P-values are computed based on a two-tailed Wilcoxon signed-rank test. (C, D) ProbeRating against the k-mer feature nearest-neighbor baseline. (E, F) ProbeRating against the FastBioseq feature nearest-neighbor baseline
In addition to the BLOSUM nearest neighbor, we further explored the idea of the nearest neighbor by incorporating two other similarity metrics: the Euclidean similarity of the k-mer frequency features used by AffinityRegression and the cosine similarity of the FastBioseq generated embedding features. These two metrics, together with BLOSUM PID, captured related but different aspects of protein sequence information, and thus gave related but different nearest neighbors. We compared ProbeRating with the resultant k-mer nearest-neighbor baseline (Fig. 3C, D) and the FastBioseq nearest-neighbor baseline (Fig. 3E, F), as also shown in Table 2. Again, ProbeRating outperformed the two baselines with P-values< on both RRMs (average SCCs for k-mer and FastBioseq feature baselines were 0.795 and 0.804) and Homeos (average SCCs for k-mer and FastBioseq feature baselines were 0.671 and 0.647) tasks. And ProbeRating was better than the two baselines on majority of the proteins.
Therefore, in general, we observed a clear advantage of ProbeRating over the three different nearest-neighbor baselines. It showed that the neural network and reconstruction approaches used inside ProbeRating were non-trivial, and that they were better than simply taking the nearest neighbor by a large margin.
3.2 ProbeRating outperformed AffinityRegression on RBP binding preference prediction task
After testing the performance over the baselines, we then sought to compare ProbeRating with the AffinityRegression method. First, we considered the RBP case. As shown in Figure 4A where proteins in the x-axis are sorted in ascending order based on AffinityRegression’s performance, ProbeRating outperformed AffinityRegression for most of the 162 RRM proteins. When focusing on the first 15 proteins (Fig. 4B), i.e. the proteins that AffinityRegression performed the poorest (lowest SCCs), we observed that ProbeRating generally agreed with AffinityRegression on the set of the hardest proteins but performed slightly better. In both plots, the blue line is almost always above the red line. If we look at Table 2 and Figure 4C, the overall average SCC of the 162 RRM proteins for AffinityRegression was 0.823, which was much lower than ProbeRating’s 0.864 (P < 0.001). Besides, AffinityRegression did have a higher mean value than the three nearest-neighbor baselines in this case (Table 2), and it was significantly better than them based on the Wilcoxon test.
Fig. 4.

Performance of ProbeRating compared to AffinityRegression on the RRM162 dataset. (A) Each dot represents the SCC between the predicted and true RNAcompete probe intensities for a protein. The solid blue line indicates the performance of ProbeRating with the FastBioseq embedded features (PR), the dashed red line indicates the original AffinityRegression with its k-mer frequency features (AR) and the dotted yellow line indicates feeding AffinityRegression with the FastBioseq embedded features (AR-F). Proteins in the x-axis are sorted in ascending order based on their original AffinityRegression’s SCCs. (B) Similar plot to (A), zoom in on the first 15 proteins. (C) Boxplot for the performance of the three methods for all 162 RRM proteins. In each box, the dashed white line denotes the mean, and the solid green line denotes the median. The significance bar represents the P-value from a two-tailed Wilcoxon signed-rank test, with *** P < 0.001
Moreover, we were wondering whether using FastBioseq features alone would lead to better performance. Since our FastBioseq was trained based on the context information of each k-mer, it may implicitly embed some sequence conservation and local structural information into the final feature vectors. To separate the effect of the features from that of the models, we fed the FastBioseq features to the AffinityRegression model replacing its original k-mer frequency features. As denoted by AR-FastBioseq in Figure 4, we compared this approach with the original AffinityRegression and our ProbeRating. As a result, AR-FastBioseq did show slightly better mean (0.827 versus 0.823 in Table 2) than AffinityRegression. But no statistical significance was detected, due to a larger variance of AR-FastBioseq than AffinityRegression (Fig. 4C). Also, AR-FastBioseq’s result was significantly worse than the ProbeRating’s result (P < 0.001).
In summary, similar to the baselines case in the last section, ProbeRating significantly outperformed the more sophisticated method AffinityRegression. A consistent advantage of ProbeRating was shown.
3.3 ProbeRating outperformed AffinityRegression on TF binding preference prediction task
Next, we asked whether the good performance of ProbeRating could generalize to TFs by considering the Homeo binding preference prediction task. This task was different from the above RRM binding preference prediction task not only because one was RBP–RNA interaction and the other was TF–DNA interaction, but also because the Homeo task worked on 8-mer DNA segments and Z-scores instead of ordinary probes and intensity scores. As mentioned earlier in Section 2.3 and in Supplementary Note, this setting has practical usage: unlike in the RRM162 case where there is only one large-scale RNAcompete assay available right now, there exist several large-scale PBM experiments with very different probe designs. 8-mer Z-score is a way to integrate the data from different sources. Thus, if our ProbeRating method could also succeed in this case, the strength of the method would be more convincing.
As a result, ProbeRating outperformed AffinityRegression on the Homeo215 dataset (in Fig. 5), just like on the previous RRM162 dataset. As shown in Figure 5A, ProbeRating was better than AffinityRegression for the majority of the Homeo domains. The mean SCC across all 215 Homeo proteins for ProbeRating was 0.772. It was again significantly better than AffinityRegression’s 0.739 with P < 0.001 (Fig. 5C and Table 2), even though AffinityRegression was significantly better than all the three baselines. When zooming in on the first 15 proteins, as shown in Figure 5B, although these proteins were also hard for ProbeRating (among the lowest SCCs across all proteins), ProbeRating generally performed better than AffinityRegression except for a few cases.
Fig. 5.

Performance of ProbeRating compared to AffinityRegression on the Homeo215 dataset. The plots here are represented in the same way as in Figure 4. In (A) and (B), proteins in the x-axis are again sorted in ascending order based on their original AffinityRegression’s SCCs. In (C), the significance bars indicate the P-values from a two-tailed Wilcoxon signed-rank test, with *** P < 0.001
Moreover, we also tested the AR-FastBioseq approach on the Homeo215 dataset. Interestingly, its mean SCC (0.747 as in Table 2) was again in the middle of ProbeRating’s and the original AffinityRegression’s mean SCCs, similar to the RRM162 case in the last section. AR-FastBioseq significantly outperformed the original AffinityRegression with P < 0.001 this time, and it was also significantly worse than ProbeRating with P < 0.001. This result showed that the FastBioseq features alone improved the performance in the AffintyRegression model, and the neural network approach building on top of that further elevated the performance in ProbeRating.
3.4 ProbeRating was compared with the binding specificity prediction method
Finally, as mentioned in Section 1, most existing methods focus on determining the NBP binding preference as a simplified summarization, like PWM or CNN filter, instead of predicting the full binding profile as AffinityRegression and ProbeRating do. Although the focuses and goals are different, the binding-specificity method Co-Evo (Yang et al., 2018) that was mentioned in Section 1 is also capable of inferring the nucleic acids preferences of an unexplored protein, and it is relatively more recent than the other methods. So, we evaluated Co-Evo on the same datasets to compare it with ProbeRating, to get a sense of where ProbeRating stands when compared with binding specificity prediction methods. As a result, we observed the SCCs of Co-Evo were much worse than ProbeRating and AffinityRegression (Table 2), which was not surprising since Co-Evo was designed to predict a PWM motif to summarize the binding preference instead of to predict the binding profile directly. The details of the Co-Evo results can be found in Supplementary Note.
4 Discussion
In this study, we introduced a new method ProbeRating to predict the binding profiles for NBPs that are experimentally unexplored. We showed that predicting the binding profile for unexplored NBPs is a critical but challenging task given is the limited data available. Thus, the task is less studied compared to the other task of directly determining the binding preference for an NBP from its experimental data. Extending the previous work of AffinityRegression, we developed a two-stage framework to tackle the task utilizing modern techniques from deep learning and word embedding. The first stage involved encoding the protein and nucleic acid sequences into distributed feature vectors. We contributed a tool FastBioseq, which essentially wrapped the famous FastText method from natural language processing to extract high-level features from biological sequences. The second stage involved recommending binding preferences for new proteins. We contributed a feedforward neural network with a non-parametric reconstruction step to leverage the training data. Our method was evaluated on the benchmark RBP and TF binding datasets. It performed well on both datasets and showed significant improvements over AffinityRegression and three baselines.
While the significant performance advancement of our method shows the advantage of using more expressive neural network models and word embedding features to study NBP–nucleic acid interactions, we see several potential improvements to this study. Here, ProbeRating propagates binding information from experimentally characterized NBPs to those unexplored ones within the same protein family. It would be interesting to investigate whether predicting for proteins from another family also works, or how similar the unexplored protein is to those already explored ones to get ProbeRating to work. As we mentioned earlier in the nearest-neighbor baselines section, the metrics to define ‘similar’ can be different, depending on what features we are using. Additionally, when investigating RBP–RNA interactions, although RNAs are known to fold themselves into secondary and tertiary structures, we do not consider this information. It is because the probes in our RNAcompete dataset were intentionally designed to be unstructured or weak structured (Ray et al., 2013). However, our two-stage framework could easily incorporate RNA structure as well as protein structure as input features when appropriate data become available in further researches. Moreover, the highly modular subnetwork structures in ProbeRating provide a lot of flexibility to be extended by other neural network models, too.
Overall, the strength of ProbeRating suggests promising capacity to the field. It is especially desired by RBPs that do not have much experimental evidence available at this moment. ProbeRating could be applied to learn binding patterns in those crucial RBP-related problems, like lncRNA regulation (Quinn and Chang, 2016; Zhao et al., 2016) or CRISPR/CAS systems (Liu et al., 2016; Wang et al., 2016). Also, similar to AffinityRegression, the prediction output from ProbeRating is the entire binding intensity profile instead of a simplified representation. So, when dealing with an unexplored NBP, the output binding profile from ProbeRating could be further fed as input to those intensively studied protein-specific methods for downstream analysis. Furthermore, besides of NBP–nucleic acid paired prediction, ProbeRating could be applied to other scenarios. For example, AffinityRegression has been used for protein–protein interaction in a tumor-related signaling pathway study (Osmanbeyoglu et al., 2017). ProbeRating could also be used in such a case to see if better performance is achieved. ProbeRating’s component FastBioseq can be used as a standalone package in other scenarios, too. It provides a general and flexible tool of biological sequence embedding for DNA, RNA and proteins.
Supplementary Material
Acknowledgements
The authors thank Wan Lam and Junwen Wang for the valuable discussions.
Funding
This work was supported by Genome Canada, and Natural Sciences and Engineering Research Council (NSERC) of Canada.
Conflict of Interest: none declared.
Contributor Information
Shu Yang, Department of Computer Science, University of British Columbia, Vancouver, BC V6T1Z4, Canada.
Xiaoxi Liu, RIKEN Center for Integrative Medical Sciences (IMS), Yokohama 230-0045, Japan.
Raymond T Ng, Department of Computer Science, University of British Columbia, Vancouver, BC V6T1Z4, Canada.
References
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. [DOI] [PubMed] [Google Scholar]
- Asgari E., Mofrad M.R. (2015) Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One, 10, e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L. et al. (2009) Meme suite: tools for motif discovery and searching. Nucleic Acids Res., 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A., Zhao K. (2009) Genomic location analysis by ChIP-seq. J. Cell. Biochem., 107, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci M. et al. (2011) Predicting protein associations with long noncoding RNAs. Nat. Methods, 8, 444–445. [DOI] [PubMed] [Google Scholar]
- Berger M.F. et al. (2006) Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol., 24, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M.F. et al. (2008) Variation in homeodomain dna binding revealed by high-resolution analysis of sequence preferences. Cell, 133, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojanowski P. et al. (2017) Enriching word vectors with subword information. Trans. Assoc. Comput. Linguist., 5, 135–146. [Google Scholar]
- Corrado G. et al. (2016) RNAcommender: genome-wide recommendation of RNA–protein interactions. Bioinformatics, 32, 3627–3634. [DOI] [PubMed] [Google Scholar]
- Dong Q. et al. (2018) Regulatory RNA binding proteins contribute to the transcriptome-wide splicing alterations in human cellular senescence. Aging, 10, 1489–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S. et al. (2019) cDeepbind: a context sensitive deep learning model of RNA–protein binding, Machine Learning in Computational Biology .
- Ghanbari M., Ohler U. (2020) Deep neural networks for interpreting RNA binding protein target preferences, Genome Res., 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M. et al. (2014) Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol., 10, e1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M. et al. (2006) Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res., 34, e117–e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y. et al. (2018) Partner-specific prediction of RNA-binding residues in proteins: a critical assessment. Proteins, 87, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazan H. et al. (2010) RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput. Biol., 6, e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J. et al. (2012) Protein–RNA interactions: new genomic technologies and perspectives. Nat. Rev. Genet., 13, 77–83. [DOI] [PubMed] [Google Scholar]
- Koo P.K. et al. (2018) Inferring sequence-structure preferences of RNA-binding proteins with convolutional residual networks, bioRxiv:418459.
- Lambert S.A. et al. (2018) The human transcription factors. Cell, 172, 650–665. [DOI] [PubMed] [Google Scholar]
- Le Q., Mikolov T. (2014) Distributed representations of sentences and documents. In Proceedings of the 31st International Conference on Machine Learning, 32, 1188–1196. [Google Scholar]
- Li S. et al. (2017) A deep boosting based approach for capturing the sequence binding preferences of RNA-binding proteins from high-throughput clip-seq data. Nucleic Acids Res., 45, e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2016) Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci. Rep., 6, 19675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C. et al. (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J., 272, 2118–2131. [DOI] [PubMed] [Google Scholar]
- Maticzka D. et al. (2014) Graphprot: modeling binding preferences of RNA-binding proteins. Genome Biol., 15, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolov T. et al. (2013) Distributed representations of words and phrases and their compositionality. In Proceedings of the 26th International Conference on Neural Information Processing Systems, 2, 3111–3119. [Google Scholar]
- Orenstein Y. et al. (2016) RCK: accurate and efficient inference of sequence- and structure-based protein–RNA binding models from RNAcompete data. Bioinformatics, 32, i351–i359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmanbeyoglu H.U. et al. (2017) Pancancer modelling predicts the context-specific impact of somatic mutations on transcriptional programs. Nat. Commun., 8, 14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.J., Yang Q. (2010) A survey on transfer learning. IEEE Trans. Knowl. Data Eng., 22, 1345–1359. [Google Scholar]
- Pan X., Shen H.-B. (2018. a) Learning distributed representations of RNA sequences and its application for predicting RNA–protein binding sites with a convolutional neural network. Neurocomputing, 305, 51–58. [Google Scholar]
- Pan X., Shen H.-B. (2018. b) Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics, 34, 3427–3436. [DOI] [PubMed] [Google Scholar]
- Pan X. et al. (2019) Recent methodology progress of deep learning for RNA protein interaction prediction. Wiley Interdiscip. Rev RNA, 10, e1544. [DOI] [PubMed] [Google Scholar]
- Park P.J. (2009) ChIPseq: advantages and challenges of a maturing technology. Nat. Rev. Genet., 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelossof R. et al. (2015) Affinity regression predicts the recognition code of nucleic acid-binding proteins. Nat. Biotechnol., 33, 1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z., Kurgan L. (2015) High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res., 43, e121–e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J.J., Chang H.Y. (2016) Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet., 17, 47–62. [DOI] [PubMed] [Google Scholar]
- Ray D. et al. (2009) Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol., 27, 667–670. [DOI] [PubMed] [Google Scholar]
- Ray D. et al. (2013) A compendium of RNA-binding motifs for decoding gene regulation. Nature, 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci F. et al. (2011) Recommender Systems Handbook, Springer, US. [Google Scholar]
- Stormo G.D. (2000) DNA binding sites: representation and discovery. Bioinformatics, 16, 16–23. [DOI] [PubMed] [Google Scholar]
- Suresh V. et al. (2015) RPI-Pred: predicting ncRNA–protein interaction using sequence and structural information. Nucleic Acids Res., 43, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak Leung R.W. et al. (2019) ENPD—a database of eukaryotic nucleic acid binding proteins: linking gene regulations to proteins. Nucleic Acids Res., 47, D322–D329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia R.R. et al. (2017) Sequence-based prediction of RNA-binding residues in proteins. Methods Mol. Biol. 1484, 205–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. et al. (2015) Design and bioinformatics analysis of genome-wide clip experiments. Nucleic Acids Res., 43, 5263–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. et al. (2016) CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem., 85, 227–264. [DOI] [PubMed] [Google Scholar]
- Weirauch M.T. et al. (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J. et al. (2016) A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues. Brief. Bioinf., 17, 88–105. [DOI] [PubMed] [Google Scholar]
- Yang S. et al. (2018) Inferring RNA sequence preferences for poorly studied RNA-binding proteins based on co-evolution. BMC Bioinformatics, 19, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. et al. (2011) Correlated evolution of transcription factors and their binding sites. Bioinformatics, 27, 2972–2978. [DOI] [PubMed] [Google Scholar]
- Yi H.-C. et al. (2018) A deep learning framework for robust and accurate prediction of ncRNA–protein interactions using evolutionary information. Mol. Ther. Nucleic Acids, 11, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H. et al. (2016) Convolutional neural network architectures for predicting DNAprotein binding. Bioinformatics, 32, i121–i127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. et al. (2019) Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Brief. Bioinf., 20, 1250–1268. [DOI] [PubMed] [Google Scholar]
- Zhao Y. et al. (2016) NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res., 44, D203–D208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.