

Abstract
Alzheimer’s disease is a highly heritable, common neurodegenerative disease characterized neuropathologically by the accumulation of β-amyloid plaques and tau-containing neurofibrillary tangles. In addition to the well-established risk associated with the APOE locus, there has been considerable success in identifying additional genetic variants associated with Alzheimer’s disease. Major challenges in understanding how genetic risk influences the development of Alzheimer’s disease are clinical and neuropathological heterogeneity, and the high level of accompanying comorbidities. We report a multimodal analysis integrating longitudinal clinical and cognitive assessment with neuropathological data collected as part of the Brains for Dementia Research study to understand how genetic risk factors for Alzheimer’s disease influence the development of neuropathology and clinical performance. Six hundred and ninety-three donors in the Brains for Dementia Research cohort with genetic data, semi-quantitative neuropathology measurements, cognitive assessments and established diagnostic criteria were included in this study. We tested the association of APOE genotype and Alzheimer’s disease polygenic risk score—a quantitative measure of genetic burden—with survival, four common neuropathological features in Alzheimer’s disease brains (neurofibrillary tangles, β-amyloid plaques, Lewy bodies and transactive response DNA-binding protein 43 proteinopathy), clinical status (clinical dementia rating) and cognitive performance (Mini-Mental State Exam, Montreal Cognitive Assessment). The APOE ε4 allele was significantly associated with younger age of death in the Brains for Dementia Research cohort. Our analyses of neuropathology highlighted two independent pathways from APOE ε4, one where β-amyloid accumulation co-occurs with the development of tauopathy, and a second characterized by direct effects on tauopathy independent of β-amyloidosis. Although we also detected association between APOE ε4 and dementia status and cognitive performance, these were all mediated by tauopathy, highlighting that they are a consequence of the neuropathological changes. Analyses of polygenic risk score identified associations with tauopathy and β-amyloidosis, which appeared to have both shared and unique contributions, suggesting that different genetic variants associated with Alzheimer’s disease affect different features of neuropathology to different degrees. Taken together, our results provide insight into how genetic risk for Alzheimer’s disease influences both the clinical and pathological features of dementia, increasing our understanding about the interplay between APOE genotype and other genetic risk factors.
Keywords: Alzheimer’s disease, dementia, APOE, beta-amyloid, neurofibrillary tangles
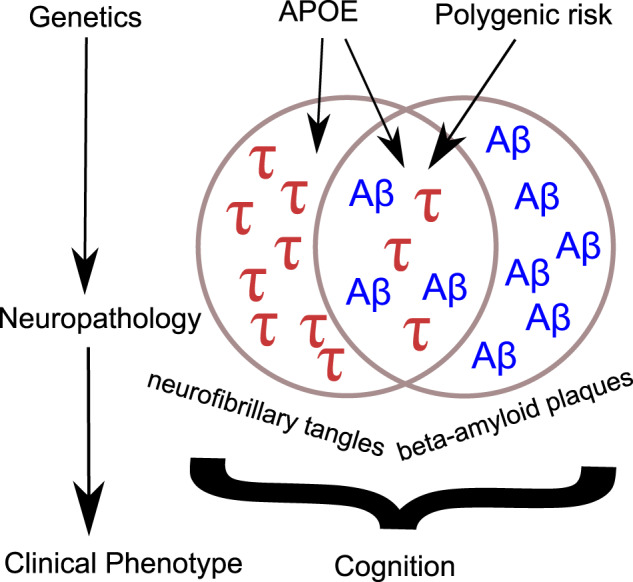
We report a multimodal analysis profiling how genetic risk factors for Alzheimer’s disease influence the development of neuropathology and clinical performance in the Brains for Dementia Research (BDR) study, highlighting a shared pathway between APOE and the development of tauopathy and beta-amyloidosis and an independent pathway associated with tauopathy.
Graphical Abstract
Graphical Abstract.
Introduction
Alzheimer’s disease is a common neurodegenerative disease characterized clinically by progressive memory and cognitive decline leading to dementia and neuropathologically by β-amyloid plaques and tau-containing neurofibrillary tangles (NFTs). The most frequent manifestation of Alzheimer’s disease is late onset Alzheimer’s disease where onset occurs after the age of 65. Late onset Alzheimer’s disease is highly heritable (Gatz et al., 2006) with the most established genetic risk factor being variants of the APOE gene. Relative to the most common genotype (ε3/ε3), the ε4 allele increases the risk of Alzheimer’s disease, with ε4 homozygosity associated with ∼20-fold increase in risk (Farrer et al., 1997). In contrast, the ε2 allele of APOE has strong protective effects (Reiman et al., 2020). Genome-wide association studies (GWAS) in large sample cohorts (Lambert et al., 2013; Marioni et al., 2018; Jansen et al., 2019; Kunkle et al., 2019) have identified additional variants in more than 40 regions of the genome which individually confer subtler effects on risk, but cumulatively account for a large proportion of genetic risk. To index an individuals’ genetic risk profile, disease-associated variants—typically including those below genome-wide significance—can be combined into a ‘polygenic risk score’ (PRS). PRSs quantify the number of genetic risk variants an individual has, weighted by their effect size, and have been shown to improve prediction models of Alzheimer’s disease (Escott-Price et al., 2015, 2019; Cruchaga et al., 2018). Of note, the Alzheimer’s disease PRS has greatest predictive power where disease status has been defined by standardized neuropathological assessment (Escott-Price et al., 2017), and is most elevated in sporadic early-onset cases (Cruchaga et al., 2018).
In addition to genetic prediction, PRSs provide a powerful mechanism to investigate how genetic risk mediates the development of symptoms, and can potentially be used to disentangle the primary causal features from the secondary consequences of disease. As well as being associated with dementia status, the Alzheimer’s disease PRS has been shown to correlate with mild cognitive impairment (Adams et al., 2015; Chaudhury et al., 2019), cognitive decline (Mormino et al., 2016; Marioni et al., 2017; Felsky et al., 2018), memory impairments (Mormino et al., 2016; Marioni et al., 2017), cortical thickness (Sabuncu et al., 2012; Corlier et al., 2018), hippocampal volume (Lupton et al., 2016; Mormino et al., 2016), cerebrospinal biomarkers (Martiskainen et al., 2014; Louwersheimer et al., 2016; Desikan et al., 2017) and neuropathology (Desikan et al., 2017; Felsky et al., 2018; Tasaki et al., 2018). The breadth of associations highlights the complexity of understanding the pathways from genetic risk to symptomatic disease. Furthermore, many of these analyses have included the APOE locus within the PRS, meaning their results may reflect APOE-specific effects rather than the consequences of a broader polygenic risk burden. To truly understand how multiple genetic risk factors combine to influence the interplay of the clinical, cognitive and neuropathological characteristics of Alzheimer’s disease, we need large, longitudinal cohorts with post-mortem tissue that can align genetics, clinical data and standardized neuropathological assessments.
A major challenge in understanding how genetic risk influences the development of Alzheimer’s disease relates to clinical and neuropathological heterogeneity, and the high level of accompanying comorbidities associated with a diagnosis of Alzheimer’s disease. The presence of the neuropathological hallmarks of Alzheimer’s disease can only be confirmed following post-mortem brain examination. Standardized sampling and staining methods, along with the introduction of a number of semi-quantitative classification schemes, each focused on a single neuropathological feature (Thal et al., 2002; Braak et al., 2003, 2006), promote consistency making it easier to harmonize data across brain banks and ultimately the reproducibility of findings across studies. It is now recognized that sporadic dementia in older people is predominantly due to multiple pathologies (Robinson et al., 2018). The most frequent comorbidity is Lewy body pathology affecting up to 50% of sporadic Alzheimer’s disease cases (Toledo et al., 2013). Another common comorbidity is the presence of inclusion bodies containing aggregates of transactive response DNA-binding protein 43 (TDP-43), particularly in the oldest old (Amador-Ortiz et al., 2007; Uryu et al., 2008; James et al., 2016). As well as influencing cognitive impairment in non-Alzheimer’s disease cases (Nag et al., 2017), these comorbidities contribute to the cognitive decline observed in Alzheimer’s disease cases beyond that associated with β-amyloid and NFT pathology (Wilson et al., 2013; Nelson et al., 2019), hence it is important to consider multiple neuropathological features simultaneously, to understand the processes that underlie cognitive performance in old age.
The paucity of comprehensive neuropathological data in large sample cohorts has limited previous genetic studies of Alzheimer’s disease-associated neuropathology. To address this gap, the Brains for Dementia Research (BDR) cohort was established in 2007 recruiting both dementia patients and unaffected controls over the age of 65 to partake in routine longitudinal assessments collecting cognitive, clinical, lifestyle and psychometric data, prior to post-mortem brain donation (Francis et al., 2018). The inclusion of standardized semi-quantitative data for a range of neuropathological features facilitates analyses into the specificity of genetic risk factors for the different abnormalities, and an assessment of their clinical contributions. In this study we report the first multimodal analysis of the BDR cohort, integrating longitudinal clinical and cognitive assessment with neuropathological data to explore how known genetic risk factors for Alzheimer’s disease influence the development of different aspects of neuropathology and cognitive performance in old age. We focus on four common neuropathological features observed in Alzheimer’s disease brain tissue: NFTs, β-amyloid plaques, Lewy bodies and TDP-43 proteinopathy. The results of this study provide insights into the neurobiological pathways to cognitive decline by refining our understanding of the complex interplay of genetic risk, clinical presentation and neuropathological burden.
Materials and methods
BDR cohort description
BDR was established in 2007 and consists of a network of six dementia research centres in England and Wales (King’s College London, Bristol, Manchester, Oxford, Cardiff and Newcastle Universities) and the associated university brain banks handling the donations (Cardiff brain donations were banked in London). Participants over the age of 65 were recruited using national and local press, TV and radio coverage, articles in charity newsletters, national magazines with an older following, BDR posters, leaflets, memory clinics, talks at carer/support groups, Women’s Institute and the University of the Third Age. There was no screening to exclude or include individuals with particular diagnoses or those carrying genetic variants associated with neurodegenerative diseases. The cohort includes individuals with and without dementia, spanning the full spectrum of dementia diagnoses. Participants underwent a series of longitudinal cognitive and psychometric assessments and registered for brain donation. An extensive description of the recruitment strategy, demographics, assessment protocols and neuropathic assessment procedures can be found in (Francis et al., 2018).
Longitudinal cognitive and clinical assessments
All assessments were conducted by a trained psychologist or research nurse. Exclusion criteria to undergo assessments included: (i) factors precluding brain donation (e.g. brain injury/trauma, major stroke), (ii) being younger than 65 for healthy controls (except where they were spouses/partners of participants with dementia), (iii) having insufficient English language skills for completing assessments and (iv) being geographically too remote from an assessment centre. Baseline assessments were conducted face-to-face (in the participant’s place of residence or a BDR centre), follow-up assessments were usually face-to-face but telephone interviews were also used for some healthy control participants. Follow-up interviews were annual for participants with cognitive impairment, and every 1–5 years (depending on age) for cognitively healthy participants. Clinical assessment was performed using the clinical dementia rating (CDR) (Morris, 1993). Cognitive assessment measures relevant to this study included the Mini-Mental State Examination (MMSE) (Folstein et al., 1975) and Montreal Cognitive Assessment (MoCA) (Nasreddine et al., 2005).
Post-mortem neuropathological assessment
After removal, the brain was examined macroscopically and digitally recorded. After slicing, the brain was comprehensively sampled according to the BDR protocol by experienced neuropathologists in each of the five network brain banks. This protocol, arrived at by consensus across the BDR network and based on the BrainNet Europe initiative (Bell et al., 2008), was used to generate a description of the regional pathology within the brain together with standardized scoring. In this study we considered five variables representing four neuropathological features: (i) Braak tangle stage which captures the progression of NFT pathology (Braak and Braak, 1991; Braak et al., 2006), (ii) Thal β-amyloid phase which captures the regional distribution of plaques (Thal et al., 2002), (iii) Consortium to Establish a Registry for Alzheimer’s disease (CERAD) stage which profiles neuritic plaque density (Mirra et al., 1991; Montine et al., 2012), (iv) Braak Lewy body stage (Braak et al., 2003) and (v) TDP-43 status (a binary indicator of the absence/presence of TDP-43 inclusions, as assessed by immunohistochemistry of the amygdala and the hippocampus and adjacent temporal cortex for phosphorylated TDP-43). All variables apart from TDP-43 were analysed as continuous variables, using their semi-quantitative nature to capture dose-dependent relationships of increasing neuropathological burden.
Genetic data
DNA extraction was performed using a standard phenol chloroform method on 100 mg of brain tissue. DNA quality was assessed using the Agilent 2200 TapeStation DNA integrity number and quantified using NanoDrop 3300 spectrometry. Genotyping was performed on the NeuroChip array which is a custom Illumina genotyping array with an extensive genome-wide backbone (n = 306 670 variants) and custom content covering 179 467 variants specific to neurological diseases (Blauwendraat et al., 2017). Genotype calling was performed using GenomeStudio (v2.0, Illumina) and quality control was completed using PLINK1.9 (Chang et al., 2015). Individuals were excluded if either (i) they had > 5% missing data, (ii) their genotype predicted sex using X chromosome homozygosity was discordant with their reported sex (excluding females with an F value > 0.2 and males with an F value < 0.8), (iii) they had excess heterozygosity [>3 standard deviation (SD) from the mean], (iv) they were related to another individual in the sample (pi hat > 0.2), where one individual from each pair of related samples was excluded considering data quality and phenotype or (v) they were classed as non-European, determined by merging the BDR genotypes with data from HapMap Phase 3 (http://www.sanger.ac.uk/resources/downloads/human/hapmap3.html), linkage disequilibrium pruning the overlapping single nucleotide polymorphisms (SNPs) such that no pair of SNPs within 1500 bp had r2 > 0.20 and visually inspecting the first two genetic principal components along with the known ethnicities of the HapMap sample to define European samples (Supplementary Fig. 1). Prior to imputation SNPs with high levels of missing data (>5%), Hardy-Weinberg equilibrium P < 0.001 or minor allele frequency <1% were excluded. The genetic data were then recoded as vcf files before uploading to the Michigan Imputation Server (Das et al., 2016) (https://imputationserver.sph.umich.edu/index.html#!) which uses Eagle2 (Loh et al., 2016) to phase haplotypes, and Minimac4 (https://genome.sph.umich.edu/wiki/Minimac4) with the most recent 1000 Genomes reference panel (phase 3, version 5). Imputed genotypes were then filtered with PLINK2.0alpha, excluding SNPs with an R2 INFO score < 0.5 and recoded as binary PLINK format. Proceeding with PLINK1.9, samples with >5% missing values, and SNPs with >2 alleles, >5% missing values, Hardy-Weinberg equilibrium P < 0.001 or a minor allele frequency of <5% were excluded. The final quality controlled imputed set of genotypes contained 6 607 832 variants.
Polygenic risk scores
GWAS results from Kunkle et al. (2019) were used to calculate an Alzheimer’s disease PRS for each individual. We choose this GWAS as it is based on clinically defined cases compared to controls. To separate the effects of APOE from other genetic variants associated with Alzheimer’s disease, we excluded the APOE region (chr19:45 116 911–46 318 605) (Kunkle et al., 2019) from the PRS calculations. We generated PRS using PRSice (v2.0) (Choi and O'Reilly, 2019) which ‘clumps’ the Alzheimer’s disease GWAS summary statistics using the BDR genotype data such that the most significant variant in each linkage disequilibrium block was retained. The PRS was then calculated in the target (BDR) dataset for each individual, as the number of reference alleles multiplied by the log odds ratio for that SNP (taken from the Kunkle et al. Alzheimer’s disease GWAS), and then summed across all retained clumped variants with an Alzheimer’s disease GWAS P-value < PT. A range of P-value thresholds (PT) were used initially, to generate multiple possible PRS, where the optimal PRS was selected as the score that explained the highest proportion of variance (Nagelkerke’s pseudo R2) in Alzheimer’s disease case control status. In this analysis, Alzheimer’s disease cases and controls were defined as Braak high (Braak tangle stages V and VI) and low (Braak tangle stages 0–II) respectively, and PRS was tested using a logistic regression model with the first 8 genetic principal components as covariates. In the BDR cohort the optimal threshold for selecting SNPs for the PRS was P < 5 × 10−8 (Supplementary Fig. 2). Prior to analysis the PRS calculated at this threshold was standardized to have a mean of 0 and SD of 1; therefore the interpretation is in units of SDs.
APOE genotyping
The APOE SNPs rs7412 and rs429358 were genotyped with TaqMan assays using standard protocols. Where APOE genotype by TaqMan assay was not available, it was generated from the NeuroChip data (n = 44). The NeuroChip array includes multiple probes to assay the two APOE SNPs; based on the optimal concordance with the TaqMan assay (91% concordant across assays) we used the probes rs7412.B3 and rs429358.T2 to determine APOE status. In all statistical analyses, APOE status was modelled as two numeric variables counting the number ε2 alleles and number of ε4 alleles an individual had. Given the rarity of ε2/ε2 genotype [only four occurrences (0.58%) in this sample], the ε2/ε2 individuals were combined with the individuals with one ε2 allele.
Statistical analysis
All statistical analyses were performed in R version 3.5.2. All analytical code is available via GitHub (https://github.com/ejh243/BDR-Genetic-Analyses).
Survival analysis
To test whether APOE and Alzheimer’s disease PRS were associated with younger age at death, we fitted Cox’s proportional hazards models using the R package survival. Three models were fitted with age at death as the outcome to test (i) APOE genotype modelled as two variables, (ii) Alzheimer’s disease PRS and (iii) APOE genotype and Alzheimer’s disease PRS simultaneously. All models included covariates for sex, BDR centre and eight genetic principal components.
Genetic analysis of neuropathology and clinical/cognitive status at death
Genetic associations between either APOE status or Alzheimer’s disease PRS and any of the continuous neuropathology variables (Braak tangle stage, Thal β-amyloid stage, CERAD stage, Braak Lewy body stage), clinical (CDR global rating) or cognitive status at death (MMSE, MoCA) were tested using a linear regression model. TDP-43 proteinopathy as a binary variable was analysed with logistic regression, but the model framework was the same. Up to four regression models were fitted for each variable. First, the effects of APOE status and Alzheimer’s disease PRS were estimated separately using Model 1 and Model 2 below.
Model 1:
Model 2:
If APOE (either variable) and PRS were significantly associated with an outcome, then a multiple regression analysis was additionally fitted testing APOE and PRS simultaneously to confirm these were independent associations (Model 3).
Model 3:
Finally, an interaction model (Model 4) between APOE and PRS was fitted to test if PRS associations differed depending on APOE genotype.
Model 4:
All analyses included age at death, sex and BDR centre as covariates and the first eight genetic principal components. Analyses for clinical or cognition measures also included a covariate that measured the time lapse between the last assessment and death.
Longitudinal clinical and cognition analyses
To test how APOE and Alzheimer’s disease PRS affected clinical status and cognitive trajectories, we fitted multi-level regression models using all available pre-mortem assessment data. A time variable was created which measured the number of days after the first visit that an assessment took place. Each cognitive variable was then tested as the dependent variable against this time variable included as a fixed effect along with covariates for age, sex, BDR centre and the first eight genetic principal components and a random effect for individual. To test for genetic effects on the cognitive trajectory, either APOE (coded as two variables) or Alzheimer’s disease PRS, was included in the model as a main effect and as an interaction with time. Models were fitted using the R packages lme4 and lmTest.
Multiple testing
In total, we tested 12 outcomes against 3 genetic variables. Our outcomes comprised five neuropathological variables, one clinical variable at death, two cognitive measures, one longitudinal clinical, two longitudinal cognitive measures and a survival analysis of age at death. Against these 12 outcomes, we tested 3 genetic variables (Alzheimer’s disease PRS and two variables to model APOE genotype). Therefore, we performed a multiple testing correction for 36 tests, reporting significant associations as those with P < 0.0014. Given the correlations between the neuropathological, clinical and cognitive variables this is likely to be a conservative approach.
Data availability
Genetic, clinical and cognitive data are available through the Dementia’s Platform UK (DPUK; https://www.dementiasplatform.uk/) platform upon application.
Results
Both tauopathy and β-amyloidosis are present at high frequencies in the BDR cohort
To profile the effects of both APOE genotype and Alzheimer’s disease PRS, our analyses were limited to BDR donors who had undergone neuropathological assessment and had NeuroChip array data (n = 693, Table 1). The participants had a mean age at death of 83.5 years (SD = 9.34 years) and 52.8% were male. Consistent with epidemiological reports, females were significantly older at death than males (mean difference = 3.84 years; P = 4.87 × 10−8). Within this cohort, 57.3% of individuals had dementia at their first assessment (i.e. at baseline), with 63.3% of the cohort affected by dementia at death. At recruitment, individuals had a mean CDR of 1.42 (SD = 1.36), a mean MMSE score of 22.3 (SD = 8.81) and a mean MoCA score of 17.2 (SD = 10.6). These scores indicate that the majority of participants only suffered mild cognitive impairment, although the full range of cognitive performance was represented in the cohort. Participants underwent a mean of 2.85 assessments (SD = 1.71) prior to death. Individuals who had at least two assessments (N = 486) were followed for a mean of 3.40 years (SD = 2.00 years) with a mean of 1.42 years between assessments (SD = 0.67 years). Our genetic analyses focused on four semi-quantitative and one indicator neuropathology variable. In 672 samples NFT pathology was quantified using Braak NFT stage (Braak and Braak, 1991; Braak et al., 2006) with a mean of 3.76 (SD = 1.90). Two variables reflecting the extent of β-amyloidosis were considered: β-amyloid distribution was measured by Thal β-amyloid phase (Thal et al., 2002) with a mean value of 3.14 (SD = 1.78) across 612 individuals and neuritic plaque density was scored using the CERAD classification (Mirra et al., 1991; Montine et al., 2012) with a mean value of 1.72 (SD = 1.26) across 634 individuals. α-Synuclein pathology was quantified using Braak Lewy body stage, where across 634 individuals the mean was 1.36 (SD = 2.26). TDP-43 status was available for 658 individuals, with 150 (22.8%) individuals classed as being TDP-43 positive.
Table 1.
Summary of BDR cohort
| % | Mean | SD | N | ||
|---|---|---|---|---|---|
| Demographics | Sex (male) | 52.8 | 693 | ||
| Age | 83.5 | 9.34 | 693 | ||
| Clinical assessments | Number of assessments | 2.85 | 1.71 | 693 | |
| Time in study (years) | 3.40 | 2.00 | 486 | ||
| Time between assessments (years) | 1.42 | 0.67 | 486 | ||
| Dementia status at first assessment | Dementia | 57.3 | 693 | ||
| MCI/inconclusive | 13.3 | 693 | |||
| No dementia | 29.3 | 693 | |||
| Dementia status at last assessment | Dementia | 63.3 | 693 | ||
| MCI/inconclusive | 14.2 | 693 | |||
| No dementia | 22.5 | 693 | |||
| Neuropathology | Braak stage tangle | 3.76 | 1.9 | 672 | |
| Thal amyloid stage | 3.14 | 1.78 | 612 | ||
| CERAD stage | 1.72 | 1.26 | 634 | ||
| Braak Lewy body stage | 1.36 | 2.26 | 597 | ||
| TDP-43 | 22.8 | 658 | |||
| Cognitive scores at first assessment | CDR | 1.42 | 1.36 | 639 | |
| MMSE | 22.3 | 8.81 | 469 | ||
| MoCA | 17.2 | 10.6 | 270 | ||
| Cognitive scores at last assessment | CDR | 1.79 | 1.3 | 639 | |
| MMSE | 19.1 | 10.3 | 469 | ||
| MoCA | 16.1 | 11 | 270 |
Genetic risk factors for Alzheimer’s disease are associated with increased mortality
To determine whether higher genetic risk for Alzheimer’s disease was associated with increased mortality we analysed survival with Cox’s proportional hazard models (Table 2). APOE genotype was modelled as two variables—the number of ε4 alleles and the number of ε2 alleles, to distinguish the hypothesized risk effects of ε4 (Corder et al., 1993; Farrer et al., 1997) from the protective effects of ε2 (Reiman et al., 2020). Analysis of APOE genetic risk found that APOE ε4 status was significantly associated with younger age at death, with each additional ε4 allele associated with 29% increased risk of death (hazard ratio = 1.29; P = 9.66 × 10−5). Alzheimer’s disease PRS was nominally associated with an increased mortality (hazard ratio = 1.11; P = 8.97 × 10−3), although this was not significant after correcting for multiple testing.
Table 2.
APOE is associated with increased mortality
| Analytical model |
APOE
|
PRS |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of ε2 alleles |
Number of ε4 alleles |
||||||||
| Hazard ratio | SE | P-value | Hazard ratio | SE | P-value | Hazard ratio | SE | P-value | |
| Model 1 | 0.835 | 0.123 | 0.142 | 1.293 | 0.066 | 9.66E−05 | |||
| Model 2 | 1.105 | 0.038 | 8.97E−03 | ||||||
| Model 3 | 0.839 | 0.124 | 0.155 | 1.292 | 0.066 | 1.00E−04 | 1.106 | 0.038 | 8.41E−03 |
APOE and Alzheimer’s disease PRS independently influence tauopathy and β-amyloidosis
The number of APOE ε4 alleles was positively associated (P < 0.00014) with all four semi-quantitative neuropathology measures (Table 3). The most significant association was with Braak NFT stage: each ε4 allele was associated with an increase in 1.16 Braak NFT stages (P = 4.16 × 10−24). Associations were also found between ε4 status and Thal β-amyloid phase (mean difference per ε4 allele = 0.981 phases; P = 3.96 × 10−20), neuritic plaque density (mean difference per ε4 allele = 0.713 stages; P = 1.03 × 10−19) and Braak Lewy body stage (mean difference per ε4 allele = 0.555 stages; P = 2.64 × 10−4). Alzheimer’s disease PRS was associated with two measures of neuropathology (Table 3): a higher polygenic burden was associated with Braak NFT stage (mean difference per SD of PRS = 0.354 stages; P = 1.36 × 10−6) and neuritic plaque density (mean difference per SD of PRS = 0.202 stages; P = 5.27 × 10−5). TDP-43 was not associated with either APOE genotype or Alzheimer’s disease PRS. Although variants in the APOE region were excluded from the PRS, we tested both APOE and PRS against Braak NFT stage and neuritic plaque density simultaneously to confirm that the identified associations were independent. The estimated effects of ε4 on both Braak NFT stage and neuritic plaque density were unaffected, while the Alzheimer’s disease PRS associations were slightly attenuated (Table 3) but remained significant. In addition to an additive model, we tested whether there was evidence for a multiplicative effect between Alzheimer’s disease PRS and APOE genotype on neuropathological burden to explore the hypothesis that in individuals with protective APOE genotypes, Alzheimer’s disease PRS is more important (i.e. has a larger effect on neuropathology). In this analysis, none of the five neuropathological variables had statistically significant differences across APOE genotype groups (P > 0.05) (Supplementary Table 1). Taken together, these results suggest that APOE status and Alzheimer’s disease PRS are independently associated with neuropathology, combining in an additive manner to influence an individual’s accumulation of tauopathy (NFTs) and β-amyloid plaques.
Table 3.
Common genetic risk factors for Alzheimer’s disease are associated with multiple aspects of neuropathology
| Analytical model | Neuropathological variable |
APOE
|
PRS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of ε 2 alleles |
Number of ε 4 alleles |
|||||||||
| P-value | Coefficient | %VarExp | P-value | Coefficient | %VarExp | P-value | Coefficient | %VarExp | ||
| Model 1 | Braak stage tangle | 0.0877 | −0.357 | 0.958 | 4.16E−24 | 1.16 | 15.1 | |||
| Thal amyloid stage | 0.00333 | −0.562 | 1.54 | 3.96E−20 | 0.981 | 13.5 | ||||
| CERAD stage | 0.0224 | −0.329 | 1.99 | 1.03E−19 | 0.713 | 13.4 | ||||
| Braak Lewy body stage | 0.988 | −0.00439 | 0.0809 | 0.000264 | 0.555 | 2.59 | ||||
| TDP-43 | 0.859 | −0.0574 | 0.00821 | 0.00158 | 0.537 | 2.58 | ||||
| Model 2 | Braak stage tangle | 1.36E−06 | 3.4 | 0.354 | ||||||
| Thal amyloid stage | 0.00288 | 1.1 | 0.201 | |||||||
| CERAD stage | 5.27E−05 | 2.95 | 0.202 | |||||||
| Braak Lewy body stage | 0.267 | 0.167 | 0.105 | |||||||
| TDP-43 | 0.315 | 0.26 | 0.104 | |||||||
| Model 3 | Braak stage tangle | 0.0885 | −0.3505 | 0.9580 | 9.40E−24 | 1.132 | 15.119 | 4.97E−06 | 0.309 | 2.465 |
| CERAD stage | 0.0224 | −0.3254 | 1.9865 | 2.02E−19 | 0.700 | 13.402 | 1.30E−04 | 0.179 | 2.192 | |
Given that the two distinct molecular pathologies—tauopathy and β-amyloidosis—that define Alzheimer’s disease are highly correlated (Supplementary Fig. 3), we wanted to establish whether APOE or Alzheimer’s disease PRS had a specific (or primary) effect on a particular aspect of neuropathology. To this end, we repeated the analysis of how Alzheimer’s disease PRS and APOE influence pathology, sequentially controlling for other neuropathology variables. This analysis revealed some interesting patterns. First, after controlling for any of the other three quantitative neuropathological variables, Braak Lewy body stage was not significantly associated with APOE ε4 (Supplementary Table 2) suggesting that the association we detected was largely driven by the fact that individuals with Lewy bodies have also NFTs and β-amyloid plaques. Second, after we controlled for Braak NFT stage, neither of the plaque measures remained significantly associated with APOE ε4 (Supplementary Table 2). In contrast, Braak NFT stage remained significantly associated with APOE ε4 status after controlling for plaque variable (adjusted for Thal phase, mean difference per APOE ε4 allele = 0.468; P = 6.44 × 10−7; adjusted for neuritic plaque density, mean difference per ε4 allele = 0.238; P = 1.82 × 10−4), albeit with an attenuated magnitude of effect. Considering the two measures of plaque burden, only Thal β-amyloid phase remained significantly associated with ε4 after controlling for neuritic plaque density (mean difference per ε4 allele = 0.265; P = 3.42 × 10−4). Neither Braak NFT stage nor neuritic plaque density remained significantly associated with Alzheimer’s disease PRS after controlling for the other measure of pathology (Supplementary Table 2). These results indicate that APOE ε4 has a specific influence on tauopathy (NFTs) as well as a shared effect on both plaque and NFT development, whereas the PRS is more generally associated with an increased burden of Alzheimer’s disease neuropathology.
Association between APOE and cognitive performance is confounded by neuropathology
We determined clinical and cognitive status at death from the final pre-mortem assessment (Table 1). Data were available from 639 individuals who had had at least one CDR assessment with a mean final score of 1.79 (SD = 1.30) measured a mean of 353 days (SD = 374 days) prior to death. In addition, 469 individuals had had at least one MMSE assessment with a mean final score of 19.1 (SD = 10.3) measured a mean of 594 days (SD = 521 days) prior to death and 270 individuals had a MoCA assessment (mean = 16.1; SD = 11.0) measured a mean of 617 days (SD = 590 days) prior to death. APOE was significantly associated with dementia severity with each ε4 allele associated with an increase in 0.492 (P = 2.14 × 10−9) in pre-mortem CDR score (Table 4). APOE was also significantly associated with lower cognitive performance in MMSE prior to death (Table 4) with each ε4 allele being associated with a decrease in 4.86 (P = 1.30 × 10−8). In contrast, Alzheimer’s disease PRS was not significantly associated with any of the measures of clinical or cognitive status prior to death. To test whether the association between APOE and clinical measures was mediated by neuropathology we repeated these analyses including Braak NFT stage as an additional covariate; this variable had the largest effect in the genetic analyses described above, and its effect additionally captured associations with plaque pathology. In this model, the associations between APOE ε4 and CDR or MMSE were attenuated and neither remained significant (Supplementary Table 3). In contrast, on retesting Braak NFT stage whilst controlling for the clinical variables in turn, we observed that APOE ε4 remained significantly associated (Supplementary Table 3). This indicates that the association between APOE and clinical variables is a consequence of an increased burden of neuropathology.
Table 4.
APOE is associated with clinical and cognitive status at death
| Analytical model | Cognitive variable |
APOE
|
PRS |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of ε 2 alleles |
Number of ε 4 alleles |
|||||||||
| P-value | Coefficient | %VarExp | P-value | Coefficient | %VarExp | P-value | Coefficient | %VarExp | ||
| Model 1 | CDR | 0.706 | −0.058 | 0.336 | 2.14E−09 | 0.492 | 9.83 | |||
| MMSE | 0.693 | 0.574 | 0.310 | 1.30E−08 | −4.859 | 10.05 | ||||
| MoCA | 0.876 | −0.299 | 0.157 | 5.00E−03 | −3.403 | 3.19 | ||||
| Model 2 | CDR | 0.034 | 0.109 | 1.82 | ||||||
| MMSE | 0.025 | −1.136 | 2.03 | |||||||
| MoCA | 0.785 | 0.191 | 0.089 | |||||||
APOE ε4 is associated with faster cognitive decline in old age, but this is driven by Alzheimer’s disease neuropathological burden
Participants had a mean of 2.85 (SD = 1.71 visits) clinical assessment visits spread over a mean of 3.40 years (SD = 2.00 years) with a mean time between visits of 1.42 years (SD = 0.67 years). Over the course of all participants' involvement in the BDR study, there was an overall decline in clinical status and cognitive performance. On average the CDR increased by a mean of 0.139 per year (P = 2.02 × 10−31), while MMSE declined by a mean of 1.07 per year (P = 3.00 × 10−29). APOE genotype was associated with worse cognitive scores at the start of the study and faster rates of decline as the study progressed (Table 5). For every ε4 allele, MMSE score was 3.19 points lower (P = 4.92 × 10−5) at the start of the study, and individuals then accumulated an additional decrease in 0.803 in their score per allele per year (P = 1.58 × 10−8). In contrast, although APOE was associated with a higher CDR score at the start of the study (mean difference per ε4 allele = 0.468; P = 4.34 × 10−8), there was no significant difference in the change in clinical status related to APOE as the study progressed. There was no significant association with MoCA scores and APOE genotype. There was no significant association between Alzheimer’s disease PRS and longitudinal clinical or cognitive profiles or clinical or cognitive status at study entry. On repeating these analyses using the participant’s age rather than time in the study, we found no significant linear associations with either cognitive status at study entry or performance as the study progressed (Table 5).
Table 5.
APOE ε4 is associated with steeper cognitive decline prior to death
| Time variable | Cognitive variable | Time |
APOE
|
Interaction (time × APOE) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of ε2 alleles |
Number of ε4 alleles |
Time × ε2 |
Time × ε4 |
||||||||
| Coefficient | P-value | Coefficient | P-value | Coefficient | P-value | Coefficient | P-value | Coefficient | P-value | ||
| Time since study entry (days) | CDR | 2.78E−04 | 2.45E−08 | 0.075 | 0.642 | 0.468 | 4.34E−08 | −1.34E−04 | 0.121 | 1.28E−04 | 9.83E−03 |
| MMSE | −1.39E−03 | 7.50E−05 | 1.209 | 0.371 | −3.195 | 4.92E−05 | −3.73E−04 | 0.529 | −2.20E−03 | 1.58E−08 | |
| MoCA | −1.33E−03 | 0.146 | −0.663 | 0.710 | −2.335 | 0.040 | 1.98E−03 | 0.151 | −3.18E−03 | 0.024 | |
| Age (years) | CDR | 2.01E−03 | 0.797 | −0.058 | 0.965 | −0.868 | 0.216 | 4.99E−04 | 0.974 | 0.017 | 0.042 |
| MMSE | −0.258 | 2.20E−04 | 4.759 | 0.675 | 2.574 | 0.689 | −0.040 | 0.766 | −0.086 | 0.268 | |
| MoCA | 0.011 | 0.904 | −4.510 | 0.783 | −5.281 | 0.616 | 5.21E−02 | 0.785 | 0.031 | 0.809 | |
Given our previous observation that genetic associations with clinical status and cognition are mediated by neuropathology, we wanted to confirm whether the longitudinal analyses were similarly affected. First, we tested whether change in clinical status was associated with neuropathology measured by Braak NFT stage, independent of genetic status (Supplementary Table 5). As expected, those with higher levels of tangle pathology at death had a more severe clinical rating, even at the start of the study (mean difference in CDR per Braak NFT stage = 0.355; P = 7.30 × 10−42) and declined quicker; each additional Braak NFT stage was associated with an additional increase in 0.0247 in CDR per year (P = 3.99 × 10−5). We observed similar results for cognitive performance measured by MMSE; at study entry, each additional Braak NFT stage was associated with a decrease in 2.58 in MMSE score (P = 7.27 × 10−26) and participants accumulated an additional decrease in 0.384 in MMSE per Braak NFT stage per year (P = 3.90 × 10−15). Repeating the APOE analysis with a covariate for the potential confounder of neuropathology found that in line with the cross-sectional analyses, the associations with both clinical severity and cognition were no longer significant after adjusting for Braak NFT stage (Supplementary Table 6). These results suggest that cognitive performance prior to death, and even many years before death, is a consequence of accumulating Alzheimer’s disease neuropathology.
Discussion
In this study, we used the longitudinal cognitive and neuropathological assessment data in the BDR cohort to investigate how genetic risk factors for Alzheimer’s disease influence the accumulation of β-amyloid plaques, tauopathy, synucleinopathy and TDP-43 proteinopathy, and progressive decline in clinical status and cognitive performance. Our results indicate that APOE ε4 status has the most dramatic influence on tauopathy (NFT burden) and that although APOE genotype is also associated with β-amyloidosis, synucleinopathy and cognition, these relationships are largely confounded by their correlation with tangle burden. Furthermore, our results indicate that APOE has a specific direct effect on NFT independent of other neuropathologies. Although this finding contradicts the predictions of the ‘amyloid cascade hypothesis’ in which tau tangle formation is considered secondary to β-amyloid pathology (Hardy and Allsop, 1991; Selkoe, 1991), it is consistent with careful neuropathologic studies that show that tauopathy can precede beta-amyloidosis, at least in some brain areas (Duyckaerts, 2011). Our results also agree with previous research showing that although the influence of APOE on tau tangles is largely mediated indirectly through neurobiological pathways associated with β-amyloid, approximately one-third of its influence on tangle development is via an alternative non-amyloid pathway (Yu et al., 2014). Our findings also support the 2-process model proposed by Mungas et al. (2014), according to which neocortical NFTs are mediated by β-amyloid deposition and medial temporal lobe NFTs and may be the consequence of a separate age-associated process.
In our analysis of pathologies that frequently co-occur with the accumulation of β-amyloid and tauopathy, we replicated the positive association between Lewy body burden and the APOE ε4 allele (Tsuang et al., 2013; Beecham et al., 2014). However, when we adjusted for either β-amyloid or NFTs, this association was attenuated, indicating that in our sample, the association may be a consequence of the higher levels of tau and β-amyloid in individuals with Lewy bodies. It should be noted that the majority of participants in our study were free of any Lewy body pathology, with 423 individuals (70.8%) having a Braak Lewy body stage of 0. Therefore these analyses may be underpowered, particularly in the context of disentangling the effects on multiple correlated neuropathology variables. In addition, we were not able to replicate associations between APOE genotype and the presence of TDP-43 proteinopathy (Josephs et al., 2014; Yang et al., 2018), although the direction of effect was consistent with previous reports. Although TDP-43 proteinopathy was not infrequent in the BDR cohort, with 22.8% participants classed as positive, our simple binary classification may have decreased our power to detect an effect. Although BDR is not limited to a particular dementia subtype, and includes unaffected controls, Alzheimer’s disease is the most common form of dementia and therefore the sample is enriched for NFT and β-amyloid pathology. To truly establish whether APOE genotype has an independent, direct effect on the common comorbidities associated with Alzheimer’s disease, such as Lewy bodies and TDP-43 proteinopathy, we will likely require a larger number of samples to detect residual effects after accounting for correlations between neuropathological variables.
As well as our examination of associations with APOE, we tested the cumulative effect of common Alzheimer’s disease-associated genetic variants on neuropathology, clinical status and cognition. Given that individual variants only confer a small amount of additional risk, we used a combined PRS to improve power. Although Alzheimer’s disease PRS was associated with both tauopathy (NFTs) and β-amyloidosis, there was no evidence of independent effects on either, suggesting that, in combination, common genetic variants have a broader, more general effect on the neuropathological burden present in Alzheimer’s disease. This contrasts with findings from a previous study testing the consequences of an Alzheimer’s disease PRS without APOE, which only reported a significant association with NFTs and not β-amyloid plaques (Felsky et al., 2018). Of note, in that study the PRS was based on an older GWAS with fewer significant association signals, and therefore our study might highlight the additional power derived using variants from the latest GWAS for Alzheimer’s disease. While leveraging multiple genetic variants into a single PRS is a powerful approach, particularly where sample sizes are small, it can be challenging to interpret shared associations. As the PRS is a harmonized variable generated in our case from seventeen genetic variants, our results could be explained by different subsets of variants being causally associated with the distinct pathologies. This explanation fits with results from previous studies that have tested individual SNPs associated with Alzheimer’s disease against multiple measures of neuropathology reporting some variants having specific effects, while others were associated with multiple aspects (Beecham et al., 2014; Mäkelä et al., 2018). Furthermore, it is likely that some genetic risk factors do not act via either plaques or tauopathy (NFTs), possibly affecting other aspects of neuropathology such as vascular disease which was not included in this study.
We found that clinical and cognitive status at study recruitment and prior to death, in addition to decline over the course of the study, are not directly associated with APOE genotype but are likely to be a consequence of neuropathological burden and in particular the accumulation of NFTs. This concurs with results from a previous study in a slightly larger cohort that focused specifically on episodic memory and non-episodic cognition (Yu et al., 2014). Alzheimer’s disease-associated cognitive decline is hypothesized to start as much as 17 years prior to death, with the rate of decline fastest in those with the most extensive neuropathology; tauopathy, β-amyloidosis, TDP-43 proteinopathy and synucleinopathy are all positively associated with decline (Boyle et al., 2017). While a strength of our study is the availability of longitudinal cognitive data, clinical data was only available for up to three years before death, limiting our ability to characterize the effects of neuropathology on cognitive trajectories. Furthermore, multiple aspects of neuropathology have been independently negatively associated with cognitive performance (Boyle et al., 2013). Although Alzheimer’s disease is characterized by β-amyloidosis and tauopathy, it is increasingly apparent that in older cohorts, there may be additional comorbidities which potentially confound this relationship (Schneider et al., 2009; James et al., 2012, 2016; Robinson et al., 2018). At present, the presence of multiple comorbidities makes it difficult to resolve cause from effect as each comorbidity may affect different domains of cognition at different times during pathogenesis. When considering the regional presence and global burden of different pathologies, there is extensive variation in the specific combination of neuropathological features that an individual develops ultimately having a unique effect on their individual cognitive performance over time (Boyle et al., 2018). The strengths of the BDR study design, collating repeated measures of cognitive performance in addition to standardized protocols for high quality neuropathological assessments in a large sample size make it an ideal dataset to ultimately disentangle the role of mixed pathologies on cognition and dementia and more extensive analyses will be possible in the future.
Our results should be considered in light of a number of limitations. First, the participants were self-selecting, which in line with many other observational cohorts introduces bias into the sample; they are from less deprived socio-economic areas and have higher levels of education than the general population. Second, consistent with the majority of genetic studies, our analysis was limited to participants of European ancestry to remove the biases associated with population stratification. Third, we only included a subset of Alzheimer’s disease and related neuropathology phenotypes, which were selected for practical reasons in that they were observed with sufficient frequency in the current sample. Analyses of rarer phenotypes will be possible with subsequent waves of the data as the overall sample size and number of cases increases. Fourth, our measures were of global cognition, rather than specific domains. As previous studies have found that different pathologies have specific effects of different cognitive domains (Yu et al., 2014), this may mean we miss some of the nuances of the relationship between neuropathology and cognition. Fifth, to aid interpretation of the analytical models we converted semi-quantitative neuropathological variables into continuous variables which assume an equal effect between all pairs of consecutive stages. This simplification may obscure some more complex patterns in the data but should enable us to pick up general correlations which were our primary interest. Finally, we did not control for severity of ischaemic brain damage or any vascular risk factors, which are common in Alzheimer’s disease cases and negatively influence cognition.
In summary, our data indicate that APOE influences Alzheimer’s disease neuropathology via two independent pathways, one where β-amyloid accumulation correlates with the development of tauopathy (NFTs), and a second pathway with direct effects on NFTs independent of β-amyloidosis. It is as a consequence of these neuropathological changes that cognitive performance is then impaired. The relationship between common genetic variants associated with Alzheimer’s disease and neuropathology is more complex, with each individual variant potentially having a different effect on neuropathology and cognition. Taken together, these results provide insights into how the symptoms of Alzheimer’s disease dementia manifest and how genetic risk factors influence the development of pathology.
Supplementary Material
Acknowledgements
We would like to gratefully acknowledge all donors and their families for the tissue provided for this study. Human post-mortem tissue was obtained from the South West Dementia Brain Bank, London Neurodegenerative Diseases Brain Bank, Manchester Brain Bank, Newcastle Brain Tissue Resource and Oxford Brain Bank, members of the Brains for Dementia Research (BDR) Network. We wish to acknowledge the neuropathologists at each centre and BDR Brain Bank staff for the collection and classification of the samples.
Funding
E.H. and J.M. were supported by Medical Research Council grant K013807. G.S. was supported by a PhD studentship from the Alzheimer’s Society. The BDR is jointly funded by Alzheimer's Research UK and the Alzheimer's Society in association with the Medical Research Council. The South West Dementia Brain Bank is part of the Brains for Dementia Research program, jointly funded by Alzheimer's Research UK and Alzheimer's Society, and is also supported by BRACE (Bristol Research into Alzheimer's and Care of the Elderly) and the Medical Research Council.
Supplementary material
Supplementary material is available at Brain Communications online.
Competing interests
The authors report no competing interests.
Glossary
- BDR =
Brains for Dementia Research;
- CDR =
clinical dementia rating;
- CERAD =
Consortium to Establish a Registry for Alzheimer’s disease;
- GWAS =
genome-wide association studies;
- MMSE =
Mini-Mental State Examination;
- MoCA =
Montreal Cognitive Assessment;
- NFT =
neurofibrillary tangle;
- PRS =
polygenic risk score;
- SD =
standard deviation;
- SNP =
single nucleotide polymorphism;
- TDP-43 =
transactive response DNA-binding protein 43
Contributor Information
Eilis Hannon, College of Medicine and Health, University of Exeter, Exeter, Devon, EX2 5DW, UK.
Gemma L Shireby, College of Medicine and Health, University of Exeter, Exeter, Devon, EX2 5DW, UK.
Keeley Brookes, School of Science & Technology, Nottingham Trent University, Nottingham, NG11 8NF, UK.
Johannes Attems, Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK.
Rebecca Sims, Division of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, CF24 4HQ, UK.
Nigel J Cairns, College of Medicine and Health, University of Exeter, Exeter, Devon, EX2 5DW, UK.
Seth Love, Bristol Medical School (THS), University of Bristol, Bristol, BS2 8DZ, UK.
Alan J Thomas, Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK.
Kevin Morgan, Human Genetics Group, School of Life Sciences, University of Nottingham, Nottingham, NG7 2RD, UK.
Paul T Francis, College of Medicine and Health, University of Exeter, Exeter, Devon, EX2 5DW, UK; Wolfson Centre for Age-Related Diseases, King's College London, London, SE1 1UL, UK.
Jonathan Mill, College of Medicine and Health, University of Exeter, Exeter, Devon, EX2 5DW, UK.
References
- Adams HH, de Bruijn RF, Hofman A, Uitterlinden AG, van Duijn CM, Vernooij MW, et al. Genetic risk of neurodegenerative diseases is associated with mild cognitive impairment and conversion to dementia. Alzheimers Dement 2015; 11: 1277–85. [DOI] [PubMed] [Google Scholar]
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol 2007; 61: 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet 2014; 10: e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE, Alafuzoff I, Al-Sarraj S, Arzberger T, Bogdanovic N, Budka H, et al. Management of a twenty-first century brain bank: experience in the BrainNet Europe consortium. Acta Neuropathol 2008; 115: 497–507. [DOI] [PubMed] [Google Scholar]
- Blauwendraat C, Faghri F, Pihlstrom L, Geiger JT, Elbaz A, Lesage S, et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol Aging 2017; 57: 247.e9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol 2013; 74: 478–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yang J, Yu L, Leurgans SE, Capuano AW, Schneider JA, et al. Varied effects of age-related neuropathologies on the trajectory of late life cognitive decline. Brain 2017; 140: 804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018; 83: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24: 197–211. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 2015; 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, Brookes KJ, Patel T, Fallows A, Guetta-Baranes T, Turton JC, et al. Alzheimer's disease polygenic risk score as a predictor of conversion from mild-cognitive impairment. Transl Psychiatry 2019; 9: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, O'Reilly PF. PRSice-2: polygenic Risk Score software for biobank-scale data. GigaScience 2019; 8: giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993; 261: 921–3. [DOI] [PubMed] [Google Scholar]
- Corlier F, Hafzalla G, Faskowitz J, Kuller LH, Becker JT, Lopez OL, et al. Systemic inflammation as a predictor of brain aging: contributions of physical activity, metabolic risk, and genetic risk. NeuroImage 2018; 172: 118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Del-Aguila JL, Saef B, Black K, Fernandez MV, Budde J, et al. Polygenic risk score of sporadic late-onset Alzheimer's disease reveals a shared architecture with the familial and early-onset forms. Alzheimers Dement 2018; 14: 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet 2016; 48: 1284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med 2017; 14: e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C. Tau pathology in children and young adults: can you still be unconditionally baptist? Acta Neuropathol 2011; 121: 145–7. [DOI] [PubMed] [Google Scholar]
- Escott-Price V, Myers AJ, Huentelman M, Hardy J. Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann Neurol 2017; 82: 311–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Myers AJ, Huentelman M, Shoai M, Hardy J. Polygenic risk score analysis of Alzheimer's disease in cases without APOE4 or APOE2 alleles. J Prev Alzheimers Dis 2019; 6: 16–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain 2015; 138: 3673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA 1997; 278: 1349–56. [PubMed] [Google Scholar]
- Felsky D, Patrick E, Schneider JA, Mostafavi S, Gaiteri C, Patsopoulos N, et al. Polygenic analysis of inflammatory disease variants and effects on microglia in the aging brain. Mol Neurodegen 2018; 13: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–98. [DOI] [PubMed] [Google Scholar]
- Francis PT, Costello H, Hayes GM. Brains for Dementia Research: evolution in a longitudinal brain donation cohort to maximize current and future value. JAD 2018; 66: 1635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006; 63: 168–74. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 1991; 12: 383–8. [DOI] [PubMed] [Google Scholar]
- James BD, Bennett DA, Boyle PA, Leurgans S, Schneider JA. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA 2012; 307: 1798–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain 2016; 139: 2983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 2019; 51: 404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol 2014; 127: 811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019; 51: 414–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45: 1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016; 48: 1443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louwersheimer E, Wolfsgruber S, Espinosa A, Lacour A, Heilmann-Heimbach S, Alegret M, et al. Alzheimer's disease risk variants modulate endophenotypes in mild cognitive impairment. Alzheimers Dement 2016; 12: 872–81. [DOI] [PubMed] [Google Scholar]
- Lupton MK, Strike L, Hansell NK, Wen W, Mather KA, Armstrong NJ, et al. The effect of increased genetic risk for Alzheimer's disease on hippocampal and amygdala volume. Neurobiol Aging 2016; 40: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä M, Kaivola K, Valori M, Paetau A, Polvikoski T, Singleton AB, et al. Alzheimer risk loci and associated neuropathology in a population-based study (Vantaa 85+). Neurol Genet 2018; 4: e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Campbell A, Hagenaars SP, Nagy R, Amador C, Hayward C, et al. Genetic stratification to identify risk groups for Alzheimer's disease. JAD 2017; 57: 275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry 2018; 8: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiskainen H, Helisalmi S, Viswanathan J, Kurki M, Hall A, Herukka SK, et al. Effects of Alzheimer's disease-associated risk loci on cerebrospinal fluid biomarkers and disease progression: a polygenic risk score approach. JAD 2014; 43: 565–73. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479–86. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Sperling RA, Holmes AJ, Buckner RL, De Jager PL, Smoller JW, et al. Polygenic risk of Alzheimer disease is associated with early- and late-life processes. Neurology 2016; 87: 481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993; 43: 2412–14. [DOI] [PubMed] [Google Scholar]
- Mungas D, Tractenberg R, Schneider JA, Crane PK, Bennett DA. A 2-process model for neuropathology of Alzheimer's disease. Neurobiol Aging 2014; 35: 301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Yu L, Wilson RS, Chen EY, Bennett DA, Schneider JA. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of Alzheimer’s disease or FTLD. Neurology 2017; 88: 653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasreddine ZS, Phillips NA, Bã©Dirian VéR, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005; 53: 695–9. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019; 142: 1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun 2020; 11: 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018; 141: 2181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabuncu MR, Buckner RL, Smoller JW, Lee PH, Fischl B, Sperling RA. The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb Cortex 2012; 22: 2653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009; 66: 200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron 1991; 6: 487–98. [DOI] [PubMed] [Google Scholar]
- Tasaki S, Gaiteri C, Mostafavi S, De Jager PL, Bennett DA. The molecular and neuropathological consequences of genetic risk for Alzheimer's dementia. Front Neurosci 2018; 12: 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Cairns NJ, Da X, Chen K, Carter D, Fleisher A, et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun 2013; 1: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 2013; 70: 223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 2008; 67: 555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Yu L, Trojanowski JQ, Chen EY, Boyle PA, Bennett DA, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 2013; 70: 1418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Yu L, White CC, Chibnik LB, Chhatwal JP, Sperling RA, et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: a community-based cohort study. Lancet Neurol 2018; 17: 773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Boyle PA, Leurgans S, Schneider JA, Bennett DA. Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol Aging 2014; 35: 819–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genetic, clinical and cognitive data are available through the Dementia’s Platform UK (DPUK; https://www.dementiasplatform.uk/) platform upon application.