

Abstract
Chemosensation is the most ubiquitous sense in animals, enacted by the products of complex gene families that detect environmental chemical cues and larger-scale sensory structures that process these cues. While there is a general conception that olfactory receptor (OR) genes evolve rapidly, the universality of this phenomenon across vertebrates, and its magnitude, are unclear. The supposed correlation between molecular rates of chemosensory evolution and phenotypic diversity of chemosensory systems is largely untested. We combine comparative genomics and sensory morphology to test whether OR genes and olfactory phenotypic traits evolve at faster rates than other genes or traits. Using published genomes, we identified ORs in 21 tetrapods, including amphibians, reptiles, birds, and mammals and compared their rates of evolution to those of orthologous non-OR protein-coding genes. We found that, for all clades investigated, most OR genes evolve nearly an order of magnitude faster than other protein-coding genes, with many OR genes showing signatures of diversifying selection across nearly all taxa in this study. This rapid rate of evolution suggests that chemoreceptor genes are in “evolutionary overdrive,” perhaps evolving in response to the ever-changing chemical space of the environment. To obtain complementary morphological data, we stained whole fixed specimens with iodine, µCT-scanned the specimens, and digitally segmented chemosensory and nonchemosensory brain regions. We then estimated phenotypic variation within traits and among tetrapods. While we found considerable variation in chemosensory structures, they were no more diverse than nonchemosensory regions. We suggest chemoreceptor genes evolve quickly in reflection of an ever-changing chemical space, whereas chemosensory phenotypes and processing regions are more conserved because they use a standardized or constrained architecture to receive and process a range of chemical cues.
Keywords: chemosensation, diversifying selection, olfaction, olfactory bulb, olfactory receptor, tetrapod
Every organism must preserve its existence and improve its fitness by first perceiving and then reacting to its surroundings. Natural selection fine-tunes sensory systems to identify relevant cues for survival and reproduction, and to ignore other signals that may interfere. No array of environmental signals is more “tangled” than the chemical landscape of the natural world. Chemical signals are affected by numerous biotic and abiotic factors (Rouyar et al. 2011; Yohe and Brand 2018). The vast diversity of odorant molecules on the planet explains why chemosensation is so ubiquitous and so important to animals (indeed, to all life forms). However, the overwhelming complexity of chemical backgrounds, chemical signals, and chemoreceptors causes olfaction to remain one of the most poorly understood senses (Hayden and Teeling 2014). Detecting adaptive signatures and gaining a functional understanding of the molecular and phenotypic basis of chemosensation are monumental challenges in comparative biology. With the advent of improved sequencing approaches and morphological imaging techniques, exploration of the convoluted “tangled bank” of chemosensory diversity is becoming tractable.
Chemosensation in the tetrapod nose is performed by two systems: the main olfactory and the accessory vomeronasal. The main olfactory system is considered to be devoted primarily to the detection of chemical cues related to diet and environment (Jørgensen 2000; Fleischer et al. 2018), while vomeronasal olfaction is associated with social chemical signaling (Liberles 2014; Stowers and Kuo 2015). The evidence for this distinction, however, comes overwhelmingly from the mammalian literature (Van Valkenburgh et al. 2014). It has become increasingly clear that these systems are not mutually exclusive (Suárez et al. 2012). The neural signaling detection mechanisms of both systems are similar: a chemical odorant molecule binds to a single-receptor-expressing neuron, triggers depolarization, and sends a signal to converge with others in the olfactory bulb, where integration and interpretation occur (Bear et al. 2016). The two systems mainly differ in the chemoreceptors they express. The primary receptors in the main olfaction are encoded by genes within the Olfactory Receptor (OR) and trace-amine associated receptor (TAAR) gene families and those in vomeronasal olfaction by vomeronasal-specific receptors (e.g., V1Rs or V2Rs) (Grus 2008; Brykczynska et al. 2013; Bear et al. 2016; Eyun et al. 2016). The focus of this study is on OR genes, owing to their homology across tetrapods (and beyond), but also because many of our findings are relevant to all chemoreceptor families.
Irrespective of gene family, chemoreceptors are encoded by genes that evolve via birth–death evolution (Nei and Rooney 2005). This process is one in which genes are constantly tandemly duplicating through time. Duplicate genes accumulate novel mutations that lead either to pseudogenization (lost function) or neofunctionalization (new function) (Pegueroles et al. 2013; Yohe, Liu, et al. 2019). In the case of chemosensory receptor genes, many duplicated receptors that accumulate neofunctional mutations evolve via diversifying selection, in which new mutations lead to new odorant detection profiles (Nei et al. 2008). The birth–death process is exceptional in ORs—duplications have led to hundreds, and sometimes thousands, of accumulated copies (Niimura 2009, 2012; Niimura et al. 2014). ORs are multigene family members that encode G-protein-coupled receptors expressed in chemosensory epithelial tissue to detect odorant molecules from the environment. ORs are short, intronless ∼900 base pair (bp) genes that evolve primarily by tandem gene duplication (Young and Trask 2002; Young et al. 2002). They are the largest gene family in the mammalian genome and compose ∼5% of the protein-coding genes (Nei et al. 2008). While most OR work has been performed on mammals (Hayden et al. 2010; Niimura et al. 2014), Classes I and II receptor genes are also present in reptiles (Brykczynska et al. 2013), birds (Steiger et al. 2008; Khan et al. 2015), and amphibians (Ji et al. 2009), as well as some fish (Zhang and Firestein 2009; Bear et al. 2016). Identifying the ligand of receptors is notoriously difficult, as the relationship of odorant to receptor is not one-to-one, and activation of odor recognition is combinatorial (Malnic et al. 1999). As a result, only a handful of receptors have had their ligands identified, and evidence is almost exclusive to model organisms (Nara et al. 2011).
The morphological structure devoted to making sense of the labyrinth of thousands of the OR-expressing sensory neurons is the olfactory bulb. It does so with astounding precision (Zou et al. 2009). In the rostral most portion of the forebrain, the olfactory bulb is organized into spheroid synaptic concentrations called glomeruli. All neurons expressing the same OR (Monahan and Lomvardas 2015), no matter the distances among them in the epithelia, will coalesce into the same glomerulus (Zou et al. 2009). In other words, hundreds of thousands of neurons deliver their sensory input into mere hundreds of cells. The number of glomeruli is correlated with the number of distinct receptors (Bressel et al. 2016), and their expression varies synchronously throughout ontogeny (Hinds and McNelly 1981). The parallels are even documented in bone, such that the number of foramina through which these bundles of olfactory sensory neurons are threaded correlate with OR repertoire size in mammals (Bird et al. 2018).
Both the molecular and morphological effectors of olfactory detection are observed from cyclostomes to mammals (Baier and Korsching 1994; Saraiva et al. 2015), suggesting over 450 million years of evolutionary conservation. Simultaneously, the individual OR genes are some of the fastest evolving within the genome. However, because the number of odor-encoding cells in the olfactory bulb is inherently governed by the receptors that their receiving sensory neurons express, we might expect synchronous patterns of evolution throughout tetrapod diversification. Here we compare the rates of evolution of OR gene repertoires and the morphological disparity of different brain regions in every major tetrapod clade. We aim in particular to quantify evolutionary rates of OR genes beyond mammals to test whether rapid evolution of OR genes is a ubiquitous tetrapod phenomenon. We compare these results to phenotypic evolution of sensory brain regions to test whether olfactory bulb morphology is evolving differently from that of other brain regions and whether olfactory bulb disparity is greater than that expected under Brownian motion. Given the tight link between OR gene repertoire and cellular morphology of the olfactory bulb, it might be predicted that rates of OR gene evolution will show significantly higher diversity than non-OR genes, and morphological disparity of the olfactory bulb should be significantly greater than that of other brain regions throughout tetrapod evolution.
Materials and Methods
Approach
We compared the ORs from 21 taxa for which genomes and soft-tissue µCT-scans were available. From the genomes, we identified ORs and chemosensory-related genes. To test whether OR genes were evolving faster than nonolfactory genes on the one hand and simulated genes on the other, we inferred gene trees and estimated rates of evolution for (1) each subfamily of ORs across species including all duplicate copies; (2) orthologs of non-OR genes; and (3) simulated genes evolved under different selection pressures across the tetrapod tree. We then used soft tissue µCT-scans of complementary specimens and reconstructed brain regions associated with sensory and nonsensory function. We modeled the disparity of the structures through time to observe whether their patterns of change were significantly different from those expected under Brownian motion.
Genome data
The following genomes were sampled: axolotl (Ambystoma mexicanum: GCA_002915635.2), 2-lined caecilian (Rhinatrema bivittatum: GCF_901001135.1), common snapping turtle (Chelydra serpentina: GCA_007922165.1), American alligator (Alligator mississippiensis: GCF_000281125.3_ASM28112v4), saltwater crocodile (Crocodylus porosus: GCF_001723895.1), Chilean tinamou (Nothoprocta perdicaria: GCF_003342845.1), greater rhea (Rhea americana: GCA_003343005.1), red jungle fowl (Gallus gallus: GCF_000002315.6), Japanese quail (Coturnix japonica: GCF_001577835.1), great cormorant (Phalacrocorax carbo: GCF_000708925.1_ASM70892v1), Okinawa rail (Gallirallus okinawae: GCA_002003005.1), zebra finch (Taeniopygia guttata: GCF_003957565.1), tuatara (Sphenodon punctatus: GCA_003113815.1_ASM311381v1), red corn snake (Pantherophis guttatus: GCA_001185365.1), green anole (Anolis carolinensis: GCF_000090745.1), ocelot gecko (Paroedura picta: GCA_003118565.1), platypus (Ornithorhynchus anatinus: GCF_004115215.1), gray short-tailed opossum (Monodelphis domestica: GCF_000002295.2), house mouse (Mus musculus: GCF_000001635.26), and common vampire bat (Desmodus rotundus: GCA_002940915.2).
OR identification
While there are several chemosensory gene families present in the genome, the OR Classes I and II genes are well known to be shared across vertebrates (Young and Trask 2002; Hayden et al. 2010) and are the focus of this study. Protein sequences of ORs from mammals (Hayden et al. 2010), as well as published sequences from squamates and birds (Steiger et al. 2008, 2009; Khan et al. 2015) were used as queries and were blasted using the protein query-translated subject BLAST version 2.10.0+ (tblastn) program (Altschul et al. 1990; Gerts et al. 2006). Hits >100 bps and a score of at least 0.2 were converted to a gff format using the blast2gff.py script within the genomeGTFtools toolkit (Mills et al. 2018). Hits were then pulled out of the genome using the getfasta program within bedtools version 2.29.2 (Quinlan 2014). Duplicate hits and containments were removed using the dedupe.sh script within BBTools bioinformatics suite (https://sourceforge.net/projects/bbmap/). Identified receptor genes were then annotated according to previously published methodology (Yohe et al. 2019). Genes with <650 bp open reading frames or genes with premature stop codons were filtered as pseudogenes. Within each of these two families are several subfamilies that receptors are binned in based on their conserved motifs of their protein-coding regions. Class I genes can be classified as either OR51, OR52, OR55, or OR56, and Class II genes can be classified as either OR1/3/7, OR2/13, OR4, OR5/8/9, OR6, OR10, OR11, OR12, or OR14. In brief, receptors were identified using the ORA version 1.9.1 (Hayden et al. 2010), a Bioperl (version 1.6.924) program that makes profile motifs of aligned amino acid sequences collected from previously published ORs. It implements HMMER version 3.1b (Eddy 2011), which uses hidden Markov models to identify and classify the gene sequences with significant hits to the sequence motif profiles. This pipeline is one of the 2 major ways that OR sequences are identified in the genome, has been used in many studies, and has been shown to be robust against false positives (Hayden et al. 2010).
Non-olfactory gene identification
To compare rates of evolution of other parts of the protein-coding genome to those of ORs, we selected 50 random loci to identify across our targeted set of species. Random genes were selected from the random gene set generator of the M. musculus genome (http://www.molbiotools.com/randomgenesetgenerator.html). If the gene was present as an ortholog in amphibians, birds, and reptiles, then we identified the corresponding RefSeq from the NCBI Ortholog catalog. Non-OR genes included a selection of kinases, opsins, transcription factors, among others. A list of the selected gene subset is listed in the Supplementary Table S1. Open reading frames were identified using the getorf function of EMBOSS version 6.6.0.0 (Rice et al. 2000).
Alignment and phylogenetic inference
For each olfactory subfamily and non-OR gene set, genes were aligned using transAlign (Bininda-Emonds 2005) that implements the FFT-NS-2 algorithm MAFFT version 7.388 for the protein alignments (Katoh and Standley 2013). A BLOSUM62 matrix was used with a gap open penalty of 1.53 and an offset value of 0.123. Sequences were inspected for misalignment and stop codons were removed from the OR alignments. ModelOMatic version 1.01 (Whelan et al. 2015) was used to estimate the best-fit codon model and nucleotide model. Gene trees were inferred using IQ-TREE version 1.6.11 (Nguyen et al. 2015).
Species tree
An ultrametric species tree was essential for the molecular evolution simulations and for the phenotype analyses. A species tree was grafted from several published phylogenetic trees. Using first a general vertebrate tree (Uyeda et al. 2017) as a backbone, an amphibian tree (Jetz and Pyron 2018), a squamate tree (Tonini et al. 2016), a turtle tree (Pereira et al. 2017) that contained a grafted bird tree (Kimball et al. 2019) and both A. mississippiensis and C. porosus binded on at divergence times from timetree.org (Kumar et al. 2017), and a mammal tree (Upham et al. 2019) were carefully grafted using the ape version 5.3 (Paradis et al. 2004) package in R version 3.6.1 (Team RC 2016). The sumtrees.py script implemented through DendroPy version 4.0.3 (Sukumaran and Holder 2010) was used to summarize the distribution of trees from vertlife.org. The tree was trimmed to match the data using the treedata() function in geiger version 2.0.6.2 (Harmon et al. 2008).
Molecular simulations
To make predictions for genes evolving under purifying or diversifying selection for this set of taxa, we simulated genes mimicking the codon frequencies and sequence length of ORs. In molecular evolution at the species level (i.e., assuming the substitution is fixed within the lineage), the strength of selection is characterized by the ω statistic, representing the ratio of rates of codon substitutions to nonamino acid-changing nucleotide substitutions (Mugal et al. 2014). Because codon substitutions change the amino acid and may have functional implications to protein function, under purifying selection, the rate of codon substitution is expected to be low (ω ≪ 1) relative to nucleotide substitutions that do not change the protein. Under diversifying or positive selection, selectively advantageous protein-coding changes may rapidly fix (ω > 1). Using the evolverNSbranchsites evolver program in paml version 4.8 (Yang 2007), we simulated 100 alignments of 900 bp each and “evolved” each sequence along the grafted species tree using codon frequencies and the estimated transition/transversion ratio (2.74592) observed from the OR56 gene family. Three evolutionary scenarios were modeled: (1) genes under strong purifying selection, in which the entire alignment (i.e., single-site class) is evolving at a rate of ω = 0.01, where the rates of codon-changing substitutions are much lower than synonymous substitutions; (2) genes under weak purifying selection, in which the entire alignment is evolving at a rate of ω = 0.1, where the rates of codon-changing substitutions are an order of magnitude higher than Scenario 1; and genes evolving under diversifying selection (i.e., positive selection), in which the 99% of the alignment evolves under strong purifying selection (ω = 0.01), and 1% of the codon sites are under positive selection (ω = 2.5). ω is a useful statistic at long time scales, but as divergence time approaches ∞, ω values approach zero (Mugal et al. 2014). Thus, at our long time scales (∼400 million years), we inferred the branch lengths of a codon substitution gene tree and compared these lengths to those of a nucleotide substitution gene tree, which contains both codon and non amino acid-changing substitutions. These values were calculated for both simulations and empirical data to provide comparable measures of evolutionary change for both simulations and observed data. A codon substitution gene tree and a nucleotide substitution gene tree were estimated using IQ-TREE under Codon+F3X4 and GTR+gamma models, respectively, for all simulations. Cumulative branch lengths were read into R and plotted with the empirical data.
Comparisons of rates of molecular evolution
To determine whether ORs were evolving at faster rates than non-OR genes and to gauge the comparable strength of selection observed from the simulations, we compared rates of nucleotide substitution to rates of codon substitution. To quantify the rates of nucleotide and codon substitution, we measured the total amount of accumulated change per gene for each of the 2 tree types by quantifying the cumulative branch lengths of each gene. To calculate the cumulative branch lengths, the diagonals of the eigenvectors of the variance–covariance matrix of each tree were quantified, which measures the height of each node (i.e., gene) that represents the rate of substitution per gene (Yohe and Dávalos 2018). These values were extracted for all observed genes and gene trees inferred from the simulated alignments. To understand the strength of selection for each of the observed groups, we tested whether slopes differed among the observed ORs (Classes I and II), non-OR genes, and the three simulation scenarios. If the slopes did not significantly differ, then that particular evolutionary scenario was determined to be a good fit for a majority of the genes within that class. To quantify differences among rates observed for we used a linear regression formula to quantify the slope of the following model:
where codon is the codon gene tree branch length and nucleotide is the nucleotide gene tree branch length for each gene (or simulated gene) i. We tested for whether there was a difference in slopes among j “classes” using class as the interaction term. Classes included Class I ORs, Class II ORs, non-OR genes, simulated genes under strong purifying selection, weak purifying selection, and diversifying selection (j = 6). Models were implemented using the lsmeans package in R (Lenth 2016). P-values were adjusted for multiple comparisons using the Tukey test. Candidate genes under diversifying selection were determined if the ratio of the branch lengths of the codon gene trees to the branch lengths of the nucleotide gene trees fell within 1 standard deviation of the mean of the ratios obtained from the gene trees inferred from diversifying selection simulations.
Specimen staining and soft tissue µCT-scanning
Most taxa in our dataset were museum specimens fixed in 10% formalin and stored in 70% ethanol. For new soft-tissue data generated within this study, specimens were stained in solutions of either 5% phosphomolybdic acid (H3PMo12O40, a.k.a., “PMA”) or 10% I2KI (Lugol’s Iodine) for 2–52 weeks, depending on the stain and specimen size. Specimens were scanned using the ultra-high-resolution Nikon H225 ST µCT-scanner at Yale University or the X-Tek HMXST Micro-CT imaging system at Harvard University. Additional µCT-scans of soft tissue were obtained from MorphoSource or from previously published studies (Fabbri et al. 2017). Supplementary Table S2 lists all specimens, details for each specimen, stains used for the respective specimen, and parameters under which the specimen was scanned. Exact species matches to their respective genome were available for each taxon, with the exception of the Okinawa rail, tinamou, and the great cormorant. As substitutes, we used Nothoprocta pentlandii (Andean tinamou), Porzana carolina (sora), and Phalacrocorax auritus (double-crested cormorant), respectively. Raw scan data were reconstructed using on-site Nikon reconstruction software and imported into VGStudio Max version 3.3 for segmentation (Volume Graphics GmbH 2014). For each scan, the following brain and sensory structures were segmented (Figure 1): olfactory bulb, optic nerves, labyrinth/semicircular canal, thalamus, cerebrum, medulla, midbrain, and cerebellum. We binned the following structures into “sensory” structures: olfactory bulb, optic nerves, and semicircular canal as these structures receive primary sensory input. The remaining segmented structures were considered “nonsensory” as they are either not involved in sensory processing or are involved at a secondary stage. Brain regions were segmented following best practices previously established by the field (Balanoff et al. 2016). Segmented regions of interest were converted into surface files and volumes of each surface were extracted within VGStudio. While the absolute values of the volumes may be susceptible to segmenting error and tissue defects (e.g., shrinkage Hedrick et al. 2018), biases should be similar across specimens. Moreover, we visually confirmed that brains in the specimens used filled the expected amount of the endocranial space.
Figure 1.

Phylogeny of taxa included in this analysis with respective total intact ORs. Note this does not include all chemoreceptors, but just those within the OR Classes I and II multigene families. Numbers on the phylogeny correspond to a subset of the segmented brain and sensory regions from the soft tissue µCT-scans. Silhouettes were obtained from phylopic.org. Silhouettes and 3D reconstructions are not to scale and are enlarged for clarity. *Exact species for both genomic and phenotype data were not available. For Rallidae, G. okinawae was used for genomic data and P. carolina was used for the soft-tissue specimen. For Phalacrocorax, P. carbo was used for genomic data and P. auritus was used for the soft-tissue specimen. For Nothoprocta, N. pentlandii was used for morphology, and N. perdicaria was used for genomic data. Scale bars are 2 mm.
Phenotypic statistical analyses
To infer the phenotypic disparity of different brain regions through time, we used the extracted volumes of each segmented brain region per species and scaled it by the total volume of all segmented brain regions for size. We calculated the disparity of the entire brain, each brain region scaled by total brain volume, and each sensory and nonsensory module using the dtt() function in the geiger package in R. The calculated morphological disparity index (MDI) is a measure computed from the average squared Euclidean pairwise distances between species (Harmon et al. 2003; Slater et al. 2010). Values less than zero indicate lower trait disparity within clades than expected under Brownian motion, and vice versa for positive MDI values. We performed 10,000 simulations to represent a null distribution of disparity of a trait evolving via Brownian motion for this group of taxa (Slater et al. 2010). Coturnix japonica and C. serpentina were removed from the morphological analyses, as these specimens were too young to reliably infer comparative volumes. We also estimated rates of trait evolution and tested whether a Brownian motion model of evolution or a model incorporating Pagel’s λ was a better fit for each respective trait using the momot package in R (Puttick et al. 2020). Pagel’s λ is a measure of phylogenetic signal that falls between 0 and 1; when λ is 1, the trait evolving under Brownian motion and when λ is 0, there is no phylogenetic signal and the trait may be evolving under a different process than expected under Brownian motion.
Results
OR identification
We observed substantial variation in the number of intact ORs across tetrapods (Figure 1). Numbers of copies for both Classes I and II ORs varied by orders of magnitude, with the snapping turtle having the largest number of Class I ORs (n = 789) and the anole having only a single intact copy. For Class II ORs, the caecilian (n = 1,552) had the largest number, while the zebra finch had the smallest (n = 7). Average GC content for alignment of different OR subfamilies ranged from 44.7 to 55.9%. Average GC content for non-OR genes ranged from 41.0 to 64.5%. Supplementary Table S3 shows the results for number of recovered genes per subfamily, alignment statistics, GC-content, and estimated best-fit models of evolution.
Rates of molecular evolution
Class II OR genes evolve at order of magnitude higher rates than non-OR genes, and both Classes I and II OR genes have numerous genes that overlap with the predicted scenarios consistent with diversifying selection (Figure 2). Figure 2A shows the outcomes of the three simulated evolutionary scenarios compared to those estimated for both ORs and non-OR genes. The linear model resulted in an intercept of 1.31 (standard error: 0.37). For the simulated evolutionary scenarios diversifying selection yielded the steepest slope (mean: 18.81; 95% confidence intervals: [18.57–19.05]), strong purifying selection (2.58 [2.41–2.76]) the lowest, and weak purifying selection in between (4.09 [3.83–4.35]). All three slopes differed from each other significantly (Figure 2B and Table 1), confirming that diversifying selection correctly simulates high rates of codon substitution relative to nucleotide substitution and can be used for comparisons to empirical data. Among the classes of genes in the genome, Class II ORs had the highest slope (4.1 [3.85–4.35]) and differed significantly from Class I and non-OR genes (Table 1). The slope of Class I genes (1.92 [1.11–2.74]) did not differ from that of non-OR genes (2.12 [1.45–2.78]; Table 1), but some outliers still reflect rates of diversifying selection. Figure 2B compares the slopes of different observed scenarios to the slopes of the simulated scenarios. For Class II OR genes (interaction term: 0.01; t-ratio = 0.032; P-value = 1; Table 1), the slope did not significantly differ from that of simulated weak purifying selection (ω = 0.1). For both Class I OR and non-OR genes, strong purifying selection (ω = 0.01) was the best fit for each class.
Figure 2.

(A) Simulated and observed gene tree branch lengths representing nucleotide substitutions per gene versus gene tree branch lengths representing codon substitutions per gene. For clarity, simulated values that were outside of observable data limits were removed from the plot (codon branch lengths >15 and nucleotide branch lengths >3). (B) Slopes of simulated (dashed lines) scenarios and observed scenarios (solid lines). Simulated points were removed. Slopes are plotted with observed points of Classes I and II ORs and non-OR genes. (C) Candidate genes under diversifying selection, determined from genes with codon to nucleotide branch ratios within 1 standard deviation of the mean of diversifying selection simulations. Colored red and blue points represent taxonomic group that genes belonged to, though diversifying selection was observed in all taxa. Only 2 non-OR genes fell within the cutoff. (D) Candidate genes under diversifying selection separated by gene family. Six OR subfamilies did not exhibit rates within our threshold. Note the change of axes limits from (A) and (C), zoomed in for clarity.
Table 1.
Interaction term estimates and standard errors for each pair of covariates are compared
| Contrast | Estimate | SE | t-ratio | P-value |
|---|---|---|---|---|
| Interactions among observed genes | ||||
| Classes I– II | −2.17 | 0.43 | −5.01 | <0.001 |
| Class I–Non-OR | −0.19 | 0.54 | −0.36 | 0.99 |
| Class II–Non-OR | 1.98 | 0.36 | 5.45 | <0.001 |
| Interactions among simulations | ||||
| Purifyingω = 0.01 to Purifyingω = 0.1 | −1.51 | 0.16 | −9.40 | <0.001 |
| Purifyingω = 0.01 to Diversifyingω = 2.5 | 16.22 | 0.15 | 107.45 | <0.001 |
| Purifyingω = 0.1 to Diversifyingω = 2.5 | 14.72 | 0.18 | 81.27 | <0.001 |
| Interactions between observed genes and simulated scenarios | ||||
| Class I—Purifyingω = 0.01 | −0.66 | 0.42 | −1.56 | 0.63 |
| Class I—Purifyingω = 0.1 | −2.17 | 0.43 | −4.97 | <0.001 |
| Class I—Diversifyingω = 2.5 | −16.89 | 0.43 | −39.03 | < 0.001 |
| Class II—Purifyingω = 0.01 | 1.51 | 0.15 | 9.79 | <0.001 |
| Class II—Purifyingω = 0.1 | 0.01 | 0.18 | 0.03 | 1 |
| Class II—Diversifyingω = 2.5 | −14.71 | 0.18 | −83.58 | <0.001 |
| Non-OR—Purifyingω = 0.01 | −0.47 | 0.35 | −1.33 | 0.77 |
| Non-OR—Purifyingω = 0.1 | −1.98 | 0.37 | −5.40 | <0.001 |
| Non-OR—Diversifyingω = 2.5 | 16.70 | 0.36 | 46.85 | <0.001 |
P-values were corrected for multiple comparisons. Purifyingω = 0.01 refers to strong purifying selection, purifyingω = 0.1 is weak purifying selection, and diversifyingω = 2.5 is diversifying or positive selection.
Candidate genes under diversifying selection
Of the 11,164 genes observed in the genome (both OR and non-OR), 1,158 of these genes had high enough ratio of rates of codon to nucleotide substitutions to be classified as candidates for genes experiencing diversifying selection (Figure 2C). Only two of these genes were non-OR genes (Celf2 in M. musculus (NM_001110228.1) and Tspan6 in O. anatinus (XM_029067969.1)). The remaining identified genes were ORs. With the exception of P. carolina and M. domestica, diversifying selection in ORs was identified in every taxon, though some taxa had higher proportions of OR genes under diversifying versus purifying selection (Table 2 and Figure 2C). Greater than 10% of the OR repertoires may be experiencing diversifying selection in over half the taxa analyzed, with 4 taxa having >20% of their repertoires experiencing diversifying selection (Table 2). A total list of gene candidates is available as a Supplementary File.
Table 2.
Number of OR genes identified as candidates under diversifying selection and the relative proportion to the total number of intact OR genes in the respective genome
| Species | Total diversifying candidates | Total intact ORs | Proportion |
|---|---|---|---|
| Ambystoma mexicanum | 3 | 47 | 0.06 |
| Rhinatrema bivittatum | 219 | 1,758 | 0.12 |
| Ornithorhynchus anatinus | 22 | 484 | 0.05 |
| Monodelphis domestica | 0 | 1,827 | 0 |
| Desmodus rotundus | 11 | 424 | 0.03 |
| Mus musculus | 43 | 1,762 | 0.02 |
| Sphenodon punctatus | 101 | 485 | 0.21 |
| Paroedura picta | 108 | 659 | 0.16 |
| Pantherophis guttatus | 55 | 439 | 0.13 |
| Anolis carolinensis | 15 | 117 | 0.13 |
| Chelydra serpentina | 360 | 1,621 | 0.22 |
| Alligator mississippiensis | 150 | 777 | 0.19 |
| Crocodylus porosus | 2 | 21 | 0.10 |
| Rhea americana | 19 | 89 | 0.21 |
| Nothoprocta perdicaria | 16 | 77 | 0.21 |
| Coturnix japonica | 2 | 36 | 0.06 |
| Gallus gallus | 7 | 69 | 0.10 |
| Porzana carolina | 0 | 74 | 0 |
| Taeniopygia guttata | 1 | 11 | 0.09 |
| Phalacrocorax carbo | 5 | 28 | 0.18 |
| Gavia stellata | 7 | 69 | 0.10 |
Phenotypic disparity
Brain volumes for total brain and brain regions are available in Supplementary Table S4. For brain and brain region volumes, MDI values did not differ significantly from what was expected under Brownian motion evolution (Table 3). Disparity among total brain volumes and associated nonsensory structures was greatest early in diversification of tetrapods, while olfactory bulb disparity steadily increased through time, though no trend was different than what was observed in Brownian motion processes. Supplementary Figures S1 and S2 show the outcomes for each brain region and associated simulations. When estimating the rates of evolution, only total brain volumes and the midbrain demonstrated significant differences from Brownian motion (Table 3). The asterisk indicates estimates for Pagel’s λ were closer to 0 for total brain and the midbrain, while they were close or at 1 for all other brain regions, including the olfactory bulb (Table 3).
Table 3.
Parameters of morphological evolution models
| Brain region | MDI | P-value | BMV | Root |
λ |
LRT P-value | AICc |
|---|---|---|---|---|---|---|---|
| Total brain* | 0.293 | 0.83 | 59,840 | 2,627 | 1e-8 | 0.03 | 7.18 |
| Sensory | −0.078 | 0.24 | – | – | – | – | – |
| Non-sensory | 0.036 | 0.50 | – | – | – | – | – |
| Olfactory bulb | 0.003 | 0.41 | 1.4e-5 | 0.11 | 1 | 1 | −2.85 |
| Semicircular canal | −0.141 | 0.16 | 2.8e-5 | 0.14 | 1 | 1 | −2.85 |
| Optic nerves | 0.303 | 0.84 | 1.7e-6 | 0.03 | 1 | 1 | −2.85 |
| Cerebrum | 0.009 | 0.42 | 1.0e-4 | 0.37 | 0.73 | 0.38 | −1.33 |
| Thalamus | 0.325 | 0.85 | 2.16e-6 | 0.05 | 0.33 | 0.21 | 0.32 |
| Cerebellum | −0.085 | 0.37 | 1.34e-5 | 0.07 | 0.85 | 0.55 | −2.12 |
| Midbrain* | 0.206 | 0.73 | 1.7e-5 | 0.12 | 0.27 | 0.03 | 6.25 |
| Medulla | 0.047 | 0.49 | 8.8e-6 | 0.13 | 1 | 1 | −2.85 |
The first 2 columns with continuous values correspond to the disparity through time analyses.
MDIs for each brain region or group of brain regions and associated P-values that demonstrate differences from the mean of 10,000 simulations of Brownian motion trait evolution. BMV is the Brownian motion covariance and root is the starting value of the trait before any change.
Discussion
We investigated the extent to which diversifying evolution of the olfactory system occurs throughout Tetrapoda. We characterized both the strength of selection in ORs—which constitute the largest gene family in the genome—and the morphological disparity of sensory and nonsensory brain regions to test whether OR genes and phenotypes display similar evolutionary trends. Our research revealed three key discoveries about olfactory systems: (1) OR genes are evolving at some of the fastest rates in the tetrapod genome; (2) olfactory bulbs evolve at similar rates to other sensory and nonsensory brain regions and do not differ from Brownian motion; and (3) olfactory bulb and OR gene evolution are decoupled.
We quantitatively demonstrate that most OR genes evolve at an order of magnitude higher rate than non-OR genes (Table 1 and Figure 2), and diversifying rates are observed ubiquitously across taxa (Figure 2C) and across multiple OR subfamilies (Figure 2D). Elevated rates of codon substitutions in chemosensory receptor genes have been incidentally reported in various studies focused on individual vertebrate genomes (e.g., Green et al. 2014; Lin et al. 2016; Mason et al. 2016; Li et al. 2018). High rates of evolution have also been reported within focused studies of chemosensory receptor genes of a specific clade (e.g., Glusman et al. 2000; Young et al. 2002; Niimura and Nei 2005; Yoder et al. 2014). However, our analysis is the first large-scale study of major vertebrate clades. We suggest the increased rate of evolution, especially in Class II genes (Table 2, Figure 2B,C), is due to the birth–death processes underlying OR gene evolution. Previous studies in mammals have found that Class II genes are under weaker evolutionary constraint and that this trend is attributed to increased rates of gene retention after duplication (Niimura et al. 2014). We also specifically propose that Class II OR subfamily 5/8/9 and OR 6 are evolving at particularly high rates in a number of divergent taxa, not just in mammals (Figure 2D). Novel duplications provide the substrate for the fixation of functional mutations (Niimura 2012), and the high rate of codon substitution relative to nucleotide substitution may indeed promote diversification of potential to bind to new odorant ligands. Simultaneously, rapid evolution puts receptors in a rather precarious position of loss of function. Previous studies have described chemosensory receptor gene families “on the verge of a functional breakdown” (Yoder and Larsen 2014). The rates of evolution are so high within these genes that if positive selection relaxes even slightly, widespread pseudogenization may occur (Yoder and Larsen 2014; Yoder et al. 2014; Hunnicutt et al. 2019). The order-of-magnitude increases in rates of molecular evolution and rates equivalent to diversifying selection of ORs that we found suggest ongoing turnover and rapid evolution over the course of 400 million years of tetrapod diversification.
Our calculated rates of molecular OR evolution stand in stark contrast to our results from olfactory morphology. Despite the allometric independence of olfactory bulb evolution (Finlay and Darlington 1995), we found that, relative to other brain regions, disparity in olfactory bulb volume does not vary significantly on the timescale of tetrapod diversification (Figure 3 and Table 3). The olfactory bulb has been shown to evolve differently within different clades and is often correlated with “olfactory ability” or some other physiological characteristic, yet the inferred patterns are inconsistent. Olfactory bulb size is smaller in aquatic versus terrestrial mammals (Gittleman 1991), but is larger in aquatic-foraging birds (Corfield et al. 2015). Nocturnality in mammals seems to predict olfactory bulb size, but the direction of the trend depends on the “ordinal-level” clade (Barton et al. 1995). There are also many instances in which olfactory bulb morphology remains highly conserved: the olfactory bulb and medulla, for example, showed among the smallest amounts of evolutionary change in 40 million years of anthropoid diversification (Smaers and Soligo 2013). With a few minor exceptions, neural projections from the olfactory bulb are highly conserved between turtles and pigeons (Reiner and Karten 1985). Indeed, the basic neural mechanism from receptor to olfactory bulb glomeruli has been maintained since the fish-mammal common ancestor (Saraiva et al. 2015). Thus, we conclude evolutionary change of brain sensory organization is consistent with that expected under Brownian motion, especially compared to the exceptional rates of change of molecular OR repertoires. It is also important to note our dataset only contains extant taxa, which may lead to some bias of the extremes observed in living taxa. We caution over-ambitious interpretation of the results and look forward to future analyses that incorporate fossil endocasts to further test our hypothesis.
Figure 3.
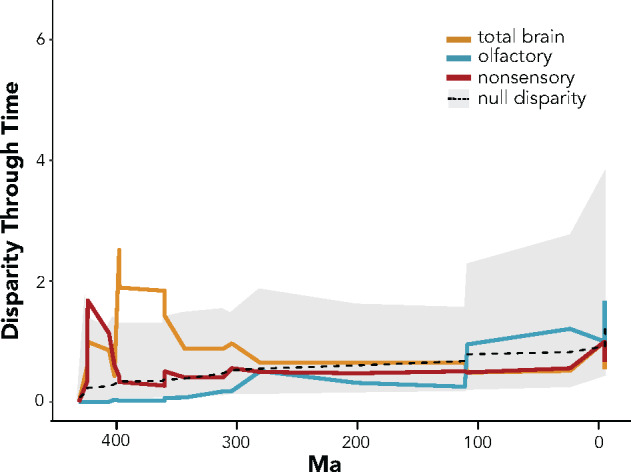
Disparity of brain volumes through time. The mustard line is the volume of the entire brain and sensory regions, blue is the olfactory bulb scaled by total brain size, and red is the regions of the brain not associated with major primary sensory input: cerebrum, thalamus, midbrain, cerebellum, and medulla. The gray region of the figure is the confidence intervals from simulations modeling the total brain volume disparity as expected under Brownian motion and the dashed line is the mean value obtained from these simulations.
The morphological and genomic disassociation of the ORs and olfactory bulbs may be a unique phenomenon of chemosensation relative to other sensory systems (Bear et al. 2016). Olfactory neurons within the olfactory epithelium stochastically express a single OR (Chess et al. 1994; Rodriguez 2013; Monahan and Lomvardas 2015). These neurons repeatedly converge until they are received by a subset of glomerular cells in the olfactory bulb (Zou et al. 2009). The neuron-glomeruli input is a hardwired neuroanatomical phenomenon that is conserved across vertebrates (Saraiva et al. 2015). With this conserved neural mechanism in place, it has been hypothesized that selection constraints on receptors are relaxed (Bear et al. 2016). Previous studies have also found the number of ORs is not necessarily connected with the number of glomeruli in the olfactory bulb (Maresh et al. 2008). Receptors may diversify and evolve independently from anatomy, as the actual expressed receptor does not strongly affect the mechanisms of sensory processing. We note, however, that there is evidence that some olfactory chamber structures, such as mammalian turbinates and cribriform plate foramina, do evolve in concert with the OR repertoire size (Garrett and Steiper 2014; Bird et al. 2018). Deeper consideration of the innervation of the olfactory and vomeronasal system with the olfactory bulb and accessory olfactory bulb is merited as recent evidence suggests a more complex and interdependent role of the two systems (Huilgol et al. 2013; Weiss et al. 2020), and this study is limited in understanding this through only studying the volume of the olfactory bulb. Our study provides the first macro-scale quantitative evidence for this hypothesis, demonstrating that OR genes evolve at higher rates than other protein-coding genes (Table 2), with some observed rates consistent with diversifying selection (Figure 2B and C), while overall chemosensory phenotype disparity remains unchanged (Figure 3).
In many ways, the signals for vision and sound (wave-forms) are less complex than the innumerable combinations of chemical odorants present in the environment (Yohe and Brand 2018), which may in part explain why, for instance, there are many more chemoreceptors in the vertebrate genome than light receptors (Nei et al. 2008; Bear et al. 2016). The impressive diversity of OR genes has evolved to deal with a highly dynamic chemical space. We caution, however, against interpreting the number of distinct intact OR genes as a direct reflection of the number of potential chemical ligands that an animal can decipher (Meister 2015). We propose that, in vertebrates, the elegant network of neurons that converges within the olfactory bulb has long worked well in making sense of a complicated chemical background. Accelerated rates of evolution in OR genes may be products of selection for increasing probability of binding affinity of relevant odorant cues, rather than for increasing the number of different potential ligands. In light of the “motor and brakes” of evolution, we submit that ORs push the limits of how an animal perceives its environment, while stable and reliable structures within the brain make it possible to understand and react to those perceptions.
Supplementary Material
Acknowledgments
The authors want to acknowledge Brandon Mercado for his assistance with µCT-scanning, the Harvard Center for Nanoscale Systems, and Greg Watkins-Colwell and Kristof Zyskowski for assistance with Yale Peabody Museum specimens.
Funding
L.R.Y. was supported by the National Science Foundation Postdoctoral Research Fellowship in Biology (NSF-DBI 1812035) and the Linnean Society of London and the Systematics Association Systematics Research Fund. M.H. was supported by the Yale Institute for Biospheric Studies Small Grants Program Doctoral Pilot Award. B.A.S.B. was supported by Yale University and the Yale Institute for Biospheric Studies.
Author Contributions
L.R.Y. designed the study, collected and analyzed all molecular data, assisted with µCT-scanning and segmenting, and wrote the manuscript. M.F. segmented the µCT scan datasets, collected the volumetric data, and edited the manuscript. M.H. assisted with obtaining and staining specimens, µCT-scanning, segmenting, and editing the manuscript. B.A.S.B. helped to conceive the study, developed staining and µCT scanning protocols, acquired, stained, and scanned specimens, segmented some datasets, and edited the manuscript.
Supplementary Material
Supplementary material can be found at https://academic.oup.com/cz.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, 1990. Basic local alignment search tool. J Mol Biol 215:403–410. [DOI] [PubMed] [Google Scholar]
- Baier H, Korsching S, 1994. Olfactory glomeruli in the zebrafish are identifiable across animals. J Neurosci 14:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanoff AM, Bever GS, Colbert MW, Clarke JA,, Field DJ et al. , 2016. Best practices for digitally constructing endocranial casts: examples from birds and their dinosaurian relatives. J Anat 229:173–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton RA, Purvis A, Harvey PH, 1995. Evolutionary radiation of visual and olfactory brain systems in primates, bats and insectivores. Philos Trans R Soc London Ser B-Biological Sci 348:381–392. [DOI] [PubMed] [Google Scholar]
- Bear DM, Lassance J-M, Hoekstra HE, Datta SR, 2016. The evolving neural and genetic architecture of vertebrate olfaction. Curr Biol 26:R1039–R1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bininda-Emonds ORP, 2005. transAlign: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinformatics 6:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird DJ, Murphy WJ, Fox-Rosales L, Hamid I, Eagle RA et al. , 2018. Olfaction written in bone: cribriform plate size parallels olfactory receptor gene repertoires in Mammalia. Proc R Soc B Biol Sci 285:20180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressel OC, Khan M, Mombaerts P, 2016. Linear correlation between the number of olfactory sensory neurons expressing a given mouse odorant receptor gene and the total volume of the corresponding glomeruli in the olfactory bulb. J Comp Neurol 524:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brykczynska U, Tzika AC, Rodriguez I, Milinkovitch MC, 2013. Contrasted evolution of the vomeronasal receptor repertoires in mammals and squamate reptiles. Genome Biol Evol 5:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess A, Simon I, Cedar H, Axel R, 1994. Allelic inactivation regulates olfactory receptor gene expression. Cell 78:823–834. [DOI] [PubMed] [Google Scholar]
- Corfield JR, Price K, Iwaniuk AN, Gutiérrez-Ibáñez C, 2015. Diversity in olfactory bulb size in birds reflects allometry, ecology, and phylogeny. Front Neuroanat 9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR, 2011. Accelerated profile HMM searches.PLoS Comput Biol 7: e1002195. [DOI] [PMC free article] [PubMed]
- Eyun SI, Moriyama H, Hoffmann FG, Moriyama EN, 2016. Molecular evolution and functional divergence of trace amine-associated receptors. PLoS ONE 11:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Mongiardino Koch N, Pritchard AC, Hanson M,, Hoffman E et al. , 2017. The skull roof tracks the brain during the evolution and development of reptiles including birds. Nat Ecol Evol 1:1543–1550. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Darlington RB, 1995. Linked regularities in the development and evolution of mammalian brains. Science 268:1578–1584. [DOI] [PubMed] [Google Scholar]
- Fleischer J, Pregitzer P, Breer H, Krieger J, 2018. Access to the odor world: olfactory receptors and their role for signal transduction in insects. Cell Mol Life Sci 75:485–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett EC, Steiper ME, 2014. Strong links between genomic and anatomical diversity in both mammalian olfactory chemosensory systems. Proc R Soc B Biol Sci 281:20132828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerts EM, Yu YK, Agarwala R, Schäffer AA, Altschul SF, 2006. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol 4:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittleman JL, 1991. Carnivore olfactory bulb size: allometry, phylogeny and ecology. J Zool 225:253–272. [Google Scholar]
- Glusman G, Bahar A, Sharon D, Pilpel Y, White J et al. , 2000. The olfactory receptor gene superfamily: data mining, classification, and nomenclature. Mamm Genome 11:1016–1023. [DOI] [PubMed] [Google Scholar]
- Green RE, Braun EL, Armstrong J, Earl D,, Nguyen N et al. 2014. Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 346:1254449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grus WE, 2008. Evolution of the vomeronasal system viewed through system-specifc genes. [DOI] [PubMed]
- Harmon LJ, Larson A, Losos JB, 2003. Tempo and mode of evolutionary radiation in iguanian lizards: a phylogenetic comparative study. Science 301:1–28. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W, 2008. GEIGER: investigating evolutionary radiations. Bioinformatics 24:129–131. [DOI] [PubMed] [Google Scholar]
- Hayden S, Bekaert M, Crider TA, Mariani S, Murphy WJ et al. , 2010. Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res 20:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden S, Teeling EC, 2014. The molecular biology of vertebrate olfaction. Anat Rec 297:2216–2226. [DOI] [PubMed] [Google Scholar]
- Hedrick BP, Yohe L, Vander LA,, Dávalos LM,, Sears K et al. , 2018. Assessing soft-tissue shrinkage estimates in museum specimens imaged with diffusible iodine-based contrast-enhanced computed tomography (diceCT. ). Microsc Microanal 24:284–291. [DOI] [PubMed] [Google Scholar]
- Hinds JW, McNelly NA, 1981. Aging in the rat olfactory system: correlation of changes in the olfactory epithelium and olfactory bulb. J Comp Neurol 203:441–453. [DOI] [PubMed] [Google Scholar]
- Huilgol D, Udin S, Shimogori T,, Saha B,, Roy A et al. , 2013. Dual origins of the mammalian accessory olfactory bulb revealed by an evolutionarily conserved migratory stream. Nat Neurosci 16:157–165. [DOI] [PubMed] [Google Scholar]
- Hunnicutt KE, Tiley GP, Williams RC, Larsen PA,, Blanco MB et al. , 2019. Comparative genomic analysis of the pheromone receptor class 1 family (V1R) reveals extreme complexity in mouse lemurs (Genus, Microcebus) and a chromosomal hotspot across mammals. Genome Biol Evol 12:3562–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetz W, Pyron RA, 2018. The interplay of past diversification and evolutionary isolation with present imperilment across the amphibian tree of life. Nat Ecol Evol 2:850–858. [DOI] [PubMed] [Google Scholar]
- Ji Y, Zhang Z, Hu Y, 2009. The repertoire of G-protein-coupled receptors in Xenopus tropicalis. BMC Genomics 10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen CB, 2000. Amphibian respiration and olfaction and their relationships: from Robert Townson (1794) to the present. Biol Rev 75:297–345. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM, 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Yang Z, Maldonado E, Li C,, Zhang G et al. , 2015. Olfactory receptor subgenomes linked with broad ecological adaptations in Sauropsida. Mol Biol Evol 32:1–12. [DOI] [PubMed] [Google Scholar]
- Kimball RT, Oliveros CH, Wang N, White ND,, Barker FK et al. , 2019. A phylogenomic supertree of birds. Diversity 11:109. [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB, 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol 34:1812–1819. [DOI] [PubMed] [Google Scholar]
- Lenth RV, 2016. Least-squares means: the R package lsmeans. J Stat Softw 1. [Google Scholar]
- Li J, Gao Y, Xie L, Deng C,, Shi P et al. , 2018. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. Proc Natl Acad Sci 115:8406–8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberles SD, 2014. Mammalian pheromones. Annu Rev Physiol 76:151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Fan S, Zhang Y, Xu M,, Zhang H et al. , 2016. The seahorse genome and the evolution of its specialized morphology. Nature 540:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnic B, Hirono J, Sato T, Buck LB, 1999. Combinatorial receptor codes for odors. Cell 96:713–723. [DOI] [PubMed] [Google Scholar]
- Maresh A, Rodriguez Gil D, Whitman MC, Greer CA, 2008. Principles of glomerular organization in the human olfactory bulb - Implications for odor processing. PLoS ONE 3:e2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason VC, Li G, Minx P, Schmitz J, Churakov G et al. , 2016. Genomic analysis reveals hidden biodiversity within colugos, the sister group to primates. Sci Adv 2:e1600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister M, 2015. On the dimensionality of odor space. eLife 4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills DB, Francis WR, Vargas S,, Larsen M,, Elemans CPH et al. , 2018. The last common ancestor of animals lacked the HIF pathway and respired in low-oxygen environments. eLife 7:e31176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K, Lomvardas S, 2015. Monoallelic expression of olfactory receptors. Annu Rev Cell Dev Biol 31:721–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, Wolf JB, Kaj I, 2014. Why time matters: codon evolution and the temporal dynamics of dN/dS. Mol Biol Evol 31:212–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara K, Saraiva LR, Ye X, Buck LB, 2011. A large-scale analysis of odor coding in the olfactory epithelium. J Neurosci 31:9179–9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Niimura Y, Nozawa M, 2008. The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet 9:951–963. [DOI] [PubMed] [Google Scholar]
- Nei M, Rooney AP, 2005. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ, 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, 2009. Evolutionary dynamics of olfactory receptor genes in chordates: interaction between environments and genomic contents. Hum Genomics 4:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, 2012. Olfactory receptor multigene family in vertebrates: from the viewpoint of evolutionary genomics. Curr Genomics 13:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Matsui A, Touhara K, 2014. Extreme expansion of the olfactory receptor gene repertoire in African elephants and evolutionary dynamics of orthologous gene groups in 13 placental mammals. Genome Res 24:1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Nei M, 2005. Comparative evolutionary analysis of olfactory receptor gene clusters between humans and mice. Gene 346:13–21. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K, 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. [DOI] [PubMed] [Google Scholar]
- Pegueroles C, Laurie S, Albà MM, 2013. Accelerated evolution after gene duplication: a time-dependent process affecting just one copy. Mol Biol Evol 30:1830–1842. [DOI] [PubMed] [Google Scholar]
- Pereira AG, Sterli J, Moreira FRR, Schrago CG, 2017. Multilocus phylogeny and statistical biogeography clarify the evolutionary history of major lineages of turtles. Mol Phylogenet Evol 113:59–66. [DOI] [PubMed] [Google Scholar]
- Puttick MN, Ingram T, Clarke M, Thomas GH, 2020. MOTMOT: models of trait macroevolution on trees (an update). Methods Ecol Evol 11:464–471. [Google Scholar]
- Quinlan AR, 2014. BEDTools: the Swiss‐army tool for genome feature analysis. Curr Protoc Bioinforma 47:11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Karten HJ, 1985. Comparison of olfactory bulb projections in pigeons and turtles. Brain Behav Evol 27:11–27. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden L, Bleasby A, 2000. EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277. [DOI] [PubMed] [Google Scholar]
- Rodriguez I, 2013. Singular expression of olfactory receptor genes. Cell 155:274–277. [DOI] [PubMed] [Google Scholar]
- Rouyar A, Party V, Prešern J, Blejec A, Renou M, 2011. A general odorant background affects the coding of pheromone stimulus intermittency in specialist olfactory receptor neurones. PLoS One 6: e26443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva LR, Ahuja G, Ivandic I, Syed AS,, Marioni JC et al. , 2015. Molecular and neuronal homology between the olfactory systems of zebrafish and mouse. Sci Rep 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GJ, Price SA, Santini F, Alfaro ME, 2010. Diversity versus disparity and the radiation of modern cetaceans. Proc R Soc B Biol Sci 277:3097–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaers JB, Soligo C, 2013. Brain reorganization, not relative brain size, primarily characterizes anthropoid brain evolution. Proc R Soc B Biol Sci 280:20130269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger SS, Fidler AE, Valcu M, Kempenaers B, 2008. Avian olfactory receptor gene repertoires: evidence for a well-developed sense of smell in birds? Proc R Soc B Biol Sci 275:2309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger SS, Kuryshev VY, Stensmyr MC, Kempenaers B, Mueller JC, 2009. A comparison of reptilian and avian olfactory receptor gene repertoires: species-specific expansion of group γ genes in birds. BMC Genomics 10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers L, Kuo T-H, 2015. Mammalian pheromones: emerging properties and mechanisms of detection. Curr Opin Neurobiol 34:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez R, García-González D, de Castro F, 2012. Mutual influences between the main olfactory and vomeronasal systems in development and evolution. Front Neuroanat 6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran J, Holder MT, 2010. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26:1569–1571. [DOI] [PubMed] [Google Scholar]
- Team RC, 2016. R: A language and environment for statistical computing.
- Tonini JFR, Beard KH, Ferreira RB, Jetz W, Pyron RA, 2016. Fully-sampled phylogenies of squamates reveal evolutionary patterns in threat status. Biol Conserv 204:23–31. [Google Scholar]
- Upham NS, Esselstyn JA, Jetz W, 2019. Inferring the mammal tree: species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol 17:1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda JC, Pennell MW, Miller ET, Maia R, Mcclain CR, 2017. The evolution of energetic scaling across the vertebrate Tree of Life. Am Nat 190:185–199. [DOI] [PubMed] [Google Scholar]
- Van Valkenburgh B, Smith TD, Craven BA, 2014. Tour of a labyrinth: exploring the vertebrate nose. Anat Rec 297:1975–1984. [DOI] [PubMed] [Google Scholar]
- VGstudio Max 3.3., 2014. Vol Graph GmbH. Heidelberg: Hexagon.
- Weiss L, Jungblut LD, Pozzi AG,, Zielinski BS,, O’Connell LA et al. , 2020. Multi-glomerular projection of single olfactory receptor neurons is conserved among amphibians. J Comp Neurol 528: 2239–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Allen JE, Blackburne BP, Talavera D, 2015. ModelOMatic: fast and automated model selection between RY, nucleotide, amino acid, and codon substitution models. Syst Biol 64:42–55. [DOI] [PubMed] [Google Scholar]
- Yang Z, 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yoder AD, Chan LM, dos RM,, Larsen PA,, Campbell CR et al. , 2014. Molecular evolutionary characterization of a V1R subfamily unique to strepsirrhine primates. Genome Biol Evol 6:213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder AD, Larsen PA, 2014. The molecular evolutionary dynamics of the vomeronasal receptor (class 1) genes in primates: a gene family on the verge of a functional breakdown. Front Neuroanat 8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohe LR, Brand P, 2018. Evolutionary ecology of chemosensation and its role in sensory drive. Curr Zool 64:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohe LR, Dávalos LM, 2018. Strength of selection on Trpc2 predicts accessory olfactory bulb form in bat vomeronasal evolution. Biol J Linn Soc 123:796–804. [Google Scholar]
- Yohe LR, Davies KT, Simmons NB, Sears KE, Dumont ER et al. , 2019. Evaluating the performance of targeted sequence capture, RNA-Seq, and degenerate-primer PCR cloning for sequencing the largest mammalian multigene family. Mol Ecol Resour 201:1–14. [DOI] [PubMed] [Google Scholar]
- Yohe LR, Liu L, Dávalos LM, Liberles DA, 2019. Protocols for the molecular evolutionary analysis of membrane protein gene duplicates. In Sikosek T, editor. Computational Methods in Protein Evolution. New York (NY: ): Springer New York; 49–62. [DOI] [PubMed] [Google Scholar]
- Young JM, Friedman C, Williams EM, Ross JA, Tonnes-Priddy L et al. , 2002. Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum Mol Genet 11:535–546. [DOI] [PubMed] [Google Scholar]
- Young JM, Trask BJ, 2002. The sense of smell: genomics of vertebrate odorant receptors. Hum Mol Genet 11:1153–1160. [DOI] [PubMed] [Google Scholar]
- Zhang X, Firestein S, 2009. Genomics of olfactory receptors Chemosensory Systems in Mammals, Fishes, and Insects. Berlin, Germany: Springer; 239–255. [Google Scholar]
- Zou D-J, Chesler A, Firestein S, 2009. How the olfactory bulb got its glomeruli: a just so story?. Nat Rev Neurosci 10:611–618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.