

Summary
Increased susceptibility of circadian clock mutant mice to metabolic diseases has led to the idea that a molecular clock is necessary for metabolic homeostasis. However, these mice often lack a normal feeding-fasting cycle. We tested whether time-restricted feeding (TRF) could prevent obesity and metabolic syndrome in whole body Cry1;Cry2 and in liver-specific Bmal1 and Rev-erbα/β knockout mice. When provided ad libitum access to food, these mice rapidly gained weight and showed genotype-specific metabolic defects. However, when fed the same diet under TRF (food access restricted to 10 hours during the dark phase) they were protected from excessive weight gain and metabolic diseases. Transcriptome and metabolome analyses showed that TRF reduced the accumulation of hepatic lipids and enhanced cellular defenses against metabolic stress. These results suggest that the circadian clock maintains metabolic homeostasis by sustaining daily rhythms in feeding and fasting, and by maintaining balance between nutrient and cellular stress responses.
Graphical Abstract
eTOC
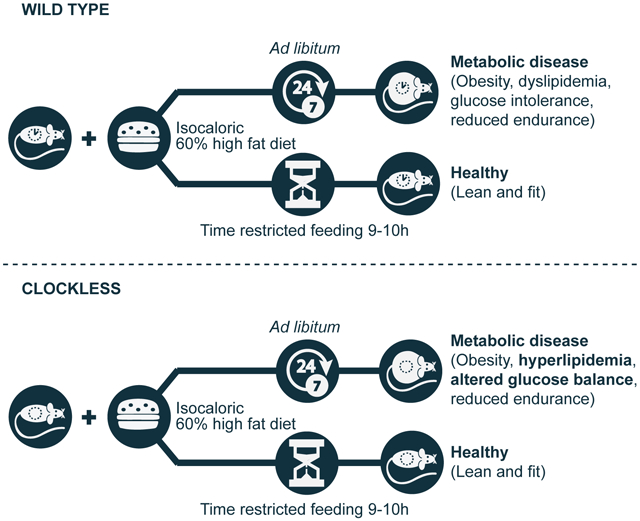
Chaix et al. show that time-restricted feeding (TRF; 10h access to food during the active phase) can improve metabolic health in mice with a compromised circadian clock. Their results point to metabolic dysfunction as secondary to disrupted behavioral rhythms (e.g. feeding-fasting), and which can be redressed by TRF.
Introduction
As nutrient availability is highly predictable on a 24-hour scale, the circadian clock modulates the expression of many metabolic enzymes in a time-of-day specific manner. In mammals, the circadian clock consists of a cell-autonomous transcription-translation feedback loop that temporally regulates the expression of a large number of genes. Heterodimers of the CLOCK (or NPAS2) and BMAL1 proteins drive the expression of their own repressors, CRY (CRY1 and CRY2) and PER (PER1, PER2, and PER3). CLOCK:BMAL1 also activates the expression of nuclear hormone receptors, namely retinoic acid–related orphan receptor (ROR) and REV-ERB (REV-ERBα and REV-ERBβ), which regulate Bmal1 gene expression (reviewed in (Hardin and Panda, 2013)). Temporally orchestrated recruitment of these transcription factors to genomic regulatory regions, as well as post-transcriptional mechanisms, account for daily oscillations of thousands of gene products in mice fed a standard diet ad libitum (ad lib) (Cho et al., 2012; Feng et al., 2011; Koike et al., 2012; Le Martelot et al., 2012; Rey et al., 2011; Vollmers et al., 2012).
The role of circadian clock components in metabolic homeostasis has been extensively studied, as many of these genes have been genetically disrupted in mice. Whole body circadian mutant mice show increased susceptibility to several metabolic diseases. Most of these diseases are also seen when clock genes are disrupted specifically in the liver, indicating that circadian clock function in liver cells is integral for metabolic health (reviewed in (Zarrinpar et al., 2016)). ChIP-seq and RNA-seq experiments have revealed that the rhythmic liver transcriptome is enriched for two major groups of transcripts implicated in cellular adaptation to nutrients. One group has a clear role in nutrient absorption, metabolism, and export, whereas the other group has been implicated in cellular stress responses (reviewed in (Asher and Sassone-Corsi, 2015; Bass, 2012; Bass and Lazar, 2016)) including the unfolded protein response, the DNA damage response, and the ER stress response. Thus, the rhythmic transcriptome contributes to cellular health by regulating metabolism and stress responses.
Circadian clock target genes are also regulated by other signals. Several circadian clock targets play key roles in metabolism and are therefore regulated by non-clock factors in response to feeding and fasting signals. In mice fed normal chow, these key metabolic regulators exhibit robust cycling (Hatori et al., 2012). In contrast, mice fed a high fat diet ad libitum (a model of diet-induced obesity (DIO) often used to mimic lifestyle that underlies metabolic diseases in humans) dampen circadian rhythms (Kohsaka et al., 2007) in many key metabolic regulators. When the DIO model is subjected to time-restricted feeding (TRF), in which mice are fed an equivalent amount of food as an ad lib cohort within a defined time period of 8-12 hours, rhythmic expression of circadian clock components and of many key metabolic regulators is sustained. In addition, TRF can prevent and even reverse many DIO-associated metabolic disorders (Chaix et al., 2014; Hatori et al., 2012; Sherman et al., 2012). The correlation between improved rhythms of circadian clock components under TRF and the health benefits raised the hypothesis that a functional circadian clock mediates TRF benefits.
TRF can drive some gene expression rhythms in circadian mutant mice. The wild type (WT) liver exhibits daily rhythms in numerous transcripts, metabolites, and proteins; rhythms that are lost in Cry1−/−Cry2−/− mice, Per1−/−;Per2−/− mice, and Bmal1−/− mice. When these mutant mice are subjected to TRF, however, daily rhythms in several, but not all, of these hepatic factors appeared (Adamovich et al., 2014; Atger et al., 2015; Vollmers et al., 2009). Conversely, WT mice under 24 h fasting show a dramatic reduction in the number of hepatic rhythmic transcripts (Vollmers et al., 2009). This dramatic effect of eating pattern on the rhythmic transcriptome (with a modest contribution from core clock components) suggests that TRF may, at least in part, compensate for the absence of a molecular clock. Many of the feeding-fasting driven daily rhythms in mutant mice, however, differ in terms of amplitude or phase of oscillation compared to WT mice (Adamovich et al., 2014; Atger et al., 2015; Vollmers et al., 2009), raising some doubt as to whether TRF can alleviate metabolic diseases in circadian mutant mice. These short-term TRF experiments are also insufficient to explain whether metabolic diseases found in circadian mutant mice result from the lack of feeding rhythms, or from the lack of an endogenous driver of rhythms (i.e., the circadian clock itself). Furthermore, when Drosophila lacking the circadian clock components cry, per, tim, or clock are subject to TRF, their cardiometabolic health does not improve, in contrast to WT controls (Gill et al., 2015). In summary, although TRF can drive daily rhythms in many genomic programs, whether it can attenuate metabolic diseases in circadian mutant vertebrates is unknown.
To test whether imposed feeding-fasting rhythms are sufficient to alleviate key metabolic diseases independent of the circadian clock, we subjected Cry1;Cry2 double knockout (CDKO) mice, or mice with liver-specific KO of Rev-erb-α and -β (Rev-erbα/βLDKO) or Bmal1 (Bmal1LKO) to a high fat diet (HFD), either ad libitum or TRF. Metabolic dysfunctions in both mutant and WT cohorts were attenuated by TRF. This suggests that the primary role of the circadian clock is to produce robust behavioral rhythms (e.g., in activity-rest and thus feeding-fasting), and that clock mutants develop metabolic diseases as a secondary consequence of their behavioral disruptions. In the absence of intrinsic behavioral rhythms (e.g., in DIO mice or clock mutants) an imposed feeding-fasting rhythm can support metabolic homeostasis.
RESULTS
Time-restricted feeding protects clock mutant mice from body weight gain on HFD without altering levels of food consumption or activity.
All circadian mutant mice fed a normal chow ad libitum develop some aspects of metabolic dysfunction which exacerbates with age. High fat diet (HFD) further accentuates metabolic dysfunctions in circadian mutant mice (Zarrinpar et al., 2016) making it easier to compare their phenotypes within a relatively uniform time-scale. To test whether a consistent daily cycle of feeding-fasting could attenuate these metabolic dysfunctions, we used three types of clock mutants 1) Cry1;Cry2 double knockout (CDKO) mice, which lacked the circadian clock in all tissues, 2) mice with liver-specific KO of Rev-erbα and Rev-erbβ (Rev-erbaLoxp;Rev-erbbLoxP;AlbCre denoted Rev-erbα/βLDKO), and 3) mice with liver-specific KO of Bmal1 (Bmal1LoxP;AlbCre, denoted Bmal1LKO). Twelve-week old male mice were fed a HFD (60% energy from fat) either ad lib (the FA groups), or via TRF, which consisted of a 9–10 h interval during the night (ZT13–22/23; the FT groups). Bmal1LKO and Rev-erbα/βLDKO mice exhibit increased hepatic steatosis and dyslipidemia on HFD (Bugge et al., 2012; Cho et al., 2012; Delezie et al., 2012; Jacobi et al., 2015), whereas CDKO mice exhibit accelerated weight gain and impaired glucose homeostasis (Barclay et al., 2013). Weekly food intake and body weight of these three mutant lines and their respective littermate controls on FA and FT regimens (12 cohorts as shown in figures 1A, 1G, 1M) were monitored for 12 weeks. The experiment was repeated with three independent mouse cohorts for all mutants, involving 256 mice in total. Rev-erbα/βLDKO mice and their WT littermates needed up to 10 h of food access to eat an equivalent amount of food as their ad lib counterparts.
Figure 1: Time-restricted feeding protects clock mutant mice from body weight gain on HFD.

A-G-M. Schematic of the experimental design depicting the 12 mice cohorts that were studied and the timing of food access relative to a 24h day.
B-H-N. Evolution of body weight in (B) Bmal1LKO (n=17-23/group), (H) Rev-erbα/βLDKO (n=16-19/group), (N) CDKO (n=24/group). Multiple t test with unequal SD and Holm-Sidak correction for multiple comparisons, * p<0.05.
C-I-O. Cumulative food consumption (kcal) in (C) Bmal1LKO (n=17-23/group), (I) Rev-erbα/βLDKO (n=16-19/group), (O) CDKO (n=24/group).
D-J-P. Body composition (g) in (D) Bmal1LKO and littermate controls (n=4-5/group), (J) Rev-erbα/βLDKO and littermate controls (n=8-11/group), (P) CDKO and littermate controls (n=8-9/group) after 12 weeks on FA or FT.
E-K-Q. Total activity during 3 days on TRF in (E) Bmal1LKO mice (n=4/group), (K) Rev-erbα/βLDKO (n=4/group) and (Q) CDKO (n=4/group). 1 way ANOVA with Holm-Sidak correction for multiple comparisons, * p<0.05.
F-L-R. Running time on a treadmill during a run-to-exhaustion assay. Each individual dot represents one mouse. Unpaired t test, * p<0.05.
As seen earlier with WT C57BL/6J mice (Chaix et al., 2014; Hatori et al., 2012), the circadian mutant mice adapted to the TRF regimen and consumed an equivalent amount of food as their ad lib cohorts. All mutant mice and their respective control cohorts exhibited weight gain under the FA condition, and TRF attenuated the weight gain to the same extent in mutants and WT controls (figures 1B, 1H and 1N). Over the 12-week experiment, Bmal1LKO mice on FA exhibited a 35.5% weight gain (26.48 ± 0.28 g to 35.88 ± 0.95 g) compared to only 17.5% for mice on FT (27.69 ± 0.39 g to 32.54 ± 0.64 g) (figure 1B). For Rev-erbα/βLDKO, weight gain on FA was 79.5% (26.09 ± 0.57 g to 46.82 ± 1.58 g) compared to only 31.3% for mice on FT (25.77 ± 0.63 g to 33.84 ± 1.61 g) (figure 1H). TRF led to a significant difference in body weight, starting 7 weeks after commencing the feeding regimen. There was no difference in body weight between Bmal1LKO and WT cohorts on FA (35.88 ± 0.95 g vs 37.71 ± 1.0 g, respectively) nor FT (32.54 ± 0.64 g vs 32.50 ± 0.86 g, respectively). Similar body weight trends were also observed in the Rev-erb groups. After 12 weeks, Rev-erbα/βLDKO and WT cohorts weighed the same on FA (46.82 ± 1.61 g vs 46.89 ± 1.39 g, respectively) and on FT (33.84 ±1.61 g vs 37.63 ± 1.26 g, respectively). From 3 weeks onwards, TRF significantly prevented body weight gain in Rev-erbα/βLKO mice, as it did in littermate controls. Finally, CDKO mice on TRF were also protected from weight gain, with 39.3% weight gain on FA (19.65 ± 0.54 g to 28.86 ± 1.31 g) compared to only 10.4% for mice on FT (19.10 ± 0.67 g to 22.57 ± 0.97 g) (figure 1N). As previously described (Ikeda et al., 2007), the CDKO animals weighed significantly less than their control littermates. The control littermates gained 63.5% of their weight on FA (25.16 ± 0.52 g to 41.13 ± 0.98 g), which was significantly higher than the 38% weight gain on FT (23.52 ± 0.68 g to 32.48 ± 0.97 g). Body composition was assessed in a subset of mice from each group at the end of the 12-week feeding protocol. All animals on TRF weighed less than their respective littermate controls on FA, and the reduced body weight was accompanied with reduced adiposity (figures 1D, 1J and 1P).
Differences in body weight in the FA versus FT feeding groups in both Bmal1LKO and WT mice could not be attributed to differences in food consumption, as their food intake was indistinguishable (figure 1C). In the Rev-erbα/β cohort, although food consumption was slightly lower in both FT groups, it was not statistically different and there was no correlation (Pearson and Spearman coefficient) between cumulative food intake and body weight at 12 weeks (figure 1I). Finally, food consumption was similar between the CDKO-FA and CDKO-FT groups, as well as for WT cohorts on FA or FT (figure 1O). Total activity (figures 1E-1K-1Q), assessed by beam breaks in metabolic cages (see figure 2), was also indistinguishable between the FA and FT groups. Furthermore, the light:dark partition of activity was similar in FA and FT (81.05 ± 16.8% vs 84.13 ± 13.8% in the dark in Bmal1LKO FA vs FT (figure S1A); 50.13 ± 6.6% vs 53.49 ± 2.7% in the dark for Rev-erbα/βLDKO FA vs FT (figure S1B); and 79.33 ± 6.4% vs 68.42 ± 13.3% in the dark for CDKO FA vs FT (figure S1C) (2-way ANOVA)). To test whether exercise capacity was compromised under FT, mice were subject to treadmill exercise. All mice on FT, with the exception of CDKO mice, ran significantly longer on a treadmill than mice on FA (figure 1F-1L-1R). Treadmill performance of the CDKO mice on FT was not significantly different from that under FA. Thus, TRF attenuated excessive body weight gain in both WT and circadian mutant mice without changing total caloric intake or activity.
Figure 2: Time-restricted feeding drives diurnal rhythms in fuel utilization.

A-E-I. Respiratory exchange ratio (RER) from metabolic cages recordings in (A) Bmal1LKO (n=4/group, 5.5 days of recording), (E) Rev-erbα/βLDKO (n=4/group, 4.5 days recording), (I) CDKO (n=4/group, 4.5 days recording).
B-F-J. Corresponding VO2 (normalized to body weight) in (B) Bmal1LKO (n=4/group), (F) Rev-erbα/βLDKO (n=4/group) and (J) CDKO (n=4/group).
C-G-K. Corresponding activity (Xt) in (C) Bmal1LKO (n=4/group), (G) Rev-erbα/βLDKO (n=4/group) and (K) CDKO (n=4/group).
D-H-L. Corresponding food consumption (g) in (D) Bmal1LKO (n=4/group), (H) Rev-erbα/βLDKO (n=4/group) and (L) CDKO (n=4/group).
Time-restricted feeding drives diurnal rhythms in fuel utilization.
The effect of TRF in reducing body weight gain in both mutant and WT controls prompted us to test whether TRF affected daily patterns of whole body fuel utilization. Bmal1LKO, Rev-erbα/βLDKO, and CDKO mice were analyzed in metabolic cages (TSE systems) for 4–5 days (after 48 h acclimation) after undergoing 6–7 weeks of the indicated feeding regimens. As described for WT mice (Chaix et al., 2014; Hatori et al., 2012; Kohsaka et al., 2007), all mutants on FA regimens displayed altered eating behavior, with caloric consumption spread almost evenly between dark and light phases (figures 2D, 2H, 2L and figures S1D-S1E-S1F). Bmal1LKO mice on FA consumed 59.97% (± 5.6) of their caloric intake during the light phase and 40.03% (± 3.6) during the dark phase (figure S1D). Light phase food consumption represented 48.89% (± 1.17) of total food intake for Rev-erbα/βLDKO mice (figure S1E) and 46.82% (± 12.7) of total food intake for CDKO mice (figure S1F).
Bmal1LKO, Rev-erbα/βLDKO, and CDKO mice on FA showed a relatively inflexible respiratory exchange ratio (RER) (figures 2A, 2E and 2I) during a 24-h day, suggesting diminished capacity to switch fuel usage between day and night. The mice on FT displayed strong diurnal rhythms of the RER with a higher RER during the fed period and lower RER during the fasting period, indicating a relatively higher usage of carbohydrates during feeding and of lipids during fasting. Interestingly, these oscillations and feeding-fasting rhythms were lost as soon as FT mice were released to FA (figures 2A, 2E and 2I, dotted lines), indicating that the imposed feeding rhythm drives the observed daily rhythm in RER in both whole-body or liver-specific circadian mutant mice, and furthermore that TRF does not lead to a learned eating rhythm in mice.
To test which aspect of whole body energy metabolism might contribute to the observed weight stabilization in FT mice, we examined the volume of oxygen (VO2) consumed throughout the 24-h day. Overall, FT mice had higher energy expenditure than FA mice, specifically during the feeding phase (figures 2B, 2F and 2J). Interestingly, when FT mice were switched to ad lib eating, they continued to exhibit increased VO2 consumption (compared to FA mice) during the first 24–48 h, supporting the hypothesis that TRF increases overall oxidative metabolism, at least transiently (Chaix et al., 2014). This increase in VO2 consumption was not associated with differences in total daily food consumption (figures 2D-2H-2L and figures S1D-S1E-S1F) nor in activity (figures 2C-2G-2K) between FA and FT cohorts. Thus, TRF promotes a rhythm in substrate utilization and is associated with increased energy expenditure in the feeding period without concomitant changes in total daily caloric intake and daily rhythms in activity. As seen earlier with a normal diet (Vollmers et al., 2009), this rhythm does not require a functional circadian clock.
Time-restricted feeding prevents whole-body fat accumulation and serum hyperlipidemia in clock-deficient mice.
Body composition analysis revealed that the percentage of fat mass (figure 3A) reduced and lean mass (figure S2A) increased when the Bmal1LKO cohort was subjected to TRF (27.0% vs 12.2%, p < 0.01 (unpaired t test) for fat mass, and 63.5% vs 75.5%, p < 0.001 (unpaired t test) for lean mass) (figures 3A and S2A). Similarly, Rev-erbα/βLDKO on TRF had significantly less fat (30.0% vs 43.2%, p < 0.01) and more lean mass (48.6% vs 58.9%, p < 0.05) than ad lib fed controls (figures 3B and S2B). Finally, CDKO-FT had significantly less fat mass (7.5% vs 39.5%, p < 0.01) and more lean mass (78.9% vs 51.4%, p < 0.05) than CDKO-FA (figures 3C and S2C). A similar trend in reduced fat mass and increased lean mass was also observed in all control littermates (figures 3A-3B-3C and figures S2A-S2B-S2C).
Figure 3: TRF prevents whole-body fat accumulation and hyperlipidemia in clock deficient mice.

A-B-C. Relative fat mass as a percent of body weight in (A) Bmal1LKO and littermate controls (n=4-5/group), (B) Rev-erbα/βLDKO and littermate controls (n=8-11/group), (C) CDKO and littermate controls (n=8-9/group). Unpaired t-test, * p<0.05, ** p<0.01, *** p<0.001.
D-E-F. Representative pictures of H&E staining of sections of (D) epididimal WAT, (E) BAT and (F) liver in the indicated genotype and feeding group.
G-H. Serum leptin (G) and adiponectin (H) levels in Bmal1LKO (n=10/group), Rev-erbα/βLDKO (n=8/group), and CDKO (n=12/group). Unpaired t-test, * p<0.05, ** p<0.01, *** p<0.001.
I-J-K. Liver triglyceride levels in (I) Bmal1LKO and littermate controls (n=12/group), (J) Rev-erbα/βLDKO and littermate controls (n=16/group), and (K) CDKO and littermate controls (n=8/group). Unpaired t-test, * p<0.05, ** p<0.01, *** p<0.001.
L-M-N. Serum triglyceride (TG) levels in (L) Bmal1LKO and littermate controls (n=8-10/group), (M) Rev-erbα/βLDKO and littermate controls (n=8-12/group), and (N) CDKO and littermate controls (n=12/group). Unpaired t-test, * p<0.05, ** p<0.01, *** p<0.001.
O-P-Q. Serum cholesterol levels in (O) Bmal1LKO and littermate controls (n=8-10/group), (P) Rev-erbα/βLDKO and littermate controls (n=8-12/group), and (Q) CDKO and littermate controls (n=12/group). Unpaired t-test, * p<0.05, ** p<0.01, *** p<0.001.
The respective wildtype cohorts for each genotype also showed similar changes in fat and lean mass upon TRF. The percentage of fat mass (figure 3A) was reduced (27.6% vs 6.7%, p < 0.001 (unpaired t test)) and the percentage of lean mass (figure S2A) was increased (60.5% vs 78.6%, p < 0.01 (unpaired t test)) in the Bmal1LWT cohort subjected to TRF (figures 3A and S2A). Similarly, Rev-erbα/βLWT on TRF had significantly less fat (35.7% vs 42.6%, p < 0.01) and more lean mass (49.6% vs 54.7%, p < 0.05) than ad lib fed controls (figures 3B and S2B). Finally, Cry control (CC) -FT had significantly less fat mass (33.8% vs 39.9%, p < 0.05) and a trend to more lean mass (56.3% vs 51.9%, ns) than CC-FA (figures 3C and S2C). This reduced fat accumulation was evident in both the adipose depots and the liver. Histological examination of the epididymal white adipose tissue (eWAT) revealed smaller adipocytes and the absence of crown-like structures in FT compared to FA mice of all genotypes (figure 3D), suggesting reduced immune infiltration and inflammation in WAT of TRF mice. H&E staining also revealed that brown adipose tissue (BAT) of TRF mice was protected from “whitening” (i.e., the presence of unilocular fat droplets characteristic of WAT) that is typically observed in FA mice of all genotypes (figure 3E). Finally, consistent with these results, leptin and adiponectin levels, two indicators of whole body adiposity, were affected by TRF. Serum leptin concentration was significantly lower in all mice on TRF (figure 3G) with a 92.0%, 83.4%, and 82.4% reduction in Bmal1LKO, Rev-erbα/βLDKO, and CDKO mice, respectively in FT compared to FA. Serum adiponectin concentrations were higher in all TRF mice (figure 3H). Thus, decreased weight gain in TRF was associated with a consistent reduction in fat mass and an increase in lean mass.
Whole body fat accumulation is often associated with fatty liver disease. Histological examination of livers from WT, Bmal1LKO, and Rev-erbα/βLDKO mice on FA showed hepatic accumulation of lipids. TRF protected all of these cohorts from hepatic steatosis (figure 3F). This reduction was confirmed by measuring triglycerides (TG) in whole-liver extracts (figures 3I-3J). Hepatic TG content was reduced by 66.1% in Bmal1LKO FT compared to FA, and by 67.1% in Rev-erbα/βLDKO. Notably, we observed very little accumulation of fat in the liver of CDKO mice under FA (figure 3F), and lower levels of TG in liver extracts (figure 3K), which did not change significantly under FT.
HFD-induced obesity is ultimately accompanied by serum dyslipidemia. Accordingly, all mice on FA showed serum hypertriglyceridemia (figures 3L-3M-3N) and hypercholesterolemia (figures 3O-3P-3Q). Nevertheless, for all mutants and their control littermates, TRF significantly protected against increases in TG (figures 3L, 3M and 3N) and cholesterol (figures 3O, 3P and 3Q) in the serum. Serum TG levels were reduced by 40% (115.4 mg/dL ± 5.8 vs 73.99 mg/dL ± 4.4) in Bmal1LKO mice FA vs FT, by 17% (49.75 mg/dL ± 2.0 vs 41.19 mg/dL ± 1.6) in Rev-erbα/βLDKO mice FA vs FT, and by 42% (96.41 mg/dL ± 9.0 vs 56.14 mg/dL ± 4.7) in CDKO FA vs FT. In TRF mice, serum cholesterol levels were reduced by 28% in Bmal1LKO mutants (232.9 ± 20.5 vs 168.3 ±8.0), 35% in Rev-erbα/βLDKO mutants (290.5 ± 14.6 vs 189.9±10.5) and 22% in CDKO mutants (165 ± 5.9 vs 128.4 ± 3.2). Taken together, these observations indicate that TRF reduces fat accumulation and supports lipid homeostasis.
As observed for hepatic steatosis, there were some genotype-specific differences in the severity of serum dyslipidemia. TG serum levels were significantly higher (56%) in Bmal1LKO mice on FA than their control littermates (115.4 mg/dL ± 5.8 vs 74.0 mg/dL ± 2.7, respectively) (figure 3L), with no difference in cholesterolemia (figure 3O). An opposite phenotype was observed in Rev-erbα/βLDKO mice, with 30% higher cholesterol than littermate controls (290.5 mg/dL ± 14.55 vs 203.3 mg/dL ± 17.87, respectively) (figure 3P). The mutant and WT controls had similarly elevated TG levels under FA (figure 3M).
Clock deficient mice on time-restricted feeding are protected from glucose intolerance and insulin resistance.
To characterize how the circadian clock and eating patterns affect glucose homeostasis, we performed intraperitoneal glucose tolerance tests (ip-GTT). Except for Bmal1LKO mice, all mice on FA showed glucose intolerance (figures 4A, 4B and 4C). Interestingly, the severity of glucose intolerance for these FA mutants (compared to controls) was variable. As quantified using the AUC above baseline, there was no difference between Rev-erbα/βLDKO and WT cohorts under FA (figure 4B insert), whereas CDKO-FA mice, despite their smaller body weight, were much more glucose intolerant than controls (figure 4C insert). Bmal1LKO mice were not glucose intolerant compared to WT (figure 4A insert). Glucose intolerance was significantly improved in all mutants and controls on TRF (figures 4A-4B-4C).
Figure 4: Clock deficient mice on time-restricted feeding are protected from glucose intolerance and insulin resistance.

A-B-C. Glucose tolerance test (ip-GTT) in (A) Bmal1LKO and littermate controls (n=8-10/group), (B) Rev-erbα/βLDKO and littermate controls (n=8-12/group), and (C) CDKO and littermate controls (n=6-8/group). Quantification of the AUC above baseline is shown in the insert. Unpaired t-test, * p<0.05, ** p<0.01, *** p<0.001.
D-E-F. Serum glucose levels in fasted (ZT22-ZT36) and refed mice (1h after IP injection of glucose (1mg/g BW) at ZT36) in (A) Bmal1LKO and littermate controls (n=4-6/group), (B) Rev-erbα/βLDKO and littermate controls (n=4-6/group), and (C) CDKO and littermate controls (n=4-6/group).
G-H-I. Corresponding serum insulin concentration.
We further measured serum glucose levels (figures 4D, 4E and 4F) and corresponding insulin levels (figures 4G, 4H and 4I) after 12 h of fasting (fasted) during the day, and 1 h after an IP injection of glucose (re-fed) to assess insulin sensitivity. In the fasted state, there were no differences in glucose levels between FA and FT groups for any of the mouse cohorts analyzed (i.e., neither in the mutants nor in the control littermates) (figures 4D, 4E and 4F, fasted). Similar trend was observed for insulin levels (figures 4G, 4H and 4I), except for Bmal1LKO mice. In the re-fed state, however, glucose levels were always lower in TRF than ad lib-fed mice, with the exception of Bmal1 cohorts (figures 4D, 4E and 4F), independently confirming the GTT results. In Rev-erbα/βLDKO, WT, and CDKO mice, there was a strong correlation between high glucose and high insulin levels in the fed state, demonstrating insulin resistance in FA. Insulin levels were lower in FT, suggesting that TRF supports insulin sensitivity.
Bmal1LKO mutants showed different results than the other models. Indeed, as mentioned previously, Bmal1LKO fed ad lib did not show glucose intolerance (figure 4A), and their insulin levels were elevated under FA (figure 4G), both in the fasted or re-fed state. Nevertheless, TRF reduced serum insulin to a level comparable to littermate controls. Additionally, insulin tolerance test showed that Bmal1LKO mutants were more insulin sensitive than controls (figure S2D). In summary, each of the three mutant models exhibited varying patterns of glucose intolerance during ad lib HFD feeding; however, TRF improved glucose tolerance in all three genotypes.
Genotype-specific hepatic metabolomic states in clock-deficient mice on TRF.
Because the liver is central to nutrient metabolism, to gain insight into the molecular pathways that are regulated by TRF, we performed untargeted metabolomics analysis of liver samples from WT (Bmal1LWT), Bmal1LKO, Rev-erbα/βLDKO, and CDKO mice in FA and FT conditions. Liver samples were collected every 4 h over a 24-h period for WT and Bmal1LKO mice, and every 3 h over a 24-h period for Rev-erbα/βLDKO and CDKO mice, representing 56 samples (see methods for details). For all genotypes and all time points, pooled liver samples from two independent mice from two independent feeding experiments were analyzed. This strategy was chosen to minimize the cost while keeping a representation of biological complexity. Up to 547 metabolites were detected, which covered all major metabolic pathways, including lipid, cholesterol, sugar, and amino acids (Table S1). We used three different strategies to statistically analyze this metabolomics dataset. In the first two approaches, time-series samples were treated as replicates to analyze the effect of TRF either within or across genotypes. In the third approach, time-series samples were treated independently and MetaCycle package was used to identify cycling metabolites in all groups (Hughes et al., 2017).
First, the effect of TRF was analyzed within genotype. Unbiased principal component analysis within each genotype are shown in figures 5A-5D. 113 metabolites were differentially abundant between FA and FT in both Bmal1LKO mice and Rev-erbα/βLDKO, whereas only 32 metabolites were differentially expressed in CDKO FA versus FT. Pathway analysis revealed that the top 5 pathways significantly altered in FA versus FT in Bmal1LKO and Rev-erbα/βLDKO were identical. They were: (i) biosynthesis of unsaturated fatty acids, (ii) TCA cycle, (iii) alanine, aspartate, and glutamate metabolism, (iv) aminoacyl-tRNA biosynthesis, and (v) glycerophospholipid metabolism. In particular, long chain fatty acids from 14 carbons to 22 carbons, both saturated and unsaturated, were elevated in Bmal1LKO and Rev-erbα/βLDKO on FA (figure 5F). This included pro-inflammatory lipids, such as arachidonate and dihomolinoleate. Although C16 palmitate and C17 margarate were also higher in CDKO-FA, longer chain fatty acids were not elevated (figure 5G). This absence of long chain fatty acid accumulation in CDKO FA was consistent with the observation that these animals did not develop fatty liver (figure 3D). In line with previously described phenotypes, some metabolites showed genotype-specific effects, specifically in CDKO mice. For instance, folate was relatively high in CDKO, whereas it was hardly detected in other mutants (figure S3A). Increased folate could be involved in protecting against hepatosteatosis (Sid et al., 2017).
Figure 5: Hepatic metabolomic footprint in clock-deficient mice on TRF.

A-B-C-D. Principal component analysis within genotype of metabolomics data obtained from liver extracts in (A) WT (n=6/group), (B) Bmal1LKO (n=6/group), (C) Rev-erbα/βLDKO (n=8/group), and (D) CDKO (n=8/group).
E. Top significant pathways obtained from comparing significantly modulated metabolites between Bmal1LKO on FA versus FT (purple) and Rev-erbα/βLDKO on FA versus FT (green).
F. Heatmap representation of the relative expression of indicated fatty acids in Bmal1LKO on FA and FT and Rev-erbα/βLDKO on FA and FT.
G. Relative levels of medium chain fatty acid in the lover of Bmal1LKO, Rev-erbα/βLDKO and CDKO on FA and FT as indicated.
H. Schematic representation of TCA cycle intermediates and their connection to glucose and lipid metabolic pathway showing liver metabolites that are significantly higher in FT (blue) or FA (red). Bar graphs of the relative levels of indicated metabolites levels are shown below.
I. Serum β-Hydroxybutyrate levels in fasted (ZT22-ZT36) and refed mice (1h after IP injection of glucose (1mg/g BW) at ZT36) in Bmal1LKO and littermate controls (n=4-6/group), Rev-erbα/βLDKO and littermate controls (n=4-6/group), and CDKO and littermate controls (n=4-6/group).
Second, to identify a TRF metabolite signature, we analyzed metabolomics results using a linear model with the two factors genotype and feeding group and a rank analysis. The two pathways that distinguished ad lib from time-restricted group were “alanine, aspartate, and glutamate metabolism” and “TCA cycle” (adj p = 0.0086 for both). Some TCA intermediates were significantly elevated (alpha-ketoglutarate and succinate) and some were significantly depleted (fumarate, malate, oxaloacetate, citrate) in FA relative to FT. Placing these intermediates on a schematic of the TCA cycle (figure 5H) revealed ad lib and time-restricted conditions give rise to the accumulation of metabolites at opposite sides of this cycle. This suggests that eating pattern modifies flux through the TCA cycle, likely by shuttling intermediates to different metabolic pathways in ad lib and time-restricted conditions. One of the products of TCA cycle is the ketone body β-HydroxyButyrate (BHBA), which was elevated during the daily fasting period in FT mice (figure 5I). Although FA mice also showed similar increase in BHBA level upon acute 12 h fast, the FT mice experienced such daily fast every day.
Recent studies suggest that NAD+ metabolism is at the crossroad of circadian regulation, gene expression, and cellular metabolism (Nakahata and Bessho, 2016; Sassone-Corsi, 2016). We found that the level of nicotinate (NA) and nicotinamide (NAM), two precursors of NAD+ through the salvage pathway, were significantly lower in FT compared to FA (figure S3C). Interestingly, both are upstream of Nampt, the enzyme controlling flux through the salvage pathway whose expression is upregulated in FT compared to FA. Together these results suggest a scenario in which the source of NAD+ may differ between TRF and ad lib fed mice, with higher flux in the NAD+ salvage pathway under TRF. Interestingly, this is reminiscent of what is observed under calorie restriction (Sato et al., 2017).
Third, to analyze the impact of TRF on the temporal profile of metabolites levels, we used Meta2Cycle (meta2d_pvalue<0.05, see methods for details) to identify cycling metabolites in each group (Results in Table S4 and figure S4). In general, except in Rev-erbα/βLDKO where the number of cycling metabolites was almost identical (25 in Rev-erbα/βLDKO–FA vs 24 in Rev-erbα/βLDKO–FT), there were more diurnal metabolites in TRF than ad lib (9 in WT-FA vs 24 in WT-FT; 7 in Bmal1LK-FA vs 33 in Bmal1LK-FT; and 8 in CDKO-FA vs 33 in CDKO-FT). These cycling metabolites were enriched in amino acids (shown in green in figure S4). These metabolites tended to peak after feeding, suggesting that the imposed feeding rhythm could drive their daily rhythms.
Liver transcriptomes reveal similarities and differences between clock mutants on TRF.
Metabolic defects in Bmal1LKO, Rev-erbα/βLDKO, and CDKO mice fed ad libitum have been linked to associated changes in gene expression (Barclay et al., 2013; Bugge et al., 2012; Cho et al., 2012; Jacobi et al., 2015; Lamia et al., 2008). We recapitulated these phenotypes in the FA condition. FT prevented these metabolic disturbances in mutant mice and their WT littermate controls. To explore the underlying hepatic gene expression changes in these mice under FA and FT conditions, we analyzed the transcriptome of liver samples from WT (Bmal1LWT), Bmal1LKO, Rev-erbα/βLDKO, and CDKO mice (see methods for details). Liver samples were collected from 2–3 mice every 3 or 4 h over a 24-h period, and polyA+ RNA-seq analysis was performed. The resulting data were analyzed using two methods. Given that the majority of hepatic transcripts (>70%) do not show diurnal variations (Hughes et al., 2009), we first analyzed the results by treating all samples in each cohort as replicates to identify genes for which the level of expression differed between groups. Second, we used MetaCycle package (Wu et al., 2016) to identify genes with a diurnal pattern of expression.
Differential expression analyses were conducted using DESeq2 (Love et al., 2014) with designs that accounted for genotype, feeding group, and time as variables (see methods for details, Table S2 for normalized read counts, and Table S3 for results). PCA analyses revealed larger gene expression differences between genotypes, while the effect of eating pattern (FA vs FT) for any given genotype was relatively modest (figure 6A). The CDKO livers under FA or FT showed the fewest differences in gene expression.
Figure 6: Liver transcriptomics unravel similarities and differences between clock mutants on TRF.

A- PCA plot of liver transcriptome data.
B-C. Schematic depicting the number of significant genes between each group as indicated.
D- Expression level (normalized read count (log2)) of some genes involved in fatty acid oxidation that are significantly lower in Bmal1LKO-FA versus WT-FA.
E- Expression level (normalized read count (log2)) of some genes involved in cholesterol metabolism that are significantly lower in Rev-erbα/βLDKO versus WT-FA.
F. Expression level (normalized read count (log2)) of enzymes involved in lipid metabolism positioned on a schematic lipid pathway representation as well as expression level of the master lipid regulators Pparα, Pparγ and Srebf1.
G. Heatmap representation of the expression levels of 366 cycling genes in WT-FA and 172 cycling genes in WT-FT and their corresponding expression in clock deficient mice as indicated.
H. Heatmap representation of the expression level of 43 cycling genes in WT-FA and WT-FT and their corresponding expression in clock deficient mice as indicated.
I. Examples of cycling genes in WT.
B,D,E: differentially expressed genes with adjusted p value<0.05.
C,F: differentially expressed genes with feeding_adjusted p value<0.01 and adjusted p interaction > 0.1.
First, because the different mutants displayed genotype-specific differences on FA, we compared WT to each mutant on FA to identify metabolic pathways that were specifically affected by each clock component with the caveat that we only used one WT cohort (Bmal1LWT). The number of genes that were differentially expressed (adjusted p ≤ 0.05) between Bmal1LKO, Rev-erbα/βLDKO, and CDKO versus WT were 2922, 5003, and 6448, respectively (figure 6B). Amongst them, 1402 genes were commonly modulated in all three mutants, suggesting that a functional clock is required for their normal expression. Examples of those include increased expression of the tumor suppressor Pten in all the mutants, and decreased expression of genes involved in lipid metabolism, such as Apoe, Fabp1, and Slc25a1 in all the mutants compared to the WT (figure S5A).
The genotype-specific differences in gene expression may explain genotype-specific metabolic defects. Functional annotation of transcripts (figure S6) that were differentially expressed between WT and Bmal1LKO livers revealed that many genes involved in lipid metabolism, particularly fatty acid oxidation (Cpt2, Acads, Acadm, Acadl, Acadvl, Hadh, Hadha, Gpd1, Gpd1l, Lpin2) were downregulated in Bmal1LKO on FA (figure 6D and S5C). Their expression changes correlated with relatively severe hyperlipidemia observed in Bmal1 mutant animals (figure 3). For instance, Gpd1 loss of function is associated with hypertriglyceridemia in infants (Joshi et al., 2014). In Rev-erbα/βLDKO-FA compared to WT-FA, many genes belonging to cholesterol homeostasis were downregulated (figure 6E and S5C). In particular, reduced expression of Fxr, the master regulator the conversion of cholesterol to bile acids and of rate-limiting enzymes of the pathway (Cyp27a1, Cyp7a1), as well as rate-limiting enzymes involved in the conversion of cholesterol to steroid hormones (Hsd11b1, Hsd17b2, Hsd17b12, Srd5a1, Fdx1) may explain hypercholesterolemia of Rev-erbα/βLDKO mice. Those changes in gene expression between Rev-erb knockout and WT animals fed HFD were in agreement with those observed when these mice were fed a normal chow in a published study {Cho, 2012 #3109} (figure S5E). The expression levels of genes encoding key regulators and enzymes of fat metabolism (figure 6F, see Pparγ, Cidec, Gpat2) were not as elevated in the CDKO-FA mice as they were in the livers of WT, Bmal1LKO and Rev-erbα/βLDKO livers under FA. Such CDKO-specific gene expression changes correlated with the lack of fatty liver diseases in CDKO mice. In summary, these genotype-specific expression differences likely contributed to genotype-specific differences in metabolic defects under FA. Many of these changes in the expression of metabolic genes are also found in circadian mutant mice fed a normal chow.
Next, we interrogated gene expression changes between FA and FT cohorts that might explain the beneficial effects of FT within each genotype (figure 6B). One simple hypothesis was that the genotype-specific gene expression changes in each mutant relative to WT, as described in the previous paragraph, were reversed by TRF. There were 257 differentially expressed genes in the livers of Bmal1LKO mice on FA versus FT (adjusted p ≤ 0.1). Among these, 68 were previously identified as altered in Bmal1LKO. There were 1505 differentially expressed genes in the livers of Rev-erbα/βLDKO mice on FA versus FT (adjusted p ≤ 0.1), (771 up in FA, 734 up in FT). Among these, 554 were previously identified as altered in Rev-erbα/βLDKO. There were 30 genes different between CDKO on FA versus FT (adjusted p ≤ 0.1); 14 were previously identified as altered in CDKO (relative to WT). We hypothesized that, within genotype, TRF could alleviate genotype-specific gene-expression differences by sustaining the expression of some transcripts to WT levels. In other words, we analyzed genes with a “favorable” profile, namely those genes for which the TRF effect was identical (either up or down) in WT and mutants (examples are shown in figure S5D). There were only 33 genes with a favorable profile in Bmal1LKO, 368 in Rev-erbα/βLDKO, and 4 in CDKO. Functional annotation of these transcripts did not explain a significant portion of TRF benefits. In summary, these analyses suggest that TRF did not correct the majority of genotype-specific gene expression changes found under ad lib feeding conditions.
TRF normalized body weight, adiposity, and metabolic profiles in all genotypes. DIO over 12 weeks time does not reach a steady state for these health parameters and TRF over the same time period does not leave legacy benefits. Therefore, gene expression patterns in these mice reflect active molecular processes that promote or prevent metabolic diseases under FA and FT. We thus hypothesized that the gene expression changes (FT vs FA) that were shared among the four genotypes could underlie TRF benefits. To isolate the generic effect of TRF regardless of the genotype, we introduced an interaction term in the design formula used for statistical analysis. There were 6178 genes different between FA versus FT (adjusted p < 0.1) in at least one genotype. Among these, 2142 genes showed a similar trend in all genotypes (i.e., they did not exhibit a significant interaction between genotype and feeding condition; adjusted p interaction > 0.1); 927 had higher expression under FA, and 1215 were increased under FT (figure 6C). Pathway enrichment analysis was carried out using Metascape on each set, and the data were manually curated to control overlapping or redundant GO terms (there were no more than 3 genes that were shared between any two clusters). Genes that were significantly upregulated in FA and reduced in FT were enriched in lipid metabolism genes (figure 6F), including β-oxidation (Acox1, Acad10, Hadh, Acaa1b), de novo lipogenesis (Fasn, Scd3, Elovl5), triglyceride synthesis (Mogat1, Gpam, Gpat12, Agpat14), and TG storage (Plin2, Cidec). Master regulators of lipid metabolism, such as Pparα, Pparγ, and Srebf1, were also higher in FA (figure 6F, insert).
Transcripts that were higher in FT relative to FA could be grouped into 5 master categories involved in the maintenance of DNA/RNA/protein integrity, or cellular defense mechanisms. These categories were: (i) DNA metabolism and transcription (83 genes, blue bars), (ii) RNA processing (114 genes, yellow bars), (iii) protein metabolism (112 genes, dark grey bars), (iv) protein folding and the unfolded protein response (94 genes, light grey bars), and (v) defense response and coagulation (113 genes, red bars) (figure 7A). (i) The DNA metabolism and transcription group included genes involved in telomere protection (Terf1), DNA damage protection (Parp9, figure 7B), nucleotide excision repair (Ercc5, Lig3, figure 7B), homologous recombination (Brca1), as well as numerous components of the general RNA polymerase II transcription machinery. (ii) The RNA processing group included genes involved in the processing of mitochondrial RNA (Pnpt1, Trmt10c), the processing of nuclear RNA (Papola (figure 7B), Cpsf2), the assembly and splicing of snRNP particles (Prmt5, Plrg1, figure 7B), and finally the processing of rRNA, as well as the export and maturation of ribosomes (Lsg1, Mphosph10). (iii) The protein metabolism group included genes involved in intracellular traffic and secretion (members of the signal recognition particle complex (SRP) and members of the signal peptidase complex (Spcs2)) (figure 7B), organelle integrity, and GPI-anchor biosynthesis (Piga, Pigf, figure 7B). (iv) The protein folding and unfolded protein response group included genes involved in protein folding, such as chaperones (4 subunits of the TCP-1 Ring Complex, Cct3 (figure 7B), and many heat shock proteins (Hsp90aa1, Hsp90b1, Cdc37l1, Hspa5, Hspe1, Dnaja1, Dnaja3, Dnaja4, Dnajb11)), as well as proteins involved in disulfide bond homeostasis/maintenance of cellular redox state, such as protein disulfide isomerase, thioredoxin, and peroxiredoxin superfamilies. This group also contained protein involved in protein degradation (proteasome subunits, E2 and E3 ubiquitine ligase, DUBs, and ERAD proteins) and regulation of the unfolded protein response (UPR), such as the main regulator XBP-1 (figure 7B). (v) The defense response and coagulation group included complement factors and their inhibitors (C6, Cfi), the serpin family of protease inhibitors, cytokine and chemokine sensors (Il1r1), and signaling molecules (Tirap, Nlrp12, figure 7B). In summary, transcripts that were elevated under FT condition in all genotypes were involved in cellular response to stress.
Figure 7: Liver function is preserved in TRF and diurnal rhythms in nutrient sensing pathway is preserved in all clock mutants on TRF.

A. Pathway enrichment analysis results of 2142 genes that are significantly higher in TRF. Related pathways are colored similarly.
B. Heatmap representation of the expression levels of the significant genes in TRF belonging to DNA metabolism and transcription (83 genes, blue bars in 7.A.) (ii) RNA processing (114 genes, yellow bars in 7.A.), (iii) Protein metabolism (112 genes, dark grey bars in 7.A.), (iv) Protein folding and unfolded protein response (94 genes, light grey bars in 7.A.), and (v) Defense response and coagulation (113 genes, red bars in 7.A.) and selected individual expression profile (below).
C-D. Representative western blot and graphical representation of the quantification (ImageJ) of the temporal expression profile of (C) phospho-ERK/ERK, GCK and active SREBP (cleaved form) and (D) the phosphorylated ribosomal protein S6 (P-S6), in the liver of clock deficient mice and control in FA versus FT as indicated.
E. Diurnal levels of the metabolite tryptophan in the liver of clock deficient mice and control in FA versus FT as indicated.
F. Diurnal levels of Gabarapl1 expression in the liver of clock deficient mice and control in FA versus FT as indicated.
The above analyses were performed without using time of tissue collection as a variable. However, as visualized with the RER, whole animal metabolism showed a clear diurnal rhythm, suggesting potential concomitant changes in the temporal pattern of gene expression. Therefore, we characterized the diurnal expression profile of liver by analyzing a time series of gene expression from WT mice under FA or FT (samples from 2 mice per time point were sequenced independently). This sampling frequency (every 3 or 4 h) and sampling duration (24 h) under light:dark cycle is sufficient to identify a representation of rhythmic transcripts (Hughes et al., 2017) to test: (i) the impact of eating pattern on daily rhythms of expression under FA and FT, and (ii) whether TRF drives rhythmic expression in mice lacking a circadian rhythm. In WT liver, we scored 366 and 172 cycling genes in FA and FT, respectively (Meta2d_pvalue < 0.01) (figure 6G). The increased number of rhythmic transcripts in the WT FA liver was partly due to increased and rhythmic expression of PPARγ, PPAR α, SREBF1, and their target genes (e.g., Lpin2, Agpat2, figure 6I) as was described recently (Eckel-Mahan et al., 2013). FA also increased the expression of PPARγ in circadian mutant mice (but did not induce rhythm) and FT reduced its overall expression (figure 6F). In WT mice, 43 genes showed rhythmic expression in both FA and FT conditions. This subset included known clock (Bmal1, Cry1, Rev-erb) and clock output genes (Nampt, Cyp7a1 figures 6H and 6I). As was reported earlier (Hatori et al., 2012), some transcripts oscillate with higher amplitudes in WT FT liver (e.g., Nampt), but others oscillate with higher amplitudes in WT FA liver (e.g., Rorc) (figure 6I). These common cyclers did not show any apparent rhythmic expression profile in the liver of circadian mutant mice under FT (figure 6H and figure S7). However, a subset of transcripts that cycled in WT FT liver also appeared rhythmic in mutant FT liver. These transcripts were most likely driven by the imposed feeding-fasting rhythm (figure 6G). Overall, TRF did not support rhythmic mRNA expression for a large number of transcripts in circadian mutant mice. However, time-series proteomics and phosphoproteomics experiments have shown that a significant proportion of the hepatic proteome shows post-translational changes in WT liver, without accompanying changes at the mRNA level (Robles et al., 2014; Robles et al., 2016). Therefore, it is likely that TRF exerts some effects at the post-transcriptional level.
Diurnal rhythms in nutrient sensing pathways in clock mutants under time-restricted feeding.
In normal conditions, the metabolic switch between anabolic and catabolic reactions follows the feeding and fasting state. In the liver, it is tightly controlled by insulin and growth factors such as FGF15/19, which are active in the fed state and downregulated in the fasted state. We analyzed phosphorylation of ERK (P-ERK) as a read-out of growth factor signaling and phosphorylation of the ribosomal protein S6 (P-S6) as a read-out of the convergence of insulin signal transduction and the nutrient-sensing pathways AMPK and mTOR (Efeyan et al., 2015). Western blot analysis showed a diurnal rhythm in P-ERK/ERK in Rev-erbβ-LWT, Rev-erbα/βLDKO and CDKO (figure 7C) (not tested in Bmal1LKO). P-S6 (figure 7D) cycled in all mice on TRF, with peak intensity around ZT18 (during feeding) and a trough around ZT3 (during fasting). Expression of glucokinase and activation of SREBP1 (cleaved form) followed the same diurnal profile in Bmal1LKO mice on FT (figure 7C). The diurnal profile of activation of P-ERK and P-S6, as well as GCK and SREBP activation, was either flat or reversed in FA mice, with high expression/activation during the light phase. The hepatic abundance of some metabolites, especially amino acids, also displayed a diurnal rhythm. For instance, tryptophan levels (figure 7E), and to a lesser extent leucine and phenylalanine, peaked just before feeding. These amino-acids are known activators of mTORC1 and matched the observed profile of P-S6 activation Finally, at the transcriptional level, when sampling time points were grouped according to subjective feeding (ZT15–ZT24) and subjective fasting (ZT3–ZT12) groups, the proportion of genes that were differentially affected between the feeding and fasting times was higher in FT, especially in the DNA (25%), RNA (26%), and protein (29%) metabolism groups, as well as for the protein folding group (36%), compared to other groups (6% for lipid genes in FA, and 12% for all significant genes). In addition, pathway enrichment analysis revealed that autophagy was significantly different between the fasted and fed state in FT, consistent with mTOR pathway activation. For instance, Atg8 homolog Gabarapl1 transcript levels, a key regulator of autophagy flux, decreased during feeding and rose with fasting (figure 7F). Thus, the abundances of metabolites, transcripts, and proteins, as well as posttranslational activation of nutrient signaling pathways, showed a consistent molecular feeding-fasting rhythm in FT that was absent in FA.
DISCUSSION
Circadian rhythms in gene functions temporally separates incompatible metabolic processes and are considered integral to metabolic homeostasis. Increased predisposition to metabolic diseases in animals with genetic disruption of the circadian clock (either in the whole organism or in specific organs) supports this thesis. Similarly, behavioral perturbations of circadian rhythms, as in shiftworkers or in animal models of shiftwork, predispose these individuals to metabolic diseases. However, an imposed daily rhythm in feeding and fasting can independently drive daily rhythms in the expression of genes mediating nutrient metabolism in mice lacking a circadian clock (Vollmers et al., 2009). As normal feeding-fasting rhythms are altered in many circadian mutant mice, both cell-autonomous circadian regulation and feeding-fasting regulated processes are disrupted in these experimental models. Therefore, we used an imposed feeding-fasting cycle to examine the relative contributions of the molecular circadian clock and feeding-fasting driven programs in metabolic homeostasis.
Here we analyzed mice lacking three different components of the circadian clock, each of which develop metabolic diseases when provided ad libitum access to food (Barclay et al., 2013; Bugge et al., 2012; Cho et al., 2012; Lamia et al., 2008). BMAL1 is an indispensable transcriptional activator (Bunger et al., 2000). Cry1 and Cry2 have overlapping functions in repressing Bmal1 function (Griffin et al., 1999; Kume et al., 1999; van der Horst et al., 1999), and in interacting with AMPK, glucocorticoid receptor (GR), and glucagon functions in the liver (Lamia et al., 2009; Lamia et al., 2008; Zhang et al., 2010). Rev-erb-α and -β have overlapping transcriptional repressor roles, both for Bmal1 and Cry, as well as non-clock repressive roles in the liver (Bugge et al., 2012; Cho et al., 2012; Delezie et al., 2012). Although metabolic phenotypes of these three mutants have been independently examined under ad libitum-fed conditions, this is the first direct comparison of these mutants fed an identical diet under ad lib and TRF paradigms. This allowed direct comparison of their disease phenotypes and assessment of their individual contributions to metabolic dysfunction.
Body weight gain and whole-body RER in mutant mice are normalized by TRF
Ad libitum feeding of HFD is a sensitizer for body weight gain and metabolic diseases and is often used to model human obesity and metabolic diseases. Therefore, it is a relevant and preferred diet for comparing different genotypes of mice that show metabolic diseases of varying severity under normal chow. As expected, WT cohorts rapidly gained weight under FA. The body weight of both Bmal1LKO and Rev-erbâ/βLDKO mice were not significantly different from that of their WT counterparts up to 12 weeks. CDKO mice are known to be smaller than their WT cohorts, yet they also disproportionately gained weight under FA conditions. Therefore, under such nutritionally sensitized conditions, circadian mutant mice were not disproportionately more prone to body weight gain. This was not because the HFD masked the effect of individual clock mutations, as the underlying transcriptome signatures and metabolic phenotypes were unique to each mutant (discussed later).
WT mice fed a normal diet ad lib usually show a diurnal rhythm in eating and a parallel rhythm in respiratory exchange ratio (RER), whereas HFD is known to blunt these rhythms. The mutant mice also showed blunted RER rhythm under FA. The RER rhythm reflects the pattern of whole-body fuel utilization, which is an overall product of a glucose utilization rhythm (peaking during the fed state), and a fat utilization rhythm (peaking during the fasted state). As direct targets of the circadian clock mediate key rate-limiting steps in both sugar and fat metabolism in the liver and other tissues, a normal circadian clock is presumed to contribute to the RER rhythm. However, under FT condition, all three mutants showed robust rhythms in RER. Individual clock components also contribute to this overall rhythm. As seen earlier with normal chow (Vollmers et al., 2009), CDKO mice consistently showed a steeper decline in RER during fasting, which reflects increased utilization of lipids as an energy source. Overall, at the whole organism level, eating pattern functions as a dominant driver of fuel-utilization rhythms. Since the RER rhythm and metabolic flexibility are considered hallmarks of health, and their dampening correlates with metabolic diseases and aging (Houtkooper et al., 2011; Riera and Dillin, 2015), an imposed feeding-fasting cycle can function as an extrinsic driver of this daily metabolic switch.
Clock mutant mice have unique metabolic defects
Under HFD ad lib feeding, individual circadian mutants showed unique signatures in the severity of specific metabolic parameters. As seen earlier for Rev-erbα/βLDKO on normal chow (Bugge et al., 2012; Cho et al., 2012) and Rev-erbα−/− on HFD (Delezie et al., 2012), Rev-erbα/βLDKO mice showed increased liver steatosis compared to littermate controls and all other mutants. Additionally, Rev-erbα/βLDKO mice display elevated serum cholesterol compared to other cohorts. Serum TGs were specifically elevated in Bmal1LKO mice (as described by (Jacobi et al., 2015)) and HFD-fed Bmal1LKO mice were more efficient at sustaining glucose homeostasis than littermate controls (as observed under normal chow (Lamia et al., 2008)). Increased glucose clearance in Bmal1LKO mice was associated with high levels of fasting insulin and increased insulin sensitivity. Finally, CDKO mice did not develop severe liver steatosis, but H&E staining of adipose tissue suggested more inflammation in these mice than in other mutants; consistent with its role as a suppressor of inflammation (Narasimamurthy et al., 2012). Relative to their smaller size, they also displayed severe glucose intolerance compared to their littermate controls.
Since the liver plays a major role in metabolism, and metabolic defects in these three mutants are often linked to altered liver physiology, we assessed liver transcriptomes in these three mutants. As seen with the distinct metabolic phenotypes, the liver transcriptomes of these three mutants were also clearly separated along two-dimensional PCA, further supporting the notion that the obesogenic HFD did not mask the individual contribution of each gene to the liver transcriptome. Closer examination of liver transcripts revealed genotype-specific changes in the expression of key genes that have been mechanistically linked to the metabolism of sugar, lipids, and cholesterol. Specific gene expression changes in cholesterol pathway regulators and rate limiting enzymes may be responsible for reduced hepatic clearance of cholesterol and associated hypercholesterolemia in Rev-erbα/βLDKO mice. Similarly, reduced expression of genes implicated in fatty acid oxidation may explain increased hepatic lipid content in Bmal1LKO and Rev-erbα/βLDKO mice.
TRF acts through multiple mechanisms to protect against nutritional challenges and bolster cellular stress responses
TRF prevented body weight gain associated with ad lib HFD feeding in all genotypes without reducing food intake or increasing activity. It is conceivable that some of the TRF benefits in the liver-specific knockout mice may have originated from extra-hepatic tissues that still have a normal circadian clock. However, body weight reduction was also found in CDKO mice that lacked a functional clock in all tissues. TRF prevented fat accumulation in adipose tissues, reducing circulating leptin and increasing adiponectin in all genotypes.
In the liver, it may be argued that TRF simply reversed gene expression changes that resulted from the loss of specific clock components, so that the FT condition reinforced a genomic program that was similar to that of WT liver under FT. However, PCA analysis of liver transcriptomes argued against this mechanism. Functional annotation of the liver transcriptome revealed two principal trends under FT: changes in macronutrient metabolism networks, and changes in cellular stress responses.
Macronutrient balance can be regulated at several levels, including synthesis, oxidation, absorption, and excretion. TRF could prevent nutritional imbalance either by acting on the same components that are dysregulated in clock mutants, or by acting at another point in the regulatory network. Mice on TRF were protected from aberrant transcriptional activation of genes involved in lipid metabolism and from increased lipid flux to de novo lipogenesis and lipid storage, all of which are associated with fatty liver disease (Fabbrini et al., 2010). Metabolomic analysis confirmed a reduction in free fatty acids in the liver under TRF conditions. It further showed that citrate was depleted in ad lib fed liver. Interestingly, an increased α-KG/citrate ratio, as observed in FA liver, drives reductive glutamine metabolism, whereby glutamine is converted to α-KG and citrate to fuel de novo lipogenesis (Fendt et al., 2013). Expression of enzymes of the TCA cycle was largely similar between FA and FT livers. As many enzymes of the TCA cycle are regulated by allosteric mechanisms indicative of the energy state of the cell, changes in TCA cycle intermediates under FA and FT reflect a change in energy state imposed by TRF, and is independent of a functioning clock.
TRF may also lessen metabolic syndrome by strengthening cellular mechanisms for coping with the stress of lipid and glucose overload. Alterations in these mechanisms are directly involved in fatty liver and glucose intolerance pathologies (Wellen and Thompson, 2010). Genes that are more expressed in TRF than ad lib-fed animals are involved in general cellular maintenance pathways, such as macromolecule quality control and repair pathways (DNA, RNA, proteins), as well as cellular defense mechanisms. Furthermore, destabilization of many of these pathways is sufficient to cause metabolic disease (Lee and Ozcan, 2014; Ozcan and Tabas, 2012; Shimizu et al., 2014).
Absolute transcriptional changes are more prominent than feeding-fasting driven gene expression rhythms under TRF
A basic paradigm of circadian regulation of metabolism is that rhythmic patterns of gene expression drive rhythms in cellular metabolism. TRF is known to further increase the number of rhythmic transcripts in WT mice fed a normal diet. However, under FA, increased expression of PPARγ and its newly acquired rhythm contributes to the increased number of rhythmic transcripts (Eckel-Mahan et al., 2013) relative to mice fed a normal diet. As seen earlier (Chaix et al., 2014), FT reduced PPARγ expression in the WT liver and reduced the number of genes exhibiting rhythmic expression patterns (Figure 6). In the knockout livers, Pparγ was expressed at a higher level under FA than under FT, but there was no apparent daily rhythm in its expression under either condition. Although FT conditions drove daily rhythms in a number of transcripts in the knockout livers, they were less pronounced than in WT liver.
Transcripts that oscillate in WT mice under FT did not display significant oscillations in clock mutants on TRF, suggesting that a larger effect of TRF in mutant mice was on levels of expression, rather than their temporal profile. However, FT might drive rhythms in post-translational protein modifications, which are known to oscillate with larger amplitudes than mRNA or metabolite rhythms (Robles et al., 2016). Proteins that exhibit strong rhythms in post-translational modification respond to feeding and fasting signals and regulate downstream effectors involved in macromolecule and organelle homeostasis. Concomitantly, we observed a diurnal rhythm in nutrient sensing pathway activation, in particular activation of the mTORC1 pathway and in amino acids known to engage this pathway (phenylalanine, tryptophan, and leucine). The mTORC1 pathway plays a crucial role in coupling cellular nutritional status to ribosome biogenesis and protein synthesis, which are some of the most energy-consuming pathways in a cell. Interestingly, mTORC1 also plays a major role in activating the integrated stress response (ISR), a physiological cellular response that allows restoration of homeostasis following exposure to a variety of stressors. One of the hallmarks of ISR activation is inhibition of general protein translation while inducing the transcription and translation of specific mRNAs, mostly encoding chaperones. Many chaperones specifically expressed in FT are integral to ISR (e.g., the TCP family). Remarkably, TCP chaperones are necessary for TRF benefits in Drosophila (Gill et al., 2015). This suggests that enforcing nutrient utilization rhythms by TRF leads to rhythmic activation of the mTORC1 and ISR pathways. This allows hepatocytes to recover from the stress of HFD feeding, thereby preserving cellular homeostasis and proper metabolic function.
Conclusion and Limitations
In conclusion, we found that TRF for up to 12 weeks is sufficient to sustain metabolic health in relatively young clock-deficient mice, suggesting that imposed feeding-fasting rhythms and associated metabolic oscillations can override otherwise compromised rhythms. However, whether TRF can exert similar protection over prolonged period or can reverse obesity and metabolic diseases in older circadian mutant animals is to be empirically determined. One of the functions of the circadian clock is the regulation of eating behavior. Although it is easy to impose a strict TRF on animals, it is premature to conclude that humans with circadian rhythm defects who may be predisposed to aberrant eating pattern can voluntarily impose a strict time restricted eating (TRE) protocol to prevent or reverse metabolic diseases. Despite the limitations of this study, these findings have important translational applications, as it opens new avenue to test the preservation of metabolic homeostasis by TRE in populations with dampened circadian rhythms, such as shiftworkers and the elderly.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Satchidananda Panda (panda@salk.edu).
Experimental Model and Subject Details
Animals and diets.
All animal experiments were carried out in accordance with the guidelines of the IACUC of the Salk Institute. Liver-specific knock out (KO) of Bmal1 and Rev-erbα/β were generated at Salk Institute by breeding AlbuminCre mice (JAX laboratory) with Bmal1LoxP (JAX laboratory, (Lamia et al., 2008)) and Rev-erbα/β double floxed mice (gift from Dr Evans, (Cho et al., 2012)) respectively. Within each cohort, Cre recombinase negative littermates mice were used as controls. Whole body clock mutant Cry1;Cry2 double KO (CDKO) were obtained from the Sancar lab and were back crossed to C57/B6 background >5 times. The genotype of the animals was confirmed by PCR. Male mice at 6–10 weeks of age were entrained to a 12h light: 12h dark cycle with normal chow food available ad libitum for 2 weeks before being randomly assigned to a 60% high fat diet (TestDiet-58Y1) ad libitum group (FA) or a high fat diet time restricted feeding group (FT). The FT group had access to food for 9-10 hours during the dark active phase, from ZT13 to ZT21/22 where ZT0 denotes light on. Food intake and body weight were monitored weekly throughout the 12 weeks experiments. The food access durations were readjusted weekly to ensure isocaloric consumption in all groups (plus or minus a maximum of one hour). The experimental setup and abbreviations used to define the 12 animal cohorts are depicted in figure 1A, 1D and 1G for Bmal1 liver KO (Bmal1LKO), Rev-erbα/β liver double KO (Rev-erbα/βLDKO), CDKO and littermate control mice Bmal1-LWT, Rev-erbα/β-LWT, CC respectively.
Method Details
Body composition
Body composition was analyzed in live mice using a body composition analyzer (EchoMRI™-100H).
Indirect calorimetry, food intake and activity
Food intake, locomotor activity, oxygen consumption and carbon dioxide production were simultaneously measured for individually housed mice with a LabMaster system (TSE Systems). Mice were acclimatized for 2-3 days, data were collected for 4-5 days and analyzed. Light and feeding conditions were kept the same as in the home cages.
Liver samples collection
After 12 weeks on the feeding paradigm, animals were sacrificed and liver samples collected every 3 hours for CDKO/CC and Rev-erbα/βLDKO/LWT mice and every 4 hours for Bmal1LWT and Bmal1LKO over 24h. This collection was repeated twice for each genotype, giving two entirely independent biological replicates at each time point for each genotype. Samples were flash frozen, ground to fine powder in liquid nitrogen and stored at −80°C for further analysis.
Hepatic triglycerides quantification
Frozen liver powder was homogenized in isopropanol and triglyceride concentration was measured using an enzymatic assay (Triglycerides LiquiColor, Stanbio). Data were normalized to liver weight.
Histology
Sections (6 μm) of formalin-fixed liver, epididimal WAT and BAT were stained with H&E and imaged under a light microscope.
Serum biochemistry
After 6-10 weeks on the feeding regimen, blood was obtained from fasted (ZT22-ZT38) or refed mice (one hour after intraperitoneal glucose injection (1 mg/g body weight) at ZT37). Triglycerides, glucose, and total cholesterol were measured using Thermo Scientific Infinity Reagents. Insulin, leptin, and adiponectin were quantified using Meso Scale Discovery immunoassays.
Glucose and insulin tolerance tests (GTT & ITT)
After 6-10 weeks on the feeding regimen, mice were fasted in paper bedding for 16 hours (ZT22-ZT38) or 3 hours (ZT13-16) for glucose or insulin tolerance tests, respectively. Glucose (1g/kg body weight) and Insulin (0.5-1U/kg body weight) were injected intraperitonally. Blood glucose level was measured using OneTouch Ultra glucose meter prior to injection and several times after injection as indicated.
Western blotting
Total liver lysates were prepared in RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 1 mM Na3VO4) supplemented with protease and phosphatase inhibitor cocktails (Complete and PhosSTOP tablets, Roche). Membranes were probed with antibodies directed against phospho-S6 (S235/236) (CST 4858), S6 (CST 2217), phospho-ERK (Sc-7383), ERK (Sc-94), GCK (Sc-7908), SREBP-1 (BD clone IgG-2A4), and Actin (ab1801).
Liver metabolomics
Liver samples were collected every 4 hours over 24 hours for WT (Bmal1LWT) and Bmal1LKO (6 time points per group) and every 3 hours over 24 hours for Rev-erbα/βLDKO and CDKO (8 time points per group) under natural housing and feeding conditions. For all the genotypes, equal weight of frozen liver powder from 2 mice obtained from two independent feeding exepriments per time point were pooled and used for detection and relative quantification of metabolites by Metabolon as described (Evans et al., 2009). Up to 547 metabolites were detected, amongst which 444 had no more than one missing value within each group. Three different approaches were used for statistical analysis of this dataset. (1) To analyze the effect of TRF within genotype, time-series samples were treated as replicates. Metabolites with >50% missing values per group were removed for all groups and other missing values were replaced by a small value (half of the minimum positive value in the original data). Data were not filtered nor normalized but log transformed. PCA and heatmaps were generated with R. Statistical and pathway enrichment analyses were performed using Metaboanalyst (Xia et al., 2015) with a list of 437 metabolites with HMDB IDs. (2) To analyze the effect of TRF across genotype, time-series samples were treated as replicates and two independent methods giving the same results were used: (2.1) we used the list of 444 metabolites presented above, a custom linear model with the 2 factors genotype and feeding group and a rank analysis using R and (2.2) a list of 319 metabolites with no missing values in any of the groups and a two-factor independent sample ANOVA analysis available in the new release of Metaboanalyst. PCA and pathway enrichment analysis were performed using Metaboanalyst (Xia et al., 2015). Both methods led to the same top 2 functional pathways. (3) To analyze the effect of TRF on the daily temporal dynamic of metabolites levels, we used MetaCycle package (Wu et al., 2016) to identify cycling metabolites in all groups independently in the list of 319 metabolites with no missing values in any of the group. Analyses were performed using the meta2d function, incorporating JTK_Cycle and Lomb-Scragle methods and Meta2d p value was computed using fisher's method. Data were not duplicated nor concatenated (Hughes et al., 2017).
Liver transcriptomics
Total RNA were prepared using Qiagen RNeasy Mini Kit and libraries were prepared using Illumina’s TruSeq Stranded mRNA HT kit according to manufacturer’s instructions. In brief, total RNA starting with 1μg was poly-A+ selected, fragmented by metal-ion hydrolysis and then converted to cDNA using Superscript II. The cDNA was then end-repaired, adenylated and ligated with Illumina sequencing adapters. Finally, the libraries were enriched by PCR amplification. Libraries were pooled into groups of 12 to a lane and sequenced using an Illumina HiSeq 2500 with 50-bp single-read chemistry. Sequenced reads were mapped to the GRCm38 genome with STAR (Dobin et al., 2013). Gene-level read counts were generated using featureCounts (Liao et al., 2014) and GENCODE gene annotation.
For Bmal1LKO, Rev-erbα/βLDKO and CDKO, RNA from 2 independent mice per time point were pooled before library preparation. For WT, 2 mice per time point were sequenced independently. Analysis of differential expression was carried out using DESeq2 (Love et al., 2014), with designs that accounted for genotype (WT(pooled duplicates), Bmal1LKO, Rev-erbα/βLDKO or CDKO), feeding group (FA, FT) and time of sacrifice (subjective fast=ZT3-ZT12; subjective fed= ZT15-ZT24). Statistical significance was assessed using a negative binomial Wald test, then corrected for multiple hypothesis testing with the Benjamini-Hochberg method. For visualizations, a regularized logarithm transformation (Love et al., 2014) was applied to the gene-level read counts. Gene expression heatmaps were generated from these transformed values following mean-centering and hierarchical clustering. Principal component analysis was performed using the 500 genes with highest variance across all samples.
To analyze the temporal dynamic of gene expression, we used MetaCycle (Wu et al., 2016). Analysis were performed using the meta2d function, incorporating JTK_Cycle and Lomb-Scragle methods and Meta2d p value was computed using fisher's method. Data were not duplicated nor concatenated (Hughes et al., 2017).
Performance assays
The treadmill exhaustion test was performed using the Exer 6M first generation treadmill (Columbus Instruments) at ZT 15. On day 1, mice were trained at low speed for 15 minutes with speed ramping up every 5 minutes. On day 2, mice were trained for 20 minutes at intermediate speed with 5 minutes of ramping. On day 3, maximal speed was reached by ramping up every 5 minutes and, when applicable, a 5° angle incline was applied to the treadmill when running time exceeded 1 hour. Mice were run until exhaustion, which was defined as the inability to continue running despite repeated stimulation by compressed air
Quantification and Statistical Analysis
Two-tailed Student’s test was used for pair-wise comparisons and ANOVA with post hoc tests was used for data with more than 2 groups (correction for multiple comparisons was done using the Tucky method). For time series, repeated measured ANOVA with post hoc tests was used to compare to the control group. Statistics were calculated using GraphPad Prism 6.0. Unless otherwise noted, throughout all figures, data are presented as mean ± SEM with statistical result of the statistical test, with *p < 0.05, **p < 0.01, ***p < 0.001. Statistical parameters, including the value of n, are noted in figure legends. Statistical significance was concluded at p < 0.05. A detailed description of the different statistical models used for metabolomics and transcriptome data analysis are provided in the method section.
Data and Software Availability
The accession number for the liver RNA-seq data is GEO: GSE102072.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phospho-S6 | CST | Cell Signaling Technology Cat# 2215L, RRID:AB_331683 |
| total S6 | CST | Cell Signaling Technology Cat# 2217, RRID:AB_331355 |
| phospho-ERK | CST | Cell Signaling Technology Cat# 4370 |
| total ERK | Santa Cruz | Santa Cruz Biotechnology Cat# sc-94, RRID:AB_2140110 |
| GCK | Santa Cruz | Santa Cruz Biotechnology Cat# sc-7908, RRID:AB_2107620 |
| SREBP-1 | BD Pharmingen | clone IgG-2A4 |
| Actin | Abcam | Abcam Cat# ab1801, RRID:AB_302617 |
| Chemicals, Peptides, Recombinant Proteins | ||
| Insulin (Humulin R) | Lilly | NDC 0002-8215-01 |
| D-Glucose | Sigma | G8270 |
| AMPure XP beads | Beckman Coulter | A63881 |
| TruSeq Stranded mRNA Library Prep | Illumina | 20020595 |
| SuperScript II | Invitrogen | 18064022 |
| Critical Commercial Assays | ||
| Glucose, serum | ThermoFisher Scientific, Infinity Glucose Hexokinase |
TR15421 |
| Cholesterol, serum | ThermoFisher Scientific, Infinity Cholesterol |
TR13421 |
| Triglycerides, serum | ThermoFisher Scientific, Infinity Triglycerides |
TR22421 |
| Triglycerides, liver | Stanbio™ Triglycerides LiquiColor™ Test (Mono) Reagents |
SB2200225 |
| Insulin, Leptin | MSD, Mouse metabolic kit | K15124C |
| Adiponectin | MSD, Mouse adiponectin kit | K152BXC |
| Deposited Data | ||
| Liver RNA seq | This study | GEO: GSE102072 |
| Experimental Models: Organisms/Strains | ||
| Cry1;Cry2 double KO (CDKO) | Gift from Dr. Sancar, Vitaterna et al., 1999 |
|
| Rev-erbα/β double floxed mice | Gift from Dr. Evans, Cho et al., 2012 | |
| Bmal1l°x | Storch et al., 2007 | Jax strain #007668; RRID:IMSR_JAX:007668 |
| Albumin-Cre | Postic et al., 1999 | Jax strain #003574; RRID:IMSR_JAX:003574 |
| Oligonucleotides | ||
| Genotyping primers | ||
| Rev-erbα - Fw: CATTACCAGTAAGTCAATGCCCAGC |
Cho at al., 2012 | |
| Rev-erbα - Rev: GAAGAGTGTGTGTTTGCCCAAGAGG |
Cho at al., 2012 | |
| Rev-erbβ - Fw: TCATCGCTCCAGTCTCCTACATTTC |
Cho at al., 2012 | |
| Rev-erbβ - Rev: GGAAGTGCTCCAACAAGGTAGTGCA |
Cho at al., 2012 | |
| Cry1_WT_Fwd1: GATTCACTGTGTGTTGAGCAAGG |
||
| Cry1_exonb_Rev1: CTTGAGTGAGTGAGTCTGCTG |
||
| Cry1_Neo_Fwd: CCTTCTATCGCCTTCTTGACGAG |
||
| Cry2_Rev: CACACTGAAATCGGCATCCAG |
||
| Cry2_Fwd1: GCAAGACAGGCTTCCCTTGG |
||
| Bmal1 - Fw: ACTGGAAGTAACTTTATCAAACTG |
Storch et al., 2007 | JAX oIMR7525 |
| Bmal1 - Rev: CTGACCAACTTGCTAACAATTA |
Storch et al., 2007 | JAX oIMR7526 |
| Cre - Fw: GAACCTGATGGACATGTTCAG |
||
| Cre - Rev: AGTGCGTTCGAACGCTAGAGC |
||
| Software and Algorithms | ||
| Fiji (ImageJ) | NIH | RRID: SCR_002285, http://fiji.sc/ |
| Prism | GraphPad | RRID: SCR_015807, https://www.graphpad.com/scientific-software/prism/ |
| R | R Foundation for Statistical Computing | RRID: SCR_001905, https://www.r-project.org |
| FastQC | Babraham Bioinformatics | RRID: SCR_014583, http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| STAR aligner | Dobin et al., 2013 | RRID: SCR_015899, https://github.com/alexdobin/STAR |
| HOMER | Heinz et al., 2010 | RRID: SCR_010881, http://homer.ucsd.edu/homer/ |
| MetaboAnalyst | Xia et al., 2016 | RRID: SCR_015539, http://www.metaboanalyst.ca |
| Metascape | Tripathi et al., 2015 | http://metascape.org |
| Other | ||
| normal chow diet | Lab Diet | 5053 |
| 60% high fat diet | TestDiet | 58Y1 |
Highlights.
TRF protects clock mutant mice from obesity without changes in activity or calories
TRF restores rhythms in feeding-fasting, metabolic and nutrient sensing pathways
TRF prevents fatty liver, dyslipidemia and glucose intolerance in clock mutant mice
TRF transcriptional program activates cellular homeostasis maintenance pathways
ACKNOWLEDGMENTS
This work was partially supported by American Federation of Aging Research (AFAR) grant M14322, and NIH grant DK115214 to S.P. and funding to research cores through NIH P30 CA014195, P30 EY019005, P50 GM085764, R24 DK080506 and the Glenn Center for Aging, Leona M. and Harry B. Helmsley Charitable Trust’s grant #2012-PG-MED002. A.C. was supported by a mentor-based postdoctoral fellowship from the American Diabetes Association (7-12-MN-64), American Heart Association Career Development Award 18CDA34110292 and has received support from the Philippe Foundation Inc., New York, and Women in Science Award from the Salk Institute. The authors would like to thank Phuong Miu and Benjamin Shifflett for technical assistance, and David O’Keefe for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interests
SP is the author of a book titled “The Circadian Code” and he collects author’s royalty.
References.
- Adamovich Y, Rousso-Noori L, Zwighaft Z, Neufeld-Cohen A, Golik M, Kraut-Cohen J, Wang M, Han X, and Asher G (2014). Circadian clocks and feeding time regulate the oscillations and levels of hepatic triglycerides. Cell metabolism 19, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, and Sassone-Corsi P (2015). Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell 161, 84–92. [DOI] [PubMed] [Google Scholar]
- Atger F, Gobet C, Marquis J, Martin E, Wang J, Weger B, Lefebvre G, Descombes P, Naef F, and Gachon F (2015). Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc Natl Acad Sci U S A 112, E6579–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay JL, Shostak A, Leliavski A, Tsang AH, Johren O, Muller-Fielitz H, Landgraf D, Naujokat N, van der Horst GT, and Oster H (2013). High-fat diet-induced hyperinsulinemia and tissue-specific insulin resistance in Cry-deficient mice. Am J Physiol Endocrinol Metab 304, E1053–1063. [DOI] [PubMed] [Google Scholar]
- Bass J (2012). Circadian topology of metabolism. Nature 491, 348–356. [DOI] [PubMed] [Google Scholar]
- Bass J, and Lazar MA (2016). Circadian time signatures of fitness and disease. Science 354, 994–999. [DOI] [PubMed] [Google Scholar]
- Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, Jager J, and Lazar MA (2012). Rev-erbalpha and Rev-erbbeta coordinately protect the circadian clock and normal metabolic function. Genes Dev 26, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, and Bradfield CA (2000). Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaix A, Zarrinpar A, Miu P, and Panda S (2014). Time-Restricted Feeding Is a Preventative and Therapeutic Intervention against Diverse Nutritional Challenges. Cell metabolism 20, 991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, et al. (2012). Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature 485, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delezie J, Dumont S, Dardente H, Oudart H, Grechez-Cassiau A, Klosen P, Teboul M, Delaunay F, Pevet P, and Challet E (2012). The nuclear receptor REV-ERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J 26, 3321–3335. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, and Sassone-Corsi P (2013). Reprogramming of the circadian clock by nutritional challenge. Cell 155, 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, and Sabatini DM (2015). Nutrient-sensing mechanisms and pathways. Nature 517, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, DeHaven CD, Barrett T, Mitchell M, and Milgram E (2009). Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Analytical chemistry 81, 6656–6667. [DOI] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, and Klein S (2010). Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology (Baltimore, Md.) 51, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, and Stephanopoulos G (2013). Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nature communications 4, 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, and Lazar MA (2011). A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331, 1315–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S, Le HD, Melkani GC, and Panda S (2015). Time-restricted feeding attenuates age-related cardiac decline in Drosophila. Science 347, 1265–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EA Jr., Staknis D, and Weitz CJ (1999). Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science 286, 768–771. [DOI] [PubMed] [Google Scholar]
- Hardin PE, and Panda S (2013). Circadian timekeeping and output mechanisms in animals. Curr Opin Neurobiol 23, 724–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori M, Vollmers C, Zarrinpar A, Ditacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JA, et al. (2012). Time-Restricted Feeding without Reducing Caloric Intake Prevents Metabolic Diseases in Mice Fed a High-Fat Diet. Cell metabolism 15, 848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, and Auwerx J (2011). The metabolic footprint of aging in mice. Scientific reports 1, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Abruzzi KC, Allada R, Anafi R, Arpat AB, Asher G, de Bekker Baldi P, Bell-Pedersen D, Blau J, et al. (2017). Guidelines for Genome-Scale Analysis of Biological Rhythms. J Biol Rhythms 32, 380–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, and Hogenesch JB (2009). Harmonics of circadian gene transcription in mammals. PLoS Genet 5, e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Yong Q, Kurose T, Todo T, Mizunoya W, Fushiki T, Seino Y, and Yamada Y (2007). Clock gene defect disrupts light-dependency of autonomic nerve activity. Biochem Biophys Res Commun 364, 457–463. [DOI] [PubMed] [Google Scholar]
- Jacobi D, Liu S, Burkewitz K, Kory N, Knudsen NH, Alexander RK, Unluturk U, Li X, Kong X, Hyde AL, et al. (2015). Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell metabolism 22, 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M, Eagan J, Desai NK, Newton SA, Towne MC, Marinakis NS, Esteves KM, De Ferranti S, Bennett MJ, McIntyre A, et al. (2014). A compound heterozygous mutation in GPD1 causes hepatomegaly, steatohepatitis, and hypertriglyceridemia. European journal of human genetics : EJHG 22, 1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, and Bass J (2007). High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell metabolism 6, 414–421. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional Architecture and Chromatin Landscape of the Core Circadian Clock in Mammals. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, and Reppert SM (1999). mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205. [DOI] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez S, Juguilon H, Panda S, Shaw RJ, et al. (2009). AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 326, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Storch KF, and Weitz CJ (2008). Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A 105, 15172–15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Martelot G, Canella D, Symul L, Migliavacca E, Gilardi F, Liechti R, Martin O, Harshman K, Delorenzi M, Desvergne B, et al. (2012). Genome-wide RNA polymerase II profiles and RNA accumulation reveal kinetics of transcription and associated epigenetic changes during diurnal cycles. PLoS Biol 10, e1001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, and Ozcan U (2014). Unfolded protein response signaling and metabolic diseases. J Biol Chem 289, 1203–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, and Bessho Y (2016). The Circadian NAD(+) Metabolism: Impact on Chromatin Remodeling and Aging. Biomed Res Int 2016, 3208429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimamurthy R, Hatori M, Nayak SK, Liu F, Panda S, and Verma IM (2012). Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc Natl Acad Sci U S A 109, 12662–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan L, and Tabas I (2012). Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annual review of medicine 63, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, and Naef F (2011). Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol 9, e1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, and Dillin A (2015). Tipping the metabolic scales towards increased longevity in mammals. Nat Cell Biol 17, 196–203. [DOI] [PubMed] [Google Scholar]
- Robles MS, Cox J, and Mann M (2014). In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet 10, e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles MS, Humphrey SJ, and Mann M (2016). Phosphorylation Is a Central Mechanism for Circadian Control of Metabolism and Physiology. Cell metabolism. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P (2016). The Epigenetic and Metabolic Language of the Circadian Clock In A Time for Metabolism and Hormones. Sassone-Corsi P, and Christen Y, eds. (Cham (CH)), pp. 1–11. [PubMed] [Google Scholar]
- Sato S, Solanas G, Peixoto FO, Bee L, Symeonidi A, Schmidt MS, Brenner C, Masri S, Benitah SA, and Sassone-Corsi P (2017). Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 170, 664–677 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman H, Genzer Y, Cohen R, Chapnik N, Madar Z, and Froy O (2012). Timed high-fat diet resets circadian metabolism and prevents obesity. FASEB J 26, 3493–3502. [DOI] [PubMed] [Google Scholar]
- Shimizu I, Yoshida Y, Suda M, and Minamino T (2014). DNA damage response and metabolic disease. Cell metabolism 20, 967–977. [DOI] [PubMed] [Google Scholar]
- Sid V, Siow YL, and O K (2017). Role of folate in nonalcoholic fatty liver disease. Canadian journal of physiology and pharmacology, 1–8. [DOI] [PubMed] [Google Scholar]
- van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, et al. (1999). Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 398, 627–630. [DOI] [PubMed] [Google Scholar]
- Vollmers C, Gill S, DiTacchio L, Pulivarthy SR, Le HD, and Panda S (2009). Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc Natl Acad Sci U S A 106, 21453–21458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers C, Schmitz RJ, Nathanson J, Yeo G, Ecker JR, and Panda S (2012). Circadian Oscillations of Protein-Coding and Regulatory RNAs in a Highly Dynamic Mammalian Liver Epigenome. Cell metabolism 16, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, and Thompson CB (2010). Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Anafi RC, Hughes ME, Kornacker K, and Hogenesch JB (2016). MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinformatics 32, 3351–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, and Wishart DS (2015). MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res 43, W251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Chaix A, and Panda S (2016). Daily Eating Patterns and Their Impact on Health and Disease. Trends Endocrinol Metab 27, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu Y, Dentin R, Pongsawakul PY, Liu AC, Hirota T, Nusinow DA, Sun X, Landais S, Kodama Y, et al. (2010). Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nature medicine 16, 1152–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the liver RNA-seq data is GEO: GSE102072.