

Abstract
A site-specific bioconjugation was developed based on direct aldol coupling using amino acid-derived organocatalysts. The functionalization exhibits fast kinetics and occurs under mild, biocompatible conditions (viz., aqueous media, moderate temperature, and neutral pH). The resulting bioconjugates were found to be stable towards abundant aldolase enzymes, as well as acidic and basic pH. The methodology was demonstrated through conjugation of a variety of small molecules, dyes, and peptides to proteins, including a single-domain antibody, which was then used for cellular imaging.
Graphical Abstract
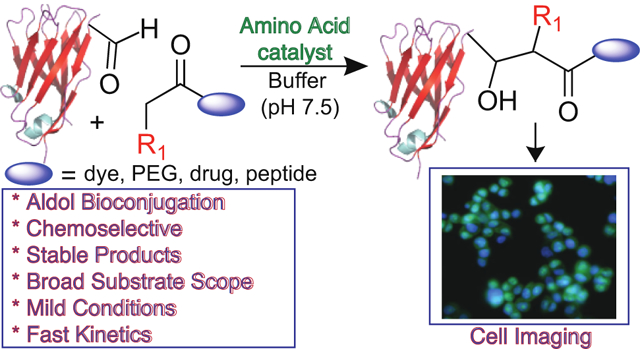
Post-translational modification of proteins to install new functionality is widely known in nature, yet similar selectivity and efficiency is currently unattainable from the available chemical methods.1,2 Single site biomolecule functionalization to generate homogeneous constructs in the presence of amino acids with diverse reactivity in both a chemo- and regioselective manner is an exceptional challenge, which is further exacerbated by the requirement for non-denaturing conditions.3 Nevertheless, bioorthogonal chemical reactions have emerged for modification of proteins with both natural and unnatural amino acids. For example, aldehydes and ketones can be readily installed onto a protein’s N-terminus by well-established methods.4 They then become key handles for further transformation, such as the commonly employed conjugations with hydrazines and hydroxylamines to generate hydrazones and oximes, respectively.5,6 However, this approach has limitations, such as slow kinetics, acidic (pH 5–6) conditions, high concentrations of harmful catalysts, cross-reactivity with metabolites, and poor stability of the resulting conjugates due to reversibility.6,7 This leads to the development of more advanced aldehyde bioconjugations, which exhibit improved stability of conjugates, such as Pictet-Spengler,8 indium-mediated allylation,9 and Wittig reactions.10
We considered that amino acids and/or peptides could be used for the conjugation of proteins by catalyzing the aldol reaction, which would lead to carbon-carbon bond formation at the site of conjugation (Figure 1).11,12 Existing bioconjugation methods utilizing the aldol reaction are limited by poor substrate scope and slow kinetics.13 While the efficiency and selectivity of the proposed amino acid-catalyzed reaction may be impeded by the presence of a variety of functional groups within complex biomolecules, we envisioned that careful use of particular amino acids or peptides would serve as highly reactive catalysts for the desired transformations under physiologically relevant conditions. The interaction between the amino group of the catalyst and the aldehyde/ketone would guide its selectivity by formation of an iminium ion-enamine complex. The ability to achieve high levels of chemo- and regioselectivity and fast reaction kinetics with aldehydes and ketones under aqueous conditions by using well-designed amino acid-derived organocatalysts suggest that they could be very attractive for the development of a new site-selective bioconjugation method leading to the modification at the N-terminus of proteins. In addition, this method has the potential to generate a chiral center at the site of bioconjugation with high stereoselectivity through judicious organocatalyst selection, which may be of therapeutic significance (see Supporting Information).
Figure 1.

(a) Proposed Aldol bioconjugation reaction using amino acid derived catalysts. (b) Key reactive intermediates
Here we report an organocatalyzed aldol bioconjugation reaction that is applicable to a wide range of peptides and proteins. The organocatalyzed aldol bioconjugation reaction is chemoselective, site-specific, rapid, and occurs under mild reaction conditions. We illustrate the diversity of this reaction for a series of expressed and commercially available proteins and antibodies.
We started our investigation with proline as catalyst because a multitude of reports have shown that proline can form an iminium ion 1 with the ketone followed by rearrangement to an enamine 2, as needed for carrying out nucleophilic attack on the electrophilic aldehyde 3 (Figure 1).14 A simple amino acid aldehyde, phenylalanine aldehyde 4, was used to optimize reaction conditions and to explore substrate scope with different ketones (5–7, Figure 2). First, we compared the rates of catalyzed versus uncatalyzed reactions between 4 and hydroxyacetone 6 (Figure 2b). No product was detected in the absence of an organocatalyst. While most aldehyde bioconjugations operate at slightly acidic pH (5.5 to 6.5),5,6,8 86% conversion occurred for the pH 7.4 phosphate buffered reaction. Furthermore, the rate was considerably faster with aqueous buffer than in neat DMSO (Figure 2c).
Figure 2.
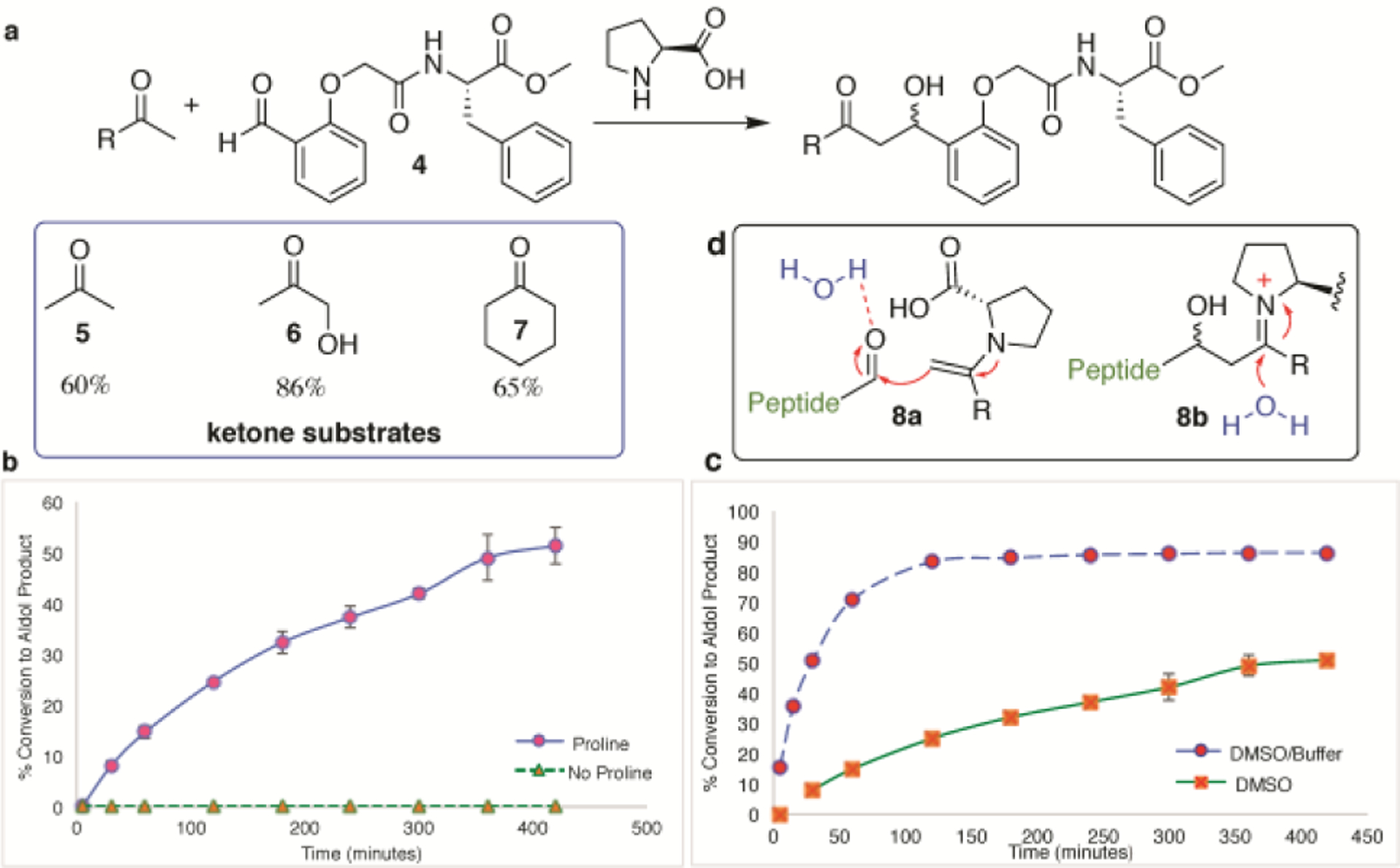
Model aldol bioconjugation between (a) phenylalanine aldehyde 4 and ketones (5-7) catalyzed by proline. (b) Comparison of uncatalyzed and proline-catalyzed (100 mM) reaction rates in DMSO at 37°C between 4 (1 equiv., 7mM) and hydroxyacetone 6 (12 equiv., 84 mM). (c) Comparison of reaction in 100% DMSO versus 1:1 sodium phosphate buffer (pH 7.4): DMSO. (d) Proposed role of water in increasing reaction kinetics.
The increase in the rate of the organocatalyzed reaction in buffer might be due to the dual role of water in activating the aldehyde for nucleophilic attack through hydrogen bonding 8a and by accelerating the hydrolysis of the iminium ion intermediate 8b formed between the aldol bioconjugate and the organocatalyst, resulting in the release of the desired bioconjugate (Figure 2d).14 Different amino acid-based organocatalysts were then screened for the reaction of either 4 or peptide aldehyde, VF 9, with 6, and the highest conversion was observed with proline (see Supporting Information). After optimizing catalyst loading and temperature, 96% product formed within 15 minutes using 100 mM proline at 60°C and pH 7.5. The optimized reaction conditions (100 mM proline, 5% DMSO, 25 mM phosphate buffer, pH 7.5 at 60°C) were then used for evaluating substrate scope (Figure 3). Unprotected peptide aldehydes 9-11, which were generated from N-terminal serine-containing peptides by sodium periodate (NaIO4) oxidation,4a were reacted with hydroxyacetone 6 resulting in high conversions to desired conjugated products 12-14 within 15 to 30 minutes. No interference was observed from the presence of other reactive amino acid side chains. In the case of lysine, self-condensation of 11 resulted in the formation of a macrocyclic imine 14a (Figure 3b). However, reaction of 14a with 6 ultimately lead to the desired aldol bioconjugate 14, presumably due to the high rate of transamination reaction of the imine 14a with proline catalyst.15
Figure 3.
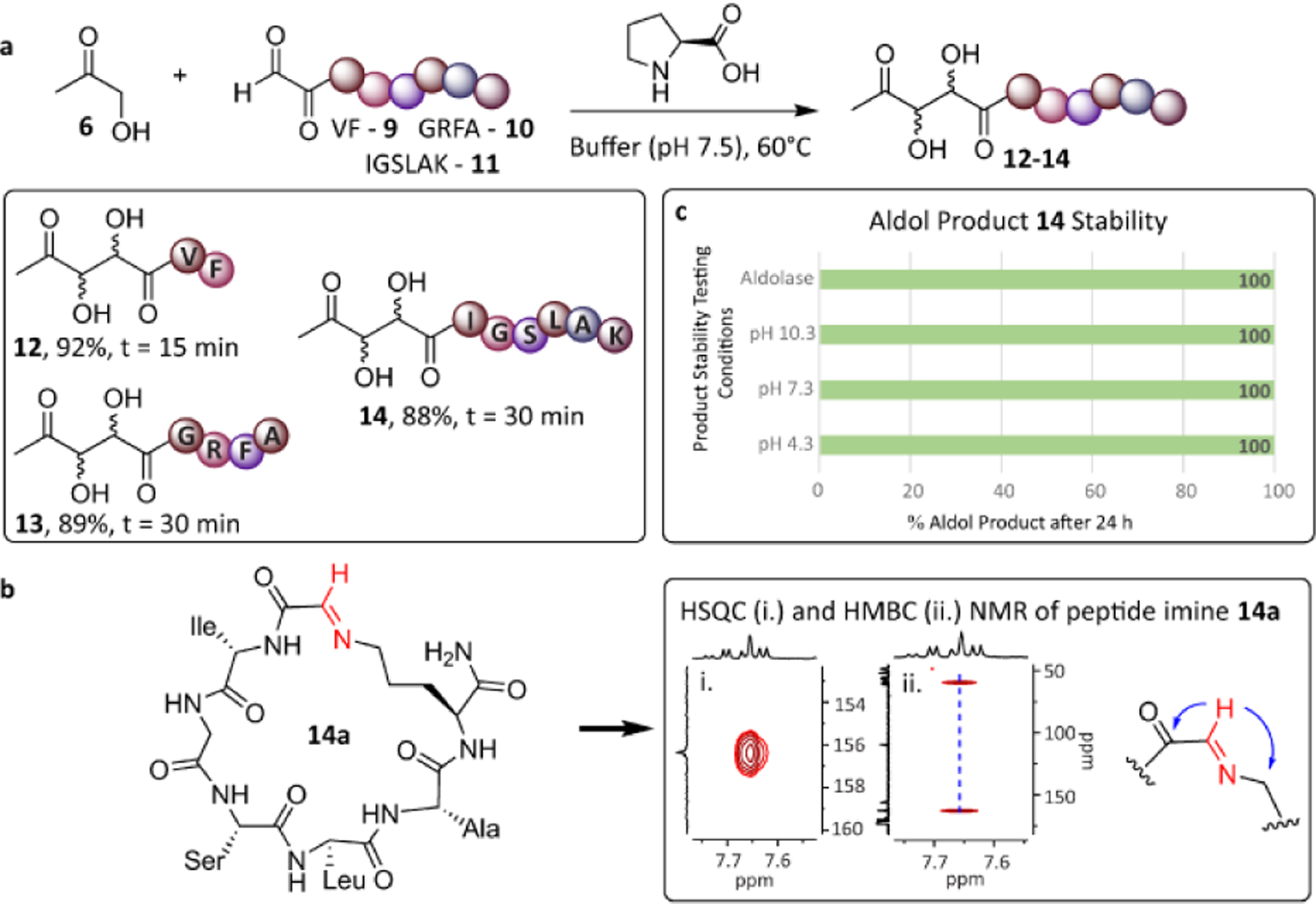
Chemoselective nature of aldol bioconjugation. (a) Reaction conditions: peptide aldehydes 9-11 (7 mM), hydroxyacetone 6 (84 mM), proline (100 mM) in 25 mM phosphate buffer (pH 7.5) and upto 15% DMSO at 60°C. (b) Peptide 11 containing Lys can form macrocyclic imine 14a, but this did not impede formation of aldol bioconjugate 14. (c) Stability of 14 with acid, base or aldolase at 37°C.
The stability of the aldol bioconjugation product was then evaluated against acidic and basic conditions, as well as aldolase enzymes (Figure 3c). As expected, incubation of 14 for 24 hours at pH 4.3, 7.3, and 10.3 at both room temperature and 37°C lead to no detectable degradation. Notably, 14 was also found to be stable towards the retro-aldol reaction catalyzed by aldolases, which are highly abundant in nature. The stability of the aldol bioconjugate towards aldolases is presumably due to the enzyme’s high substrate specificity.16
We next explored the aldol bioconjugation reaction with horse heart myoglobin (Figure 4). The N-terminal glycine residue was converted to an aldehyde using the well-known pyridoxal 5′-phosphate (PLP) reagent.4b Hydroxyacetone 6 was then reacted with myoglobin aldehyde 15 at 37°C in buffer using proline resulting in bioconjugated myoglobin 16 with 80% conversion within 30 minutes as determined by LC-MS. The analysis of the resulting myoglobin bioconjugate 16 using CD and UV showed that the structure of modified protein 16 remained intact after the bioconjugation (see Supporting Information). Two control reactions were then performed using (1) unmodified myoglobin (i.e., no aldehyde handle) and (2) no organocatalyst. In both cases, no bioconjugation product was observed after 24 h.
Figure 4.

Organocatalyzed protein modification. (a) Ligation of N-terminal myoglobin aldehyde 15 (0.15mM) with different ketones (6, 17, 18 (10 mM)) using either proline (b, c) or O-tBu-Thr (d, e) (100 mM) as catalysts in 25 mM phosphate buffer (pH 7.5) and upto 15% DMSO at 37°C. Deconvoluted ESI-MS spectra of (b) unconjugated myoglobin aldehyde 15, (c) myoglobin hydroxyacetone bioconjugate 16 generated by reaction with 6, (d and e) myoglobin-peptide bioconjugates 19 (60%) and 20 (40%) generated by reaction with peptide ketones GIRVF 17 and ACF 18.
Peptide ketones, GIRVF 17 and ACF 18, were generated from reaction of PLP with peptides, AGIRVF and AACF, respectively. These were then reacted with myoglobin aldehyde 15 resulting in desired protein bioconjugation products, 19 (66%) and 20 (83%), respectively when using O-tBu-Thr as organocatalyst in place of proline. Peptide ketones gave high conversion to bioconjugated products with O-tBu-Thr as compared to proline (see Supporting Information). Further, reaction of myoglobin aldehyde with drug, FITC and biotin ketones (see Supporting Information) demonstrated the flexibility of this approach with various ketones which is in contrast to known bioconjugation techniques, where reaction works with only one type of ketones.13
Antibody-drug conjugates (ADCs) are utilized for targeted drug delivery, and the synthesis of homogeneous conjugate is a critical for both the safety and efficacy of this therapy.17 We aimed to conjugate a peptide ketone, GIRVF 17, to a single domain antibody G110. Single domain antibodies, also referred to as nanobodies, have been shown to be equally specific as normal antibodies, and in some cases more bioactive.18,19 We chose a nanobody for conjugation because of their small size, robust stability, and simpler expression and purification in E. coli cells. The bioconjugation of 17 to nanobody G110 containing an N-terminal aldehyde 21 was catalyzed by O-tBu-Thr and produced homogeneous ADC 22 with 66% conversion (Figure 5).
Figure 5.
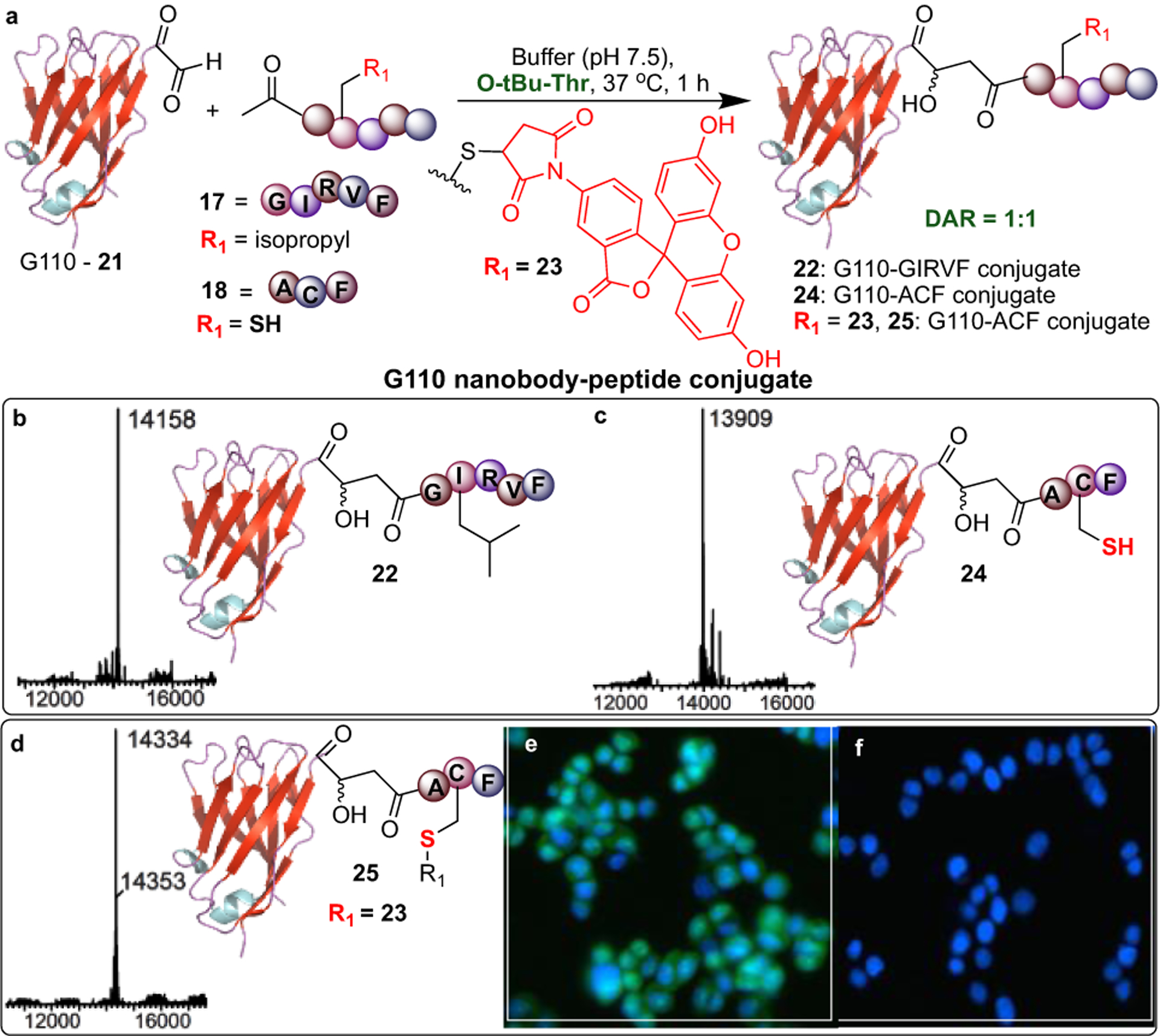
Nanobody drug conjugates (a) Synthesis of nanobody-drug conjugates (NDCs) at 1:1 drug-to-nanobody ratio (DNR). Reaction conditions: G110 21 (0.15 mM), ketones 17 or 18 (10 mM) O-tBu-Thr (100 mM) in 25 mM phosphate buffer, pH 7.5 and upto 15% DMSO at 37°C. Deconvoluted ESI-MS spectra of (b) NDC 22 (66%), (c) NDC 24 (83%), and (d) dual labeled NDC 25 (99%). (e) Fluorescent imaging with 25 indicates that these conjugates bind to cell surface receptors. (f) Control reaction: cells treated with dye but no nanobody or nanobody lacking dye do not fluoresce.
The consistent nanobody-peptide conjugate ratio is in contrast to some other bioconjugation techniques generating heterogeneous mixtures of ADCs with variable drug to antibody ratio (DAR).20 The mild conditions required for this bioconjugation method maintained the structurally critical disulfide bond in G110, which was confirmed by reaction with a maleimide dye 23 that resulted in no cysteine modification. While traditional ADCs use a variety of linkers to attach drug molecules to antibodies, our method has the capability of being linker-free.
Modification of proteins in the presence of free cysteines is particularly challenging due to the strong nucleophilicity of the thiol group. To assess compatibility of our approach with free cysteines, we synthesized peptide ketone ACF 18 and conjugated it to 21, which formed nanobody-peptide conjugate 24 (83% conversion, Figure 5c). Cys remained unreacted and allowed for further ligation with a maleimide dye 23 generating multiply labeled G110 25 (Figure 5d). Fluorescence microscopy showed that this labeled G110 nanobody 25 can bind to an EGF receptor-expressing breast cancer cell line, MDA-MB468, which confirms the stability of aldol bioconjugates in cellular assays (Figure 5e). Thus, this method can be used when the chemistry for Cys modification is not compatible or for applications that require specific labeling at multiple locations through orthogonal methods.
In all the above experiments, a peptide or protein with an electrophilic N-terminal aldehyde was conjugated to a small molecule containing a nucleophilic ketone, yet these roles can be reversed. A ketone was generated on protein α-lactalbumin 26 containing glutamic acid at the N-terminus, followed by reaction with 2-pyridine carboxyaldehyde (2-PCA) 27 and peptide aldehyde VF 9 using O-tBu-Thr as organocatalyst, yielded aldol bioconjugates 28 (50%) and 29 (57%), respectively (Figure 6). Notably, the resulting aldol bioconjugate 28 further underwent dehydration to the more stable α,β-unsaturated enone 30 in 1 h. The condensation product was only observed with 2-PCA 27 presumably due to stabilization of the resulting double bond by conjugation with the aromatic ring of 2-PCA. Both aldehyde and ketone moieties can be easily installed at the N-terminus of proteins by using well know PLP4b and Rapoport’s salt (RS)4c transamination methods. Further, reaction with other peptide ketones and aldehydes (see Supporting Information) demonstrated the applicability of this approach for modification of a broad range of different proteins with varying amino acids at the N-terminus under mild conditions.
Figure 6.

Protein ketone with aldehydes 2-PCA 27 and VF 9 (a) Formation of α-lactalbumin conjugates 29 and 30. (b) ESI-MS of α-lactalbumin conjugates 29 and 30.
We note that the ease of utilizing amino acids or peptides as catalysts makes them particularly attractive for routine modification of biomolecules for application in chemical biology, medicine and for generating peptide-polymer conjugates in material science. Here, we demonstrated an organocatalyzed bioconjugation strategy that proceeds in aqueous media under mild temperature and pH conditions while being able to tolerate a wide variety of reactive functional groups and leads to formation of a stable C-C bond at the site of conjugation. Additionally, this method demonstrates excellent selectivity and broad scope for modification of a vast majority of proteins with different amino acids at the N-terminus by converting them into either aldehyde or ketone handles. Importantly, these amino-acid based organocatalysts catalyze the bioconjugation reaction efficiently, independent of the nature of proteins as an electrophile or nucleophile. No such role-reversal was reported for the existing bioconjugation approaches. Furthermore, the compatibility of this reaction with free cysteine groups was utilized for dual labeling of proteins for cellular imaging. Thus, this method has the potential for synthesizing multiple conjugated biomolecules for various biophysical studies and drug delivery.
These organocatalysts have the potential of generating chiral centers at the site of bioconjugation, which is of high importance in the bioconjugation of proteins involved in drug discovery since stereoselectivity is crucial for the active binding conformation.12 A full investigation of these organocatalysts in catalyzing protein bioconjugation using a variety of different chemistries in a stereoselective manner is currently underway in our laboratory.
Supplementary Material
Footnotes
Supporting Information
Supporting figures, experimental procedures, and analytical data for new compounds (PDF). The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interests.
REFERENCES
- (1).McKay CS; Finn MG Chem. Biol 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zheng M; Zheng L; Zhang P; Li J; Zhang Y Molecules 2015, 20, 3190–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shih H-W; Prescher JA J. Am. Chem. Soc 2015, 137, 10036–10039. [DOI] [PubMed] [Google Scholar]
- (4).(a) Geoghegan KF; Stroh JG Bioconjugate Chem. 1992, 3, 130–146; [DOI] [PubMed] [Google Scholar]; (b) Witus LS; Francis M Curr. Protoc. Chem. Biol 2010, 2, 125–134; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Witus LS; Netirojjanakul C; Palla KS; Muehl EM; Weng CH; Iavarone AT Francis MB J. Am. Chem. Soc 2013, 135, 17223–17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dirksen A; Dirksen S; Hackeng TM; Dawson PE J. Am. Chem. Soc 2006, 128, 15602–15603. [DOI] [PubMed] [Google Scholar]
- (6).Dirksen A; Hackeng TM; Dawson PE Angew. Chem. Int. Ed 2006, 45, 7581–7584. [DOI] [PubMed] [Google Scholar]
- (7).(a) Wendeler M; Grinberg L; Wang X; Dawson PE; Baca M Bioconjugate Chem. 2014, 25, 93–101; [DOI] [PubMed] [Google Scholar]; (b) Kalia J; Raines RT Angew. Chem. Int. Ed. Engl 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sasaki T; Kodama K; Suzuki H; Fukuzawa S; Tachibana K Bioorg. Med. Chem. Lett 2008, 18, 4550–4553. [DOI] [PubMed] [Google Scholar]
- (9).Alam J; Keller TH; Loh T-P Chem. Commun 2011, 47, 9066–9068. [DOI] [PubMed] [Google Scholar]
- (10).Han MJ; Xiong DC; Ye XS Chem. Commun 2012, 48, 11079–11081. [DOI] [PubMed] [Google Scholar]
- (11).Tang Z; Yang Z-H; Cun L-F; Gong L-Z; Mi A-Q; Jiang Y-Z Org. Lett 2004, 6, 2285–2287. [DOI] [PubMed] [Google Scholar]
- (12).Nguyen LA; He H; Pham-Huy C Int. J. Biomed. Sci 2006, 2, 85–100. [PMC free article] [PubMed] [Google Scholar]
- (13).Alam J; Keller TH; Loh TJ Am. Chem. Soc 2010, 132, 9546–9548; [DOI] [PubMed] [Google Scholar]; (b) Wang P; Zhang S; Meng Q; Liu Y; Shang L; Yin Z Org. Lett 2015, 17, 1361–1364. [DOI] [PubMed] [Google Scholar]
- (14).Raj M; Singh VK Chem. Commun 2009, 6687–6703. [DOI] [PubMed] [Google Scholar]
- (15).Byeon J-Y; Limpoco FT; Bailey RC Langmuir, 2010, 26, 15430–15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Galkin A; Li Z; Li L; Kulakova L; Pal LR; Dunaway-Mariano D; Herzberg O Biochemistry 2009, 48, 3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Agarwal P; Bertozzi CR Bioconjugate Chem. 2015, 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Harmsen MM; De Haard HJ Appl. Microbiol. Biotechnol 2007, 77, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Siontorou CG Int. J. Nanomedicine 2013, 8, 4215–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim MT; Chen Y; Marhoul J; Jacobson F Bioconjugate Chem. 2014, 25, 1223–1232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.