

ABSTRACT
Excessive inflammation may lead to irreparable injury and even death, but the key mediators and underlying mechanisms remain unclear. Our recent findings indicate that SQSTM1/p62 (sequestosome 1), a well-known macroautophagy/autophagy receptor, is a lethal inflammatory mediator of sepsis and septic shock. The release of SQSTM1 occurs during tissue damage or microbial invasion through two main ways: one is passive and the other is active. Passive release occurs in the context of GSDMD-mediated pyroptosis. Active SQSTM1 secretion requires two basic steps: the first step is the expression and phosphorylation of SQSTM1 mediated by STING1/STING/TMEM173, and then the unconventional secretion of SQSTM1 by secretory lysosomes. After release, the extracellular SQSTM1 binds to membrane receptor INSR to activate glycolysis, leading to subsequent production of pro-inflammatory cytokines in a transcription factor NFKB-dependent manner. Functionally, genetic deletion or pharmacological inhibition of the SQSTM1-INSR pathway limits tissue damage, systemic inflammation, organ failure, and death in experimental sepsis models in mice. Moreover, the activation of the SQSTM1-INSR pathway is related to the severity of sepsis in patients. These findings highlight a pathological role of extracellular SQSTM1 in infection, inflammation, and immunity.
KEYWORDS: Autophagy, DAMP, immunometabolism, inflammasome, INSR, sepsis, SQSTM1, STING1, TLR4
Sepsis is now defined as a life-threatening organ dysfunction caused by the dysregulated host response to infection. This pathological condition represents a major medical challenge in the intensive care unit, and it is involved in various infections, including the current SARS-CoV-2 virus infection. During sepsis, the unrestricted activation of the innate immune system by pathogen-associated molecular patterns (PAMPs) and danger/damage-associated molecular patterns (DAMPs) can lead to severe and continuous inflammation, which is characterized by excessive release of various cytokines, collectively referred to as a “cytokine storm”. However, the key immune mediator of sepsis is still unknown. Recently, we have demonstrated that one of the intracellular autophagy receptors, SQSTM1/p62 (sequestosome 1), can be released into the extracellular space by macrophages and monocytes, acting as a lethal inflammatory mediator in septic shock by activating an INSR (insulin receptor)-dependent pathway (Figure 1) [1]. Our findings not only establish the new role of extracellular SQSTM1 in immunity, but also propose potential strategies for the treatment of sepsis.
Figure 1.
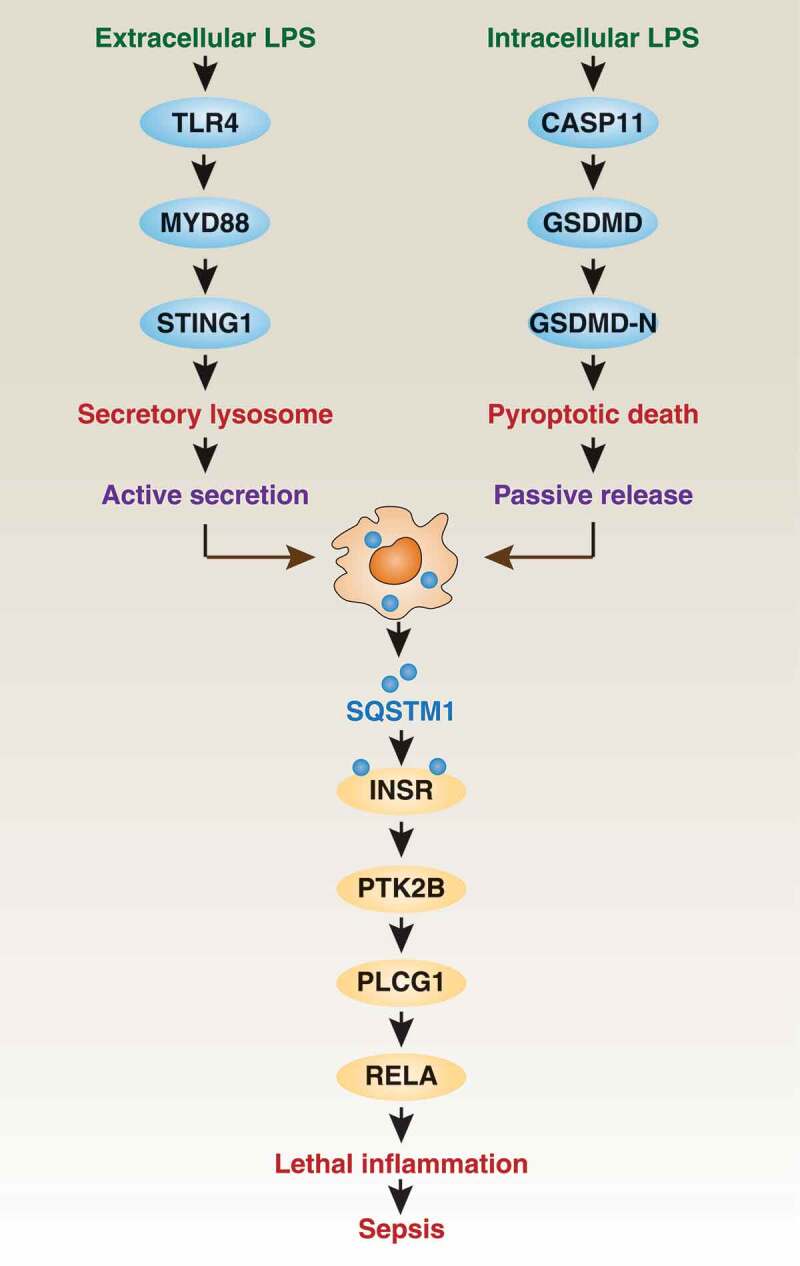
Extracellular SQSTM1 is an inflammatory mediator in sepsis. SQSTM1 can be actively secreted or passively released in macrophages in response to extracellular or intracellular LPS, respectively. Once released, the extracellular SQSTM1 acts as an inflammatory mediator by activating the INSR-dependent NFKB pathway during sepsis
Mechanism of SQSTM1 release
SQSTM1 is a ubiquitin-binding protein, which is involved in the regulation of various physiological and pathological processes, especially autophagy (a lysosome-mediated degradation mechanism that maintains cell homeostasis under stress). As an autophagy receptor, intracellular SQSTM1 functions as a bridge connecting cargo (e.g., aggregate proteins or damaged organelles) and Atg8/LC3 family members (autophagosome markers), thus driving subsequent lysosomal enzyme-mediated cargo degradation in autolysosomes. Interestingly, an elevated serum SQSTM1 level has recently been observed in patients with steatosis and lobular inflammation, indicating a potential extracellular role of SQSTM1 in immunity. Because Gram-negative bacterial infection is the main cause of clinical sepsis, we first investigated whether lipopolysaccharide (LPS, the main component of the outer membrane of Gram-negative bacteria) can trigger the expression and release of SQSTM1 in macrophages and monocytes. Using the classic membrane receptor TLR4 (toll like receptor 4) and adaptor protein MYD88 (MYD88 innate immune signal transduction adaptor), nontoxic LPS induces the expression and subsequent secretion of SQSTM1 in a secretory lysosome-dependent manner, but does not rely on classical autophagy machinery (e.g., ATG5 [autophagy related 5] or ATG7 [autophagy related 7]). RAB7A (RAB7A, member RAS oncogene family) and MCOLN1/TRPML1 (mucolipin 1) are important regulators of LPS-induced SQSTM1 secretion caused by lysosomal exocytosis. Other autophagy receptors, such as NBR1 (NBR1 autophagy cargo receptor), CALCOCO2/NDP52 (calcium-binding and coiled-coil domain 2), and OPTN (optineurin), are not secreted by LPS-activated macrophages. In addition, LPS-induced SQSTM1 secretion requires SQSTM1 phosphorylation at Ser403, and this process is positively regulated by STING1/STING/TMEM173 (stimulator of interferon response cGAMP interactor 1)-dependent TBK1 (TANK binding kinase 1) activation. Together, these in vitro findings reveal the signaling mechanism of SQSTM1 secretion in macrophages activated by LPS.
The pore-forming protein GSDMD (gasdermin D) becomes an effector of pyroptosis (a type of regulated cell death in immune cells) after being cleaved by inflammatory caspases to produce N-terminal GSDMD (GSDMD-N). In addition to TLR4, intracellular LPS can bind cytoplasmic CASP11 (caspase 11) to active inflammasomes, and then release cytokines or DAMPs by GSDMD-N-mediated pyroptosis. Passive release of SQSTM1 in macrophages is also observed in CASP11- and non-CASP11-mediated inflammasome activation (e.g., CASP1 [caspase 1] or CASP8 [caspase 8]). These findings indicate that SQSTM1 is a DAMP during GSDMD-N-mediated pyroptotic cell death after inflammasome activation.
Function of extracellular SQSTM1
Subsequently, we explored how extracellular SQSTM1 affects the immunometabolic reprogramming of macrophages, which is an important pathological event in patients with sepsis. First, we observed that the recombinant SQSTM1 protein (rSQSTM1) increases the extracellular acidification rate (ECAR, an indicator of glycolysis), and inhibits the oxygen consumption rate (OCR, an indicator of oxidative phosphorylation). Accordingly, rSQSTM1-induced glycolysis is related to increased glucose uptake and lactic acid production. Second, we evaluated the effect of rSQSTM1 on macrophage polarization. After rSQSTM1 treatment, macrophages appear as the M1 (pro-inflammatory phenotype), but not M2 (anti-inflammatory phenotype) state. In contrast, the loss of glycolytic enzyme genes prevents rSQSTM1-mediated M1 production, indicating that glycolysis is required for macrophage M1 polarization. Third, we found that the receptor INSR (but not TLR) is responsible for the immunometabolic activity of rSQSTM1 by activating the classic inflammatory transcription factor NFKB (nuclear factor kappa B). Mechanistically, rSQSTM1-mediated phosphorylation of INSR causes subsequent phosphorylation of PLCG1 (phospholipase C gamma 1) and PTK2B/PYK2 (protein tyrosine kinase 2 beta), leading to the phosphorylation of RELA/p65 (RELA proto-oncogene, NF-kB subunit) during NFKB activation. Collectively, these studies highlight the unique inflammatory pathway in macrophages driven by the SQSTM1-INSR axis.
SQSTM1 as a mediator of septic death
Finally, we investigated whether blocking the SQSTM1-INSR axis can prevent experimental sepsis (induced by cecal ligation and puncture, or infection by Escherichia coli or Streptococcus pneumoniae) and endotoxemia in mice. As with administration of anti-SQSTM1 monoclonal antibodies, conditional knockout of Insr or Sqstm1 in myeloid cells in mice also reduces septic death, systemic inflammation, and hyperlactatemia, as well as multiorgan failure. In addition, direct injection of rSQSTM1 can also cause septic-like symptoms in mice. Consistent with these animal studies, the activation of the SQSTM1-INSR axis is related to the severity of sepsis in patients, including the sequential organ failure assessment/SOFA score and disseminated intravascular coagulation/DIC score.
In summary, our findings uncover a mechanism by which extracellular SQSTM1 drives immunometabolism in macrophages and monocytes, and identify INSR as being responsible for SQSTM1 activity in innate immunity. Our research also indicates that targeting the SQSTM1-INSR axis may prevent septic death.
Funding Statement
This work was supported by the National Institute of General Medical Sciences [GM131919].
Disclosure statement
No potential conflicts of interest were disclosed.
Reference
- [1].Zhou B, Liu J, Zeng L, et al. Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signaling. Nat Microbiol. 2020; 5: 1576–1587 . doi: 10.1038/s41564-020-00795-7 [DOI] [PMC free article] [PubMed] [Google Scholar]