

ABSTRACT
Striking age-related changes occur in the human immune system, beginning in the sixth decade of life. Age is a non-modifiable, universal risk factor that results in the dysregulation of many cellular homeostatic processes. The decline in immune cell macroautophagy/autophagy and the increased generation of proinflammatory cytokines during agingfuels the development of diseases in the elderly. We reported that higher Th17 inflammation during aging was secondary to dysregulation in T cell autophagy. However, the mechanism underlying lower anti-CD3 and anti-CD28 activation-induced T cell autophagy during aging remain unknown. Our data fuel the speculation that dysregulation of the glutathione (GSH) system might cause the decline in T cell autophagy in aging, additionally provoked by reactive oxygen species signaling emanating from the mitochondria.
KEYWORDS: Aging, autophagy, glutathione, membrane potential, mitochondria, oxidative stress
Autophagy is a catabolic cellular recycling process that is required for cellular homeostasis. The decline in autophagy with age is implicated in aging-associated diseases. Decline in global immune cell autophagy and the increased generation of proinflammatory cytokines during aging fuels the development of cognitive decline, malignancies and autoimmunity in the elderly. We reported that higher production of proinflammatory Th17 cytokines differentiate lean, normoglycemic older adults from younger adults, and it is secondary to the aging T cell’s inability to upregulate autophagy, coupled with altered cellular bioenergetics. In vitro treatment with metformin of CD4+ T cells from older adults enhances autophagy and redirects fuel utilization to lower inflammation [1]. Interestingly, genetic inhibition of autophagy, but not PINK1 mediated mitophagy, promotes Th17 cytokine production in CD4+ T cells from younger adults, and recapitulates the respiratory profiles of T cells from older adults. However, the mechanism underlying lower anti-CD3/anti-CD28 activation-induced T cell autophagy during aging remains unknown. Our data fuel the speculation that dysregulation of the glutathione (GSH) system might be the elemental cause of decline in T cell autophagy in aging, additionally provoked by reactive oxygen species (ROS)-induced ROS signaling (RIR) emanating from the mitochondria.
Autophagy silencing in younger adults’ CD4+ T cells via siRNA for ATG3 (autophagy related 3), a protein required for the formation of the lipidated form of MAP1LC3 (microtubule associated protein 1 light chain 3; LC3-II), recapitulates the bioenergetic phenotype of aging T cells. This establishes a link between ATG3/autophagy and bioenergetics, and indicates regulation by a common denominator. Closer examination of our data suggests the common regulator may be the GSH system. T cell activation induces aerobic glycolysis to meet biosynthetic and redox regulatory needs. GSH, a predominant antioxidant, is oxidized to GSSG by GPX (glutathione peroxidase) as well as directly by free radical species. GSH restoration after clearance of hydrogen peroxide (H2O2) is catalyzed by GSR (glutathione reductase) in the presence of nicotinamide adenine dinucleotide phosphate (NADPH). Glycolysis provides electrons for H2O2 metabolism via the pentose cycle to regenerate NADPH. Thus, NADPH and GSH are needed to preserve redox-sensitive active sites on cytosolic enzymes in the necessary reduced state. As we described, aging T cells have an active respiratory chain, which can be a potent source of ROS. ROS can seep into the cellular environment and can become uncontrolled as the cells fail to upregulate glycolysis. Proteins are regulated by oxidants via disulfide bonds and inter-protein complex formation. An accepted mechanism for LC3 lipidation is that LC3 binds to ATG7 before its transfer to ATG3, subsequently followed by conjugation of LC3 to phosphatidylethanolamine (PE). The catalytic thiol groups of ATG3 and ATG7 are in close proximity to allow the transfer of LC3. Autophagy activation makes the interaction between the proteins transient, thus exposing the catalytic thiols of ATG3 and ATG7, making them susceptible to oxidation. Catalytic thiol availability on both ATG3 and ATG7 would promote intermolecular disulfide bond formation, and a single catalytic thiol availability within ATG3 or ATG7 would promote s-glutathiolation. It was shown elsewhere that addition of GSSG in a cell-free system inhibits LC3 lipidation by s-glutathiolation, causing unbound ATG3 and ATG7 to accumulate due to catalytic cysteine oxidation consistent with enhanced s-glutathiolation, thus preventing autophagy (Figure 1). Absence of autophagy can promote cellular stress signaling including that of the Th17 regulatory transcription factor STAT3. STAT3 translocation to mitochondria can drive oxidative phosphorylation dependence, thus completing a vicious loop. Metformin’s action of promoting autophagy probably through replenishing cellular GSH and restoring redox balance could release the brake on continuous cysteine oxidation of ATG3 and ATG7, and prevent the s-glutathiolation of LC3, disrupting the cycle promoted by oxidative stress.
Figure 1.
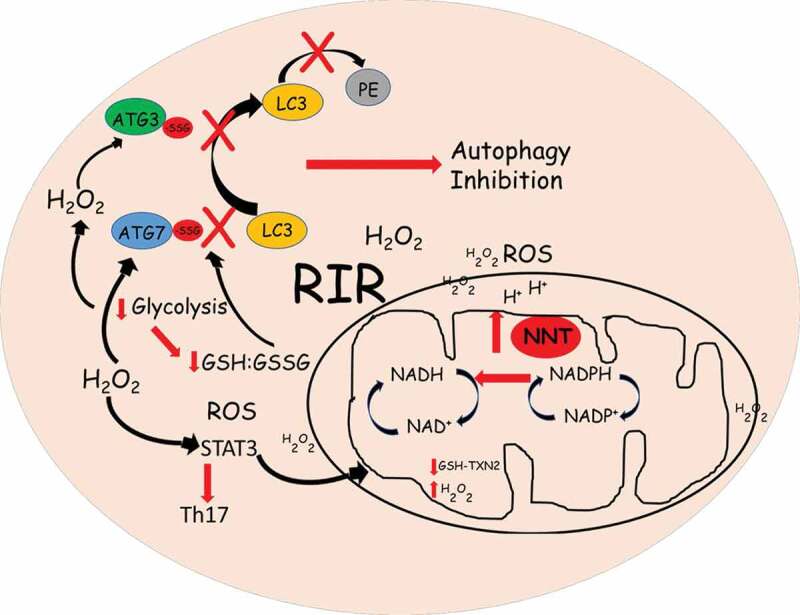
Model: lower activation-induced glycolysis together with mitochondrial redox imbalance promote altered GSH:GSSG and RIR signaling, resulting in s-glutathiolation and catalytic thiol oxidation of proteins to inhibit autophagy in aging CD4+ T cells
Another mechanism that could be implicated in aging T cell inflammation is uncontrolled ROS within the mitochondria. Although defective mitophagy does not independently generate Th17 in our study, mitochondrial involvement in Th17 cytokine production cannot be ignored. An exuberant electron transport chain can promote oxidative stress within the mitochondria and can induce an RIR cascade outside the mitochondria in early phases of cellular activation. It is estimated that the steady-state concentration of O·2− in the mitochondrial matrix is five to tenfold higher than in the cytosol in basal conditions. Further ROS buildup occurs in T cells during pathophysiological conditions such as aging, as evidenced by our data. Mitochondrial matrix antioxidant systems, GSH and TXN2 (thioredoxin 2) depend on the reductive power of NADPH, generated by the inner mitochondrial membrane protein NNT (nicotinamide nucleotide transhydrogenase), to keep mtROS under control. NNT couples the reaction of proton return to the matrix, to the transfer of hydride from NADH to NADP+. NNT is thought to account for most of the mitochondrial NADPH. We observed higher NNT expression in T cells from older adults, perhaps due to the need for higher activity to quench the rapidly building oxidative stress.
It was intriguing to find that, although the basal and maximal oxygen consumption rates are higher in T cells from older adults, there is a modest decline in the membrane potential. Similar to anti-CD3-/anti-CD28-stimulated cells, unstimulated T cells also have a lower membrane potential, raising speculations regarding the activity of NNT in aging T cells. NNT activity would be impaired if the membrane potential collapses, and NADPH production would be abolished. However, we speculate that lower membrane potential necessitates higher NNT expression as a compensatory mechanism to account for the declining activity of the enzyme. Another potential explanation, reported elsewhere, is that in situations of pathological workload-induced reductions in membrane potential, NNT function reverses and hydride transfer occurs from NADPH to NAD+ to regenerate NADH, which is consumed by the ETC as protons are pumped into the intermembrane space. This would deprive mitochondria of NADPH, GSH, TXN2 and damage mitochondrial proteins. H2O2 leakage into the extramitochondrial space could occur as the result of mitochondria being incapable of quenching ROS generated within, thus initiating a RIR cascade (Figure 1). Although our study was unable to pinpoint mtROS involvement in Th17 generation, it is not possible to conclude definitively that mtROS are uninvolved in the generation of a proinflammatory milieu in aging T cells. These insights will require experimental validation.
Funding Statement
This work was supported by The College of Health Sciences, Merrimack College Faculty Development Grant (LPB), and NIH R01DK108056 (BSN).
Disclosure statement
All authors have no conflicts of interest to declare.
Reference
- [1].Bharath LP, Agrawal M, McCambridge G, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020 Jul 7 [Epub 2020 May 12]; 32(1):44-55.e6. PubMed PMID: 32402267; PubMed Central PMCID: PMC7217133. DOI:: 10.1016/j.cmet.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]