

ABSTRACT
Macroautophagy/autophagy is suppressed by MTOR (mechanistic target of rapamycin kinase) and is an anticancer target under active investigation. Yet, MTOR-regulated autophagy remains incompletely mapped. We used proteomic profiling to identify proteins in the MTOR-autophagy axis. Wild-type (WT) mouse cell lines and cell lines lacking individual autophagy genes (Atg5 or Ulk1/Ulk2) were treated with an MTOR inhibitor to induce autophagy and cultured in media with either glucose or galactose. Mass spectrometry proteome profiling revealed an elevation of known autophagy proteins and candidates for new autophagy components, including CALCOCO1 (calcium binding and coiled-coil domain protein 1). We show that CALCOCO1 physically interacts with MAP1LC3C, a key protein in the machinery of autophagy. Genetic deletion of CALCOCO1 disrupted autophagy of the endoplasmic reticulum (reticulophagy). Together, these results reveal a role for CALCOCO1 in MTOR-regulated selective autophagy. More generally, the resource generated by this work provides a foundation for establishing links between the MTOR-autophagy axis and proteins not previously linked to this pathway.
Abbreviations: ATG: autophagy-related; CALCOCO1: calcium binding and coiled-coil domain protein 1; CALCOCO2/NDP52: calcium binding and coiled-coil domain protein 2; CLIR: MAP1LC3C-interacting region; CQ: chloroquine; KO: knockout; LIR: MAP1LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MEF: mouse embryonic fibroblast; MLN: MLN0128 ATP-competitive MTOR kinase inhibitor; MTOR: mechanistic target of rapamycin kinase; reticulophagy: selective autophagy of the endoplasmic reticulum; TAX1BP1/CALCOCO3: TAX1 binding protein 1; ULK: unc 51-like autophagy activating kinase; WT: wild-type.
KEYWORDS: ATG5, CALCOCO1, MAP1LC3, mass spectrometry proteomics, MTOR, reticulophagy, selective autophagy, ULK1, ULK2
Introduction
Macroautophagy/autophagy, the cellular process of “self-eating”, is critical for cellular metabolism and has been implicated in many diseases, including cancer [1]. Components of the molecular machinery of autophagy were first identified in yeast, many of which are shared with the cytoplasm-to-vacuole (Cvt) pathway, an autophagy-like delivery system to move specific hydrolases to the vacuole (reviewed in [2]). Since the early genetic screens in yeast, conserved and novel autophagy related (Atg) genes have also been identified in higher eukaryotes [3]. Selective autophagy in mammals evokes mechanisms that are similar to the Cvt pathway, utilizing selective autophagy receptors or adapters to connect specific cargo with LC3 autophagy proteins, specifically, MAP1LC3 (microtubule associated protein 1 light chain 3) and GABARAP (GABA type A receptor-associated protein) isoforms, for autolysosomal degradation (reviewed in [4–6]). However, many molecular features of selective autophagy remain undefined, especially in mammalian systems. Here, we demonstrate a proteomic profiling approach for identifying new proteins of autophagy.
MTOR in complex 1 (MTORC1) is a growth factor- and nutrient-sensing kinase, whose role in autophagy is conserved from yeast to humans [7]. Yeast Tor suppresses the activity of the autophagy-initiating kinase Atg1, so that conditions that inhibit Tor, such as starvation or the Tor inhibitor rapamycin, also induce autophagy [8,9]. In mammals, there are at least two orthologs of Atg1, ULK1 (unc 51-like autophagy activating kinase 1) and ULK2 with partially redundant functions in autophagy [10,11]. The relatively mild phenotype of the ulk1–/– mouse [10] compared to the neonatal lethality of the ulk1/2–/– mouse [11], suggests that ULK2 can compensate for most types of autophagy, except for the selective autophagy of mitochondria (mitophagy) in reticulocytes, which is attributed to ULK1 [10]. As in yeast, nutrient starvation, rapamycin, or rapamycin analogs (rapalogs) are commonly used to inhibit MTOR, thus activating ULK1/2 and autophagy, although with mechanistic differences [12]. In some cell types, rapalogs are incomplete inhibitors of MTORC1, whereas next-generation MTORC kinase inhibitors, such as torin1 or MLN0128, are more complete inhibitors of MTORC1 and more potent inducers of autophagy [13,14]. Recently, we showed that a combination of a rapalog and a dual MTOR-PI3K inhibitor potently induced both autophagy and mitophagy [15], leading to the idea that MTOR inhibitors may in some cases induce selective autophagy. This notion is supported by studies showing that torin1 induces autophagy of ribosomes (ribophagy) [16] and the endoplasmic reticulum (ER) (reticulophagy) [17].
The ER is a dynamic and functionally-diverse organelle that includes nuclear envelope domains, which are sheet-like, and peripheral domains, which are either tubular structures or sheets [18]. The ER network reaches deep into the cytoplasmic regions of the cell, where it interacts with and supports the functions of other organelles. For example, the mitochondria-associated ER membranes (MAMs) are regions of inter-organelle communication that are important for autophagy [19] and are increased by MTOR inhibitors or glucose starvation [15]. Proper ER function is maintained in part by selective autophagy (reticulophagy), and the list of reticulophagy receptors is growing (reviewed in [20,21]). Known reticulophagy receptors, some of which mediate selective autophagy of specific sub-ER domains, include RETREG1/FAM134b (reticulophagy regulator 1), SEC62 (SEC62 homolog, preprotein translocation factor), RTN3 (reticulon 3), CCPG1 (cell cycle progression 1), and TEX264 (testis expressed gene 264) [17,22–25]. The complex structure and multi-functional nature of the ER make it likely that additional unidentified reticulophagy receptors exist.
Here we identified a new component of the molecular machinery of autophagy, CALCOCO1 (calcium binding and coiled-coil domain 1), using a combination of mass spectrometry proteomics, cell biology, and biochemistry. The specific approach used in our mass spectrometry proteomics screen was designed based on our analysis of a recent study in yeast and then extended to homologous mammalian systems. We validated that CALCOCO1 is an autophagy-associated protein across multiple mammalian cell lines, show that it interacts physically with known autophagy proteins, and demonstrate that loss of CALCOCO1 decreased reticulophagy, but increased bulk autophagy. Together, these results show that CALCOCO1 functions in selective autophagy. More broadly, the proteomic data sets generated by this work provide a resource for discovering additional proteins that function in or are regulated by autophagy.
Results
A yeast proteomics screen revealed autophagy proteins
To determine the feasibility of a proteomics approach to identify proteins that function in autophagy, we examined the publicly-available multi-omic “Y3K” data set [26], which included two autophagy-related strains with single-gene deletions of either atg1 (atg1Δ yeast) or atg5 (atg5Δ yeast). In the Y3K study, knockout (KO) and wild type (WT) yeast were cultured in biological triplicate under two media conditions: (i) a fermentation growth condition with high glucose (+ Glucose) and (ii) a respiration growth condition with low glucose (– Glucose) (Figure 1A). Although the design of the Y3K study focused on mitochondrial function, nutrient deprivation, such as glucose limitation, inhibits Tor or MTOR and activates autophagy [27,28]. atg1Δ and atg5Δ yeast showed distinct elevation of Atg8, Atg34, and Atg39, known autophagy-associated proteins, when cultured in autophagy-inducing low-glucose conditions (Figure 1B), but not when cultured in high-glucose conditions (Figure 1C). The elevation of Atg39 was also a relatively unique feature of the atg1Δ and atg5Δ yeast strains compared to all other strains in the Y3K study (Figure 1D). Atg39 (Ylr312c) was only recently discovered as an autophagy receptor that localizes to the perinuclear ER and induces nucleophagy [29], a specific type of reticulophagy. Together, these analyses support the idea that mass spectrometry-based proteome abundance profiling of cell lines with both genetic disruption of autophagy and environmental induction of autophagy could be used as an approach to discover novel autophagy-related proteins (Figure 1E).
Figure 1.
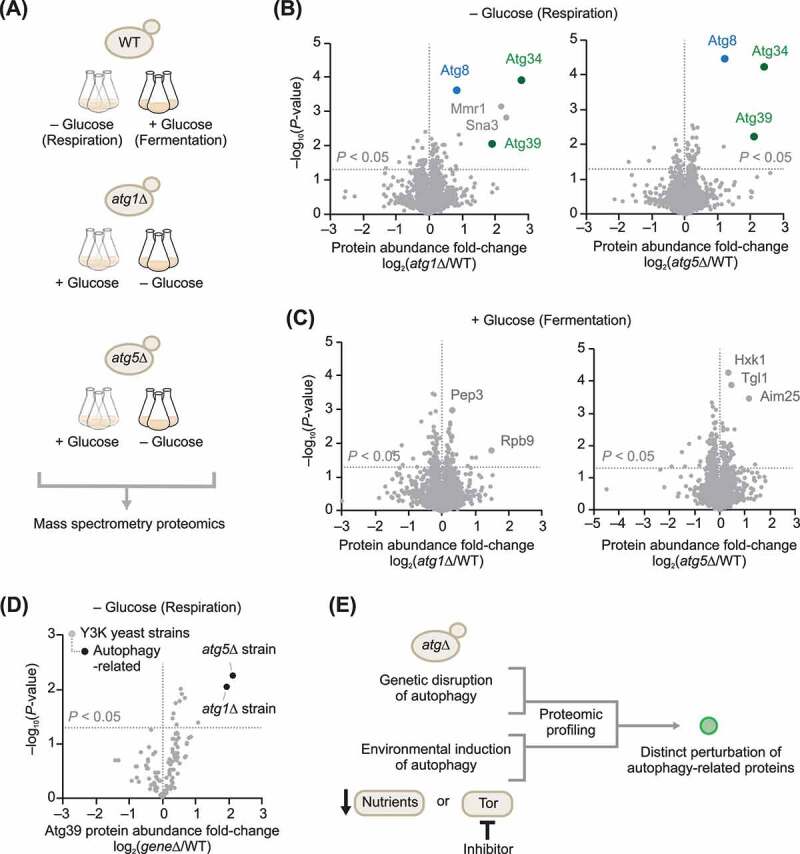
Select autophagy receptors are elevated in atgΔ yeast. (A) Scheme of the autophagy-related yeast knockout cultures that were part of the Y3K study [26]. (B) Relative protein abundances in atg1Δ or atg5Δ yeast compared to WT (mean, n = 3, respiration culture condition). P-values for the protein abundance changes were determined by a two-sided Student’s t-test. For panels B–D, the original raw data are from the Y3K data set [26]. (C) Relative protein abundances in atg1Δ or atg5Δ yeast compared to WT (mean, n = 3, fermentation culture condition). (D) Relative abundance of Atg39 across all geneΔ yeast strains in the Y3K study [26] (mean, n = 3, respiration culture condition) versus statistical significance. (E) General strategy for the discovery of autophagy-related proteins based on this work
A mammalian proteomics screen reveals candidate autophagy proteins
Based on the above yeast studies, we hypothesized that loss of Atg1 or Atg5 in mammalian cells cultured under autophagy-inducing conditions would distinctly elevate currently undefined mammalian autophagy components. To test this, we performed mass spectrometry proteomic profiling of mouse embryonic fibroblast (MEF) cells lacking the mammalian homologs of Atg1 or Atg5: ulk1–/– single-knockout MEFs, ulk1/2–/– double-knockout (DKO) MEFs [10,11], and atg5–/– single-knockout MEFs [30] (Figure 2A). We included single ulk1 KO and ulk1/2 DKO MEFs because both ULK1 and ULK2 are orthologs of yeast Atg1, downstream targets of MTOR [28], and have redundant functions in autophagy [10,11]. A comparison of ulk1 KO and ulk1/2 DKO MEF datasets is expected to reveal the shared and distinct roles of these related kinases. We included atg5 KO MEFs to increase confidence that perturbations observed in both ulk1/2 DKO and atg5 KO MEFs are due to defects in autophagy, and not to autophagy-independent functions of these proteins [31,32]. The three knockout MEF lines and their corresponding WT MEF lines were cultured in media with glucose (10 mM) or galactose (10 mM) and the ATP-competitive MTOR kinase inhibitor MLN0128 (abbreviated here as “MLN”; also known as INK128, TAK-128, and sapanisertib). We included MLN0128 to potently induce autophagy in both metabolic conditions, and because of its clinical relevance as an investigational drug for multiple malignancies (www.clinicaltrials.gov). We included galactose media as a nutrient stress condition to drive cells toward mitochondrial respiration [33], similar to the low-glucose high-glycerol condition used in the above yeast studies.
Figure 2.
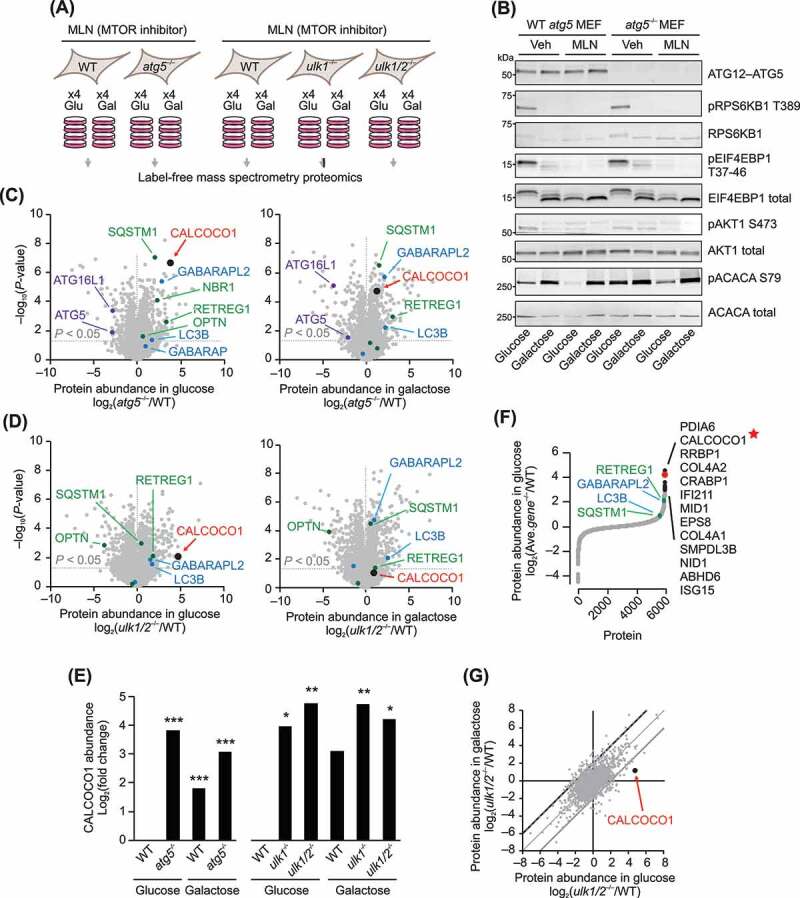
Select autophagy receptors are elevated in atg–/– MEFs. (A) Summary of the mouse embryonic fibroblast (MEF) tissue culture conditions for the mass spectrometry proteomic profiling experiment. (B) Protein immunoblot analyses with the indicated antibodies of samples from the MEF cultures analyzed by mass spectrometry, plus a set a parallel MEFs cultured without MLN (Vehicle, Veh). (C) Relative protein abundances in atg5–/– MEFs compared to WT (mean, n = 4) cultured in media containing either glucose or galactose. Proteins were quantified by label-free mass spectrometry. P-values for the protein abundance changes were determined by a two-sided Student’s t-test. ATG5 and ATG16L1 proteins shown in purple font; Atg8 homologs are shown in blue; known selective autophagy receptors in green; CALCOCO1 in red. (D) Relative protein abundances in ulk1/2–/– MEFs compared to WT (mean, n = 4) cultured in media containing either glucose or galactose. (E) Relative abundance of mouse CALCOCO1 in atg5–/–, ulk1–/–, and ulk1/2–/– MEFs compared to WT in glucose and galactose media. P-values: * <0.05; ** <0.01; *** <0.001. (F) Average protein abundance changes in atg5–/–, ulk1–/–, and ulk1/2–/– MEFs compared to WT (mean, n = 4) in the glucose media condition. (G) Protein abundance changes in ulk1/2–/– MEFs compared to WT (mean, n = 4) in glucose media compared to galactose media
Immunoblot analyses of samples from the cultures used in the proteomics experiments confirmed: (i) loss of ATG5 protein or ULK1 protein in the corresponding knockout cell lines, (ii) inhibition of MTORC1 by MLN as indicated by decreased phosphorylation of RPS6KB1/S6K1 (ribosomal protein S6 kinase, polypeptide 1) and EIF4EBP1/4EBP1 (eukaryotic translation initiation factor 4E binding protein 1), (iii) inhibition of MTORC2 by MLN as indicated by decreased phosphorylation of AKT1/PKB (thymoma viral proto-oncogene 1), and (iv) activation of PRKAA/AMPK (protein kinase AMP-activated) in galactose media as indicated by increased phosphorylation of ACAC/ACC (acetyl-Co A carboxylase) (Figure 2B and S1A). The proteomics datasets (Dataset S1 and S2) also revealed expected changes in autophagy-related proteins. For example, both ATG5 and its physical interaction partner ATG16L1 (autophagy related 16-like 1) (ref [34].) significantly decreased in the atg5–/– MEF line (Figure 2C). Furthermore, in a striking similarity to the yeast proteomics results, the protein abundances of known autophagy receptors (reviewed in [35,36]) and known Atg8 homologs were significantly increased in atg5–/–, ulk1–/–, and ulk1/2–/– MEFs (Figure 2C,Figure 2D and S1B). Elevated autophagy receptors included RETREG1, SQSTM1/p62 (sequestosome 1), OPTN (optineurin), and NBR1 (NBR1 autophagy cargo receptor). Elevated Atg8 homologs included GABARAPL2 and MAP1LC3B. Together, these results benchmark the quality of the proteomics dataset and suggest the potential to discover new autophagy-related proteins.
Examination of the data set for potential new autophagy-related proteins led us to CALCOCO1, as it was one of the most significantly elevated proteins in the atg5–/– and ulk1/2–/– MEFs (Figure 2C–Figure 2E). Moreover, averaging the protein abundance fold-changes across all three knockout lines revealed CALCOCO1 as the second most elevated protein (Figure 2F and Dataset S3). The top hit in this analysis, PDIA6 (protein disulfide isomerase associated 6), was recently linked to autophagy [37]. Furthermore, while the underlying mechanism remains unclear, we were intrigued by the observation that the magnitude of CALCOCO1 elevation was more pronounced in glucose media conditions compared to galactose (Figure 2G, S1C, and S1D), suggesting the potential for metabolic regulation of CALCOCO1. Together, these proteomics analyses nominated CALCOCO1 as a potential autophagy-related protein.
CALCOCO1 is regulated by autophagy induced by MTOR inhibition
To test the hypothesis that CALCOCO1 is an autophagy-regulated protein, we first examined CALCOCO1 in the KO and WT MEF cells used in the proteomics study under the same metabolic conditions, with and without MLN0128. We found that CALCOCO1 abundance decreased in WT MEFs treated with MLN0128, similar to the expression of the reticulophagy receptor RETREG1 (Figure 3A and S2A). Importantly, levels of both CALCOCO1 and RETREG1 were stable or slightly increased in autophagy-deficient MEFs, most robustly in the atg5–/– MEFs in glucose-containing media, strongly supporting the hypothesis that CALCOCO1 is regulated by MTOR in an autophagy-dependent manner.
Figure 3.
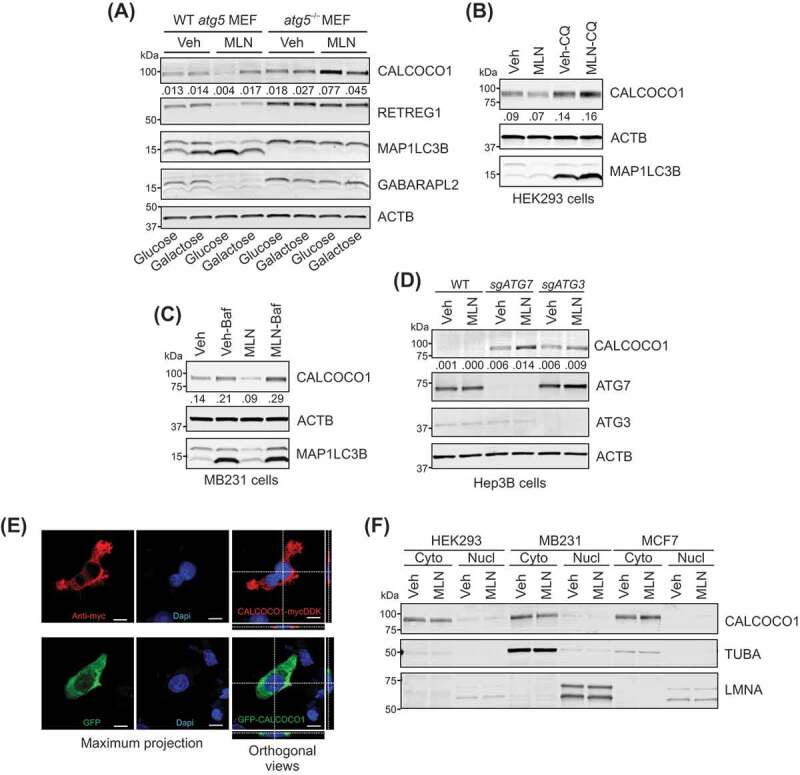
CALCOCO1 is regulated by MTOR and autophagy. (A) Protein immunoblot analyses with antibodies as shown, in WT and atg5–/– MEFs, in glucose– or galactose-containing media. MEFs were treated with vehicle (Veh) or 100 nM MLN0128 (MLN) for ~17 h. The ratio of CALCOCO1:ACTB is shown. (B) Immunoblot analyses of HEK293 cells treated for 24 h with Veh, 100 nM MLN, and 100 µM chloroquine (CQ) as labeled. The ratio of CALCOCO1:ACTB is shown. (C) Immunoblot analyses of MB231 cells treated for 24 h with Veh, 100 nM MLN, and 50 nM bafilomycin A1 (Baf) as labeled. The ratio of CALCOCO1:ACTB is shown. (D) Immunoblot analyses of WT, sgATG7 KO, and sgATG3 KO Hep3B cells, treated for 24 h with Veh or 100 nM MLN0128. The ratio of CALCOCO1:ACTB is shown. (E) Images of MB231 cells. Top: Immunofluorescent images of CALCOCO1-MYCDDK using anti-MYC (9E10) antibody and DAPI . Bottom: Images of GFP-CALCOCO1. Single-color maximum projection images are shown on the left. Dual-color orthogonal views from Z-stack images are shown on the right. Scale bar: 10 µm. (F) Immunoblot analyses of cytoplasmic and nuclear fractions from HEK293, MB231, and MCF7 cells treated for 24 h with Veh or 100 nM MLN
Inhibition of MTOR with MLN0128 also caused a progressive decline of CALCOCO1 levels in HEK293 cells, similar to the decrease observed in the autophagy protein SQSTM1 (Fig. S2B). The decrease in CALCOCO1 could not be explained by MLN0128 inhibition of protein synthesis [38], as CALCOCO1 abundance was relatively stable in cells treated with the translation inhibitor cycloheximide (CHX), with a half-life of approximately 24 h (Fig. S2B). To confirm that autophagy degrades CALCOCO1, we inhibited autophagy pharmacologically or genetically in multiple human cell lines. We chose human embryonic kidney HEK293 cells, and Hep3B hepatocellular carcinoma cells, which we have shown are capable of mounting a strong autophagy response to starvation and/or MTOR inhibition [13,15,39]. We also included two breast cancer cell lines, MB231 cells, a model of triple-negative breast cancer, and estrogen receptor-positive MCF7 cells [40], because of reports that CALCOCO1 expression may correlate with tumorigenic potential in breast cancer [41–43]. These reports were based in part on CALCOCO1’s reported role as a transcription coactivator for nuclear receptors [44–47], which led to our choice of both estrogen-positive and -negative breast cancer cell lines. We found that CALCOCO1 protein levels were increased in HEK293, MB231, and MCF7 cells when chloroquine (CQ) or bafilomycin A1 (Baf) inhibited autophagy (Figure 3B,Figure 3C and S2C), two drugs that inhibit the lysosome, but through different mechanisms [48]. Furthermore, CALCOCO1 expression, which was nearly undetectable in Hep3B cells, elevated significantly in cells that were gene-edited to disrupt two different autophagy genes, ATG7 or ATG3, and further increased with MLN0128 treatment (Figure 3D). These data demonstrate that CALCOCO1 is regulated by MTOR and autophagy in multiple cell lines.
The finding that CALCOCO1 expression was sensitive to MTOR inhibition was interesting because several reports suggest there is a positive correlation between CALCOCO1 expression and tumorigenicity [41–43]. These studies were primarily in breast cancer, a tumor type for which MTOR inhibitors are either FDA-approved or under clinical investigation [49]. To investigate whether CALCOCO1 has a role in anti-growth effects associated with MTOR inhibitors, we used CRISPR/Cas9 gene editing to delete CALCOCO1 in MB231 cells (Fig. S3A), a cell line in which CALCOCO1 expression levels were regulated by autophagy induced by MTOR inhibition (Figure 3C). MLN0128 treatment of MB231 control cells efficiently inhibited proliferation, cell migration, and wound healing when compared to the vehicle (Fig. S3B-S3D). None of these, however, were altered by loss of CALCOCO1 following treatment with either vehicle or MLN (Fig. S3B-S3D), consistent with the idea that CALCOCO1 does not have an essential role in the regulation of cell growth in these cells. Interestingly, in an assay for tumor sphere formation, there were ~50% fewer mammospheres in CALCOCO1 KO cells compared to control cells, a defect that persisted when spheres were passaged for a second time (Fig. S3E). These data suggest that CALCOCO1 may have a role in the maintenance of cancer stem cells. Future work is warranted to determine if this defect can be attributed to autophagy and to examine its significance in vivo.
The data that autophagy degrades CALCOCO1 led us to question CALCOCO1’s cellular location, particularly as descriptions often label CALCOCO1 as a transcription coactivator for nuclear receptors [44–47]. Despite this putative nuclear function, exogenous CALCOCO1 fusion proteins are primarily cytosolic [46,50], data that are consistent with the Human Protein Atlas, showing that CALCOCO1 is a ubiquitously expressed cytoplasmic protein that also associates with vesicles (www.proteinatlas.org/ENSG00000012822-CALCOCO1). In agreement with these reports, we found that CALCOCO1 fused to either a carboxyl terminus MYCDDK epitope tag or an amino terminus green fluorescent protein (GFP), was primarily in the cytoplasmic compartment (Figure 3E and S2D). Moreover, endogenous CALCOCO1 predominantly localized to the cytoplasmic fraction of three different cell lines, HEK293, MB231, and MCF7 cells, which was not changed by MTOR inhibition (Figure 3F and S2E), arguing against a localization artifact due to the epitope tags on either terminus or due to overexpression. Our data, together with existing data, argue that CALCOCO1 localizes to both the nucleus and the cytoplasm and that lysosomes degrade cytoplasmic CALCOCO1 in an autophagy-dependent manner.
Conserved and unique features of CALCOCO1
While previous experimental evidence does not link CALCOCO1 to autophagy, protein sequence analyses show that CALCOCO1 is part of the calcium-binding coiled-coil (CALCOCO) domain family (Figures 4 and Figures 5A), which includes two proteins that are selective autophagy receptors: CALCOCO2 and TAX1BP1/CALCOCO3 (TAX1 binding protein 1). CALCOCO2 and TAX1BP1 have been implicated in xenophagy [51,52], the selective autophagic clearance of bacteria, and mitophagy [53], suggesting that CALCOCO1 may have a similar function.
Figure 4.
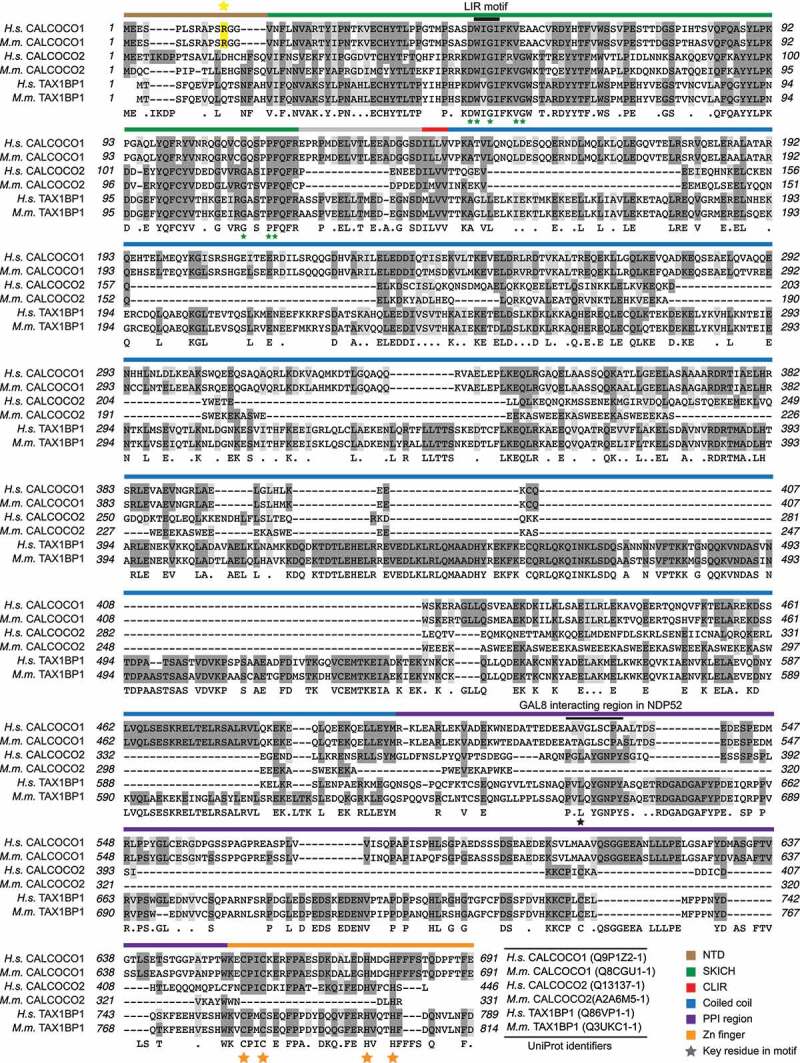
CALCOCO family sequence alignment. Sequence analysis of human and mouse CALCOCO1, CALCOCO2, and TAX1BP1. Key domains are depicted by colored bars. See the color key. LIR motif is marked with a black bar. Arginine 12 (R12) is highlighted in yellow
Figure 5.
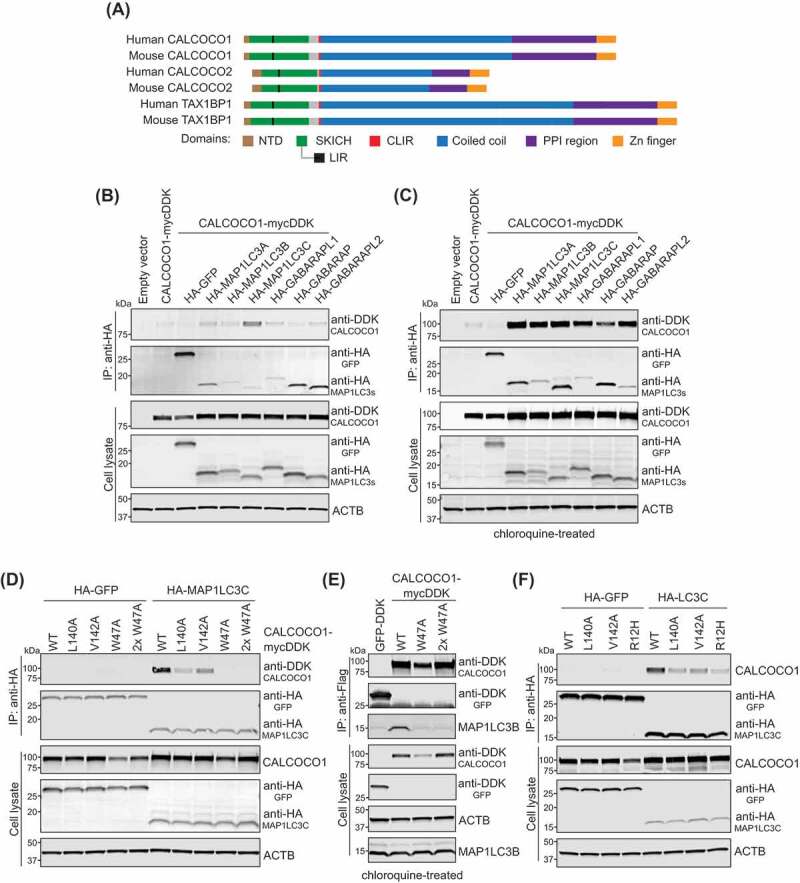
CALCOCO1 interacts with MAP1LC3C and other LC3/GABARAP family members. (A) Cartoon of domains and motifs found in human and mouse CALCOCO family members. (B) Immunoprecipitation (IP) analyses of CALCOCO1-MYCDDK and HA-tagged MAP1LC3/GABARAP orthologs in HEK293 cells. HA-tagged protein complexes were immunoprecipitated with anti-HA antibody, resolved by SDS-PAGE, and probed with anti-HA or anti-DDK antibody. Expressions of tagged proteins and ACTB in cell lysates are shown with antibodies as described. (C) IP analyses of CALCOCO1-MYCDDK and HA-tagged MAP1LC3/GABARAP orthologs expressed in HEK293 cells that were treated overnight with chloroquine (CQ) to inhibit lysosomal flux. HA-tagged protein complexes were IP’d with anti-HA antibody and probed with anti-DDK or mixed antibodies against HA, MAP1LC3A/B, and MAP1LC3C. Expressions of tagged proteins and ACTB in cell lysates are shown with antibodies as described. (D) IP analyses of HA-MAP1LC3C or HA-GFP co-expressed with wild type (WT) CALCOCO1-MYCDDK, CLIR mutants L140A and V142A, and LIR mutant W47A in HEK293 cells. CALCOCO1W47A was transfected at 2 different concentrations to match WT expression levels. HA-tagged protein complexes were immunoprecipitated with anti-HA antibody, and probed with anti-HA or anti-CALCOCO1 antibody. Expressions of tagged proteins and ACTB in cell lysates are shown with antibodies as noted. (E) IP analyses of CALCOCO1-MYCDDK with endogenous MAP1LC3B. WT CALCOCO1-MYCDDK, 2 concentrations of the CALCOCO1W47A, and GFP-DDK (control) were expressed in HEK293 cells, then treated for 18 h with 100 nM MLN0128 and 100 uM CQ. CALCOCO1-MYCDDK complexes were immunoprecipitated with anti-Flag® M2 affinity gel (MilliporeSigma, A2220) and probed with anti-DDK or MAP1LC3B antibodies. Expressions of CALCOCO1-MYCDDK, GFP-DDK, ACTB and MAP1LC3B in cell lysates are shown with antibodies as noted. (F) IP analyses of HA-MAP1LC3C or HA-GFP co-expressed with WT CALCOCO1-MYCDDK, CLIR mutants (L140A and V142A), and R12H mutant in HEK293 cells. HA-tagged protein complexes were immunoprecipitated with anti-HA antibody and probed with anti-HA or anti-CALCOCO1 antibody. Expressions of tagged proteins and ACTB in cell lysates are shown with antibodies, as noted
Primary sequence analysis of CALCOCO1 revealed that its protein domain structure is similar to that of CALCOCO2 and TAX1BP1, but CALCOCO1 also contains distinct features in regions predicted to impact protein function (Figures 4 and Figures 5A). Important with regards to autophagy, CALCOCO1 possesses a SKICH domain, which is essential for selective autophagy in TAX1BP1 [54], a non-canonical MAP1LC3C interacting region (CLIR), which is important for xenophagy in both CALCOCO2 and TAX1BP1 [51,52], and a more traditional LC3 interacting region (LIR), which in TAX1BP1 mediates its interaction with MAP1LC3C and GABARAP [55] (Figures 4 and Figures 5A). The N-terminal SKICH, CLIR, and LIR domains are highly conserved across CALCOCO1, TAX1BP1, and CALCOCO2, supporting the hypothesis that all three proteins function in some form of selective autophagy as receptors. However, these proteins diverge in the primary sequences of their coiled-coil domains and the domain linking the coiled-coil to the C-terminal zinc finger, which we denoted as the “protein-protein interaction (PPI)” domain because it is known to mediate an important protein-protein interaction between LGALS8/LGAL (galectin 8) and CALCOCO2 [56,57] (Figure 4). The primary sequence divergence of the PPI domain in particular, including the specific motif known to mediate the CALCOCO2-LGALS8 interaction (Figure 4), suggests that CALCOCO1 has unique protein interaction partners that could mediate forms of selective autophagy distinct from those of TAX1BP1 and CALCOCO2.
CALCOCO1 interacts with MAP1LC3C and other LC3 family members
To test the hypothesis that the conserved N terminus of CALCOCO1 mediates a function in autophagy via physical interaction with known autophagy proteins, we co-expressed CALCOCO1-MYCDDK with HA-tagged members of the MAP1LC3/GABARAP family in HEK293 cells, followed by immunoprecipitation (IP) of the HA-tagged LC3 proteins and immunoblotting for CALCOCO1-MYCDDK. Under these conditions, CALCOCO1 associated strongest with MAP1LC3C (Figure 5B), data supported by immunofluorescence of MB231 cells transfected with CALCOCO1 and MAP1LC3C, which showed overlapping signals in regions of discrete puncta (Fig. S4A). These data are consistent with the conserved CLIR domain in CALCOCO1, which mediates the binding of MAP1LC3C to CALCOCO2 or TAX1BP1. However, they do not explain the regulation and autophagy dependence of CALCOCO1 in MEFs (Figures 2 and Figures 3A), because to our knowledge, Map1lc3c is not in the mouse genome. CALCOCO1 also has a conserved LIR motif (Figures 4 and Figures 5A), which suggests that CALCOCO1 may also interact with other LC3 family members, similar to TAX1BP1 [55]. Furthermore, CALCOCO1 has a reported interaction with GABARAPL2 (https://thebiogrid.org/), which was one of two LC3 homologs significantly increased in our autophagy-deficient MEFs (Figures 2C,Figure 2D,Figures 3F and Figures 3A). We reasoned that if we inhibited autophagy with CQ, we might capture more transient CALCOCO1-interacting proteins in flux. In support of this hypothesis, the interaction between CALCOCO1, MAP1LC3B, GABARAPL2, and all LC3 family members was greatly enhanced in cells treated with CQ (Figure 5C), consistent with a functional LIR motif.
To determine the importance of the CLIR and LIR motifs for CALCOCO1’s interaction with MAP1LC3C, we introduced alanine (A) mutations in the CLIR domain, targeting leucine 140 and valine 142 (L140A, V142A), and in the LIR domain, targeting tryptophan 47 (W47A). In co-IP experiments, the interaction between CALCOCO1 and MAP1LC3C was significantly weakened by either of the CLIR mutations and abolished by the W47A LIR mutation (Figure 5D). Immunoprecipitation of WT CALCOCO1, but not the CALCOCO1W47R LIR mutant, also pulled out endogenous MAP1LC3B and GABARAPL2 from lysates of cells treated with MLN0128 and bafilomycin A1 (Figure 5E and S4B). These data demonstrate that whereas the CLIR motif may be important for CALCOCO1’s binding to MAP1LC3C, it is the LIR motif that is essential for its interaction with MAP1LC3C, MAP1LC3B, and GABARAPL2.
We next looked for evidence of the importance of the N terminus of CALCOCO1 in human pathology. Interestingly, in a subset analysis of coding variants of CALCOCO1 and 16 other genes in a panel of women with breast cancer, an arginine to histidine (R12H) mutation in CALCOCO1 was one of only two variants that correlated with a small but significant risk of breast cancer [41], a finding attributed to CALCOCO1’s putative role as a transcriptional coactivator of the estrogen receptor. In the context of childhood cancers, genomic profiling identified the same R12H mutation in a patient with lymphoblastic leukemia [58]. R12 and the upstream amino acids are unique to CALCOCO1 when compared to CALCOCO2 and TAX1BP1 (Figure 4), but R12 is highly conserved within CALCOCO1 across mammalian species. We found that the R12H mutation significantly decreased the association of CALCOCO1 with MAP1LC3C, similar to mutations in the CLIR domain (Figure 5F and S4C), suggesting those patient tumors harboring this mutation may have a defect in CALCOCO1-mediated autophagy. Further studies are warranted to determine the relevance of CALCOCO1 and its association with LC3s in tumor biology. We conclude that the N terminus of CALCOCO1 is important for its binding to MAP1LC3C and other members of the LC3 family.
CALCOCO1s role in selective autophagy
For insight as to the type(s) of selective autophagy that requires CALCOCO1 under our conditions, we reexamined the proteomic results in yeast and MEFs. Both studies uncovered known reticulophagy receptors, consistent with increased ER remodeling under glucose starvation and/or MTOR inhibition (Figures 1 and Figures 2). We then performed gene ontology (GO) enrichment analysis of the most significantly increased proteins in our datasets, focusing on GO cellular components (CC), which annotate the known cellular location of proteins. In each of the three KO MEFs cultured in glucose conditions, we found a significant enrichment in ER, Golgi, and plasma membrane components, most profoundly in the ulk1/2–/– MEFs (Fig. S5). In galactose medium, these same components were enriched to a lesser degree only in the ulk1/2–/– MEFs and were insignificant in the ulk1–/– and atg5–/– MEFs (Fig. S5). A complete list of the enrichment categories can be found in Tables S1-S6. The more pronounced enrichment of ER proteins in glucose conditions correlated with the pattern we observed with CALCOCO1 in Figure 2, suggesting that CALCOCO1 may have a role in ER remodeling, or reticulophagy, induced by inhibition of MTOR. In support of this premise, we were able to capture GFP-CALCOCO1 in close proximity to the ER using 3D structured illumination microscopy (3D-SIM) (Fig. S6A).
To investigate the importance of CALCOCO1 in reticulophagy, we deleted CALCOCO1 in HEK293 cells using CRISPR/Cas9 technology and measured reticulophagy using the GSTLSCSGFP-cb5 assay [59]. Cb5, terminology referring to the 35 carboxyl-terminus amino acids of rat CYB5A (cytochrome b5 type A), is a known ER-specific targeting sequence [60], which we placed at the C terminus of a GST-GFP fusion protein that has a lysosome-specific cleavage sequence (LSCS) between the two proteins. We have previously shown that GSTLSCSGFP-cb5 localizes specifically to the ER and is degraded by autophagy, producing an autolysosomal GST-fragment which can be purified from cell lysates or light membrane fractions (LMFs) and measured by western blotting [59]. Although we sometimes observed more than one fragment in these cells, only the bottom fragment proved to be lysosomal (Fig. S6B). MLN treatment consistently caused an increase in the lower GST fragment in control cells, which reduced by approximately 50% in sgCALCOCO1 KO cells (Figure 6A,B). We found endogenous CALCOCO1 in both the high-speed supernatant (HSS), and LMF fractions (Figure 6B), consistent with an association between CALCOCO1 and membrane-bound vesicles. Reticulophagy induced by glucose starvation or tunicamycin, similarly reduced by ~50% in sgCALCOCO1 KO cells, while reticulophagy in galactose media remained unchanged (Figure 6B).
Figure 6.
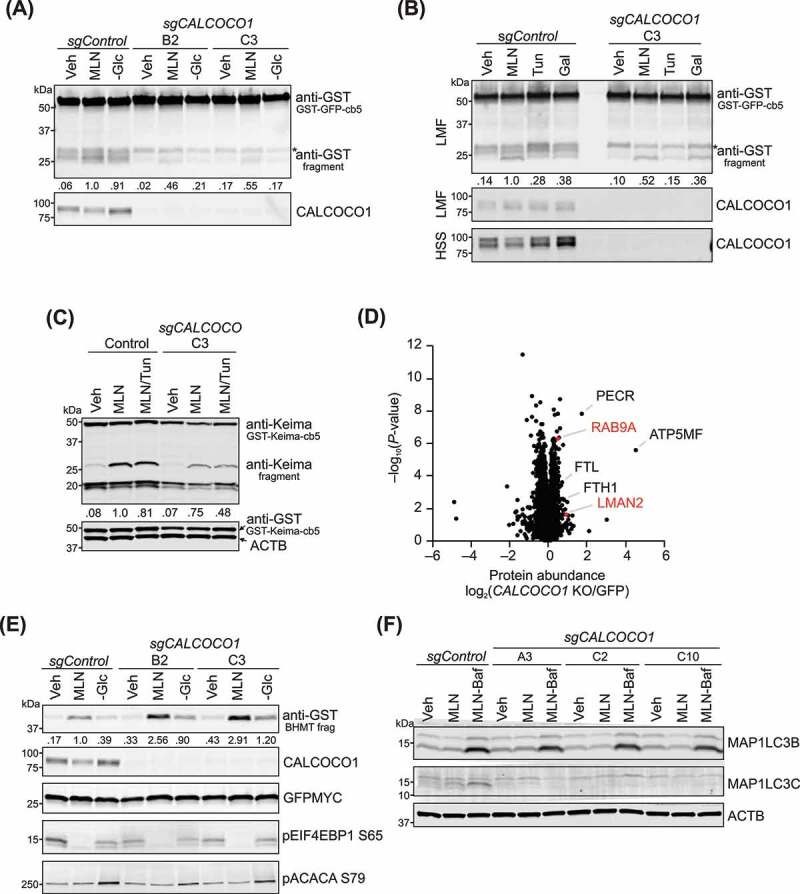
CALCOCO1 has a role in reticulophagy. (A) GSTLSCSGFP-cb5 reticulophagy assay in HEK293 control and 2 sgCALCOCO1 KO cell lines, treated with Veh, 100 nM MLN, or glucose (Glc) starved. GST-tagged proteins/peptides in cell lysates were captured by glutathione agarose affinity purification. Ratio of the bottom fragment to the full-length fusion protein is shown, normalized to MLN in control cells. CALCOCO1 in cell lysates is shown. “B2” and “C3” represent different targets for guide RNA. *nonspecific fragment (B) GSTLSCSGFP-cb5 reticulophagy assay in HEK293 control and sgCALCOCO1 KO cells, treated with Veh, MLN, 10 ug/ml tunicamycin (Tun), or with galactose (Gal). Top panel. GST-tagged proteins/peptides in the LMF. Ratio of the bottom fragment to the full-length fusion protein is shown, normalized to MLN in control cells. Bottom panel. Immunoblot analyses of CALCOCO1 in LMF and high-speed supernatant (HSS) fractions. *nonspecific fragment (C) Immunoblot analysis of GST-Keima-cb5 assay in HEK293 cells, treated with Veh, MLN, and 5 ug/ml Tun as labeled. Ratio of 25 kD fragment to the full-length fusion protein is shown, normalized to MLN in control cells. (D) Relative protein abundances in sgCALCOCO1 KO HEK293 cells compared to sgGFP (GFP) control WT cells (mean, n = 5). Cells were treated for 24 h with 100 nM MLN0128. Proteins were quantified by label-free mass spectrometry. P-values for the protein abundance changes were determined by a two-sided Student’s t-test. (E) GST-BHMT autophagy assay in control and sgCALCOCO1 KO clones, with treatments as in Figure 6A. Ratio of BHMT fragment to GFP, normalized to MLN in control cells, is shown. Immunoblots were probed with antibodies as described. (F) Immunoblot analysis of WT MB231 and 3 sgCALCOCO1 KO cell lines, with antibodies, as shown. Cells were treated for 48 h with Veh, 100 nM MLN, and 20 nM bafilomycin A1, as shown
As a second approach to measure reticulophagy, we replaced the GFP protein in GSTLSCSGFP-cb5 with Keima, an acid-stable fluorescent protein [61]. The processing of Keima fusion proteins produces an acid-stable form of ~25 kD [16], which we confirmed was lysosomal (Fig. S6C). Using this approach, we found that the increase in processed Keima observed in MLN-treated control cells modestly decreased by ~25% in sgCALCOCO1 KO cells (Figure 6C), consistent with the GSTLSCSGFP-cb5 reticulophagy assay, although to a smaller degree. Keima proteins emit green or red signals at neutral or acidic pH, respectively, allowing a determination of autophagic flux. Similar to processed Keima, MLN induced discrete red puncta in both control and sgCALCOCO1 KO cells (Fig. S6D). Although there was considerable cell-to-cell variation in the size and number of puncta in both conditions, there was a trend toward fewer puncta in KO cells.
The relatively modest decrease of MTOR-induced reticulophagy in sgCALCOCO1 KO cells, suggests that CALCOCO1 may have a discreet role in reticulophagy induced by MTOR inhibition, rather than a global one. In support of this hypothesis, ER mass did not significantly differ in sgCALCOCO1 KO cells compared to control cells (Fig. S6E), based on the relative ratio of ER to cell area measurements using ER-TrackerTM Red, as previously described [25]. Together, these data are consistent with the hypothesis that CALCOCO1 supports a subset of reticulophagy induced by MTOR inhibitors. It also raises the possibility that CALCOCO1 has additional selective autophagy targets independent of reticulophagy. To begin to explore this hypothesis, we performed a mass spectrometry proteomics analysis of MLN-treated HEK293 cells with CRISPR-mediated deletion of CALCOCO1 compared to sgGFP control cells under the same conditions. Next, we anticipated the elevation of protein autophagy substrates degraded by CALCOCO1-dependent pathways in CALCOCO1 KO cells. Indeed, in concert with the above reticulophagy assay results, we observed a significant (P < 0.05) increase in select ER-localized proteins, RAB9A (RAB9A, member RAS oncogene family), and LMAN2 (lectin, mannose binding 2), under the conditions used for this mass spectrometry screen (Figure 6D and Dataset S4). Interestingly, we also observed a significant (P < 0.05) increase in select mitochondrial proteins, such as ATP5MF (ATP synthase membrane subunit f), and peroxisomal proteins, such as PECR (peroxisomal trans-2-enoyl-CoA reductase), and in FTL (ferritin light chain) and FTH1 (ferritin heavy chain 1), suggesting that CALCOCO1 might mediate selective autophagy of additional sub-organellar compartments (e.g., sub-mitochondrial mitophagy or ferritophagy) in addition to its role in reticulophagy.
To determine whether the deletion of CALCOCO1 caused a defect in nonselective autophagy, we used the GST-BHMT cargo assay as previously described [39,59] in HEK293 control and gene-edited sgCALCOCO1 KO clones that we treated as in Figure 6A. In contrast to our findings with reticulophagy, the amplitude of bulk autophagy increased in CALCOCO1 KO cells that were MLN-treated or starved of glucose compared to control cells (Figure 6E). In concordance with increased autophagy in the GST-BHMT assay, levels of SQSTM1 decreased slightly faster in sgCALCOCO1 KO cells (Fig. S6F), although we interpret the latter with caution, as SQSTM1 is an autophagy receptor for ubiquitinated cargo. There was no difference in the accumulation of MAP1LC3B-II in HEK293 cells at short time points (Fig. S6G) or in MB231 WT and sgCALCOCO1 KO cells that we treated for 48 h with MLN and 20 nM bafilomycin A1 (Figure 6F). Although we were not able to detect endogenous MAP1LC3C at the shorter time points, we successfully captured MAP1LC3C and demonstrated lysosomal flux under the latter conditions, consistent with prolonged 48 h serum starvation used in previous MAP1LC3C studies [62]. Interestingly, the accumulation of MAP1LC3C-II observed in control cells was abolished in all three sgCALCOCO1 KO cell lines (Figure 6F). These surprising data support the importance of the CALCOCO1-MAP1LC3C interaction and suggest that CALCOCO1 supports MAP1LC3C-dependent selective autophagy in human cells.
Discussion
Proteomic profiling approach for discovering autophagy proteins
We used high-resolution mass spectrometry proteomics to analyze autophagy-deficient MEFs treated with an MTOR kinase inhibitor to identify new autophagy substrates and autophagy-related proteins that are regulated by MTOR. We focused on CALCOCO1 in our initial investigations. However, the datasets contain a wealth of hits that future studies can mine. These include proteins commonly perturbed in atg5–/– and ulk1–/–/ulk2–/– MEFs, and those that are uniquely ATG5, ULK1, or ULK1/2-dependent. Our GO enrichment analyses highlight key differences between the KO MEFs for future exploration. Notably, several of our top candidate proteins are upregulated in human cancer. Among these are PDIA6, whose expression is significant in breast and non-small cell lung cancer (NSCLC) [37,63]; CRABP1 (cellular retinoic acid binding protein 1), a poor prognostic factor in prostate and triple-negative breast cancer [64,65]; and RRBP1 (ribosome binding protein 1), which is increased in colon, breast, and lung cancers and is predictive of poor prognosis [66–68]. Interestingly, RRBP1, an integral ER protein, and its interacting partner on the mitochondrial outer membrane protein, SYNJ2BP (synaptojanin 2 binding protein), were recently shown to facilitate MAM formation [69]. Importantly, MAMs are sites of autophagosome formation [19] and are increased by MTOR inhibitors and glucose deprivation [15], raising the interesting possibility that RRBP1 has a potential role in autophagy at MAMs.
ISG15 (ISG15 ubiquitin like modifier), which was among the most significantly increased proteins in all three KO MEF lines, is another compelling hit because our tissue culture conditions should not have invoked IFN signaling. ISG15 is a ubiquitin-like protein with distinct roles as a ubiquitin-like modifier and as a soluble protein [70,71]. ISG15-modifications regulate protein translation through ISGylation of newly synthesized polyribosome-bound proteins, EIF4E2 (eukaryotic translation initiation factor 4E family member 2), and EIF2AK2 (eukaryotic translation initiation factor 2 alpha kinase 2), all of which inhibit translation [71]. We hypothesize that MTOR, a master regulator of translation, also regulates ISG15 function. ISG15 is associated with autophagy through ISGylation of BECN1 (Beclin 1) [72] and PRKN (parkin RBR E3 ubiquitin ligase) [73]. ISG15 also has a role in the selective removal of cell aggregates [74] and in maintaining mitochondrial function [75], both of which may involve autophagy. In sum, the proteomic data sets are a valuable resource for those studying MTOR and autophagy.
CALCOCO1 is a selective autophagy receptor with a role in reticulophagy
We have defined a new role for CALCOCO1 as a selective autophagy receptor for reticulophagy induced by MTOR inhibition. The finding that CALCOCO1 mediates a fraction of the reticulophagy in these conditions does not weaken its potential importance in this role. We found that CALCOCO1 interacts with LC3 family members, with a preference for human MAP1LC3C. CALCOCO1’s non-canonical CLIR domain partially mediates this interaction; however, the LIR region is essential. Interestingly, MAP1LC3C has been found at the ER, where it cooperates with TECPR2 (tectonin beta-propeller containing protein 2) in the ER export and trafficking of COP-II vesicles [76]. As with CALCOCO1, TECPR2 interacts with MAP1LC3C through a canonical LIR motif. Importantly, Stadel et al showed that MAP1LC3C depletion caused an expansion of ER-sheets [76], confirming the important role of MAP1LC3C in ER maintenance. Our data showing the importance of CALCOCO1 for MAP1LC3C lipidation, suggest that MAP1LC3C localization to both autophagosome and ER membranes is likely to be impaired by loss of CALCOCO1. It remains to be determined what role CALCOCO1 may play in ER vesicle trafficking. Remarkably, although reticulophagy became impaired in sgCALCOCO1 KO cells, the amplitude of bulk autophagy was increased, raising the possibility that CALCOCO1 facilitates a switch between reticulophagy and autophagy. To our knowledge, this phenomenon has not been described for other selective autophagy receptors; therefore, it deserves further investigation. As with its homologs CALCOCO2 and TAX1BP1, CALCOCO1 is likely a selective autophagy receptor for other types of selective autophagy, as suggested by our proteomics analysis of cells lacking CALCOCO1.
We found that under conditions of lysosome inhibition, we could capture CALCOCO1 in association with the other LC3 proteins, including endogenous MAP1LC3B and GABARAPL2. This observation is important because we selected CALCOCO1 as a candidate autophagy receptor from MEFs, which do not express MAP1LC3C. Both GABARAPL2 and MAP1LC3B were enriched in the proteomic datasets, suggesting that they are potential candidates to interact with CALCOCO1 in mice. There are several unanswered questions from these data. Does CALCOCO1, in association with GABARAPL2, MAP1LC3B, or other LC3 proteins, support reticulophagy in rodents? Or does mouse CALCOCO1 mediate autophagy selection and degradation of different substrates? These questions remain open for future investigation.
In the proteomics data, CALCOCO1 had a distinct metabolic profile, as it was more elevated in autophagy-deficient MEFs in glucose-containing medium, compared to galactose medium. This outcome was remarkable considering the high degree of correlation between most hits in the two mediums, and the dominant nature of MLN0128 in both conditions. GO enrichment analyses demonstrated a similar bias for ER components in glucose conditions, which was most striking in the ulk1/2–/– MEFs, suggesting that regulation of reticulophagy induced by MTOR inhibitors is through the MTOR-ULK1/2 signaling pathway. We found that CALCOCO1 deletion had little effect on reticulophagy in cells cultured in galactose medium, consistent with the idea that reticulophagy and CALCOCO1 are more active in glucose conditions. We found it interesting that one of the first roles attributed to CALCOCO1 was as an activator of the key metabolic enzyme PGM (phosphoglucomutase) [77], which reversibly catalyzes the conversion of glucose-6-phosphate to glucose-1-phosphate in the glycogen pathway. In galactose medium, PGM1 activity is important for the ultimate conversion of galactose to glucose-6-phosphate as a substrate for glycolysis, suggesting that CALCOCO1’s role in PGM activation may take precedence when galactose is the predominant carbon source. It is tempting to speculate on the signal(s) that may control CALCOCO1’s preference for reticulophagy or regulation of PGM1 in glucose or galactose mediums, respectively. One possibility is that CALCOCO1 is regulated by glycosylation, based on reports that galactose promotes protein glycosylation much more potently than glucose [78]. This possibility may have clinical relevance, as D-galactose dietary supplementation effectively increase total protein glycosylation and relieve some symptoms in patients with PGM1-CDG, a congenital disorder of glycosylation [79]. Alternatively, phosphorylation, which we discuss in more detail below, may also play a role, as phosphorylation events are key regulators of glycogenesis and glycogenolysis. Further studies are warranted to determine the signals that regulate CALCOCO1 activity in autophagy and metabolism.
In 2003, CALCOCO1 was described as a transcriptional coactivator of nuclear steroid hormone receptors [44] and found to be a component of the p160/steroid receptor coactivator (SRC) complex, along with NCOA2/GRIP1 (nuclear receptor coactivator 2) and CTNNB1 (catenin beta 1) [47]. The presence of a C-terminal zinc finger domain, which can function in DNA-binding, suggests that CALCOCO1 is a nuclear protein. We found little evidence, however, that CALCOCO1 was in the nucleus of either MB231, MCF7 cells, or HEK293 cells, nor that inhibition of MTOR regulates its localization, supporting the idea that CALCOCO1 has another cytoplasmic function. Importantly, our localization data do not exclude CALCOCO1 from a role in the nucleus but suggest that CALCOCO1 functions in both the nucleus and the cytoplasm. Under our conditions, CALCOCO1 expression levels did not appear to correlate with estrogen signaling, with little difference between CALCOCO1 levels in estrogen receptor-positive (MCF7) and negative (MB231) breast cancer cells. Additional studies are needed to determine how CALCOCO1 is transcriptionally regulated. We chose to study CALCOCO1 in breast cancer cells because of reports that CALCOCO1 expression is increased in breast cancer [41–43], and because of the clinical relevance of MTOR inhibitors for this tumor type. Allosteric MTOR inhibitors are second-line therapy for hormone receptor-positive, HER2 (human epidermal growth factor 2)-negative breast cancer, and other next-generation MTOR inhibitors such as MLN0128, are under clinical investigation for other types of breast cancer. Our data showing that CALCOCO1 supports mammosphere formation suggests CALCOCO1 is important for the maintenance of cancer stem cells, a role often attributed to autophagy [80]. Future studies will be important to determine if CALCOCO1-mediated selective autophagy is a mechanism that mediates cancer stem cell survival.
A previous report showed that a CALCOCO1R12H mutation in human breast tumors correlated with an increased risk of breast cancer [41]. We have found that the R12H mutation decreased the interaction between CALCOCO1 and MAP1LC3C, suggesting that this mutation impairs reticulophagy or other cellular functions that depend on this interaction. R12 is preceded by three serine (S) residues, S4, S7, and S11, which are potential phosphorylation sites. S11 and S4 (www.phosphosite.org) were curated from a large proteogenomic study of 105 human breast tumors, which included basal, luminal A, luminal B, and HER2-positive subtypes [81], and from a human cancer cell phosphoproteomic study [82], respectively. The multi-functional roles of CALCOCO1 suggest that phosphorylation and other post-translational modifications could control CALCOCO1’s role in selective autophagy. Determination of the role of kinases, including the MTOR-ULK1/2 axis, in the regulation of CALCOCO1, warrant future studies.
In sum, this study offers a rich database of potential autophagy-regulated proteins to be used as a resource by the autophagy community. In addition, we have defined a new role for CALCOCO1 as a selective autophagy receptor for reticulophagy. There are many unanswered questions regarding the regulation of CALCOCO1 and its roles in selective autophagy, metabolism, and transcription regulation that warrant future investigation. Future studies will undoubtedly clarify the role of CALCOCO1 in cancer and normal physiology.
Materials and methods
Cell culture and experimental conditions
All cells were cultured at 37°C, in 5% CO2, in custom DMEM prepared in house with 4 mM glutamine, 1 mM sodium pyruvate, and 10% fetal calf serum (FCS; Thermo Fisher Scientific, 16000044). FCS in experiments conducted with glucose starvation or galactose was dialyzed extensively against 25 mM HEPES (Thermo Fisher Scientific, J16924AP), pH 7.3, 50 mM NaCl, using Spectra/Por membrane with MWCO 3500 (Spectrum Chemical, 888–11563). MEF cells used in mass spectrometry proteomics were treated for ~17 h in DMEM with 10 mM glucose or 10 mM galactose, with 100 nM MLN0128 (LC laboratories, I-3344). We chose 10 mM glucose for this metabolic experiment, as opposed to the higher, more “diabetic-like” 25 mM glucose. Other cell lines were cultured in DMEM with 25 mM glucose, unless otherwise noted. Chloroquine (CQ; MilliporeSigma, C6628), bafilomycin A1 (Baf; MilliporeSigma, B1793), and tunicamycin (MilliporeSigma, T7765) were used at times and concentrations as described. HEK293 sgControl (sgGFP) and sgCALCOCO1 KO cells used in mass spectrometry proteomics were treated for 24 h with 100 nM MLN0128.
Cell lines used in this study
atg5–/– and matched WT MEFs were a gift from Noboru Mizushima (University of Tokyo) [30]. ulk1–/–, ulk1/2–/– and matched WT MEFs were a gift from Craig Thompson (Memorial Sloan Kettering Cancer Center) [10,11]. HEK293 (human embryonic kidney, sex unknown, ATCC CRL-1573, RRID:CVCL_0045). Hep3B cells (human hepatocellular carcinoma, male, ATCC HB-8064, RRID:CVCL_0326). MDA-MB231 cells (human mammary breast, female, ATCC HTB-26, RRID:CVCL_0062). MCF7 cells (human mammary breast, female, ATCC HTC-22, RRID:CVCL_0031).
All cells were tested routinely for mycoplasma (Lonza, LT07-218). MEFs that were originally found to be mycoplasma-positive were treated prior to the experiments described herein, with Plasmocin (InvivoGen, anti-mpt) and/or BM Cyclin (MilliporeSigma, 10799050001) until they were confirmed to be free of contamination.
Cell transfections
HEK293 cells were transfected by the calcium phosphate method [83], using sterile filtered BES-buffered saline (BBS; 25 mM BES; Sigma-Aldrich, 14853, 140 mM NaCl, 0.75 mM Na2HPO4), pH 7, and 125 mM CaCl2. MB231 cells were transfected with Lipofectamine 3000 (Invitrogen, L3000015), following the manufacturer’s recommended protocol.
CRISPR/Cas9 gene editing
ATG3 and ATG7 genes in Hep3B cells were targeted for gene-editing using the protocol, design tools, and lentiCRISPRv2 one vector system (Addgene, 52961; deposited by Fen Zhang) [84,85]. Five potential target sites were identified for each gene, using the guide design tool at http://crisper.mit.edu. sgRNA target with PAM sequence chosen for ATG3 is in exon 3: 5ʹ-tagtccaccactgtccaacatgg-3ʹ, and for ATG7 is in exon 2: 5ʹ-gctgccagctcgcttaacattgg-3ʹ. Virus particles were prepared at the viral vector core, Cincinnati Children’s Hospital, Cincinnati, OH. A GFP sequence was used as a non-editing control: 5ʹ-gagctggacggcgacgtaaa-3ʹ. Hep3B cells were transduced with viral particles and selected with puromycin (Thermo Fisher Scientific, A1113803). Pooled cells were tested for efficacy of target deletion, and then subjected to limited dilution for single-cell clonal selection. Insertions/deletions were confirmed by sequencing. A similar approach was used for CALCOCO1 in HEK293 and MB231 cells, except for the oligo design. Four sgRNA oligos for CALCOCO1 were chosen based on target sequences identified by an optimized sgRNA algorithm in the Brunello library [86].
Antibodies
Primary antibodies used in this study include: Abcam antibodies for CALCOCO1/CoCoA (ab70564); ABD SeroTech antibodies GAPDH (HCA272); BD Biosciences antibodies for SQSTM1 (610832) and RPS6KB1 (611261); Cell Signaling Technology antibodies for ATG3 (3415), ATG5 (8540), ATG7 (8558), MAP1LC3A/B (12741), MAP1LC3C (14723), ULK1 (6439 and 8054), p-EIF4EBP1 T37/46 (2855), p-EIF4EBP1 S65 (9456), EIF4EBP1 (9644) p-AKT1 S473 (4060), AKT1 (9272) p-RPS6KB T389 (9234), p-ACACA S79 (11818), ACACA (3662) DYKDDDDK-tag (14793), MYC-tag (2276), ACTB (beta-Actin; 3700), LMNA (lamin A/C; 4777); Novus antibodies for MAP1LC3B (NB100-2220); Protein Tech antibodies for RETREG1 (21537-1-AP), GABARAPL2 (18724-1-AP), MAP1LC3C (18726-1-AP); Santa Cruz Biotechnology antibodies for CALCOCO1 (sc-515670), HA-tag (sc-805), GST-tag (sc-138), TUBA (alpha-tubulin; sc-5286); MilliporeSigma antibody for Flag M2 (F1804). Secondary antibodies include LiCOR IRDye 800 CW goat anti-rabbit (926–32211), IRDye 680 RD goat anti-mouse (926–68070); Invitrogen Alexa Fluor 488 goat anti-rabbit (A11088), Alexa Fluor 568 goat anti-mouse (A11004). The anti-HA 12CA5 and anti-MYC 9E10 monoclonal antibodies were purified from 12CA5 and 9E10 hybridomas.
Recombinant proteins
GST-BHMT-IRES-GFP and GSTLSCSGFP-cb5 have been previously described [39,59] (Addgene, 104442 and 104453; deposited by Carol Mercer). GST-RFP-cb5 and GST-Keima-cb5 (Addgene, 137754 and 137755; deposited by Carol Mercer) were made by PCR of RFP or Keima from existing plasmids, using sequence-specific primers with recognition sites for NotI and XbaI restriction enzymes (RE). Purified and RE-digested DNA products were subcloned into GSTLSCSGFP-cb5 acceptor vector. All sequences were confirmed by Sanger sequencing.
HA-tagged MAP1LC3A, MAP1LC3B, MAP1LC3C, GABARAPL1, GABARAP, and GABARAPL2 (Addgene, 137756, 137757, 137758, 137759, 137760, and 137761; deposited by Carol Mercer) were originally cloned from either a human full-length cDNA library (Panomics, NA) or a Jurkats cDNA library, using sequence-specific primers for NCBI reference sequences NM_032514, NM_022818, NM_001004343, NM_031412, NM_007278, and NM_007285, with recognition sites for NotI and XbaI restriction enzymes, and subcloned into pRK5 vectors (kindly provided by George Thomas [IDIBELL]) with an in-frame HA sequence upstream of NotI. HA-GFP (Addgene, 137763; deposited by Carol Mercer) was made in a similar manner. A non-tagged MAP1LC3C (Addgene, 137762; deposited by Carol Mercer) was made by placing MAP1LC3C between NruI and XbaI RE sites in a pRK5 vector. GFP-DDK (Addgene, 137764; deposited by Carol Mercer) was created by placing DYKDDDDK (Flag) with stop codon between NotI and XbaI in pRK5 vector. All sequences were confirmed by Sanger sequencing.
CALCOCO1-MYCDDK in pCMV6-Entry vector was purchased from OriGene (RC201375). GFP-CALCOCO1 and CALCOCO1-HA (Addgene, 137765 and 137766; deposited by Carol Mercer) were created by subcloning CALCOCO1 into pRK5 vectors using techniques described above. L140A, V142A, W47A, and R12H mutations of CALCOCO1 (Addgene, 137767, 137768, 137769, and 137770; deposited by Carol Mercer) were introduced by quick-change PCR mutagenesis. Sense and antisense primers were designed to anneal to the target sequence with the mutated codon(s) in the middle of the primer. Primers were between 25 and 45 bases in length, with a Tm greater than or equal to 78°C (Tm = 81.5 + 0.41[%GC]-[675/N]-%mismatch). PCR was performed using Platinum Pfx DNA polymerase (Invitrogen, 11708), and an annealing temperature of 55°C or less for 18 cycles. DpnI (NEB, R0176) was added to the PCR product to digest template, and the resulting product was transformed into PX5-alpha or PX1-blue competent cells (Protein Express, 961–015 or 961–325). All sequences were confirmed by Sanger sequencing.
Mammalian proteomics liquid chromatography-mass spectrometry
Pelleted cells were lysed in 6 M guanidine hydrochloride (Sigma-Aldrich, C0267-100G) by boiling, and protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23227). 100 µg of protein was aliquoted from each sample and precipitated with 90% methanol. The sample was centrifuged at 12,000 g for 5 min, and the supernatants were discarded. The pellets were resuspended in 8 M urea, 10 mM tris(2-carboxyethyl)phosphine (TCEP; VWR, K831-10G), 40 mM chloroacetamide (CAA; Sigma-Aldrich, G4504-1KG) and 100 mM tris pH 8 for reduction and alkylation of the cysteine residues. Resolubilized samples were then diluted to 1.6 M urea (Sigma-Aldrich, U5378-1KG) using 50 mM tris pH 8 and digested overnight (at room temperature) with sequencing-grade modified trypsin (Promega, V5113), 1:50 enzyme to protein. For CALCOCO1 KO experiments, resolubilized samples were first pre-digested for 4 h (at room temperature) with sequencing-grade LysC (Wako Chemicals, 125–05061), 1:50 enzyme to protein. Samples were then diluted to 1.5 M urea using 100 mM tris pH 8 and digested overnight (at room temperature) with sequencing-grade modified trypsin (Promega, V5113), 1:50 enzyme to protein. Following digestion, peptides were acidified with TFA then de-salted using Strata X polymeric reversed-phase 10 mg/mL desalting columns (Phenomenex, 8B-S100-AAK) equilibrated with 1 mL 100% ACN followed by 1 mL 0.2% formic acid (Thermo Fisher Scientific, 28905). Samples were washed with 1 mL 0.2% formic acid then eluted into clean tubes with 1 mL 80% ACN. Eluate was dried and reconstituted in 0.2% formic acid, and peptide concentration was measured using the Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific, 23275).
2 µg of peptides were loaded onto a reverse-phase nano-LC column (Waters BEH C18 130Ǻ 1.7 µm) that was packed in-house using 35 cm x 75 µm ID, 360 µm OD fused-silica capillary tubing with polyimide coating and a laser-pulled electrospray tip. The chromatography system comprised an UltiMate 3000 UPLC system (Thermo Fisher Scientific, ULTIM3000RSLCNANO) and a column heater set to 55°C. Mobile phase A was 0.2% formic acid in water, and mobile phase B was 70% ACN, 0.2% formic acid. Samples were separated over a 90-min gradient, including time for column re-equilibration, at a flow rate of 325 nL/min. For CALCOCO1 KO experiments, samples were separated over a 135-min gradient including time for column re-equilibration, at a flow rate of 300 nL/min.
Samples were analyzed using an orbitrap Fusion Lumos (Q-OT-qIT, Thermo Fisher Scientific) with an electrospray ionization source. Precursor scans were collected from 300 to 1350 m/z and 60k resolution using a 1e6 AGC target. Precursors selected for MS/MS analysis were isolated at 0.7 Th with the quadrupole mass filter then fragmented via HCD with a collision energy of 25. The maximum inject time for MS/MS analysis was 15 ms with an AGC target of 3e4. Precursors of charge state 1 and those greater than 8 were excluded from selection, dynamic exclusion was set to 5 s with a mass tolerance of 25 ppm, and scans events were performed in top speed mode with a cycle time of 2 s.
Mammalian proteomics data analysis
The raw data were processed using MaxQuant (Version 1.5.5.1), including the Andromeda search algorithm and a target-decoy database of human proteins with isoforms (Uniprot). The following parameters were set: precursor search tolerance at 4.5 ppm; product mass tolerance at 0.5 Da; fixed modification for carbamidomethylation of cysteine residues; variable modification for the oxidation of methionine and N-terminal acetylation; and a maximum of 2 missed cleavages. Peptide spectral match (PSM) false discovery rate (FDR) and protein FDR were both set to 1%. Proteins were quantified within MaxQuant using MaxLFQ, a label-free, intensity-based method that does not require additional chemicals or metabolic labeling. The LFQ minimum ratio count was set to 1 the match between runs feature was used, and MS/MS spectra were not required for LFQ comparisons.
CALCOCO1 protein sequence analysis
CALCOCO family protein sequences were downloaded from UniProt and aligned using MUSCLE [87]. Protein sequences aligned included Homo sapiens CALCOCO1 (UniProt ID Q9P1Z2-1), Homo sapiens CALCOCO2 (Q13137-1), Homo sapiens CALCOCO3 (Q86VP1-1), Mus musculus CALCOCO1 (Q8CGU1-1), Mus musculus CALCOCO2 (A2A6M5-1), and Mus musculus CALCOCO3 (Q3UKC1-1).
The N-terminal domain (NTD) was defined as sequence N-terminal to the conserved SKICH domain. The conserved SKICH domain and the conserved CLIR domain were defined based on a previously defined protein crystal structure of CALCOCO2/NDP52 (calcium binding and coiled coil domain 2) [51]. The conserved LIR motif was identified by searching for a conserved (E/D)Wxx(L/I) motif, which was found to be DW(I/V)GI across the human and mouse CALCOCO proteins. The coiled-coil domain and the zinc finger domain were defined based on their alignment with those previously defined for CALCOCO2 [56]. The LGALS8-interacting motif was previously defined for CALCOCO2 [56]. Because of the location of the LGALS8-interacting motif, which mediates a protein-protein interaction, we defined the sequence between the coiled-coil domain and the zinc finger domain as the “protein-protein interaction” (PPI) region.
Gene list enrichment
For each of the six conditions (atg5−/– KO, ulk1−/– KO, and ulk1/2−/– DKO in galactose and glucose), we created lists of significantly differentially abundant proteins that were either up or down when compared to WT control cells: log2(fold-change) > 0.5 or < – 0.5 and P-value < 0.1. We mapped proteins to Entrez IDs and performed gene list enrichment of GO: Cellular Component terms (lists of proteins associated with a cellular compartment) using the geneListEnrichment function in the CLEAN R package https://github.com/uc-bd2k/CLEAN. GO categories with less than 10 proteins or greater than 1000 proteins were discarded and the full list of ~6000 proteins measured in the study was used as background for gene list enrichment. Each list of differentially abundant proteins was tested for significant overlap with each GO term using a Fisher Exact test. Again, P-values are reported as – log10(p-value).
Immunoprecipitation studies
Cells were transfected with WT or mutant CALCOCO1-MYCDDK, HA-MAP1LC3s, HA-GABARAPs or HA-GFP and harvested in CHAPS extraction buffer (40 mM HEPES, pH 7.5, 120 mM NaCl, 1 mM EDTA (MilliporeSigma, EX05505), 10 mM NaPPi (Sigma-Aldrich, 71501) 10 mM beta-glycerolphosphate (Sigma-Aldrich, G9422), 50 mM NaF (Sigma-Aldrich, S7920), 0.3% CHAPS (Sigma-Aldrich, 220201)), supplemented with 0.2 mM PMSF (Sigma-Aldrich, 78830). Total protein measurements were performed by the Bradford method (Bio-Rad, 5000006). Equal amounts of protein were diluted at least 1:4 in CHAPS extraction buffer, before precipitation with Pierce Protein G agarose (Thermo Scientific, 20398) pre-coupled to an anti-HA (12CA5) or anti-Flag M2 antibody for 2–4 h at 4°C. Beads were washed with CHAPS wash buffer (CHAPS EB without NaCl), and bound proteins were eluted in SDS-PAGE sample buffer, resolved by SDS-PAGE and visualized by western blotting.
GST-BHMT autophagy assay
The GST-BHMT assay was performed as previously described [39,59]. In brief, cells expressing GST-BHMT-IRES-GFPmyc (Addgene, 104442, deposited by Carol Mercer) were treated as described, with leupeptin (11 µM; Bachem, N-1000) and E64d (6 µM; Bachem, N-1650) added at the beginning of each treatment to stabilize the lysosomal fragment. Cells were harvested in cold extraction buffer (EB; 50 mM TRIS-HCl pH 8, 120 mM NaCl, 5 mM NaPPI, 10 mM NaF, 30 mM para-nitrophenylphosphate (Thermo Scientific Pierce, PI034045), 1 mM benzamidine (Thermo Fisher Scientific, AC401791000), 0.2% NP-40 (Thermo Fisher Scientific, 18-607-275) and fresh 0.2 mM PMSF) with 1% Triton X-100 (EB-TX; Thermo Fisher Scientific, BP151). Total protein was determined by the Bradford method; and GST-BHMT was precipitated with Pierce glutathione agarose (Thermo Fisher Scientific, 16100) from cell lysates (300–400 µg). Bound GST-proteins were washed once in EB with 500 mM NaCl, and 1–2 times in wash buffer (50 mM MOPS (Sigma-Aldrich, 69947) pH 7.5, 10 mM NaF, 30 mM beta-gycerolphosphate, 1 mM DTT, 0.1% Triton X-100, 10% glycerol). Precipitates were eluted in SDS-PAGE sample buffer, resolved by SDS-PAGE, followed by western blotting. Whole-cell lysates (typically 25 µg) were resolved by SDS-PAGE, followed by western blotting, probing for antibodies as described. Western blotting for GFP-MYC was used to normalize expression of the GST-BHMT-IRES-GFPmyc plasmid. An autophagy index was calculated as we have done previously [15], taking the ratio of the GST-BHMT fragment to GFP, normalized to the value of the positive control, MLN0128.
GSTLSCSGFP-cb5 and GST-Keima-cb5 reticulophagy assays
The GSTLSCSGFP-cb5 reticulophagy assay was performed as previously described [59]. Cells transfected with GSTLSCSGFP-cb5 were treated as described, with leupeptin (11 µM) and E64d (6 µM) added at the beginning of each treatment. Cell lysates were collected as described for the GST-BHMT assay, or were fractionated for light membrane fractions (LMFs) as we have done previously [15,39]. All steps were performed on ice or at 4°C. In brief, PBS-washed cells were collected in PBS (137 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4) and centrifuged at 1000 g for 5 min; the cell pellet was suspended in 500 µL homogenization buffer (HB; 10 mM TRIS pH 7.5, 300 mM sucrose, 0.2 mM EDTA, 0.2 mM PMSF) and disrupted by Dounce homogenization for 18–20 strokes. The homogenate was centrifuged at 540 g, and the supernatant was collected. The homogenization step was repeated with the low-speed pellet, and the combined supernatant was centrifuged at 12,000 g for 20 min to separate the LMF in the pellet from the high-speed supernatant (HSS). The LMF fraction was homogenized in EB-TX; LMF protein was determined by the Bradford method; and GST-tagged proteins were precipitated with glutathione agarose from LMF extract (typically 100 µg), as described above. 10 µg of LMF extract was resolved by SDS-PAGE, followed by western blotting. The ratio of the GST-fragment to full-length GST-GFP was used to normalize for expression differences of the exogenous reporter protein.
For the biochemical GST-Keima-cb5 assay, treatments did not include leupeptin or E64d because of the lysosomal stability of the Keima protein, and cells were harvested in EB-TX. Protein concentration was determined by Bradford method, resolved on SDS-PAGE, and probed with anti-Keima antibody. The Keima to GST-Keima-cb5 ratio was used to normalize for expression differences of the reporter protein.
For live-cell imaging of GST-Keima-cb5 expressing cells, 320,000 MB231 cells were seeded on 35 mm coverslip dishes (MatTek Corporation, P35G-0.170-14-C), transfected with GST-Keima-cb5, and treated as described. Z-stack images of live cells were acquired with a Zeiss LSM710 confocal with a Zeiss Observer.Z1 stand and a Zeiss Plan-Apochromat 63x/1.4 objective. Keima fluorescence was measured from 600–740 nm upon excitation with a 458 nm or 561 nm laser, respectively. Images were analyzed with Fiji ImageJ (https://imagej.net/Fiji). The experiment was performed a minimum of 3 times.
ER-trackerTM Red staining and analysis
ER-TrackerTM Red (Invitrogen, E34250) was prepared and used according to manufacturer recommendations. MB231 cells were seeded on 35 mm coverslip dishes (MatTek Corporation, P35G-0.170-14-C) and loaded with a final concentration of 0.5 uM ER-TrackerTM for 15 min. Live cells were imaged using the Keyence BZ-9000 microscope, 60x oil objective. Stained images were subject to background subtraction, thresholding and quantitation using Fiji ImageJ as previously described [25]. Cells were delineated by outlining to determine the internal area of each cell. The percentage of ER:cell was calculated, averaged by cell line and treatment condition. Data were plotted as ER percentage, with error bars representing 1 standard deviation (SD).
Mammospheres
Complete MammoCultTM medium (STEMCELL Technologies, 05620) was prepared according to the manufacturer’s recommendation. The mammosphere protocol followed the guidelines of MammoCultTM Technical Bulletin #29936. Briefly, MB231 cells were grown in a monolayer to ~80% confluence. Cells were washed twice with warm Hank’s Balanced Salt Solution (HBSS) (Thermo Fisher Scientific, 14170112), scraped, resuspended in 10 mL MammoCultTM medium, and centrifuged for 3 min at 500 g. The cell pellet was resuspended in 2 mL MammoCultTM medium and cells were counted using the Countess II cell counter (Thermo Fisher Scientific, AMQAF1000). 5,000 cells per well were seeded in triplicate in 6 well ultra-low adherent plates in MammoCultTM medium and grown for 7 d at 37°C, 5% CO2. On day 7, the number of mammospheres in each well of a size greater than 60 µm was determined by light microscopy. To passage spheres, tumor spheres were collected in a 50 mL conical tube and centrifuged for 5 min at 350 g. The supernatant was removed, 800 µL of warm Trypsin-EDTA (Thermo Fisher Scientific, 25200056) was added to each pellet, and cells were triturated against the wall of the tube for 2 min and the reaction stopped with 5 mL of cold HBSS with 2% bovine serum albumin (BSA; Sigma-Aldrich, A9647). Cells were centrifuged for 5 min at 350 g, and cell pellet was resuspended in 500 µL MammoCultTM medium. Viable cells were counted by Trypan Blue (Thermo Fisher Scientific, 15250061) exclusion assay as above. 5,000 cells per well were seeded in triplicate in 6 well ultra-low adherent plates, grown for 7 d, and counted as described above. The experiment was repeated a minimum of 3 times.
Proliferation assay
Cell proliferation assays were performed using the CellTiter-Glo® Luminescent Cell Viability assay (Promega, G7570) following the manufacturer’s protocol. In brief, cells were trypsinized, counted, and dilutions were made for each WT and sgCALCOCO1 KO cell line. 4,000 cells/well were plated in sextuplicate in white-sided 96-well plates. Wells on a parallel titration plate were seeded in quadruplicate with cell numbers from 4,000 to 16,000 to confirm linearity. After 24 h, the titration plate was assayed for luminescence (Promega GloMAX), and the average of the 4,000 cell wells was used as the day 0 reading. On day 0, cells were treated with Veh or 100 nM MLN0128, and assayed for luminescence at 24, 48, and 72 h. Relative light units (RLUs) were averaged, and fold-change determined relative to day 0.
Scratch assay
Scratch assays were performed using the IncuCyte® scratch wound cell migration assay (Essen Biosciences). Briefly, 45,000 MB231 cells (WT and sgCALCOCO1 KO clones) were seeded in 9 replicate wells for each condition and cell type on 96-well ImageLock plates (Essen Biosciences, 4379) and allowed to settle overnight. The next day, a 700–800 µm wound was made in each well using a 96-pin WoundMakerTM (Essen Biosciences, 4493). Wells were washed twice with DMEM, and fresh media was added with vehicle or 100 nM MLN0128. Images of each well were taken every 2 h, using the IncuCyte® Zoom, 10x objective. Data acquisition and analyses were performed per recommended protocol.
Nuclear and cytoplasmic extracts
HEK293, MB231, and MCF7 cells were treated for 24 h with vehicle or 100 nM MLN0128 and harvested for nuclear and cytoplasmic extracts using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78833). Briefly, cells were harvested with Trypsin-EDTA, centrifuged at 500 g for 5 min, washed in PBS, and centrifuged at 500 g for 2–3 min. The cytoplasm and nuclear proteins were extracted with cytoplasmic reagents I and II (CER1, CER2), and nuclear extraction reagent (NER), as recommended.
Microscopy
MB231 cells were seeded on 15 mm coverslips, grown to ~70% confluency and treated as described. Cells were washed with PBS, fixed with either 4% paraformaldehyde (PFA) or ice-cold methanol, rinsed x3 with PBS, permeabilized with 0.3% Triton X-100 (PBS), and blocked with 1% bovine serum albumin (BSA) and 0.1% Triton X-100 in PBS. Primary antibodies for immunofluorescence, anti-HA (12CA5; 1:4000 dilution); anti-MAP1LC3C (1:250); anti-MYC (9E10; 1:1000), were diluted in block and incubated for 1 h at 37°C in a humidity chamber. Cells were rinsed x3 with PBS, incubated with Alexa Fluor 488 or Alexa Fluor 568 secondary antibody (1:1000), rinsed, dried and cover-slipped with mounting media with DAPI. 6–12 z-stack images were captured for each treatment, using a Zeiss Axio Observer Z1 inverted microscope, 63x oil immersion lens (1x zoom), connected to a Zeiss LSM710 confocal, and analyzed with Fiji ImageJ (https://imagej.net/Fiji). GFP and RFP proteins were cover-slipped with mounting media with DAPI (Abcam, ab104139), and imaged by confocal microscopy as described above. High-resolution images were collected with the support of Dr. Birgit Ehmer, using a Nikon SIMe superresolution microscope with a Nikon Ti2e inverted stand and a Nikon SR HP Spo TIRF 100x oil lens. Imaging for ER-TrackerTM Red used the Keyence BZ-9000 microscope, 60x oil objective.
Statistical analysis
Statistical analyses of IP and ER-tracker Red utilized GraphPad Prism 8. Statistical significance was determined using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons tests to compare multiple treatments. Data are presented as mean ±SD. P-values for mass spectrometry abundance changes were determined by a two-sided Student’s t-test. Specific details of the proteomics and GO analyses can be found under the “Mammalian proteomics data analysis” and “Gene list enrichment” sections, respectively.
Supplementary Material
Acknowledgments
We acknowledge Drs. Noboru Mizushima and Craig Thompson for generously sharing the atg5–/–, ulk1–/–, ulk1/2–/– and matched MEFs, and Drs. Heeseon An and Wade Harper for sharing the mito-Keima plasmid, which we used to make GST-Keima-cb5. We thank Dr. Birgit Ehmer (UC Dept. of Cancer Biology Center for Biological Microscopy) for her knowledge and kind assistance with confocal and SIM microscopy. We are indebted to Dr. Maria Czyzyk-krzeska for discussions on MAP1LC3C, her generosity with MAP1LC3C resources, and for critically reading the manuscript.
Funding Statement
This work was supported by the Ride Cincinnati Foundation (PI: CAM, Co-PI: HET), and by the National Institute of General Medical Sciences, 1R35GM131795 (PI: DJP).
Highlights
Proteomic profiling approach for discovering autophagy proteins in yeast or mammals
Resource of nutrient- or MTOR- regulated proteins linked to autophagy
CALCOCO1 functions in autophagy
CALCOCO1 interacts with MAP1LC3C and supports reticulophagy
Data deposition
Raw data for the MEF proteomics screen is available on the PRoteomics IDEntifications database (PRIDE): project accession PXD014990.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Levine B, Kroemer G.. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yang Z, Klionsky DJ.. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yang Z, Klionsky DJ.. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Farre JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17:537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lebovitz CB, DeVorkin L, Bosc D, et al. Precision autophagy: will the next wave of selective autophagy markers and specific autophagy inhibitors feed clinical pipelines? Autophagy. 2015;11:1949–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017;41:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Noda T. Regulation of autophagy through torc1 and mtorc1. Biomolecules. 2017;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kamada Y, Funakoshi T, Shintani T, et al. Tor-mediated induction of autophagy via an apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kamada Y, Yoshino K, Kondo C, et al. Tor directly controls the atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kundu M, Lindsten T, Yang CY, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cheong H, Lindsten T, Wu J, et al. Ammonia-induced autophagy is independent of ulk1/ulk2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dunlop EA, Tee AR. Mtor and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. [DOI] [PubMed] [Google Scholar]
- [13].Thomas HE, Mercer CA, Carnevalli LS, et al. Mtor inhibitors synergize on regression, reversal of gene expression, and autophagy in hepatocellular carcinoma. Sci Transl Med. 2012;4:139ra184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Thoreen CC, Kang SA, Chang JW, et al. An atp-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mtorc1. J Biol Chem. 2009;284:8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thomas HE, Zhang Y, Stefely JA, et al. Mitochondrial complex i activity is required for maximal autophagy. Cell Rep. 2018;24(2404–2417):e2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].An H, Harper JW. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol. 2018;20:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].An H, Ordureau A, Paulo JA, et al. Tex264 is an endoplasmic reticulum-resident atg8-interacting protein critical for er remodeling during nutrient stress. Mol Cell. 2019;74:891–908.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].English AR, Zurek N, Voeltz GK. Peripheral er structure and function. Curr Opin Cell Biol. 2009;21:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at er-mitochondria contact sites. Nature. 2013;495:389–393. [DOI] [PubMed] [Google Scholar]
- [20].Grumati P, Dikic I, Stolz A. Er-phagy at a glance. J Cell Sci. 2018;131(17). [DOI] [PubMed] [Google Scholar]
- [21].Wilkinson S. Er-phagy: shaping up and de-stressing the endoplasmic reticulum. Febs J. 2019;286(14):2645–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. [DOI] [PubMed] [Google Scholar]
- [23].Fumagalli F, Noack J, Bergmann TJ, et al. Translocon component sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol. 2016;18:1173–1184. [DOI] [PubMed] [Google Scholar]
- [24].Grumati P, Morozzi G, Holper S, et al. Full length rtn3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife. 2017;6:e25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Smith MD, Harley ME, Kemp AJ, et al. Ccpg1 is a non-canonical autophagy cargo receptor essential for er-phagy and pancreatic er proteostasis. Dev Cell. 2018;44(217–232):e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stefely JA, Kwiecien NW, Freiberger EC, et al. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat Biotechnol. 2016;34:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Iwama R, Ohsumi Y. Analysis of autophagy activated during changes in carbon source availability in yeast cells. J Biol Chem. 2019;294:5590–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kim J, Kundu M, Viollet B, et al. Ampk and mtor regulate autophagy through direct phosphorylation of ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mochida K, Oikawa Y, Kimura Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–362. [DOI] [PubMed] [Google Scholar]
- [30].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. [DOI] [PubMed] [Google Scholar]
- [31].Li TY, Sun Y, Liang Y, et al. Ulk1/2 constitute a bifurcate node controlling glucose metabolic fluxes in addition to autophagy. Mol Cell. 2016;62:359–370. [DOI] [PubMed] [Google Scholar]
- [32].Bestebroer J, V’Kovski P, Mauthe M, et al. Hidden behind autophagy: the unconventional roles of atg proteins. Traffic. 2013;14:1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Marroquin LD, Hynes J, Dykens JA, et al. Circumventing the crabtree effect: replacing media glucose with galactose increases susceptibility of hepg2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. [DOI] [PubMed] [Google Scholar]
- [34].Mizushima N, Kuma A, Kobayashi Y, et al. Mouse apg16l, a novel wd-repeat protein, targets to the autophagic isolation membrane with the apg12-apg5 conjugate. J Cell Sci. 2003;116(116):1679–1688. [DOI] [PubMed] [Google Scholar]
- [35].Mancias JD, Kimmelman AC. Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol. 2016;428(428):1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Birgisdottir AB, Lamark T, Johansen T. The lir motif - crucial for selective autophagy. J Cell Sci. 2013;126:3237–3247. [DOI] [PubMed] [Google Scholar]
- [37].Bai Y, Liu X, Qi X, et al. Pdia6 modulates apoptosis and autophagy of non-small cell lung cancer cells via the map4k1/jnk signaling pathway. EBioMedicine. 2019;42:311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saxton RA, Sabatini DM. Mtor signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. [DOI] [PubMed] [Google Scholar]
- [39].Mercer CA, Kaliappan A, Dennis PB. Macroautophagy-dependent, intralysosomal cleavage of a betaine homocysteine methyltransferase fusion protein requires stable multimerization. Autophagy. 2008;4:185–194. [DOI] [PubMed] [Google Scholar]
- [40].Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Haiman CA, Garcia RR, Hsu C, et al. Screening and association testing of common coding variation in steroid hormone receptor co-activator and co-repressor genes in relation to breast cancer risk: the multiethnic cohort. BMC Cancer. 2009;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].von der Heyde S, Wagner S, Czerny A, et al. Mrna profiling reveals determinants of trastuzumab efficiency in her2-positive breast cancer. PLoS One. 2015;10:e0117818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Muranen TA, Greco D, Fagerholm R, et al. Breast tumors from chek2 1100delc-mutation carriers: genomic landscape and clinical implications. Breast Cancer Res. 2011;13:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim JH, Li H, Stallcup MR. Cocoa, a nuclear receptor coactivator which acts through an n-terminal activation domain of p160 coactivators. Mol Cell. 2003;12:1537–1549. [DOI] [PubMed] [Google Scholar]
- [45].Kim JH, Stallcup MR. Role of the coiled-coil coactivator (cocoa) in aryl hydrocarbon receptor-mediated transcription. J Biol Chem. 2004;279:49842–49848. [DOI] [PubMed] [Google Scholar]
- [46].Yang CK, Kim JH, Stallcup MR. Role of the n-terminal activation domain of the coiled-coil coactivator in mediating transcriptional activation by beta-catenin. Mol Endocrinol. 2006;20:3251–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang CK, Kim JH, Li H, et al. Differential use of functional domains by coiled-coil coactivator in its synergistic coactivator function with beta-catenin or grip1. J Biol Chem. 2006;281:3389–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Steelman LS, Martelli AM, Cocco L, et al. The therapeutic potential of mtor inhibitors in breast cancer. Br J Clin Pharmacol. 2016;82:1189–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wiemann S, Arlt D, Huber W, et al. From orfeome to biology: A functional genomics pipeline. Genome Res. 2004;14:2136–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].von Muhlinen N, Akutsu M, Ravenhill BJ, et al. Lc3c, bound selectively by a noncanonical lir motif in ndp52, is required for antibacterial autophagy. Mol Cell. 2012;48:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tumbarello DA, Manna PT, Allen M, et al. The autophagy receptor tax1bp1 and the molecular motor myosin vi are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog. 2015;11:e1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase pink1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang Y, Wang G, Huang X, et al. Crystallographic and modelling studies suggest that the skich domains from different protein families share a common ig-like fold but harbour substantial structural variations. J Biomol Struct Dyn. 2015;33:1385–1398. [DOI] [PubMed] [Google Scholar]
- [55].Whang MI, Tavares RM, Benjamin DI, et al. The ubiquitin binding protein tax1bp1 mediates autophagasome induction and the metabolic transition of activated t cells. Immunity. 2017;46:405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Thurston TL, Wandel MP, von Muhlinen N, et al. 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kim BW, Hong SB, Kim JH, et al. Structural basis for recognition of autophagic receptor ndp52 by the sugar receptor galectin-8. Nat Commun. 2013;4:1613. [DOI] [PubMed] [Google Scholar]
- [58].Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. [DOI] [PubMed] [Google Scholar]
- [59].Dennis PB, Mercer CA. The gst-bhmt assay and related assays for autophagy. Methods Enzymol. 2009;452:97–118. [DOI] [PubMed] [Google Scholar]
- [60].Zhu W, Cowie A, Wasfy GW, et al. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. Embo J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- [61].Katayama H, Kogure T, Mizushima N, et al. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. [DOI] [PubMed] [Google Scholar]
- [62].Mikhaylova O, Stratton Y, Hall D, et al. Vhl-regulated mir-204 suppresses tumor growth through inhibition of lc3b-mediated autophagy in renal clear cell carcinoma. Cancer Cell. 2012;21:532–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ramos FS, Serino LT, Carvalho CM, et al. Pdia3 and pdia6 gene expression as an aggressiveness marker in primary ductal breast cancer. Genet Mol Res. 2015;14:6960–6967. [DOI] [PubMed] [Google Scholar]
- [64].Liu RZ, Garcia E, Glubrecht DD, et al. Crabp1 is associated with a poor prognosis in breast cancer: adding to the complexity of breast cancer cell response to retinoic acid. Mol Cancer. 2015;14:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Choi N, Park J, Lee JS, et al. Mir-93/mir-106b/mir-375-cic-crabp1: A novel regulatory axis in prostate cancer progression. Oncotarget. 2015;6:23533–23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pan Y, Cao F, Guo A, et al. Endoplasmic reticulum ribosome-binding protein 1, rrbp1, promotes progression of colorectal cancer and predicts an unfavourable prognosis. Br J Cancer. 2015;113:763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liang X, Sun S, Zhang X, et al. Expression of ribosome-binding protein 1 correlates with shorter survival in her-2 positive breast cancer. Cancer Sci. 2015;106:740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tsai HY, Yang YF, Wu AT, et al. Endoplasmic reticulum ribosome-binding protein 1 (rrbp1) overexpression is frequently found in lung cancer patients and alleviates intracellular stress-induced apoptosis through the enhancement of grp78. Oncogene. 2013;32:4921–4931. [DOI] [PubMed] [Google Scholar]
- [69].Hung V, Lam SS, Udeshi ND, et al. Proteomic mapping of cytosol-facing outer mitochondrial and er membranes in living human cells by proximity biotinylation. Elife. 2017;6:e24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hermann M, Bogunovic D. Isg15: in sickness and in health. Trends Immunol. 2017;38:79–93. [DOI] [PubMed] [Google Scholar]
- [71].Han HG, Moon HW, Jeon YJ. Isg15 in cancer: beyond ubiquitin-like protein. Cancer Lett. 2018;438:52–62. [DOI] [PubMed] [Google Scholar]
- [72].Xu D, Zhang T, Xiao J, et al. Modification of becn1 by isg15 plays a crucial role in autophagy regulation by type i ifn/interferon. Autophagy. 2015;11:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Im E, Yoo L, Hyun M, et al. Covalent isg15 conjugation positively regulates the ubiquitin e3 ligase activity of parkin. Open Biol. 2016;6:160193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nakashima H, Nguyen T, Goins WF, et al. Interferon-stimulated gene 15 (isg15) and isg15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (hdac6) and sqstm1/p62. J Biol Chem. 2015;290:1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Baldanta S, Fernandez-Escobar M, Acin-Perez R, et al. Isg15 governs mitochondrial function in macrophages following vaccinia virus infection. PLoS Pathog. 2017;13:e1006651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Stadel D, Millarte V, Tillmann KD, et al. Tecpr2 cooperates with lc3c to regulate copii-dependent er export. Mol Cell. 2015;60:89–104. [DOI] [PubMed] [Google Scholar]
- [77].Takahashi K, Inuzuka M, Ingi T. Cellular signaling mediated by calphoglin-induced activation of ipp and pgm. Biochem Biophys Res Commun. 2004;325:203–214. [DOI] [PubMed] [Google Scholar]
- [78].Sasaoka N, Imamura H, Kakizuka A. A trace amount of galactose, a major component of milk sugar, allows maturation of glycoproteins during sugar starvation. iScience. 2018;10:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Morava E. Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable cdg. Mol Genet Metab. 2014;112:275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Boya P, Codogno P, Rodriguez-Muela N. Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development. 2018;145:dev146506. [DOI] [PubMed] [Google Scholar]
- [81].Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhou H, Di Palma S, Preisinger C, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12:260–271. [DOI] [PubMed] [Google Scholar]
- [83].Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: A highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- [84].Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for crispr screening. Nat Methods. 2014;11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale crispr-cas9 knockout screening in human cells. Science. 2014;343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Doench JG, Fusi N, Sullender M, et al. Optimized sgrna design to maximize activity and minimize off-target effects of crispr-cas9. Nat Biotechnol. 2016;34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Edgar RC. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.