

ABSTRACT
Microglial activation-induced neuroinflammation is closely associated with the development of Parkinson disease (PD). Macroautophagy/autophagy regulates many biological processes, but the role of autophagy in microglial activation during PD development remains largely unclear. In this study, we showed that deletion of microglial Atg5 caused PD-like symptoms in mice, characterized by impairment in motor coordination and cognitive learning, loss of tyrosine hydroxylase (TH) neurons, enhancement of neuroinflammation and reduction in dopamine levels in the striatum. Mechanistically, we found that inhibition of autophagy led to NLRP3 (NLR family pyrin domain containing 3) inflammasome activation via PDE10A (phosphodiesterase 10A)–cyclic adenosine monophosphate (cAMP) signaling in microglia, and the sequential upregulation of downstream IL1B/IL-1β in turn increased the expression of MIF (macrophage migration inhibitory factor [glycosylation-inhibiting factor]), a pro-inflammatory cytokine. Inhibition of NLRP3 inflammasome activation by administration of MCC950, a specific inhibitor for NLRP3, decreased MIF expression and neuroinflammatory levels, and rescued the loss of TH neurons in the substantial nigra (SN). Interestingly, we found that serum MIF levels in PD patients were significantly elevated. Taken together, our results reveal an important role of autophagy in microglial activation-driven PD-like symptoms, thus providing potential targets for the clinical treatment of PD.
Abbreviations: ATG: autophagy related; cAMP: cyclic adenosine monophosphate; cKO: conditional knockout; NOS2/INOS: nitric oxide synthase 2, inducible; IL1B: interleukin 1 beta; ITGAM/CD-11b: integrin alpha M/cluster of differentiation molecule 11B; MAP1LC3: microtubule-associated protein 1 light chain 3; MIF: macrophage migration inhibitory factor (glycosylation-inhibiting factor); NLRP3: NLR family pyrin domain containing 3; PBS: phosphate-buffered saline; PD: parkinson disease; PDE10A: phosphodiesterase 10A; SN: substantial nigra; TH: tyrosine hydroxylase; TNF: tumor necrosis factor; WT: wild type.
KEYWORDS: Autophagy, cytokine, microglia, neuroinflammation, parkinson disease
Introduction
Parkinson disease (PD) is one of the most common progressive neurodegenerative diseases and is characterized by the loss of dopaminergic neurons in the substantia nigra (SN) and loss of dopaminergic nerve terminals and dopamine content in the striatum [1–5]. Multiple lines of evidence show that chronic neuroinflammation induced by microglia is closely linked with the progress of PD, thus representing a potential therapeutic target [6–9]. However, whether defects in genes regulating microglia-induced inflammation play a causative role in PD needs to be clarified. Microglia are cells originating from the yolk sac that populate the central nervous system (CNS) during embryogenesis. Activated microglia are also observed in neurodegenerative diseases other than PD, i.e.; Alzheimer disease (AD) and stroke [8,10–12]. The activation status of microglia can be generally divided into 2 types: M1 and M2. M1 microglial activation leads to pro-inflammatory effects by via production of pro-inflammatory cytokines such as TNF/TNF-α (tumor necrosis factor), IL6 (interleukin 6) and IL1B (interleukin 1 beta). M2 microglial activation plays anti-inflammatory roles by phagocytosing cells debris or damaged neurons and secreting anti-inflammatory cytokines such as IL4 and IL10. Notably, microglia can transform back and forth into each of these types. However, until now, the regulatory mechanisms for these transformations have remained poorly understood.
Recent studies have revealed that autophagy plays a critical role in immunity, balancing beneficial and detrimental effects. Autophagy dysfunction is associated with multiple inflammation-related diseases [13–18]. In the central nervous system, autophagy defects cause neurodegeneration in mice [19–21]. However, studies have mainly focused on neuronal cells, rather than microglia. As microglia-induced inflammation plays a critical role in the development of neurodegenerative diseases, the role of microglial autophagy in microglial activation has gained increasing attention. In Drosophila, autophagy regulates the activation of Manf (Mesencephalic astrocyte-derived neurotrophic factor) in immunoreactive cells, which are cells similar to microglia in vertebrates [22]. In mammalian cells, microglial autophagy has been demonstrated to be critical for microglial activation in vitro, and inhibition of microglial autophagy causes increased M1 microglial activation, leading to upregulation of pro-inflammatory cytokines [23–25]. Our previous study demonstrated that autophagy inhibited MAVS (mitochondrial antiviral signaling protein) activation in microglia. Moreover, inhibition of ABL1/c-Abl-MAVS signaling prevents 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced microglial activation and dopaminergic neuron loss [26]. However, whether defects in microglial autophagy directly induce neurodegenerative diseases has not been characterized.
In this study, we showed that loss of microglial Atg5 function caused impairment in motor coordination and cognitive learning in mice. Mechanistically, defects in microglial autophagy increased neuroinflammation by activating the NLRP3 inflammasome via PDE10A-cAMP signaling, suggesting that microglial autophagy might be a potential target for the treatment of microglia-driven inflammatory brain diseases.
Results
Loss of microglial Atg5 causes defects in motor coordination and cognitive learning
Atg5 (autophagy related 5) is one of the core components of autophagy-related machinery. However, the cellular expression pattern of Atg5 in the central nervous system has not been characterized due to the unavailability of ATG5 antibodies in mouse brain tissue. To address this issue, we used the fluorescent RNAScope probes, a sensitive in situ hybridization (ISH) technique, to analyze Atg5 expression in sectioned brain tissue. As shown in Figure 1A, Atg5 signal could be detected in Cx3cr1 (a specific marker for microglia)-positive cells, Gfap (a specific marker for astrocyte) -positive cells and Rbfox3 (a specific marker for neuron)-positive cells, indicating that Atg5 is expressed in most cells of the adult mouse brain. Previous studies have shown that autophagy defects in neurons are linked with the development of neurodegeneration [19–21], but the role of microglial autophagy has yet to be clarified. To investigate the effect of microglial autophagy in neurological disorders, we generated a mouse strain that had a microglia-specific deletion of the Atg5 gene by crossing Atg5 floxed mice with Itgam/CD-11b-Cre mice. As shown in Figure 1A,B, compared to Atg5 wild type (WT) mice, the level of Atg5 in the microglia from Atg5 conditional knockout (cKO) mice was dramatically decreased, with no change in astrocytes and neurons. In addition, we cultured primary microglia, astrocytes and neurons from Atg5 WT and Atg5 cKO mice and found that Atg5 knockout only occurred in primary microglia, but not in primary astrocytes and neurons (Fig. S1A). Furthermore, we injected a pAAV-EF1a-DIO-mCherry virus (a Cre enzyme-initiated virus) into the substantial nigra (SN) of Itgam-Cre mice. As shown in Fig. S1B, mCherry was only present in IBA1-positive cells, but not in GFAP-positive cells, indicating microglial specificity of Itgam-Cre in the mouse brain.
Figure 1.
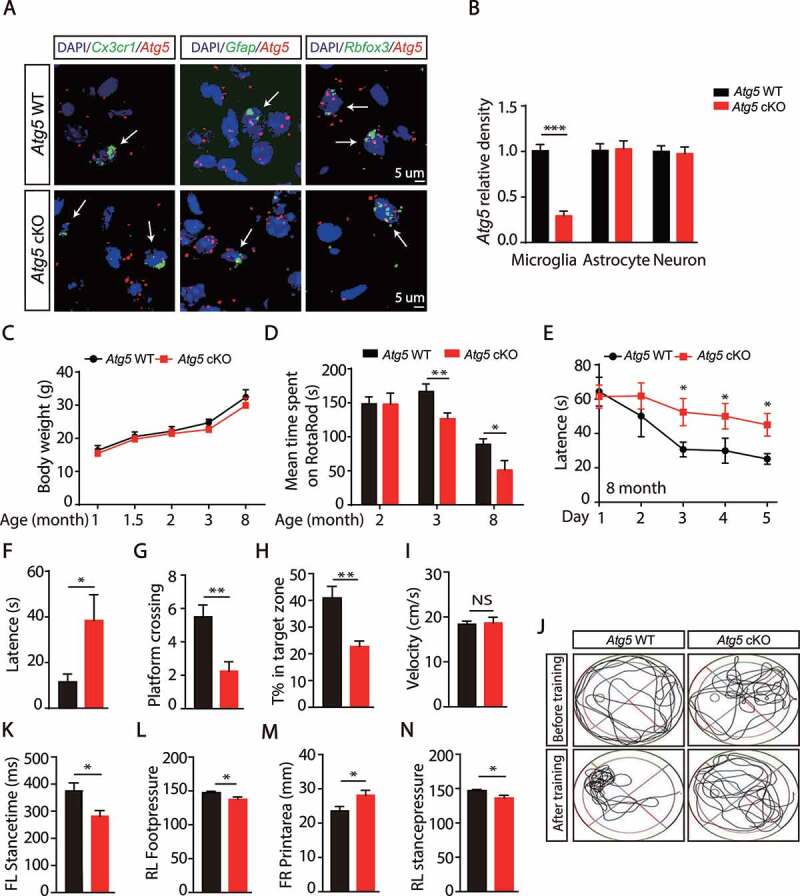
Loss of microglial Atg5 causes motor coordination and cognitive learning defects. (A) ISH analyses for Atg5 expression in the SN were performed by using RNAScope Atg5 probe (Red) together with Cx3cr1 probe (Green), Gfap probe (Green) or Rbfox3 probe (Green) in WT and Atg5 cKO mice. The nuclei were labeled with DAPI (Blue). Scale bar: 5 µm. (B) Quantitative data of Atg5 levels as indicated. The Atg5 density was quantified using the Image J software. N > 20. (C) The body weight of WT (N = 12) or Atg5 cKO mice (N = 8) in different age time as shown. (D) The mean time spent on Rotarod of different month old WT (N = 10) or Atg5 cKO mice (N = 8). *means p < 0.05. **means p < 0.01. (E) Morris water-maze analysis of 8-month-old mice on 5 consecutive days. (F) Results of the probe trial of WT (N = 8) or Atg5 cKO mice (N = 8) at day 7. Data are shown as mean ± SEM. *means p < 0.05. (G) Data are given as the times of crossing platform, which is hidden in this trail. **means p < 0.01. (H) Data are given as the percentage of the time of mice spent in the target quadrant, where the platform previously located. **means p < 0.01. (I) The mean velocity of WT (N = 8) or Atg5 cKO mice (N = 8). Data are shown as mean ± SEM. *means p < 0.05. (J) Representative routes of WT or Atg5 cKO mice from trails. (K-N) Gait analysis of 8-month-old WT (N = 6) or Atg5 cKO mice (N = 8). Data are shown as mean ± SEM. *means p < 0.05
Next, we checked whether deletion of Atg5 in microglia impaired development and behavior in mice. We found that Atg5 cKO mice were fertile and developed normally (Figure 1C). Interestingly, we found that deletion of microglial Atg5 significantly impaired motor coordination in 3- and 8-month-old Atg5 cKO mice, but not in 2-month-old mice (Figure 1D and S2A). The defect in motor coordination in Atg5 cKO mice was not due to changes in grip strength (Fig. S2B). In addition, both forced swimming and tail suspension tests showed no difference between Atg5 WT mice and Atg5 cKO mice (Fig. S2C and S2D). Water maze analysis revealed that there were no differences between Atg5 WT mice and Atg5 cKO mice with respect to velocity, acquisition of platform location, platform zone crossing, or time in target zone in 3-month-old mice (Fig. S2E-S2J), suggesting deletion of microglial Atg5 only impaired motor coordination, but not learning and memory activity in 3-month-old mice. Interestingly, we observed a significant defect in learning and memory activity in 8-month-old Atg5 cKO mice (Figure 1E–J), suggesting that the learning and memory defect is age-dependent. Additionally, we found that Atg5 cKO mice showed gait abnormalities at 8 months old (Figure 1K–N), which were not observed at 3 months. Together, these data suggest that autophagy deficiency in microglia causes age-dependent motor coordination and cognitive learning defects in mice.
Deletion of Atg5 in microglia or knockdown microglial Atg5 in the SN induces neurotoxicity and neuroinflammation
To analyze the correlation between motor defects and loss of TH-positive neurons in the SN, we counted TH-positive neurons in Atg5 WT and Atg5 cKO mice in 2, 3 and 8-month-old mice. As shown in Figure 2A,B, the microglial Atg5 defect significantly decreased TH-positive neurons in 3- and 8-month-old Atg5 cKO mice, suggesting that impaired motor coordination contributes to the degeneration of TH neurons. Since we observed detectable PD-like behavior defects in Atg5 cKO mice from 3 months old, we then examined the levels of dopamine and its metabolites in the striatum. As shown in Figure 2C, dopamine (DA) levels were dramatically reduced in 3 -month-old Atg5 cKO mice, along with a trend toward decreased levels of the metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) (Fig. S3A). However, there was no difference in the level of the metabolite homovanillic acid (HVA) or the ratio of (DOPAC+HVA):DA (Fig. S3B and S3C). In addition, we found that there was a significant increase in SNCA/α-synuclein levels in 3-month-old Atg5 cKO mice, which was further aggravated in 8-month-old mice (Fig. S3D and S3E), indicating an accumulation of SNCA in the brain. Furthermore, we found that the mRNA levels of Snca in 3- and 8-month-old Atg5 cKO mice were significantly increased (Fig. S3F and S3G). In addition, dramatic enhancement of pro-inflammatory cytokines including Il1b, Nos2/Inos (nitric oxide synthase 2, inducible) and Tnf were observed in both 3- and 8-month-old Atg5 cKO mice (Figure 2D,E). Taken together, these results suggest that autophagy deficiency in microglia induces neurotoxicity and neuroinflammation in the brain.
Figure 2.
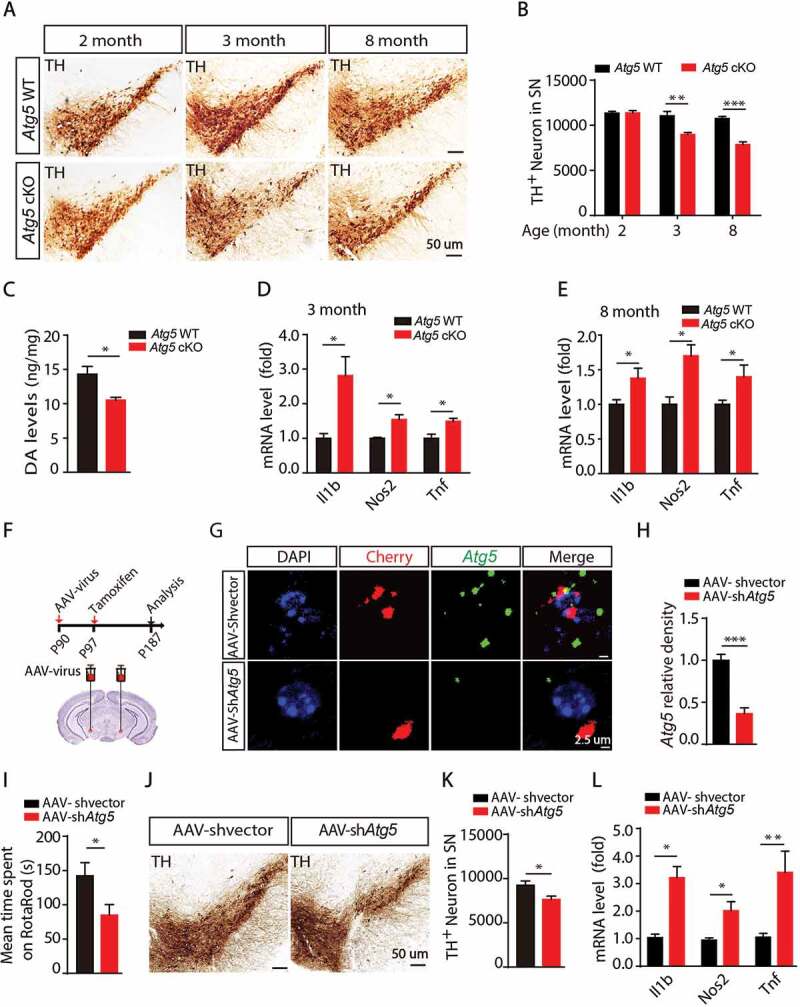
Deletion of Atg5 in microglia or knockdown of Atg5 in the SN induces neuronal damage and neuroinflammation. (A) Immunohistochemical staining for TH in the SN from Atg5 WT group mice and Atg5 cKO group mice at different age as indicated. (B) Quantitative data of TH-positive neurons in the SN as indicated (N = 4 for each group). (C) The levels of DA in the striatum from 3-month-old Atg5 WT and Atg5 cKO mice. *means p < 0.05. (D) Expression of Il1b, Nos2 and Tnf in the striatum from 3-month-old Atg5 WT and Atg5 cKO mice. *means p < 0.05. (E) Expression of Il1b, Nos2 and Tnf in the striatum from 8-month-old Atg5 WT and Atg5 cKO mice. *means p < 0.05. (F) Model of knockdown microglial Atg5 in the SN by using AAV-virus. (G) ISH analyses for Atg5 expression in the microglia in the SN in AAV-shvector and AAV-shAtg5 injection group mice. Scale bar represents 2.5 µm. (H) Quantitative data of Atg5 levels as indicated. N > 20. (I) The mean time spent on Rotarod of AAV-shvector injection group mice (N = 8) and AAV-shAtg5 injection group mice (N = 9). *means p < 0.05. (J) Immunohistochemical staining for TH in the SN in AAV-shvector injection group mice (N = 6) and AAV-shAtg5 injection group mice (N = 6) as indicated. (K) Quantitative data of TH-positive neurons in the SN in AAV-shvector injection group mice (N = 6) and AAV-shAtg5 injection group mice (N = 6). *means p < 0.05. (L) Expression of Il1b, Nos2 and Tnf in the SN from 2 groups of mice as indicated. *means p < 0.05. **means p < 0.01
To exclude the effect of peripheral leukocytes in Itgam-Cre mice, we injected pAAV-EF1a-DIO-mCherry-shAtg5 virus into the SN of Cx3cr1-CreER mice, which excludes the effect of peripheral leukocytes [27,28]. First, 3 shRNAs targeting mouse Atg5 were designed and tested in NIH3T3 cells, then shAtg5 #3 was chosen for the present studies (Fig. S4A). The specification of AAV-virus was further determined in brain, and as shown in Fig. S4B, mCherry signals were mainly present in IBA1-positive cells, but not in GFAP-positive cells, suggesting that the AAV virus only infected microglia in vivo.
A scramble virus or pAAV-EF1a-DIO-mCherry-shAtg5 was injected into the SN of 3-month-old Cx3cr1-CreER mice, and 1 week later tamoxifen was injected. Three months later, behavior and other neurological parameters were examined (Figure 2F). Using RNAscope probes, we found that AAV-shAtg5 dramatically decreased the expression level of Atg5 in microglia (Figure 2G,H). As expected, we found that knockdown of microglial Atg5 significantly impaired motor coordination (Figure 2I). Compared to the AAV-shvector virus injection group, there was a significant increase of protein levels and mRNA levels of Snca in AAV-shAtg5 virus injection group (Fig. S4H-S4J). Consistently, the numbers of TH-positive neurons in the SN were also significantly reduced (Figure 2J,K). Furthermore, increased levels of pro-inflammatory cytokines, including Il1b, Nos2, Tnf, were observed in the SN in the AAV-shAtg5 virus injection group (Figure 2L). Together, these results suggest that knockdown of microglial Atg5 in the SN also causes motor coordination defects, TH-positive neuronal loss and neuroinflammation.
PDE10A is involved in the activation of NLRP3 inflammasome
To further define the underlying mechanism of autophagy deficiency-induced PD-like symptoms in mice, we analyzed the protein expression profile in the striatum (Figure 3A). In 3-month-old mice, we identified a total of 5447 and 5477 proteins in Atg5 WT and cKO mice, respectively, among which microglial Atg5 deficiency resulted in 180 significantly upregulated and 39 downregulated proteins (Fig. S5A). Furthermore, the significantly altered proteins in the striatum of 3-month-old mice could be classified into 5 functional categories, including PD, dopaminergic synapses, amyotrophic lateral sclerosis (ALS), renin secretion and synaptic vesicle cycle (Fig. S5B). In 8-month-old mice, we identified a total of 5267 proteins in WT mice and 5058 proteins in Atg5 cKO mice. Compared to the WT mice, there were 93 significantly upregulated and 22 downregulated genes in microglial Atg5 KO mice. In 8-month-old mice, 6 functional groups were enriched, including cell meiosis, Hippo signaling pathway, Fc gamma R-mediated phagocytosis, glutamatergic synapses and synaptic vesicle cycle (Fig. S5B). Together, these proteomics profiles indicate that several neurodegenerative-related pathways were dysregulated in Atg5 cKO mice, which may have contributed to the development of PD-like symptoms.
Figure 3.
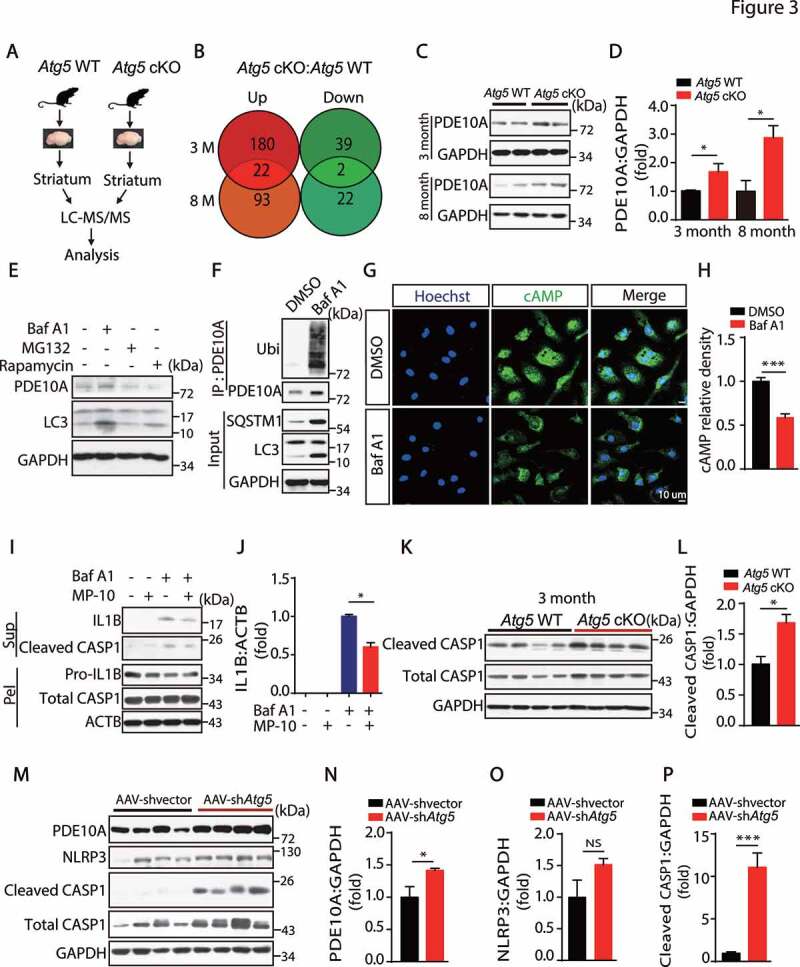
PDE10A is involved in the activation of NLRP3 inflammasome. (A) The model of LC-MS/MS analysis. (B) The data shows the numbers of upregulated and downregulated genes in 3-month and 8-month WT and Atg5 cKO mice. (C) Immunoblotting analysis of PDE10A and GAPDH in the striatum from 3-month and 8-month-old Atg5 WT mice and Atg5 cKO mice as indicated. (D) Quantitative data of PDE10A levels in the striatum in 3-month and 8-month-old Atg5 WT and Atg5 cKO mice. (E) Immunoblotting analysis of PDE10A, LC3 and GAPDH in the primary microglial cells treated with Baf A1 (50 nM, 6 h), MG132 (10 µM, 6 h) and Rapamycin (20 nM, 6 h). Experiments were carried out for 3 independent times. (F) Primary microglial cells were treated with Baf A1 (50 nM, 6 h). Cell lysates were immunoprecipitated with anti-PDE10A antibody coated gel and then blotted for Ubiquitin, PDE10A, SQSTM1, LC3 and GAPDH. (G) Immunofluorescent staining of cAMP in the primary microglial cells treated with or without Baf A1 (50 nM, 6 h). Scale bar represents 10 µM. N = 50. (H) Quantitative data of cAMP levels as indicated. (I) Immunoblotting analysis of cleaved CASP1 and IL1B levels in the supernatant, Pro-IL1B, total CASP1 and ACTB levels in the pellets from the primary microglial cells treated with Baf A1 (50 nM, 6 h) and MP-10 (5 µM, pre-treatment for 1 h) as indicated. Experiments were carried out 3 independent times. (J) Quantitative data of IL1B levels as indicated. (K) Immunoblotting analysis of cleaved CASP1, total CASP1 and GAPDH in the striatum from 3-month-old Atg5 WT and Atg5 cKO mice as shown. (L) Quantitative data of cleaved CASP1 levels in the striatum in 3-month-old Atg5 WT and Atg5 cKO mice. *means p < 0.05. (M) Immunoblotting analysis of PDE10A, NLRP3, cleaved CASP1, total CASP1 and GAPDH in the SN from AAV-shvector injection group mice and AAV-shAtg5 injection group mice as indicated. (N-P) Quantitative data of PDE10A, NLRP3 and cleaved CASP1 levels. *means p < 0.05. ***means p < 0.001
Multiple lines of evidence implicate dysfunction of the immune system and neuroinflammation in the development of PD [9,26,29,30]. Among the significantly altered proteins, 22 were increased, while 2 were downregulated in both 3- and 8-month-old Atg5 cKO mice (Figure 3B and Table S2). Interestingly, we identified that PDE10A levels were significantly increased in 3-month and 8-month-old Atg5 cKO mice in the proteomic list. Additionally, we and studies conducted by others have reported that the PDE family of proteins is involved in microglial activation and neuroinflammation [31–34]. As shown in Figure 3C,D, deletion of Atg5 in microglia dramatically increased PDE10A protein levels in 3- and 8-month-old Atg5 cKO mice, with no change of its mRNA levels (Fig. S5C). To test whether PDE10A was involved in autophagy defect-induced microglial activation, we first measured the levels of the substrate of PDE10A, cAMP, which is critical for microglial activation, especially activation of the NLRP3 inflammasome [35,36]. As expected, we found that cAMP levels were significantly reduced in Atg5 cKO mice (Fig. S5D). Next, we sought to examine whether autophagy regulates the degradation of PDE10A in microglia. We treated the primary microglial cells with bafilomycin A1 (Baf A1, an autophagy inhibitor), MG132 (a proteasome degradation inhibitor) or rapamycin (an autophagy stimulator), and found that Baf A1 treatment increased PDE10A level, while there was no difference in PDE10A protein expression upon MG132 treatment. In contrast, rapamycin treatment significantly reduced PDE10a levels (Figure 3E). Furthermore, we found that Baf A1 treatment dramatically increased the ubiquitination levels of PDE10A in microglial cells (Figure 3F), suggesting that autophagy processes regulate PDE10A degradation in microglia. Consistently, we found that Baf A1 treatment largely decreased cAMP levels in microglia (Figure 3G,H). To test whether PDE10A carries a direct role in microglial activation, we treated primary microglial cells with MP-10, a specific inhibitor for PDE10A, and found MP-10 treatment significantly inhibited either autophagy inhibition- or nigericin-induced NLRP3 activation (Figure 3I, and S5E).
Importantly, we found that cleaved CASP1/caspase-1 levels were significantly increased in 3-month-old Atg5 cKO mice in vivo (Figure 3K,L). As expected, the levels of cleaved CASP1 were dramatically increased in 8-month-old Atg5 cKO mice (Fig. S5F and S5G), suggesting that deletion of Atg5 in microglia caused NLRP3 inflammasome activation in vivo. Consistently, we found that there was a significant enhancement of PDE10A levels in the SN of the AAV-shAtg5 virus injection group, together with increased expression of cleaved CASP1 (Figure 3M–P). Together, these results suggest that autophagy regulates NLRP3 inflammasome activation by modulating levels of PDE10A in microglial cells.
Microglial Atg5 defects causes an increase of MIF in a NLRP3 inflammasome-dependent manner
Among the 24 significantly altered proteins in both 3- and 8-month-old Atg5 cKO mice, we found that an important pro-inflammatory cytokine, MIF, was elevated (Figure 4A–C). As shown in Figure 4D–F, we confirmed that protein levels of MIF were significantly elevated in the striatum of Atg5 cKO mice, while APOE was not significantly altered.
Figure 4.
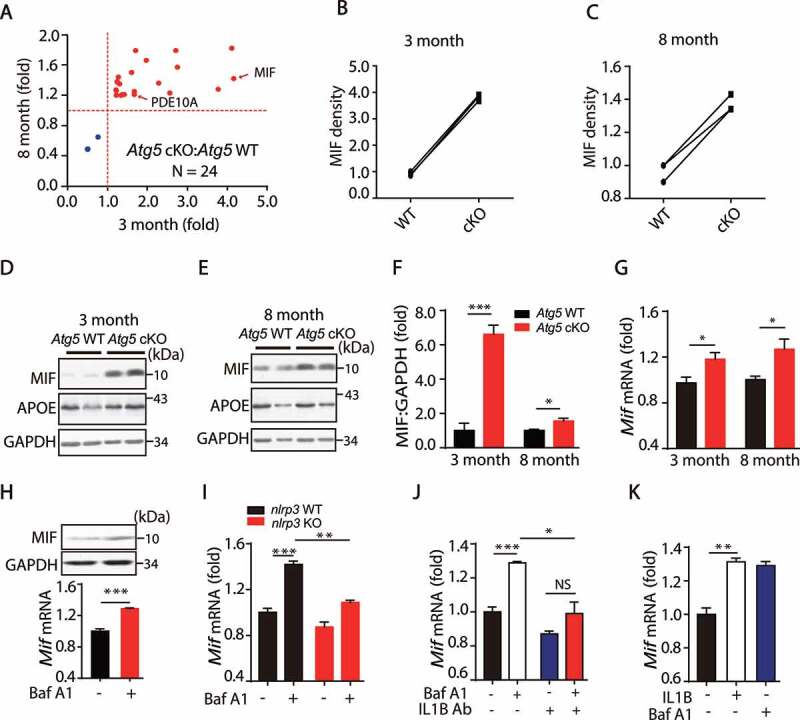
Microglia Atg5 defects causes an increase of MIF in a NLRP3 inflammasome dependent manner. (A) The 24 significantly changed proteins from 3 month and 8-month-old WT or Atg5 cKO mice. (B and C) The relative levels of MIF in 3 month and 8 month WT and Atg5 cKO mice. (D and E) Immunoblotting and quantitative analysis of MIF, APOE and GAPDH in the striatum from 3-month and 8-month-old Atg5 WT and Atg5 cKO mice. *means p < 0.05. ***means p < 0.001. (F) Quantitative data of MIF levels in the striatum in 3-month and 8-month-old Atg5 WT and Atg5 cKO mice. (G) Expression of Mif in the striatum in 3-month and 8-month-old Atg5 WT and Atg5 cKO mice. *means p < 0.05. Error bars are mean ± SEM. (H) Primary microglial cells treated with Baf A1 (50 nM, 6 h), and expressions of MIF and GAPDH levels were detected (Upper). The Expression of Mif in the primary microglial cells treated with Baf A1 (50 nM, 6 h) (Down). (I) Expression of Mif in the primary microglial cells from WT and nlrp3 KO mice treated with Baf A1 (50 nM, 6 h) as indicated. (J) Primary microglial cells were treated with IL1B-neutralizing antibody (1 µg), then treated with Baf A1 (50 nM, 6 h), the expressions of Mif mRNA levels were detected. (K) The expressions of Mif mRNA levels from primary microglial cells were treated with IL1B (1 ng/mL, 6 h) and Baf A1 (50 nM, 6 h). Error bars are mean ± SEM. Experiments were carried out in triplicate, at least 3 independent times
It has been reported that MIF functions as an important pro-inflammatory cytokine in neuronal system diseases [37–40]. Recently, MIF was reported as a DNAase enzyme involved in neurotoxicity following stroke [41]. Here, we found the MIF levels were significantly increased in Atg5 cKO mice. We also observed that transcriptional levels of Mif were significantly increased in Atg5 cKO mice (Figure 4G), suggesting that autophagy might regulate MIF levels in a transcription-dependent manner. To confirm this, we treated the primary microglia with Baf A1 to block the autophagy process. As shown in Figure 4H, Baf A1 treatment induced a significant increase in Mif/MIF at both mRNA and protein levels. Since we found that autophagy inhibition increased the expression levels of both IL1B and MIF in microglia, we hypothesized that autophagy inhibition-mediated NLRP3 inflammasome activation is important for MIF induction. To test this, isolated nlrp3 KO microglia were treated with Baf A1. As shown in Figure 4I, nlrp3 KO dramatically abolished the upregulation of Mif induced by autophagy inhibition. Furthermore, we found that IL1B-neutralized antibody also dramatically reduced Mif levels in microglia (Figure 4J). Consistently, exogenous cytokine IL1B elevated Mif mRNA levels in microglia (Figure 4K). Together, these results indicate that NLRP3 inflammasome activation upregulates Mif levels in a transcription-dependent manner in microglia.
Inhibition of NLRP3 activation rescues Atg5 defect-induced microglial activation and neuronal loss
To study the regulatory mechanism in vivo, 2-month-old WT and Atg5 cKO mice were treated with MCC950, a specific inhibitor for NLRP3 [42,43], every other day for 1 month (Figure 5A). As shown in Figure 5B,C, MCC950 administration significantly inhibited the expression level of cleaved CASP1, suggesting that activation of the NLRP3 inflammasome was blocked by MCC950. Furthermore, we found that inhibition of NLRP3 completely rescued Atg5 knockout-induced enhancement of MIF levels (Figure 5B,D), suggesting that enhancement of MIF in Atg5 cKO mice was NLRP3 inflammasome-dependent.
Figure 5.
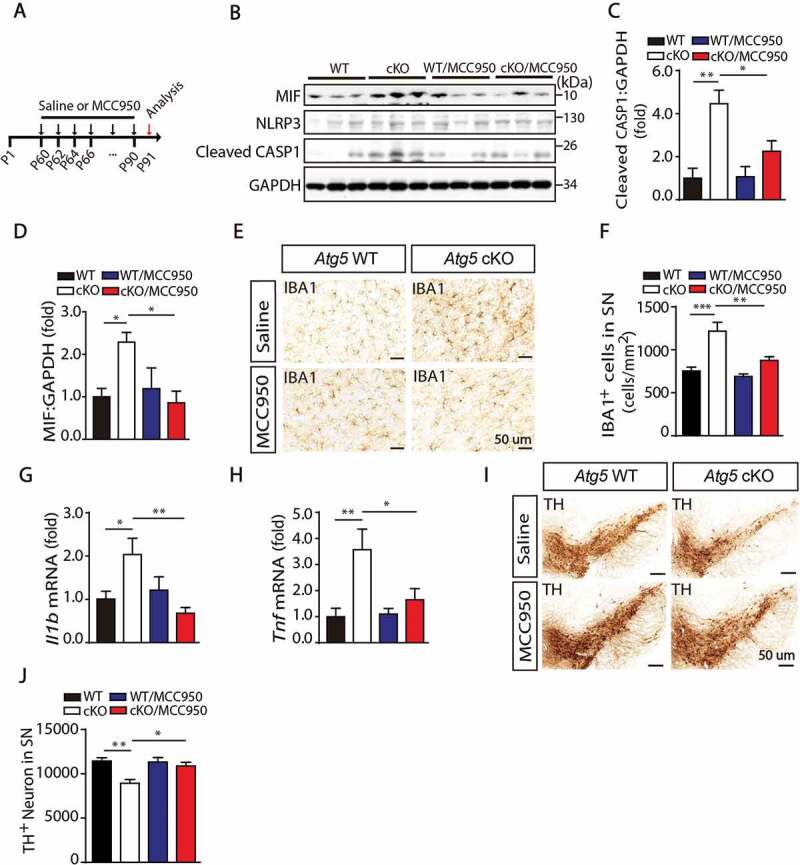
Inhibition of NLRP3 activation rescued Atg5 defect induced microglial activation and neurons loss. (A) Model of administration of MCC950 to block NLRP3 activation and check the microglial activation and neurons loss. Two-month-old Atg5 WT and cKO mice received intraperitoneal injections of sterile PBS (control) or MCC950 (10 mg/kg in PBS) every second day for 1 month. (B) Immunoblotting analysis of MIF, NLRP3, cleaved CASP1 and GAPDH in the SN from Atg5 WT group, Atg5 cKO group, Atg5 WT with MCC950 group and Atg5 cKO with MCC950 group mice. (C and D) Quantitative data of cleaved CASP1 and MIF levels in the SN from these 4 groups of mice as indicated. (E) Immunohistochemical staining for IBA1 in the SN from these 4 groups of mice. (F) Quantitative data of IBA1 positive cells in the SN from these 4 groups of mice as indicated (N = 6 or N = 8). (G and H) Expression of Il1b and Tnf in the SN from these 4 groups of mice as indicated. (I) Immunohistochemical staining for TH in the SN in these 4 groups of mice as indicated. (J) Quantitative data of TH-positive neurons in the SN in these 4 groups of mice as indicated (N = 4 for each group)
To examine whether microglial activation was involved in this process, we counted microglia stained with IBA1 in the SN. As shown in Figure 5E,F, there was a significant increase in the number of microglia in Atg5 cKO mice, which was inhibited by MCC950 treatment. Accordingly, in vivo NLRP3 inhibition also attenuated the expression of Tnf and Il1b induced by Atg5 cKO (Figure 5G,H). Next, we tested whether inhibition of NLRP3 inflammasome activation by administration of MCC950 could restore the loss of TH-positive neurons in Atg5 cKO mice. As shown in Figure 5I,J, we found that there was a significant reduction in TH-positive neurons in Atg5 cKO mice, compared to WT mice in age matched groups. Importantly, treatment with MCC950 rescued the loss of TH-positive neurons. Together, these results suggest that inhibition of NLRP3 activation rescued Atg5 defect-induced MIF enhancement, microglial activation and neuron loss in vivo.
Serum MIF is elevated in PD patients
Although the activation of NLRP3 inflammasome was reported to be involved in many neurodegenerative diseases, the downstream cytokine IL1B is almost undetectable in the serum of PD patients, although it can be detected in peripheral blood mononuclear cell (PBMC) supernatants and cerebral spinal fluid (CSF) [44]. As we found that NLRP3/IL1B elevated MIF levels in microglia, we hypothesized that serum MIF might be a clinical biomarker for PD diseases. We collected serum samples from 92 PD patients and 87 age/sex-matched controls. Interestingly, there was a dramatic increase in serum MIF levels in PD patients (Figure 6A,B). In addition, we analyzed the correlations between MIF levels and 21 other parameters and found that serum MIF levels were significantly correlated with sex and age (Figure 6C,D). Together, our results suggest that the serum MIF levels might be a potential clinical biomarker for the diagnosis of PD.
Figure 6.
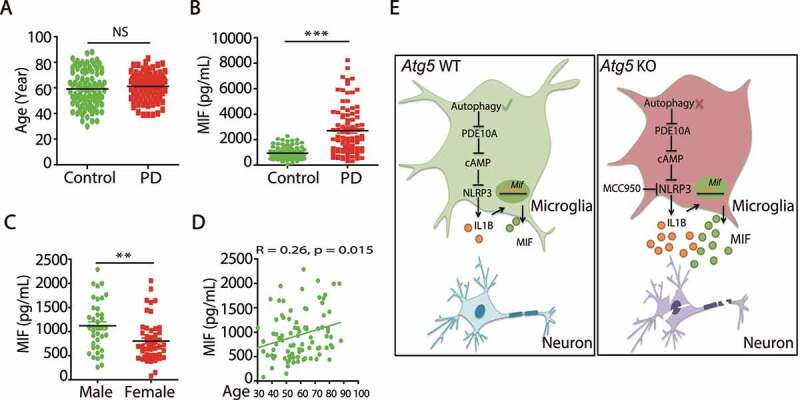
Serum MIF is elevated in PD patients. (A) The ages of control people (N = 87) and PD patients (N = 92). (B) ELISA of MIF levels in the serum from control people (N = 87) and PD patients (N = 92). ***means p < 0.001. (C) The MIF levels in the serum of control male (N = 38) and female people (N = 49). **means p < 0.01. (D) The correlation of serum MIF levels with age in control people (N = 87). (E) Schematic model of molecular mechanisms underlying microglial autophagy deficiency impaired locomotion and cognition in mice
In summary, our present work showed that microglial autophagy deficiency impaired locomotion and cognition in mice through accelerating inflammasome activation, indicating that microglial autophagy is critical for microglial activation and neuroinflammation in the development of PD (Figure 6E).
Discussion
Microglia activation-mediated neuroinflammation plays an important role in PD, but the regulatory mechanism requires clarification. In this study, we found that microglial autophagy regulated microglial activation in the development of PD: (1) microglial Atg5 deficiency caused dysfunction of motor coordination and cognitive learning in an age-dependent manner; (2) autophagy inhibition elevated PDE10A levels and aggravated NLRP3 activation in microglia; (3) IL1B, a downstream cytokine of the NLRP3 inflammasome, increased Mif levels in a transcription-dependent manner in microglia; (4) inhibition of NLRP3 activation rescued Atg5 defect-induced microglia activation and neuronal loss; (5) serum MIF levels were significantly increased in PD patients, suggesting a useful diagnostic and therapeutic target in PD.
Almost 30 years ago, activated microglia was observed in the SN pars compacta of PD patients in a postmortem study [45]. Since then, many clinical and animal studies demonstrated that microglial activation is critical in PD development. Recently, Ejlerskov et al. reported that lack of neuronal IFNB-IFNAR (interferon-α/β receptor) causes PD-like dementia [46], providing direct evidence for the initial effects of immune dysfunction in neurological diseases. Injection of LPS (lipopolysaccharides) in the SN induces PD-like pathogenesis and symptoms in mice through activating neuroinflammation [47]. Similarly, injection of poly (I:C), a TLR3 (toll-like receptor 3) agonist, renders adult rats susceptible to low dose 6-hydroxydopamine-mediated neurotoxicity, which could be effectively inhibited by systematic administration of IL1 receptor antagonist (IL1-RA) [48]. Accordingly, here we reported a new avenue through which microglial autophagy deficiency could trigger microglial activation and spontaneously produce a model of age-dependent murine PD. Along with others, we argue that neuroinflammation is a critical initiation step for dopaminergic neuron degeneration and PD development.
Autophagy is a conserved biological process in eukaryotes, and dysfunction of autophagy has been linked with multiple human diseases [14–16]. Defects in the autophagy-related Atg5 or Atg7 genes in neurons causes neurodegeneration in mice [19,20], and upregulation of SQSTM1/p62 expression in brain could mitigate cognitive deficits in AD mouse models [49]. Importantly, rapamycin-induced autophagy activation has been shown to extend lifespan in mice [50], suggesting that autophagy confers a protective role in aging and age-related diseases. However, the above-mentioned studies mainly focused on neurons themselves without deciphering the role of autophagy in microglia in the development of neurological diseases. Here, we defined the role of autophagy in microglia during the development of PD. Targeting microglial autophagy might be an effective way to regulate neuroinflammation in the treatment of neurodegenerative diseases.
The activation of the NLRP3 inflammasome was involved in multiple neuronal system diseases, including PD, AD, ALS and stroke [51–57]. However, the regulatory mechanism for activation requires further exploration. Here, we found that microglial autophagy defects increased PDE10A levels in microglia, and inhibition of PDE10A by MP-10 significantly decreased Baf A1- and nigericin-induced NLRP3 activation.Therefore, besides direct NLRP3 inhibition, specific PDE10A inhibitors could be potential therapeutic agents in PD treatment.
To our knowledge, this is first report demonstrating that IL1B, as the main downstream cytokine of NLRP3 inflammasome, increased mRNA and protein levels of MIF in brain. Most importantly, we found that MIF levels were significantly increased in the serum of PD patients and correlated with PD development. IL1B could be an essential marker of neuroinflammation, but serum IL1B is almost undetectable due to trace concentrations (0–0.854 pg/mL) [58]. Therefore, serum MIF levels could be a potential biomarker for the diagnosis of PD and may represent a therapeutic target for treatment.
In summary, our results reveal that microglial autophagy deficiency led to increased neuroinflammation and PD-like symptoms in mice. Furthermore, we showed that PDE10A was involved in NLRP3 activation, which further upregulates MIF levels, suggesting that microglial autophagy plays a protective role in the development of microglial activation-driven PD. This represents a potential treatment method for PD patients.
Materials and methods
Mice
Mice with microglial deletion of Atg5 were generated by crossing the Atg5 locus floxed mice (provided by Prof. Noboru Mizushima, The University of Tokyo, Tokyo, Japan) [20] with Itgam/CD11b-Cre mice (purchased from the European Mouse Mutant Archive and gifted by Dr. Hailong Dong, The Fourth Military Medical University, Xi’an, China) [59]. Cx3cr1-CreER mice were purchased from the Jackson Laboratory. Mice with nlrp3 KO were provided by Prof. Rongbin Zhou (University of Science and Technology of China, Hefei, China) [36]. All animals were maintained in the Animal Care Facility at our institute. All animal experiments were approved by the Institutional Animal Care and Use Committee at Beijing Institute of Basic Medical Sciences.
Tamoxifen (Sigma, T5648) was dissolved in corn oil (Sigma, C8267). Adult mice were intragastric administrated with a total dose of 20 mg tamoxifen in 3 consecutive days.
RNA in situ hybridization
In situ hybridization (ISH) experiments were performed on adult mouse brain sections (15 µm). RNAscope fluorescence of Atg5 probes (469711) together with Cx3cr1 probes (314221), Gfap (313221) or Rbfox3/Neun probes (313311) was performed according to the manufacturer’s protocol (Advanced Cell Diagnostics).
Motor coordination test
Motor coordination tests were performed as previously described [60]. Briefly, performance of mice was measured using an accelerating Rota-rod (Panlab, LE8200, Energia, Cornella, Spain). Mice were trained on the Rota-rod at 10 rpm, 3 times per day (at 1 h intervals) for 2 d. Then, mice were tested on the rod with speed accelerating from 4 to 40 rpm over a period of 300 s. The average latency to fall from the rod over 3 consecutive trials performed at 1 h intervals was analyzed.
Grip strength test
Grip strength was measured using a Grip Strength Meter according to the manufacturer’s instructions (Bioseb). Briefly, mice were held by the tail and put on a grid, then brought to an almost horizontal position. The grid was smoothly moved horizontally until mice released the grid. The average of 3 consecutive measurements was analyzed.
Forced swim test
Mice were transferred to a clear glass cylinder (height = 45 cm, diameter = 20 cm) filled with 2 L water (20°C). Cumulative immobility time was recorded during the last 4 min of a 6 min test. After the experiment, mice were dried gently using paper towels and a heat lamp.
Tail suspension test
Mice were suspended on a bar (50 cm above the floor) by the tail using tape for 6 min. Cumulative immobility time during the last 4 min were recorded.
Morris water maze test
Learning and memory were measured using the Morris Water Maze test with a tracking system as previous reported [61]. Briefly, mice were trained in the water maze (22°C) with a flag placed on a visible platform and no spatial cues for 60 s on the first day. Then, mice were trained in the water maze for 90 s with the platform hidden and spatial cues present 3 times per day from day 2–6, with different starting locations for each trial. The daily average latency to find the platform was recorded for each mouse. On day 7, the platform was removed, and mice were placed into the water maze from the farthest distance from the previous platform zone, and tested for 90 s. The latency to first cross the platform zone, the cumulative times of crossing the platform zone, and the total time in the platform zone and each quadrant were recorded.
Gait analysis
Locomotion was recorded in 3- and 8-month-old Atg5 WT and Atg5 cKO groups using a TreadSan system (Clever Sys. Inc), as previous reported [62].
Adeno-associated virus delivery of ShAtg5 in SN
The Atg5 knockdown fragment was ligated to a pAAV-EF1a-DIO-mCherry plasmid (Obio Technology [Shanghai] Corp., Ltd., Y6122). The titers of adeno-associated virus (AAV) particles were between 1 × 1013 and 2 × 1013 units/ml (Obio Technology [Shanghai] Corp., Ltd.). Mice were anesthetized by intraperitoneal injection of 0.7% pentobarbital sodium and then fixed in a stereotaxic apparatus. A total of 2 × 109 units of scramble virus or virus targeting mouse Atg5 in a 1 µl volume was slowly injected into the SN (AP: −3.3 mm; ML: ±1.3 mm; DV: −4.7 mm from bregma) at a rate of 0.2 µl/min using a 5 µl Hamilton syringe.
Stereological assessment
All procedures were performed as previously described [26,63]. Briefly, 40 µm coronal sections were cut throughout the entire brain, and every fourth section was analyzed using Stereo Investigator software (MicroBrightfield).
Striatal DA and its metabolites measurement
The levels of DA and its metabolites dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) in the striatum were analyzed using high performance liquid chromatography with an electrochemical detector (HPLC-ECD) (Model 5600A Coul Array Detector System ESA) as previously described [64].
Cell preparation and stimulation
Microglial cells were prepared from neonatal mice (age 1–3 d) as described previously [26,65]. For inducing immune activation, 2 × 105 cells were plated into 12-well plates and cultured overnight, and medium was exchanged with opti-MEM (Thermo Fisher, 31,985,070) prior to cell stimulation.
Immunohistochemistry and immunofluorescence
All procedures were performed as previously described [26,63]. Briefly, mice were perfused with saline and brains were fixed with 4% paraformaldehyde (w:v) for one week. Fixed mouse brains were cryoprotected in 30% sucrose. Coronal sections were cut throughout the whole brain and sections were stained with TH (Pel-Freez Biologicals, P40101; 1:1000), GFAP (Sigma, G3893; 1:1000) or IBA1 (WAKO, NCNP24; 1:500). Primary microglial cells were fixed in 4% paraformaldehyde (w:v) for 30 min at room temperature, then washed 3 times with phosphate-buffered saline (PBS; 8 g/l NaCl [Sigma, 746398], 0.2 g/l KCl [Sigma, 746436], 1.44 g/l Na2HPO4 [Sigma, 71649], 0.24 g/l KH2PO4 [Sigma, P0662]) and blocked with 10% goat serum (Abcom, ab7481) in PBS containing 0.2% Triton X-100 (Sigma, V900502). Then, cells were incubated with cAMP antibody (Abcam, ab134901; 1:400) overnight at 4°C. Alexa Fluor 488-conjugated secondary antibody (Invitrogen, A32731; 1:500) was added for 1 h at room temperature, then cells were washed 3 times with PBS.
Immunoprecipitation and immunoblot analysis
Coimmunoprecipitation and immunoblotting were performed as described previously [66]. Samples were fractionated by SDS-PAGE and transferred to nitrocellulose (GE Amersham, 10600002). Immunoblots were probed with the following primary antibodies and visualized by ECL (Thermo Fisher, 32106): NLRP3 (Adipogen, AG-20B-0014; 1:1000), CASP1/caspase-1 (Adipogen, Ag-20B-0042; 1:1000), IL1B/IL1β (R&D, AF401; 1:1000), SNCA/α-synuclein (Abcam, ab1903; 1:1000), PDE10A (Santa Cruz Biotechnology, sc-515023; 1:1000), MIF (Abclonal, A1391; 1:1000), APOE (Abclonal, A10963; 1:1000), LC3 (Cell Signaling Technology, 2775; 1:1000), SQSTM1 (MBL, PM045; 1:1000), ATG5 (MBL, M153-3; 1:1000), Ubiquitin (Cell Signaling Technology, 3933; 1:1000), GAPDH (Cwbiotech, CW0100A; 1:1000).
Elisa
Human MIF was measured in serum according to manufacturer’s instructions (R&D, DMF00B). cAMP levels in mouse brain exactions were measured according to the manufacturer’s instructions (Enzo, ADI-901-066).
Quantitative RT-PCR
Total RNA was extracted from cells or brains using Trizol reagent (Invitrogen, 15596018). cDNA synthesis was performed using a one-step first strain cDNA synthesis kit (Transgen Biotech, AT341). Quantitative Real-time PCR was performed with primers for Tnf/Tnf-α, Nos2, Il1b/Il1β, Mif, Snca/α-synuclein, Pde10a and Gapdh (Table. S1). RT-PCR reactions were performed using 2× SYBR Green PCR master mix (Transgen Biotech, AQ131) and an Agilent Mx3005P RT-PCR system. The mRNA levels were normalized to Gapdh expression levels.
Protein preparation, digestion, identification, abundance level and bioinformatics analysis
All procedures for preparation, digestion, identification and abundance were performed as previously described [67]. Briefly, the striatum from Atg5 WT (N = 3) and Atg5 cKO mice (N = 3) were dissected, homogenized, exacted and digested. An LC-MS/MS system was used to analyze 3 independently digested peptide samples from each striatum. The protein abundance in each group (triplicates of each sample) was analyzed by PEAKS software (version 7.5, Bioinformatics Solutions Inc.) using a label-free strategy. The following search parameters were chosen: precursor ion and MS/MS tolerances: 15 ppm and 0.05 Da; enzyme specificity: trypsin; maximum missed cleavages: 2; fixed modification: carbamidomethyl (C, +57.02); and variable modification: oxidation (M, + 15.99). The false discovery rate (FDR) was controlled at ≤1% at both the peptide and protein level. A protein with at least 1 unique peptide and at least 2 spectra was considered to be identified. Peptide features and proteins were considered to have significant changes if the P value was <0.05. To interpret the biological implications of the proteins identified, the unique identifiers were used as an input for functional category analyzes using ClueGO v2.1.7, a Cytoscape plug-in (http://www.ici.upmc.fr/cluego/) as previously described [67].
Statistical analysis
All values are expressed as the mean ± SEM. Statistical analysis was performed using t-tests for 2 groups, and one-way ANOVA for multiple groups (GraphPad Software). P value < 0.05 was considered significant. Error bars represent SEM.
Supplementary Material
Acknowledgments
We sincerely thank Dr. Hailong Dong (The Fourth Military Medical University, Xian, China) for providing us the Itgam-Cre mice, Dr Rongbin Zhou (University of Science and Technology of China, Heifei, China) for providing nlrp3 KO mice.
Funding Statement
This work was supported by grants from the National Nature Science Foundation of China (81870839; 81630026; 81430023 and 81701187), the National Key Plan for Scientific Research and Development of China (2016YFC1306000), the National Major Project of Support Program (AWS17J013), the Beijing Nature Science Foundation (7161009) and Key Field Research and Development Program of Guangdong Province (2018B03033700).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Surmeier DJ, Sulzer D.. The pathology roadmap in Parkinson disease. Prion. 2013;7:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mu L, Sobotka S, Chen J, et al. Alpha-synuclein pathology and axonal degeneration of the peripheral motor nerves innervating pharyngeal muscles in Parkinson disease. J Neuropathol Exp Neurol. 2013;72:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dexter DT, Jenner P.. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–144. [DOI] [PubMed] [Google Scholar]
- [4].Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord. 2009;23:295–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Del Tredici K, Rub U, De Vos RA, et al. Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol. 2002;61:413–426. [DOI] [PubMed] [Google Scholar]
- [6].Depboylu C, Stricker S, Ghobril JP, et al. Brain-resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T-lymphocytes in the MPTP mouse model of Parkinson disease. Exp Neurol. 2012;238:183–191. [DOI] [PubMed] [Google Scholar]
- [7].Schiess MC, Barnes JL, Ellmore TM, et al. CSF from Parkinson disease patients differentially affects cultured microglia and astrocytes. BMC Neurosci. 2010;11:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang XJ, Yan ZQ, Lu GQ, et al. Parkinson disease IgG and C5a-induced synergistic dopaminergic neurotoxicity: role of microglia. Neurochem Int. 2007;50:39–50. [DOI] [PubMed] [Google Scholar]
- [9].De Virgilio A, Greco A, Fabbrini G, et al. Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun Rev. 2016;15:1005–1011. [DOI] [PubMed] [Google Scholar]
- [10].Banati RB, Gehrmann J, Czech C, et al. Early and rapid de novo synthesis of Alzheimer beta A4-amyloid precursor protein (APP) in activated microglia. Glia. 1993;9:199–210. [DOI] [PubMed] [Google Scholar]
- [11].Noelker C, Morel L, Lescot T, et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep. 2013;3:1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schwab C, Steele JC, McGeer PL.. Neurofibrillary tangles of Guam parkinson-dementia are associated with reactive microglia and complement proteins. Brain Res. 1996;707:196–205. [DOI] [PubMed] [Google Scholar]
- [13].Wilson CM, Magnaudeix A, Yardin C, et al. Autophagy dysfunction and its link to Alzheimer’s disease and type II diabetes mellitus. CNS Neurol Disord Drug Targets. 2014;13:226–246. [DOI] [PubMed] [Google Scholar]
- [14].Li L, Zhang X, Le W. Autophagy dysfunction in Alzheimer’s disease. Neurodegener Dis. 2010;7:265–271. [DOI] [PubMed] [Google Scholar]
- [15].Wang F, Jia J, Rodrigues B. Autophagy, metabolic disease, and pathogenesis of heart dysfunction. Can J Cardiol. 2017;33:850–859. [DOI] [PubMed] [Google Scholar]
- [16].Yang DJ, Zhu L, Ren J, et al. Dysfunction of autophagy as the pathological mechanism of motor neuron disease based on a patient-specific disease model. Neurosci Bull. 2015;31:445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].El-Khider F, McDonald C. Links of autophagy dysfunction to inflammatory bowel disease onset. Dig Dis. 2016;34:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today. 2014;19:963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- [20].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- [21].Friedman LG, Lachenmayer ML, Wang J, et al. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J Neurosci. 2012;32:7585–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stratoulias V, Heino TI. MANF silencing, immunity induction or autophagy trigger an unusual cell type in metamorphosing Drosophila brain. Cell Mol Life Sci. 2015;72:1989–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Su P, Zhang J, Wang D, et al. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. 2016;319:155–167. [DOI] [PubMed] [Google Scholar]
- [24].Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Han HE, Kim TK, Son HJ, et al. Activation of autophagy pathway suppresses the expression of iNOS, IL6 and cell death of LPS-stimulated microglia cells. Biomol Ther (Seoul). 2013;21:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cheng J, Liao Y, Xiao L, et al. Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ. 2017;24:276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bruttger J, Karram K, Wortge S, et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity. 2015;43:92–106. [DOI] [PubMed] [Google Scholar]
- [29].Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. [DOI] [PubMed] [Google Scholar]
- [30].Mehta SH, Tanner CM. Role of neuroinflammation in Parkinson disease: the enigma continues. Mayo Clin Proc. 2016;91:1328–1330. [DOI] [PubMed] [Google Scholar]
- [31].Zhao S, Yang J, Wang L, et al. NF-kappaB upregulates type 5 phosphodiesterase in N9 microglial cells: inhibition by sildenafil and yonkenafil. Mol Neurobiol. 2016;53:2647–2658. [DOI] [PubMed] [Google Scholar]
- [32].Mizuno T, Kurotani T, Komatsu Y, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46:404–411. [DOI] [PubMed] [Google Scholar]
- [33].Ghosh M, Garcia-Castillo D, Aguirre V, et al. Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia. 2012;60:1839–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cheng H, Wu Z, He X, et al. siRNA-mediated silencing of phosphodiesterase 4B expression affects the production of cytokines in endotoxin-stimulated primary cultured microglia. Exp Ther Med. 2016;12:2257–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee GS, Subramanian N, Kim AI, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yan Y, Jiang W, Liu L, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73. [DOI] [PubMed] [Google Scholar]
- [37].Matsuura T, Sun C, Leng L, et al. Macrophage migration inhibitory factor increases neuronal delayed rectifier K+ current. J Neurophysiol. 2006;95:1042–1048. [DOI] [PubMed] [Google Scholar]
- [38].Chalimoniuk M, King-Pospisil K, Metz CN, et al. Macrophage migration inhibitory factor induces cell death and decreases neuronal nitric oxide expression in spinal cord neurons. Neuroscience. 2006;139:1117–1128. [DOI] [PubMed] [Google Scholar]
- [39].Zis O, Zhang S, Dorovini-Zis K, et al. Hypoxia signaling regulates macrophage migration inhibitory factor (MIF) expression in stroke. Mol Neurobiol. 2015;51:155–167. [DOI] [PubMed] [Google Scholar]
- [40].Inacio AR, Ruscher K, Leng L, et al. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J Cereb Blood Flow and Metab. 2011;31:1093–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang Y, An R, Umanah GK, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016;6308:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhai Y, Meng X, Ye T, et al. Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates diabetic encephalopathy in db/db mice. Molecules. 2018;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dzamko N, Geczy CL, Halliday GM. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience. 2015;302:89–102. [DOI] [PubMed] [Google Scholar]
- [45].McGeer PL, Itagaki S, Boyes BE, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. [DOI] [PubMed] [Google Scholar]
- [46].Ejlerskov P, Hultberg JG, Wang J, et al. Lack of neuronal IFN-beta-IFNAR causes lewy body- and Parkinson’s disease-like dementia. Cell. 2015;163:324–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tanaka S, Ishii A, Ohtaki H, et al. Activation of microglia induces symptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. J Neuroinflammation. 2013;10:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Deleidi M, Hallett PJ, Koprich JB, et al. The Toll-like receptor-3 agonist polyinosinic: polycytidylicacid triggers nigrostriatal dopaminergic degeneration. J Neurosci. 2010;30:16091–16101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Caccamo A, Ferreira E, Branca C, et al. p62 improves AD-like pathology by increasing autophagy. Mol Psychiatry. 2016;6:865–873. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [50].Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Satoh M, Tabuchi T, Itoh T, et al. NLRP3 inflammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin Sci. 2014;126:233–241. [DOI] [PubMed] [Google Scholar]
- [52].Tan MS, Yu JT, Jiang T, et al. The NLRP3 inflammasome in Alzheimer’s disease. Mol Neurobiol. 2013;48:875–882. [DOI] [PubMed] [Google Scholar]
- [53].Hutton HL, Ooi JD, Holdsworth SR, et al. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology. 2016;21:736–744. [DOI] [PubMed] [Google Scholar]
- [54].De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Perera AP, Kunde D, Eri R. NLRP3 inhibitors as potential therapeutic agents for treatment of inflammatory bowel disease. Curr Pharm Des. 2017;23:2321–2327. [DOI] [PubMed] [Google Scholar]
- [56].Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bugyei-Twum A, Abadeh A, Thai K, et al. Suppression of NLRP3 inflammasome activation ameliorates chronic kidney disease-induced cardiac fibrosis and diastolic dysfunction. Sci Rep. 2016;6:39551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chibana H, Kajimoto H, Ueno T, et al. Interleukin-1beta is associated with coronary endothelial dysfunction in patients with mTOR-inhibitor-eluting stent implantation. Heart Vessels. 2017;32:823–832. [DOI] [PubMed] [Google Scholar]
- [59].Boillee S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. [DOI] [PubMed] [Google Scholar]
- [60].Wu R, Chen H, Ma J, et al. c-Abl-p38alpha signaling plays an important role in MPTP-induced neuronal death. Cell Death Differ. 2016;23:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bromley-Brits K, Deng Y, Song W. Morris water maze test for learning and memory deficits in Alzheimer’s disease model mice. J Vis Exp. 2011;53:2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Simjee SU, Jawed H, Quadri J, et al. Quantitative gait analysis as a method to assess mechanical hyperalgesia modulated by disease-modifying antirheumatoid drugs in the adjuvant-induced arthritic rat. Arthritis Res Ther. 2007;9:R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shin JH, Ko HS, Kang H, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cui YQ, Jia YJ, Zhang T, et al. Fucoidan protects against lipopolysaccharide-induced rat neuronal damage and inhibits the production of proinflammatory mediators in primary microglia. CNS Neurosci Ther. 2012;18:827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. [DOI] [PubMed] [Google Scholar]
- [66].Yuan Z, Kim D, Shu S, et al. Phosphoinositide 3-kinase/Akt inhibits MST1-mediated pro-apoptotic signaling through phosphorylation of threonine 120. J Biol Chem. 2010;285:3815–3824. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [67].Hu H, Bienefeld K, Wegener J, et al. Proteome analysis of the hemolymph, mushroom body, and antenna provides novel insight into honeybee resistance against varroa infestation. J Proteome Res. 2016;15:2841–2854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.