

ABSTRACT
There are 100 trillion diverse bacterial residents in the mammalian gut. Commensal bacterial species/strains cooperate and compete with each other to establish a well-balanced community, crucial for the maintenance of host health. Pathogenic bacteria hijack cooperative mechanisms or use strategies to evade competitive mechanisms to establish infection. Moreover, pathogenic bacteria cause marked environmental changes in the gut, such as the induction of inflammation, which fosters the selective growth of pathogens. In this review, we summarize the latest findings concerning the mechanisms by which commensal bacterial species/strains colonize the gut through cooperative or competitive behaviors. We also review the mechanisms by which pathogenic bacteria adapt to the inflamed gut and thrive at the expense of commensal bacteria. The understanding of bacterial adaptation to the healthy and the inflamed gut may provide new bacteria-targeted therapeutic approaches that selectively promote the expansion of beneficial commensal bacteria or limit the growth of pathogenic bacteria.
KEYWORDS: Gut microbiota, commensal bacteria, pathogenic bacteria, intestinal inflammation
1. Introduction
The gut microbiota is a vast community of commensal microorganisms that reside in the gastrointestinal (GI) tract. These microorganisms are from multiple kingdoms, including bacteria, fungi, and viruses, each comprising numerous different members of microbes, with high diversity. For example, the kingdom Bacteria within the gut microbiota comprises over 1,000 different bacterial species. Each species or strain of gut commensal bacteria performs unique biological functions, and therefore, their balance is vital for the maintenance of GI homeostasis. The perturbation of this balance – so-called gut dysbiosis – triggers or exacerbates various GI diseases, such as inflammatory bowel disease (IBD).1 Each bacterial species or strain uses a wide variety of mechanisms to adapt to the gut microenvironment and to stabilize its colonization. Importantly, the colonization of each bacterium is influenced by the presence of other bacteria, through both cooperative and competitive behaviors. In this review, we will discuss the mechanisms by which gut bacteria adapt to the gut microenvironment, both in the steady state and in disease.
2. Bacterial adaptation in the steady-state gut
Commensal bacteria have evolved various strategies to adapt to the gut environment, mainly through the acquisition of available nutrients. In this section, we will summarize the mechanisms by which commensal bacteria sense and adapt to the gut environment, through their metabolic landscape. Also, metabolic cooperation and competition between bacterial strains will be discussed. Some examples of the strategies used by pathogenic bacteria to establish infection at the expense of competing commensal bacteria will be described Figure 1.
Figure 1.
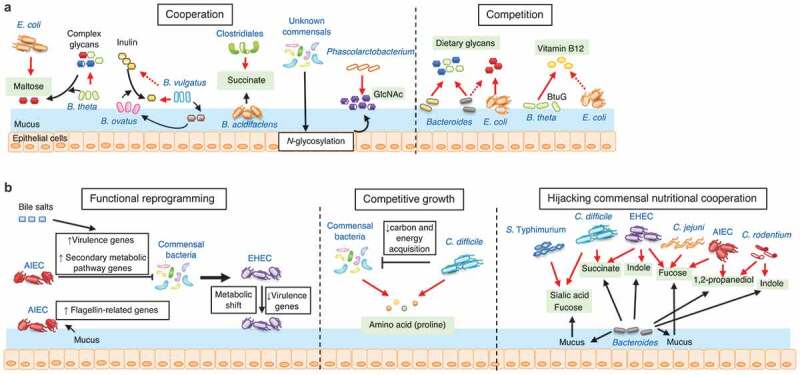
Mechanisms for the adaptation of commensal and pathogenic bacteria in the steady-state gut
(A) Commensal bacterial species or strains cooperate or compete with other bacteria to establish the colonization of each bacterial strain. Cooperation: Gut resident B. thetaiotaomicron (B. theta) liberates maltose from dietary glycans, which, in turn, promotes the growth of E. coli. B. ovatus degrades dietary inulin and generates inulin derivatives. B. vulgatus, which fails to break down inulin, can be fed by the inulin derivatives. The expanded population of B. vulgatus supports the growth of B. ovatus. Clostridiales owe the successful establishment of their colonization to succinate produced by other bacteria, such as B. acidifaciens. The colonization by gut commensals increases the N-glycosylation of host epithelial cell or mucosal proteins. Phascolarctobacterium uses N-acetylglucosamine (GlcNAc) released from host proteins. Competition: Bacteroides species compete with each other for dietary glycans; whereas, they do not compete for glycans with metabolically unrelated species, such as E. coli. In contrast, B. thetaiotaomicron uses the lipoproten BtuG to compete with E. coli for vitamin B12. (B) Functional reprogramming: When exposed to the gut microenvironment, pathogenic bacteria reprogram their functions to establish a stable colonization. Adherent–invasive E. coli (AIEC) modulate the expression of virulence and metabolic pathway genes through the sensing of gut metabolites (e.g., bile salts) or mucus. Enterohemorrhagic E. coli (EHEC) alters the expression of virulence or metabolic pathway genes in the presence of commensal microbiota, thereby changing its behavior in the gut. Competitive growth: C. difficile competes for proline with commensal bacteria. C. difficile infection results in the reduction of carbon and energy acquisition genes in commensals, allowing the pathogen to occupy the nutrient niche. Hijacking commensal nutritional cooperation: Several pathogenic bacteria harbor strategies to use the products generated by commensal bacteria for their growth. The gut colonization of Bacteroides species results in the generation of metabolic by-products, such as sialic acid, fucose, succinate, indole, and 1,2-propanediol. These metabolites are often used for the cooperative growth of other commensal bacteria. Pathogens can hijack nutritional cooperation systems and use these metabolites for their own growth to establish infection.
2.1. Environmental sensing by commensal bacteria
Nutritional and geographical adaptation are two of the most critical mechanisms that commensal bacteria use to colonize and grow in the gut. In the gut lumen, plant-derived indigestible complex polysaccharides, which are provided through the food consumed by the host, are the major nutrient source for resident bacteria. On the other hand, in close proximity to the mucosa, host-derived polysaccharides, such as mucus glycans, provide an alternate nutrient source. The ability to digest plant- or host-derived polysaccharides is a key factor contributing to the spatial adaptation to the gut environment by commensal bacteria. For example, commensal Bacteroides species possess unique gene clusters called polysaccharide utilization loci (PULs).2 PULs contain various genes associated with the use of a wide variety of plant- or host-derived complex glycans, such as enzymes that breakdown complex glycans, as well as transporters for generated simple sugars.3,4 PULs determine the niche adaptation of Bacteroides species in the gut. Based on the available polysaccharides in their surroundings, Bacteroides species selectively express corresponding PULs to adapt to the environmental niche immediately.3 In commensal B. fragilis, a unique class of PULs – commensal colonization factors (CCF) – is critical for the adaptation to the gut.5 The ccf genes are upregulated at the colonic surface, thereby enabling B. fragilis to penetrate the colonic mucus and stably localize at the bottom of the crypts.5 The expression of ccf genes in B. fragilis regulates the biosynthesis of capsular polysaccharides (PS).6 CCF-dependent regulation of PS induces the development of PS-specific IgA responses by the host.6 IgA binding to B. fragilis capsular PS increases the adherence to intestinal epithelial cells, thereby enhancing colonization stability at the mucosal niche.6 In addition to CCF-mediated transcriptional regulation, the synthesis of capsular PS in B. fragilis is influenced by PS locus promoter orientation.7 PS locus promoters contain DNA segments that undergo reversible inversions.8 PS locus promoters are therefore placed in the correct (i.e., ON) or incorrect (i.e., OFF) orientation that regulates the transcription of PS biosynthesis genes.7 Although the factors driving the promoter orientations of B. fragilis in the gut are incompletely understood, the environmental factors, such as the presence of complex microbiota, influence the promoter orientations.7 Capsular PS enhance the stability of the bacterium at the mucosal niche,6 and also elicit host immune responses, which may in turn influence bacterial colonization.9–11 Therefore, PS promoter orientations may be important for the adaptation of bacteria – commensals and potential pathogens – in the gut environment. In this context, a significantly higher proportion of gut B. fragilis strains had the PSA promoter oriented OFF in individuals with IBD compared to healthy individuals.12 Thus, the sensing of the surrounding environment (nutritional or spatial) triggers the expression of appropriate genes and facilitates the adaptation of commensal bacteria to the specific gut environment. In-depth studies of the spatial adaptation mechanisms deployed by commensal bacteria are ongoing. Using hybrid selection RNA sequencing, Donaldson et al. discriminated the transcriptomic profile of the commensal B. fragilis in the mucosal niche from that in the gut lumen.13 B. fragilis colonized in the mucosal niche is significantly more metabolically active than B. fragilis localized to the lumen, as evidenced by the upregulation of numerous genes for protein synthesis, including genes that likely encode acetylglucosamine-6-sulfatase (BF3086) and cyclomaltodextrinase (BF3134), which are important for mucin glycan foraging.13 In the colonic mucus of gnotobiotic mono-colonized mice, mutant B. fragilis (∆BF3086 and ∆BF3134) showed significantly lower levels than wildtype bacteria. Therefore, B. fragilis deploys a specific genetic program at distinct sites within the gut.13
2.2. Metabolic cross talk between commensal bacteria
2.2.1. Cooperation (cross feeding)
Different species or strains of commensal bacteria in the gut harbor distinct nutritional preferences that are used to adapt to the gut environment. The specific nutritional habit is, therefore, a vital determinant of the balance between each species or strain within the gut microbiota. Some commensal bacteria develop a mutualistic relationship with other bacteria and cooperatively establish the colonization of each bacterial strain Figure 1a. This phenomenon is termed cross feeding.14,15 For example, Escherichia coli and Bacteroides thetaiotaomicron both colonize the steady-state gut, but each has a different capability in the use of glycans for their growth. E. coli lacks glycoside hydrolase activity for dietary glycan, whereas B. thetaiotaomicron is a glycophile, and thus, is able to break down a broad array of dietary glycans. In E. coli mono-colonized mice, the expression of genes for maltose utilization is increased when the mice are co-colonized with B. thetaiotaomicron. This observation indicates that E. coli can be fed by B. thetaiotaomicron, which liberates maltose from dietary glycans.16 Tuncil et al. found that human gut Bacteroides species, such as the closely related species B. ovatus and B. thetaiotaomicron, have cognate polysaccharide utilization loci for degrading a number of dietary glycans. However, they do not simultaneously use all of these glycans. Each species displays variable and sometimes opposite rank orders for some glycans, thereby maintaining their stable coexistence in a competitive environment.17 Rakoff-Nahoum et al. discovered a dedicated cross-feeding enzyme system in the gut symbiont B. ovatus, which extracellularly digests and liberates considerable amounts of inulin breakdown products to benefit other species that cannot use inulin, such as B. vulgatus. In return, B. vulgatus can benefit B. ovatus by detoxifying inhibitory substances and producing growth-promoting factors, thereby supporting the growth of B. ovatus.18 Moreover, some commensal bacterial strains rely entirely on the metabolic functions of cooperative partners to establish their colonization in the gut. Kim et al. showed that Clostridiales bacteria fail to colonize germ-free (GF) mice, whereas Clostridiales successfully establish colonization if the GF mice are pre-colonized with some commensal bacteria, such as B. acidifaciens.19 The pre-colonized B. acidifaciens produces succinate, which enhances the growth of Clostridiales.19 This metabolic cross talk between B. acidifaciens and Clostridiales is essential for the maturation of the gut microbiota from neonate to adult. Likewise, the colonization of the gut microbiota alters the N-glycosylation of the host mucus or epithelial proteins, which, in turn, foster the expansion of Phascolarctobacterium, which can use host-derived glycans as nutrients.20
2.2.2. Competition
In addition to cooperative behaviors, bacterial species and strains in the gut can also compete with each other to maintain abundance at the expense of other bacteria. Nutritional (metabolic) competition is one of the major regulatory forces of the gut bacterial community Figure 1a. For example, many members of the genus Bacteroides share the same mechanism for digesting multiple diet- and host-derived glycans, whereby they compete with each other for nutrients in the gut lumen.21 Likewise, Lee et al. reported that Bacteroides colonize the gut in a species-specific and saturable manner. Single Bacteroides species mono-associated mice are resistant to colonization by the same, but not different, species (e.g., E. coli).5 CCF is a determinant for the interspecies niche competition, as CCF deletion in B. fragilis results in colonization defects and reduced horizontal transmission. In addition to carbohydrates, other nutrients, such as vitamins, are also essential for commensal fitness. B. thetaiotaomicron, unlike E. coli, is replete with conserved genes encoding proteins for three functional vitamin B12 acquisition systems.22,23 In particular, BtuG, a surface-exposed lipoprotein essential for efficient vitamin B12 transport in B. thetaiotaomicron, binds vitamin B12 with femtomolar affinity and removes vitamin B12 from the host’s own vitamin B12 collecting protein, thus enhancing its fitness. When B. thetaiotaomicron, with and without BtuG, is placed in a gnotobiotic mouse model, the B. thetaiotaomicron lacking functional BtuG is outcompeted by the bacteria with functional BtuG, suggesting that BtuG-mediated capture of vitamin B12 is a crucial factor for the fitness of this strain.
2.3. Establishing infection by pathogenic bacteria
Similar adaptation mechanisms are used by pathogenic bacteria to establish an infection in the gut Figure 1b. Adherent–invasive E. coli (AIEC) exhibits the expression of genes related to flagella formation when it senses the gut environment, such as the presence of bile salts and mucus.24 The expression of flagellin-related genes, in turn, promotes the penetration of AIEC into the mucus layer and transit to the epithelial surface.24 As flagellin is a critical factor for AIEC pathogenesis,25 this is an essential adaptation mechanism that enables AIEC to establish disease-causing colonization. Bile salts can also modulate AIEC expression of other virulence genes and secondary metabolic pathway genes, conferring the fitness advantage to AIEC over competing indigenous bacteria.26 Enterohemorrhagic E. coli (EHEC) reprograms its gene transcription when it is cultured in cecal content isolated from human microbiota–associated rats.27 EHEC shifts its metabolism from glycolytic to anaplerotic.27 The ex vivo gut environment inhibits the expression of various virulence genes involved in attaching and effacing lesion formation by EHEC.27 The downregulation of virulence factors may be necessary to promote faster growth of the pathogen population, as virulence expression is costly.28 EHEC may occupy the spatial or nutritional niches, forcing out commensal competitors by growing faster. On achieving sufficient robustness in the gut, they may turn on the virulence to establish infection. Moreover, Clostridioides difficile uses amino acids, particularly proline, available in the gut lumen to grow and subsequently establish infection.29 In the presence of commensal bacteria, especially metabolically associated Clostridia, C. difficile competes for proline, which limits its growth.30 The absence of competitors, as occurs in germ-free mice, in patients with diarrhea, and with antibiotic treatment, enhances the availability of free proline in the gut lumen, thereby fostering the growth of C. difficile.29 Of note, once C. difficile has established infection, it induces a shift in the transcriptional activities of the commensal microbiota, particularly a minority subset of species.31 As a result, genes associated with carbon and energy acquisition are greatly reduced in the gut microbiota, which, in turn, allows C. difficile to occupy a nutrient niche and sustain infection.31
In addition to the nutritional competition between pathogens and commensals, pathogens often use nutrient sources offered by commensal bacteria to help them adapt to the gut environment (i.e., nutritional cooperation). Ng et al. found that Salmonella enterica serovar Typhimurium (S. enterica ser. Typhimurium) and C. difficile use sialic acid and fucose, liberated from the host glycans by symbiotic Bacteroides, as carbohydrate sources.32 Likewise, EHEC and C. difficile use succinate generated by the B. thetaiotaomicron–colonized murine gut.33 Citrobacter rodentium, a mouse pathogen that models EHEC and enteropathogenic E. coli (EPEC) in humans, uses 1,2-propanediol produced by B. thetaiotaomicron and converts it to propionate, which, in turn, promotes the expression of LEE1 virulence genes.34 Campylobacter jejuni, a non-saccharolytic bacterium, can catabolize l-fucose released by commensal B. vulgatus to scavenge enough carbohydrate nutrient for efficient proliferation and colonization.35 EHEC senses the fucose released from the mucus by B. thetaiotaomicron, and modulates its metabolism by way of the two-component signal transduction system FusKR, optimizing growth and decreasing competition with commensal E. coli for carbon sources.36 Fucose also promotes AIEC proliferation through propanediol dehydratase (PduC), suggesting a capacity for intestinal adaptation by catabolizing nutrients liberated from the host glycans.37 Furthermore, EHEC and C. rodentium use indole generated by commensal B. thetaiotaomicron through the histidine kinase CpxA to downregulate LEE virulence gene expression during colonization in the colonic lumen, while the repression is lifted at the mucosa due to lower indole concentrations as the result of host cell absorption.38,39
3. Bacterial adaptation in the inflamed gut
Pathogens and pathobionts are enriched with genes linked to flagella, secretion systems, adhesins, and proteins involved in biofilm formation. All facilitate the invasion of the mucus barrier and adherence to the intestinal epithelium, which have been regarded as key determinants of the inflammatory response.40 Gut inflammation caused by pathogens significantly alters the gut microenvironment, which, in turn, impacts the fitness of bacteria in the gastrointestinal tract, thereby shaping the structure of the resident microbial community. For example, inflammation results in the elevation of certain nutrients that selectively promote the growth of pathogenic bacteria. Also, metabolic by-products released by inflamed host cells support the proliferation of some, most likely pathogenic, bacteria. Moreover, inflammation elicits metabolic reprogramming (e.g., transcriptional regulation, horizontal gene transfer) and, as a result, pathogens acquire functions that enable them to adapt to the inflammatory milieu. In this section, we will discuss microbial adaptation mechanisms in the inflamed gut Figure 2.
Figure 2.
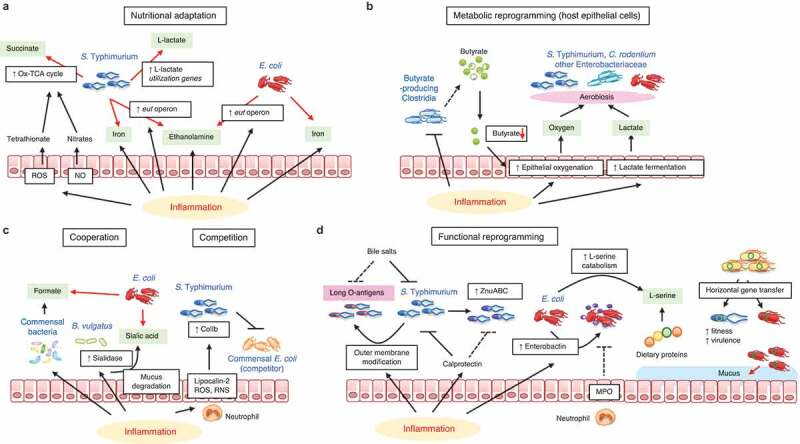
Pathogenic bacteria adaptations in the inflamed gut
(A) Nutritional adaptation: Intestinal inflammation induces reactive oxygen species (ROS) and nitric oxide (NO), which lead to the generation of tetrathionate and nitrates, respectively. These electron acceptors induce the oxidative TCA cycle (Ox-TCA cycle) in S. enterica ser. Typhimurium, conferring the growth advantage to this pathogen over commensals. Inflammation likewise induces the transcription of the eut operon in S. enterica ser. Typhimurium and pathogenic E. coli, enabling the pathogens to grow on ethanolamine, which is induced during inflammation. (B) Metabolic reprogramming (host epithelial cells): Intestinal inflammation shifts the metabolism of intestinal epithelial cells (IECs). In the inflamed gut, the reduction of butyrate-producing Clostridia and subsequent decrease in luminal butyrate levels lead to epithelial oxygenation and lactate fermentation. The resulting elevation of oxygen and lactate, produced by IECs, promotes the aerobiotic growth of Enterobacteriaceae pathogens, such as S. enterica ser. Typhimurium and C. rodentium. (C) Cooperation or Competition: Intestinal inflammation promotes gut dysbiosis. The dysbiotic microbiota alters its metabolic function, thereby generating formate. Likewise, inflammation changes sialidase activity in B. vulgatus, increasing sialic acid release from mucin glycans. Upregulated formate and sialic acid, in turn, support the growth of E. coli in the inflamed gut. S. enterica ser. Typhimurium ColIb production is induced by inflammatory mediators, such as lipocalin-2, ROS, and reactive nitrogen species (RNS). ColIb limits the growth of commensal E. coli and promotes the competitive fitness of S. enterica ser. Typhimurium. (D) Functional reprogramming: Intestinal inflammation induces antimicrobial responses that limit the growth of the resident bacteria. Pathogenic bacteria have evolved strategies to evade such antimicrobial responses through functional reprogramming. S. enterica ser. Typhimurium modifies its outer membranes in the inflamed gut. The very long O-antigens chains of S. enterica ser. Typhimurium inhibit the bactericidal effect of bile salts. Also, S. enterica ser. Typhimurium expresses the high-affinity zinc transporter ZnuABC to resist the inflammation-induced calprotectin-mediated zinc chelation. Pathogenic E. coli secretes enterobactin to dampen the activity of myeloperoxidase (MPO), and thus evades immune-mediated killing. Pathogenic E. coli increases transcription of l-serine catabolism genes in the inflamed gut, thus gaining a growth advantage over commensal competitors by using dietary protein–derived l-serine. Also, some pathogenic bacteria acquire novel functions through horizontal gene transfer to adapt better to the surrounding environment (e.g., growth on host mucus glycans).
3.1. Using inflammatory niches for the growth of pathogens
3.1.1. Pathogens survive and proliferate in the inflamed gut
Enteropathogenic bacteria are prone to survive and proliferate in the inflamed gut. As described in Figure 2a, during Salmonella-induced gastroenteritis, mucosal inflammation creates a niche that favors the expansion of the pathogen population over the commensal bacteria. The host inflammatory response in the inflamed gut generates by-products of reactive nitrogen, which can react to form nitrates, which, in an anaerobic environment, are used as respiratory electron acceptors of S. enterica ser. Typhimurium to enhance the growth of this pathogen.41 Inflammation-derived electron acceptors induce a complete oxidative TCA cycle in S. enterica ser. Typhimurium. This oxidative central metabolism enables S. enterica ser. Typhimurium to use the microbiota-derived fermentation product succinate as a nutrient to compete with the microbiota.42 Iron is an essential trace element that plays a crucial role in the proliferation and virulence of pathogens and pathobionts, as well as in the growth of commensal bacteria.43,44 Inflammation causes tissue disruption, which leads to the release of iron. Both gram-negative (e.g., E. coli, Pseudomonas aeruginosa, Klebsiella pneumoniae) and gram-positive (e.g., Staphylococcus aureus) bacteria acquire iron by synthesizing and secreting diverse siderophores.45 Siderophores display a stronger affinity to iron than host iron-binding proteins; hence, bacterial pathogens can acquire iron and gain a growth advantage in the inflamed gut.46,47
3.1.2. Pathogens change cellular metabolism in the inflamed gut
The cellular metabolism of intestinal epithelial cells (IECs) is altered during inflammation, which, in turn, affects the growth of certain pathogenic bacteria in the gut Figure 2b. For instance, Salmonella-induced colitis drives a depletion of butyrate-producing Clostridia. Due to the lack of butyrate, epithelial oxygenation is increased, causing oxygen to leak from the tissue into the lumen.48 The increased oxygen level in the epithelial cells supports the growth of Salmonella through high-affinity terminal oxidases in aerobic conditions.48 Also, C. rodentium can use aerobic respiration in the inflamed gut to gain a growth advantage over commensal bacteria.49 Cevallos et al. found that dextran sodium sulfate (DSS)-induced colitis increased epithelial oxygenation and the bioavailability of luminal oxygen, thereby driving the expanded growth of E. coli among intestinal microorganisms through aerobic respiration.50 Gills et al. also reported that inflammation alters the metabolism of IECs. In inflammation, IECs display increased lactate fermentation, which leads to the elevation of luminal lactate.51,52 Salmonella uses lactate as an electron donor in conjunction with oxygen as the terminal electron acceptor to support its colonization and overgrowth in the gut.51,52 During C. rodentium infection, the IECs shift to cholesterol and carbon metabolism, triggering aerobic glycolysis and activating cholesterol biogenesis and efflux, while dampening central carbon metabolism, in particular, the production of mitochondrial cardiolipins. This coincides with increased mucosal oxygen levels and a reduction in colon-associated anaerobic commensals, thus supporting the expansion of mucosa-associated Enterobacteriaceae.53,54
3.1.3. Pathogen cooperation and competition in the inflamed gut
Bacterial cooperation or competition with other bacteria are key elements for defining the bacterial fitness in the inflamed gut Figure 2c. DSS-induced intestinal inflammation is often accompanied by an alteration of the gut microbiota (i.e., dysbiosis). Metagenomic sequencing has revealed that bacterial formate oxidation and oxygen respiration are overrepresented metabolic pathways in the dysbiotic microbiome. E. coli uses microbiota-derived formate through oxygen respiration to enhance its fitness in the inflamed gut Figure 2c. 55 Colicins are bacterial protein toxins that show potent activity against sensitive strains in vitro. S. enterica ser. Typhimurium produces colicin Ib (ColIb), a narrow-spectrum protein toxin active against related Enterobacteriaceae, such as colicin-sensitive commensal E. coli.56 The inflammatory environment in the gut provides unique conditions that potentiate the effects of colicins. Neutrophils migrate into the lumen of the inflamed gut and release iron-depleting agents and reactive oxygen and nitrogen species. This significantly induces the genes for ColIb production in S. enterica ser. Typhimurium and its corresponding ColIb-surface receptor CirA in commensal E. coli. Hence, S. enterica ser. Typhimurium produces ColIb, which confers a growth advantage over commensal E. coli in the inflamed gut. In contrast, in the absence of gut inflammation, ColIb production does not confer a competitive advantage to S. enterica ser. Typhimurium.56 Abnormal mucus regulation is a hallmark of intestinal inflammation, linked to the expansion of pathogens. During DSS-induced colitis, the increased release of sialic acids from mucin is caused by the expansion of B. vulgatus, which produces a sialidase. Sialic acids are, in turn, incorporated into the bacterial capsule of Enterobacteriaceae, such as E. coli, thus enhancing fitness Figure 2c 57,58
3.2. Microbial functional reprogramming of pathogens in the inflamed gut
Inflammation-driven functional reprograming is crucial for allowing pathogens to gain the edge in the inflamed gut. As shown in Figure 2d, Salmonella-induced colitis increases the luminal concentration of total bile acid. To resist the elevated bile concentration during colitis, S. enterica ser. Typhimurium modifies its outer membranes (i.e., very long O-antigen chains) through regulation of FepE.59 This enables S. enterica ser. Typhimurium to adapt better to the inflamed gut than commensal bacteria, which are susceptible to bile salts. Also, S. enterica ser. Typhimurium expresses genes that are required to tolerate the host antimicrobial responses. Salmonella-induced colitis releases calprotectin, an antimicrobial protein secreted from dead neutrophils in the intestinal lumen, which can inhibit bacterial growth by sequestering essential micronutrient metals (e.g., zinc). S. enterica ser. Typhimurium expresses a high-affinity zinc transporter (ZnuABC) in the inflamed gut.60 ZnuABC gives S. enterica ser. Typhimurium a significant fitness advantage over commensal bacteria by overcoming calprotectin-mediated zinc chelation Figure 2d. Likewise, S. enterica ser. Typhimurium upregulates the transcription of l-lactate utilization genes to use lactate in the gut lumen as an electron donor.52 S. enterica ser. Typhimurium and AIEC increase transcription of the eut operon to use intestinal ethanolamine, which is secreted from intestinal epithelial cells during inflammation.61,62 This selective use of ethanolamine gives the pathogens a competitive edge over commensal bacteria. As discussed earlier, iron is of critical importance to pathogen fitness in several species of Proteobacteria (e.g., E. coli). Iron metabolism is vital for efficient bacterial colonization and presentation in the gut and proliferation in the disseminated bloodstream.63–66 During intestinal inflammation, AIEC overexpress the genes encoding propanediol utilization (pdu operon) and iron acquisition (yersiniabactin, chu operon), thereby promoting intestinal inflammation.37 Also, it has been shown that enterobactin, a catecholate siderophore secreted by E. coli, dampens the activity of myeloperoxidase released from neutrophils in the inflamed gut, thus giving the growth advantage to E. coli over other commensals.67 Kitamoto et al. found that pathogenic Enterobacteriaceae, such as AIEC LF82 and C. rodentium, shift their metabolism from carbohydrate to amino acid catabolism in the inflamed gut.68 In particular, l-serine catabolism is vital for the competitive fitness of these pathogens over commensal bacteria. Intriguingly, l-serine catabolism does not control the fitness of these pathogens in the absence of inflammation, suggesting that l-serine–dependent growth is a selective strategy used by pathogenic Enterobacteriaceae during inflammation Figure 2d.
In addition to transcriptional regulation, some bacteria acquire novel functions through horizontal gene transfer (HGT) to adapt better to the surrounding environment. Gut inflammation can boost HGT between pathogenic and commensal bacteria. For example, in S. enterica ser. Typhimurium–induced colitis, commensal E. coli acquires colicin-plasmid p2 from S. enterica ser. Typhimurium, thereby increasing its fitness by evading colicin Ib–mediated killing.69 Also, S. enterica ser. Typhimurium transfers its prophage SopEΦ to other bacteria in the inflamed gut; thereby enhancing their virulence and fitness.70 Moreover, EHEC strains can carry multiple phage-borne nanS-p alleles.71 As nanS-p genes are responsible for the cleavage of mucin O-acetyl residues, EHEC that express phage-borne esterases can gain a growth advantage over competing strains by adapting to mucosal niches. Thus, interbacterial HGT (i.e., plasmid, bacteriophage) is a key functional reprogramming mechanism that some pathogens deploy in the inflamed gut.
3.3. Commensal bacteria resilience after inflammation
Although the adaptation to the inflammatory environment is a central strategy for pathogenic bacteria to thrive in the gut at the expense of commensal bacteria, the commensal bacteria also harbor similar mechanisms. The avoidance of inflammation-induced growth suppression is often used by commensal bacteria to boost their resilience after inflammation. For example, during inflammation-associated iron limitation, pathogens capture iron using siderophores. Human commensal B. thetaiotaomicron does not produce siderophores. However, B. thetaiotaomicron uses xenosiderophores – siderophores enterobactin and salmochelin produced by members of the Enterobacteriaceae family – for iron acquisition to survive during colitis.72 Using RNA-seq analysis, Zhu et al. determined that xusABC genes are upregulated in B. thetaiotaomicron during Salmonella-induced gut inflammation. The XusABC system is required for B. thetaiotaomicron to capture xenosiderophores produced by enterobacteria.72 Indeed, a mutant B. thetaiotaomicron lacking the xusABC locus is defective for xenosiderophore-mediated iron uptake in vitro.72 Likewise, commensal bacteria are capable of self-modification to assist colonization during gut inflammation. For instance, commensal Bacteroidetes can modify their lipopolysaccharide structure, resulting in increased resistance to antimicrobial peptides and resilience during gut inflammation.73
4. Targeting bacterial adaptation strategies to treat disease
Thus far, we have discussed the mechanisms by which commensal and pathogenic bacteria colonize the healthy and the inflamed gut. Notably, pathogenic bacteria use unique metabolic adaptation mechanisms to gain a growth advantage over commensal bacteria or to avoid host antimicrobial immunity. We can target these pathogen-specific strategies to develop potential treatments that selectively suppress the growth of pathogenic bacteria without influencing the beneficial commensal bacteria.
The most straightforward approach is to supply the disease-causing bacteria with metabolic competitors. Various probiotic bacterial strains, such as lactic acid–producing bacteria, have been used to treat bacteria-driven diseases, such as IBD.74 Likewise, the transplantation of healthy fecal microorganisms (so-called fecal microbiota transplantation [FMT]) has shown promise in treating C. difficile infection and IBD.75,76 These bacterial therapies provide metabolically associated bacteria that can directly compete with disease-causing bacteria for nutrient or colonizing niches. In addition, probiotic bacteria and beneficial bacteria contained in healthy microbiota can elicit host protective immunity or anti-inflammatory immunity, which, in turn, helps to combat pathogens and promote the recovery from pathogen-caused inflammatory damage.77–79
Targeting the specific inhibitors that impede the metabolic pathways used by disease-causing bacteria is a rational approach to the treatment of disease. Tungstate has been identified as a specific inhibitor of the molybdenum cofactor-dependent microbial respiratory pathways.80 This pathway, which is used by pathogenic Enterobacteriaceae, such as E. coli, only during the period of inflammation, can be inhibited to attenuate colitis and colon cancer caused by pathogenic Enterobacteriaceae.80,81 Similarly, targeting pathogen-specific nutrients is a selective treatment that prevents colonization by pathogenic bacteria and subsequent disease development. As mentioned, the increase in luminal sialic acids during DSS-induced inflammation promotes the growth of E. coli. The oral administration of sialidase inhibitors effectively suppresses the expansion of E. coli and subsequently ameliorates DSS-induced colitis in mice.57 Likewise, l-serine catabolism promotes the growth of pathogenic Enterobacteriaceae, such as AIEC LF82 and C. rodentium in the inflamed gut.68 Since most of the l-serine available in the gut lumen, and used by the disease-causing pathogens, is supplied by diet, depriving dietary proteins of l-serine can inhibit the expansion of pathogenic Enterobacteriaceae, thereby ameliorating the disease they cause.68 Thus, inhibition of pathogen-specific metabolic pathways can selectively “starve” the disease-causing bacteria. However, targeting such pathways may not be sufficient to treat the disease caused by those bacteria. As such metabolic pathways are mainly used by pathogenic bacteria to gain a competitive fitness advantage over other resident bacteria,68 pathogenic bacteria can survive without these metabolic benefits in the absence of competitors. In this regard, providing suitable competitors (e.g., probiotic treatment, FMT) alongside pathogen metabolic inhibition may more effectively treat the disease caused by those bacteria. Alternatively, supplying diet preferred by competing commensals is an option to promote the indigenous competitors instead of providing competing bacteria exogenously.
In addition to targeting pathogen-specific metabolic pathways, inhibiting pathogen localization may be another option. As discussed earlier, pathogenic bacteria localize in close proximity to the host mucosa to evade nutritional competition with commensals in the gut lumen and to gain access to unique nutrient sources available at the mucosal niche (e.g., host-derived mucus glycans).82–85 Therefore, targeting molecules responsible for the colonization of pathogen-specific niches (e.g., mucosa adhesion molecules, flagellar proteins required for mucus penetration, type 3 secretion system) can force pathogens to stand in the same ring with their metabolic competitors.
5. Conclusion
The recent advances in next-generation sequencing and mass spectrometry technologies highlight the metabolic landscape in the development of the gut bacterial ecosystem. As discussed, the bacterial metabolic pathways, which are the key mechanisms that regulate the bacterial community, can be targeted for the editing of the gut microbial community. However, it is noteworthy that bacteria can change their metabolic and nutritional preferences according to the surrounding microenvironment, such as inflammation. The bacterial environment-dependent metabolic flexibility, therefore, needs to be considered before editing the microbiota. For example, the deprivation of key nutrients by modulating the diet can efficiently reduce opportunistic growth and infection by pathogenic bacteria in the steady-state gut. However, the same strategy is ineffective once inflammation has developed, as pathogenic bacteria reprogram their metabolic requirements. Thus, the same treatments, namely dietary interventions, may not be equally effective for different patients, or even for the same individuals who are at distinct stages of disease (e.g., grade of inflammation). On the one hand, metabolic flexibility benefits pathogenic bacteria, enabling the avoidance of metabolic limitations. On the other hand, metabolic flexibility ensures the safety of dietary interventions. Certain metabolic pathways only operate in pathogenic bacteria in certain disease conditions (e.g., inflammation); control and regulation of these pathways has no impact on the bacterial ecosystem in the healthy gut. For example, the l-serine–deficient diet does not affect the fitness of pathogenic E. coli and C. rodentium in the healthy gut, whereas it effectively limits their growth in the inflamed gut.68 Thus, this dietary treatment only impacts the gut microbial community during inflammation, not in the steady state. Clearly, the understanding of bacterial metabolic pathways and their flexibility in disease conditions will advance the development of personalized, disease-specific microbiota editing strategies.
Biography
Y.G., S.K., and N.K. wrote the manuscript.
Funding Statement
This work was supported by the National Institutes of Health grants DK108901, DK119219, AI142047, DK125087 (to N.K.), National Natural Science Foundation of China grant 31700127 (to Y.G.), the Department of Defense CA191087, the University of Michigan Clinical and Translational Science Awards Program UL1TR002240, the Prevent Cancer Foundation, and the University of Michigan Center for Gastrointestinal Research Pilot Feasibility Project P30 DK034933 (to S.K.).
Disclosures
The authors declare no competing interests.
References
- 1.Nagao-Kitamoto H, Kamada N.. Host-microbial Cross-talk in Inflammatory Bowel Disease. Immune Netw. 2017;17(1):1–12. doi: 10.4110/in.2017.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terrapon N, Lombard V, Gilbert HJ, Henrissat B. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics. 2015;31:647–655. [DOI] [PubMed] [Google Scholar]
- 3.Lapébie P, Lombard V, Drula E, Terrapon N, Henrissat B. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat Commun. 2019;10(1):2043. doi: 10.1038/s41467-019-10068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 2014;506(7489):498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013;501(7467):426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, Conner ME, Earl AM, Knight R, Bjorkman PJ, et al. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science. 2018;360(6390):795–800. doi: 10.1126/science.aaq0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Troy EB, Carey VJ, Kasper DL, Comstock LE. Orientations of the Bacteroides fragilis capsular polysaccharide biosynthesis locus promoters during symbiosis and infection. J Bacteriol. 2010;192(21):5832–5836. doi: 10.1128/JB.00555-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krinos CM, Coyne MJ, Weinacht KG, Tzianabos AO, Kasper DL, Comstock LE. Extensive surface diversity of a commensal microorganism by multiple DNA inversions. Nature. 2001;414(6863):555–558. doi: 10.1038/35107092. [DOI] [PubMed] [Google Scholar]
- 9.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 10.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107(27):12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332(6032):974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blandford LE, Johnston EL, Sanderson JD, Wade WG, Lax AJ. Promoter orientation of the immunomodulatory Bacteroides fragilis capsular polysaccharide A (PSA) is off in individuals with inflammatory bowel disease (IBD). Gut Microbes. 2019;10(5):569–577. doi: 10.1080/19490976.2018.1560755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson GP, Chou WC, Manson AL, Rogov P, Abeel T, Bochicchio J, Ciulla D, Melnikov A, Ernst PB, Chu H, et al. Spatially distinct physiology of Bacteroides fragilis within the proximal colon of gnotobiotic mice. Nat Microbiol. 2020;5(5):746–756. doi: 10.1038/s41564-020-0683-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doebeli M. A model for the evolutionary dynamics of cross-feeding polymorphisms in microorganisms. Population Ecology. 2002;44:59–70. doi: 10.1007/s101440200008. [DOI] [Google Scholar]
- 15.Pacheco AR, Moel M, Segrè D. Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems. Nat Commun. 2019;10(1):103. doi: 10.1038/s41467-018-07946-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Limenitakis JP, Fuhrer T, Geuking MB, Lawson MA, Wyss M, Brugiroux S, Keller I, Macpherson JA, Rupp S, et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun. 2015;6:8292. doi: 10.1038/ncomms9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuncil YE, Xiao Y, Porter NT, Reuhs BL, Martens EC, Hamaker BR, Walter J, Ruby EG. Reciprocal prioritization to dietary glycans by gut bacteria in a competitive environment promotes stable coexistence. mBio. 2017;8(5):e01068–17. doi: 10.1128/mBio.01068-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533(7602):255–259. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim YG, Sakamoto K, Seo SU, Pickard JM, Gillilland MG 3rd, Pudlo NA, Hoostal M, Li X, Wang TD, Feehley T, et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science. 2017;356(6335):315–319. doi: 10.1126/science.aag2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagao-Kitamoto H, Leslie JL, Kitamoto S, Jin C, Thomsson KA, Gillilland MG 3rd, Kuffa P, Goto Y, Jenq RR, Ishii C, et al. Interleukin-22-mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat Med. 2020;26(4):608–617. doi: 10.1038/s41591-020-0764-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Degnan PH, Barry NA, Mok KC, Taga ME, Goodman AL. Human gut microbes use multiple transporters to distinguish vitamin B₁₂ analogs and compete in the gut. Cell Host Microbe. 2014;15(1):47–57. doi: 10.1016/j.chom.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wexler AG, Schofield WB, Degnan PH, Folta-Stogniew E, Barry NA, Goodman AL. Human gut bacteroides capture vitamin B(12) via cell surface-exposed lipoproteins. Elife. 2018;7:e37138. doi: 10.7554/eLife.37138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevrin G, Massier S, Chassaing B, Agus A, Delmas J, Denizot J, Billard E, Barnich N. Adaptation of adherent-invasive E. coli to gut environment: impact on flagellum expression and bacterial colonization ability. Gut Microbes. 2020;11(3):364–380. doi: 10.1080/19490976.2017.1421886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imai J, Kitamoto S, Sugihara K, Nagao-Kitamoto H, Hayashi A, Morhardt TL, Kuffa P, Higgins PDR, Barnich N, Kamada N. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019;12(3):632–643. doi: 10.1038/s41385-019-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delmas J, Gibold L, Faïs T, Batista S, Leremboure M, Sinel C, Vazeille E, Cattoir V, Buisson A, Barnich N, et al. Metabolic adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci Rep. 2019;9(1):2175. doi: 10.1038/s41598-019-38628-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh T, Singh PK, Das S, Wani S, Jawed A, Dar SA, Harel J. Transcriptome analysis of Escherichia coli O157: h7grown in vitro in the sterile-filtrated cecal content of human gut microbiota associated rats reveals an adaptive expression of metabolic and virulence genes. Microbes Infect. 2015;17(1):23–33. doi: 10.1016/j.micinf.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Diard M, Garcia V, Maier L, Remus-Emsermann MN, Regoes RR, Ackermann M, Hardt WD. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature. 2013;494(7437):353–356. doi: 10.1038/nature11913. [DOI] [PubMed] [Google Scholar]
- 29.Battaglioli EJ, Hale VL, Chen J, Jeraldo P, Ruiz-Mojica C, Schmidt BA, Rekdal VM, Till LM, Huq L, Smits SA, et al. Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Sci Transl Med. 2018;10:464. doi: 10.1126/scitranslmed.aam7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez CA, McNeely TP, Nurmakova K, Beavers WN, Skaar EP. Clostridioides difficile proline fermentation in response to commensal clostridia. Anaerobe. 2020;63:102210. doi: 10.1016/j.anaerobe.2020.102210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenior ML, Leslie JL, Young VB, Schloss PD, Green Tringe S. Clostridium difficil alters the structure and metabolism of distinct cecal microbiomes during initial infection to promote sustained colonization. mSphere. 2018;3(3). doi: 10.1128/mSphere.00261-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502(7469):96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtis MM, Hu Z, Klimko C, Narayanan S, Deberardinis R, Sperandio V. The gut commensal Bacteroides thetaiotaomicron exacerbates enteric infection through modification of the metabolic landscape. Cell Host Microbe. 2014;16(6):759–769. doi: 10.1016/j.chom.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connolly JPR, Slater SL, O’Boyle N, Goldstone RJ, Crepin VF, Ruano-Gallego D, Herzyk P, Smith DGE, Douce GR, Frankel G, et al. Host-associated niche metabolism controls enteric infection through fine-tuning the regulation of type 3 secretion. Nat Commun. 2018;9(1):4187. doi: 10.1038/s41467-018-06701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garber JM, Nothaft H, Pluvinage B, Stahl M, Bian X, Porfirio S, Enriquez A, Butcher J, Huang H, Glushka J, et al. The gastrointestinal pathogen Campylobacter jejuni metabolizes sugars with potential help from commensal Bacteroides vulgatus. Commun Biol. 2020;3(1):2. doi: 10.1038/s42003-019-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, Sperandio V. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492(7427):113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis. 2014;20(11):1919–1932. doi: 10.1097/MIB.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 38.Kumar A, Sperandio V, Casadevall A. Indole signaling at the host-microbiota-pathogen interface. mBio. 2019;10(3). doi: 10.1128/mBio.01031-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexeev EE, Lanis JM, Kao DJ, Campbell EL, Kelly CJ, Battista KD, Gerich ME, Jenkins BR, Walk ST, Kominsky DJ, et al. Microbiota-derived indole metabolites promote human and murine intestinal homeostasis through regulation of interleukin-10 receptor. Am J Pathol. 2018;188(5):1183–1194. doi: 10.1016/j.ajpath.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards LA, Bajaj-Elliott M, Klein NJ, Murch SH, Philips AD, Fessler MB. Bacterial-epithelial contact is a key determinant of host innate immune responses to enteropathogenic and enteroaggregative Escherichia coli. PLoS One. 2011;6(10):e27030. doi: 10.1371/journal.pone.0027030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez CA, Rivera-Chávez F, Byndloss MX, Bäumler AJ, Payne SM. The periplasmic nitrate reductase NapABC supports luminal growth of salmonella enterica serovar typhimurium during colitis. Infect Immun. 2015;83(9):3470–3478. doi: 10.1128/IAI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spiga L, Winter MG, Furtado de Carvalho T, Zhu W, Hughes ER, Gillis CC, Behrendt CL, Kim J, Chessa D, Andrews-Polymenis HL, et al. An oxidative central metabolism enables salmonella to utilize microbiota-derived succinate. Cell Host Microbe. 2017;22(3):291–301.e6. doi: 10.1016/j.chom.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Motta JP, Allain T, Green-Harrison LE, Groves RA, Feener T, Ramay H, Beck PL, Lewis IA, Wallace JL, Buret AG. Iron sequestration in microbiota biofilms as a novel strategy for treating inflammatory bowel disease. Inflamm Bowel Dis. 2018;24(7):1493–1502. doi: 10.1093/ibd/izy116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buret AG, Motta JP, Allain T, Ferraz J, Wallace JL. Pathobiont release from dysbiotic gut microbiota biofilms in intestinal inflammatory diseases: a role for iron? J Biomed Sci. 2019;26(1):1. doi: 10.1186/s12929-018-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson BR, Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Siderophores in iron metabolism: from mechanism to therapy potential. Trends Mol Med. 2016;22(12):1077–1090. doi: 10.1016/j.molmed.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hider RC, Kong X. Chemistry and biology of siderophores. Nat Prod Rep. 2010;27(5):637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 47.Golonka R, Yeoh BS, Vijay-Kuma M. The iron tug-of-war between bacterial siderophores and innate immunity. J Innate Immun. 2019;11(3):249–262. doi: 10.1159/000494627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rivera-Chávez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, et al. Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of salmonella. Cell Host Microbe. 2016;19(4):443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez CA, Miller BM, Rivera-Chávez F, Velazquez EM, Byndloss MX, Chávez-Arroyo A, Lokken KL, Tsolis RM, Winter SE, Bäumler AJ. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science. 2016;353(6305):1249–1253. doi: 10.1126/science.aag3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cevallos SA, Lee JY, Tiffany CR, Byndloss AJ, Johnston L, Byndloss MX, Bäumler AJ, Ehrt S. Increased epithelial oxygenation links colitis to an expansion of tumorigenic bacteria. mBio. 2019;10:5. doi: 10.1128/mBio.02244-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gillis CC, Hughes ER, Spiga L, Winter MG, Zhu W, Furtado de Carvalho T, Chanin RB, Behrendt CL, Hooper LV, Santos RL, et al. Dysbiosis-associated change in host metabolism generates lactate to support salmonella growth. Cell Host Microbe. 2018;23(1):54–64.e6. doi: 10.1016/j.chom.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gillis CC, Winter MG, Chanin RB, Zhu W, Spiga L, Winter SE, Raffatellu M. Host-derived metabolites modulate transcription of salmonella genes involved in L-lactate utilization during gut colonization. Infect Immun. 2019;87:4. doi: 10.1128/IAI.00773-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hopkins EGD, Roumeliotis TI, Mullineaux-Sanders C, Choudhary JS, Frankel G, Rappuoli R. Intestinal epithelial cells and the microbiome undergo swift reprogramming at the inception of colonic citrobacter rodentium infection. mBio. 2019;10:2. doi: 10.1128/mBio.00062-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berger CN, Crepin VF, Roumeliotis TI, Wright JC, Carson D, Pevsner-Fischer M, Furniss RCD, Dougan G, Dori-Bachash M, Yu L, et al. Citrobacter rodentium subverts ATP flux and cholesterol homeostasis in intestinal epithelial cells in vivo. Cell Metab. 2017;26(5):738–752.e6. doi: 10.1016/j.cmet.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hughes ER, Winter MG, Duerkop BA, Spiga L, Furtado de Carvalho T, Zhu W, Gillis CC, Büttner L, Smoot MP, Behrendt CL, et al. Microbial respiration and formate oxidation as metabolic signatures of inflammation-associated dysbiosis. Cell Host Microbe. 2017;21(2):208–219. doi: 10.1016/j.chom.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nedialkova LP, Denzler R, Koeppel MB, Diehl M, Ring D, Wille T, Gerlach RG, Stecher B, Galán JE. Inflammation fuels colicin Ib-dependent competition of Salmonella serovar Typhimurium and E. coli in enterobacterial blooms. PLoS Pathog. 2014;10(1):e1003844. doi: 10.1371/journal.ppat.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang YL, Chassard C, Hausmann M, von Itzstein M, Hennet T. Sialic acid catabolism drives intestinal inflammation and microbial dysbiosis in mice. Nat Commun. 2015;6:8141. doi: 10.1038/ncomms9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouchet V, Hood DW, Li J, Brisson JR, Randle GA, Martin A, Li Z, Goldstein R, Schweda EK, Pelton SI, et al. Host-derived sialic acid is incorporated into Haemophilus influenzae lipopolysaccharide and is a major virulence factor in experimental otitis media. Proc Natl Acad Sci U S A. 2003;100(15):8898–8903. doi: 10.1073/pnas.1432026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crawford RW, Keestra AM, Winter SE, Xavier MN, Tsolis RM, Tolstikov V, Bäumler AJ, Nassif X. Very long O-antigen chains enhance fitness during Salmonella-induced colitis by increasing bile resistance. PLoS Pathog. 2012;8(9):e1002918. doi: 10.1371/journal.ppat.1002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, Restrepo NA, Hosking MP, Edwards RA, Battistoni A, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11(3):227–239. doi: 10.1016/j.chom.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Bäumler AJ. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108(42):17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ormsby MJ, Logan M, Johnson SA, McIntosh A, Fallata G, Papadopoulou R, Papachristou E, Hold GL, Hansen R, Ijaz UZ, et al. Inflammation associated ethanolamine facilitates infection by Crohn’s disease-linked adherent-invasive Escherichia coli. EBioMedicine. 2019;43:325–332. doi: 10.1016/j.ebiom.2019.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yilmaz B, Li H. Gut microbiota and iron: the crucial actors in health and disease. Pharmaceuticals (Basel). 2018;11(4):98. doi: 10.3390/ph11040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA, Galán JE. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4(2):e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vogel-Scheel J, Alpert C, Engst W, Loh G, Blaut M. Requirement of purine and pyrimidine synthesis for colonization of the mouse intestine by Escherichia coli. Appl Environ Microbiol. 2010;76(15):5181–5187. doi: 10.1128/AEM.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiang SL, Mekalanos JJ. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol. 1998;27(4):797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- 67.Singh V, Yeoh BS, Xiao X, Kumar M, Bachman M, Borregaard N, Joe B, Vijay-Kumar M. Interplay between enterobactin, myeloperoxidase and lipocalin 2 regulates E. coli survival in the inflamed gut. Nat Commun. 2015;6:7113. doi: 10.1038/ncomms8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kitamoto S, Alteri CJ, Rodrigues M, Nagao-Kitamoto H, Sugihara K, Himpsl SD, Bazzi M, Miyoshi M, Nishioka T, Hayashi A, et al. Dietary L-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat Microbiol. 2020;5(1):116–125. doi: 10.1038/s41564-019-0591-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stecher B, Denzler R, Maier L, Bernet F, Sanders MJ, Pickard DJ, Barthel M, Westendorf AM, Krogfelt KA, Walker AW, et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc Natl Acad Sci U S A. 2012;109(4):1269–1274. doi: 10.1073/pnas.1113246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diard M, Bakkeren E, Cornuault JK, Moor K, Hausmann A, Sellin ME, Loverdo C, Aertsen A, Ackermann M, De Paepe M, et al. Inflammation boosts bacteriophage transfer between Salmonella spp.. Science. 2017;355(6330):1211–1215. doi: 10.1126/science.aaf8451. [DOI] [PubMed] [Google Scholar]
- 71.Saile N, Schwarz L, Eißenberger K, Klumpp J, Fricke FW, Schmidt H. Growth advantage of Escherichia coli O104: h4strains on 5-N-acetyl-9-O-acetyl neuraminic acid as a carbon source is dependent on heterogeneous phage-Borne nanS-p esterases. Int J Med Microbiol. 2018;308(4):459–468. doi: 10.1016/j.ijmm.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 72.Zhu W, Winter MG, Spiga L, Hughes ER, Chanin R, Mulgaonkar A, Pennington J, Maas M, Behrendt CL, Kim J, et al. Xenosiderophore utilization promotes bacteroides thetaiotaomicron resilience during colitis. Cell Host Microbe. 2020;27(3):376–388.e8. doi: 10.1016/j.chom.2020.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. Gut microbiota. antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science. 2015;347(6218):170–175. doi: 10.1126/science.1260580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martín R, Miquel S, Ulmer J, Kechaou N, Langella P, Bermúdez-Humarán LG. Role of commensal and probiotic bacteria in human health: a focus on inflammatory bowel disease. Microb Cell Fact. 2013;12:71. doi: 10.1186/1475-2859-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sadowsky MJ, Khoruts A. Faecal microbiota transplantation is promising but not a panacea. Nat Microbiol. 2016;1:16015. doi: 10.1038/nmicrobiol.2016.15. [DOI] [PubMed] [Google Scholar]
- 76.Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol. 2016;13(9):508–516. doi: 10.1038/nrgastro.2016.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Vrese M, Schrezenmeir J. Probiotics, prebiotics, and synbiotics. Adv Biochem Eng Biotechnol. 2008;111:1–66. [DOI] [PubMed] [Google Scholar]
- 78.Isolauri E, Rautava S, Salminen S. Probiotics in the development and treatment of allergic disease. Gastroenterol Clin North Am. 2012;41(4):747–762. doi: 10.1016/j.gtc.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 79.Nermes M, Kantele JM, Atosuo TJ, Salminen S, Isolauri E. Interaction of orally administered Lactobacillus rhamnosus GG with skin and gut microbiota and humoral immunity in infants with atopic dermatitis. Clin Exp Allergy. 2011;41(3):370–377. doi: 10.1111/j.1365-2222.2010.03657.x. [DOI] [PubMed] [Google Scholar]
- 80.Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Büttner L, de Lima Romão E, Behrendt CL, Lopez CA, et al. Precision editing of the gut microbiota ameliorates colitis. Nature. 2018;553(7687):208–211. doi: 10.1038/nature25172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu W, Miyata N, Winter MG, Arenales A, Hughes ER, Spiga L, Kim J, Sifuentes-Dominguez L, Starokadomskyy P, Gopal P, et al. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J Exp Med. 2019;216(10):2378–2393. doi: 10.1084/jem.20181939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamada N, Seo SU, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. [DOI] [PubMed] [Google Scholar]
- 83.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336(6086):1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, McHargue JW, Hightower GA, Smith JT, Autieri SM, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76(3):1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Le Bouguénec C, Schouler C. Sugar metabolism, an additional virulence factor in enterobacteria. Int J Med Microbiol. 2011;301(1):1–6. doi: 10.1016/j.ijmm.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]