

ABSTRACT
Hepatocellular carcinoma (HCC) is one of the most aggressive malignant diseases and requires more effective prevention and treatment strategies. Mutations or overexpression of endoplasmic reticulum (ER) proteins have been frequently identified in a solid tumor, suggesting that ER proteins play an important role in tumor development. SEC61G, a component of Sec61 complex located in the membrane of the human ER, has been revealed a potential relevance in glioblastoma multiforme. Analyses from TCGA database showed that SEC61G was overexpressed in HCC. Additionally, the expression of SEC61G mRNA was associated with the survival time of HCC patients. We verified that the higher expression of SEC61G in HCC tissues than paracancerous tissues. Moreover, knockdown of SEC61G inhibited cell proliferation and induced cell apoptosis in vitro. Besides, SEC61G was required for cell migration and invasion, conferring a potential role for SEC61G in tumor transfer. Taken together, our results revealed the role of SEC61G in HCC cells. Further detailed understanding of the signaling networks underlying SEC61G involvement in HCC cells would make SEC61G as a viable therapeutic target for pharmaceutical intervention of HCC.
KEYWORDS: Hepatocellular carcinoma (HCC), SEC61G, Sec61 complex
1. Introduction
In the solid tumor microenvironment, hypoxia and nutrient deficiency of tumor cells unavoidably cause tumor cells to persistent endoplasmic reticulum (ER) stress, with the accumulation of unfolded or misfolded proteins in ER [1–5]. Under conditions of hypoxia, ER stress response pathways are utilized by tumors to maintain tumor cell proliferation [6–10]. The unfolded protein response (UPR) is an intracellular signaling pathway activated by the accumulation of unfolded proteins in the ER which in turn improves the capacity of ER to process misfolded proteins and then opens up the anti-apoptotic signal pathway [11,12]. Nevertheless, the UPR can commit cells to apoptosis when survival mechanisms are exhausted [13]. It is generally considered that the UPR is activated in hepatocellular carcinoma (HCC), the most common primary liver tumors with poor prognosis, and is capable to result in tumor cell anti-apoptosis and drug resistance [14,15], hinting that a growth advantage could be caused by the genetic change of the ER-based protein folding machinery which is an apparent metabolic characteristic of HCC.
The heterotrimeric Sec61 complex is formed by SEC61 α, β, and γ. It localizes in the membrane of the ER and is critical for the transport of newly synthesized and nascent precursor polypeptides into the ER [16]. In addition to the SEC61 complex, the complete protein translocase formed by SEC62 and SEC63 also participates in protein proper folding, post-translational modification, and transport [17]. In tumor cells, genetic changes of ER-resident chaperones have been revealed, hinting that ER proteins are critical for tumor development. Especially, it has been reported that SEC62 and SEC63 are frequently mutated and/or overexpressed in kinds of cancers [18–20]. However, there are few studies about the role of SEC61 in tumorigenesis. SEC61G is one of the 3 subunits of the Sec61 complex. It has been reported that SEC61G is remarkably overexpressed in glioblastoma multiforme and is critical for tumor cell growth and involved in ER stress [21]. Notably, SEC61G is found to be up-regulated in HCC drinkers without HBV and also in alcohol-treated MHCC-97 L and L-02 cell lines [22], suggesting the potential role of SEC61G in HCC. However, the function of SEC61G in HCC has not been fully clarified.
In this study, SEC61G was revealed overexpressed in HCC samples, followed by an investigation of the effect of knockdown of SEC61G on the proliferation, apoptosis, migration, and invasion of HCC cells in vitro. The findings of this study will provide a better understanding of the molecular mechanism underlying HCC, as well as lead to improved diagnosis or prognosis of this disease.
2. Materials and methods
2.1. Retrieval and analysis of the cancer genome atlas (TCGA) data
The gene expression data and clinical data of 369 HCC patients and 160 nontumor controls were obtained from TCGA database (https://cancergenome.nih.gov/). GEPIA (http://gepia.cancer-pku.cn/) is a visualization website based on TCGA data and contains differential gene expression between tumor and non-tumor samples, and analysis of the association between gene expression and overall survival or clinical tumor stages [23]. Based on GEPIA, the expression of SEC61G between HCC tumor and non-tumor samples were analyzed. According to the median value of the expression of the SEC61G, all the samples were divided into high-SEC61G and low-SEC61G groups. Then, the Kaplan-Meier survival curve was performed to evaluate the association between SEC61G expression and survival time, followed by log rank test.
2.2. Immunohistochemistry (IHC) assay
Tissue microarray (TMA) (Cat No. HLivH180Su14) paraffin blocks of HCC were purchased from Shanghai Outdo Biotech Company (Shanghai, China). A total of 85 pairs of tumor and paracancerous tissues were subjected to IHC assay. For IHC, each TMA slide was firstly stained with a rabbit anti-SEC61G antibody (dilution, 1:50; 11,147-2-AP; PTG, Chicago, IL) and then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG secondary antibody (dilution, 1: 200; GB23303; Servicebio, Inc., Woburn, MA). After rinsing, the color was developed using 3, 3’-diaminobenzidine (DAB, Servicebio, Inc.). Sections were counter-stained with hematoxylin. A microscope (400× magnification; XSP-C204, COIC, Chongqing, China) was used to photograph the TMA slide. Images were then captured by a panoramic viewer (3DHISTECH Kft.; Budapest, Hungary), and the data were analyzed using Quant Center (3DHISTECH). Immunohistochemistry score (H-SCORE) = ∑(PI × I) = (percentage of cells of weak intensity × 1) + (percentage of cells of moderate-intensity × 2) + (percentage of cells of strong intensity × 3), where PI represents the percentage of positive cells in all cells in the section, and I is for coloration intensity [24,25].
2.3. Cell culture
HepG2 cells (ATCC, Rockville, MD, USA)) and 7402 cells (Shanghai cell bank of Academy of Chinese Sciences, Shanghai, China) were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, South Logan, UT) supplemented with 10% fetal bovine serum (Gibico BRL, Ground Island, NY, USA), 100 U/ml penicillin (Sigma, St Louis, MO, USA) and 0.1 mg/ml streptomycin (Sigma) in a 37°C, 5% CO2 incubator. Cells in the logarithmic phase were used in all subsequent cell experiments. Notably, HepG2 cell line was authenticated by Short Tandem Repeat (STR) profiling by ATCC, and 7402 cell line was STR-authenticated by Shanghai Biowing Applied Biotechnology Co., Ltd (Shanghai, China). STR authentication results confirmed that both two cell lines were not contaminated by other cell lines.
2.4. Small interfering RNA (siRNA) transfection
Four siRNAs against SEC61G, including si-SEC61G-1: 5’-GCAAUAGGAUUUGCUAUAATT-3’ (forward) and 5’-UUAUAGCAAAUCCUAUUGCTT-3’ (reverse), si-SEC61G-2: 5’-CUUAGAGAUUGGUGAACAATT-3’ (forward) and 5’-UUGUUCACCAAUCUCUAAGTT-3’ (reverse), si-SEC61G-3: 5’-GCACUAAACCUGAUAGAAATT-3’ (forward) and 5’-UUUCUAUCAGGUUUAGUGCTT-3’ (reverse), and si-SEC61G-4: 5’-CAUUGUUGGUGGCUGAAUATT-3’ (forward) and 5’-UAUUCAGCCACCAACAAUGTT-3’ (reverse), as well as a scrambled siRNA used as negative control (NC) were synthetized at Oligobio Co. Ltd in Beijing. When the cells reached the logarithmic phase, the medium was replaced with fresh complete culture medium 2 hours before transfection. Cell transfection was then performed using Lipofectamine 2000 (Invitrogen, California, USA) according to the manufacturer’s instructions. The medium was replaced with fresh complete culture medium 6 hours after transfection. Cells were collected 48 hours after transfection and the transfection efficiency of siRNAs was examined using quantitative PCR (Q-PCR).
2.5. Q-PCR
Total RNA was extracted from different transfected cells using a Trizol RNA extraction kit. We then used the reverse transcription kit to reversely transcribe RNA into cDNA. Real-time Q-PCR was then performed to detect the expression of SEC61G as mentioned in a previous study [26].
2.6. Cell counting kit-8 (CCK8) assay
Cells were collected and counted and then cell suspension was prepared. Then, 100 μl cell suspension was taken and seeded into a 96-well plate according to the standard of 1500 cells per hole. The proliferation of HepG2 or 7402 cells in si-SEC61G and NC group was detected respectively. Cell viability was measured every 24 hours. Briefly, 10 μl CCK8 reagent (Solarbio, Beijing, China) was added to each hole and incubated at 37°C for 1.5 hours. OD value was detected at 450 nm by an enzyme labeling instrument (Infinite® M1000 PRO, TECAN, Switzerland).
2.7. Colony formation assay
After 48 hours of transfection, cells were detached with 0.25% trypsin. The cell suspension was prepared and the cell number was counted. Then, 500 cells were seeded into a 60 mm dish containing 5 ml culture medium preheated at 37°C for 0.5 hours. The dish was rotated gently to make the cells disperse evenly. The dish was incubated at 37°C, 5% CO2 incubator for 1–2 weeks with frequent observation. Cell culture was terminated when visible clones emerged. The supernatant was removed and cells were washed carefully twice with PBS. Cells were fixed with 4% paraformaldehyde for 0.5 hours. Paraformaldehyde was removed and then cells were dyed with 0.1% crystal violet for 0.5 hours. The dish was washed gently with running water and then left the dish to dry naturally. The number of clones was counted directly by the naked eye. The size and number of clones were compared finally.
2.8. Cell apoptosis
The culture medium was removed and replaced with a serum-free culture medium after 48 hours of transfection. Cells were collected and centrifuged in a centrifugal tube at 1000 rpm for 5 min after 24 hours of starvation. Cells were resuspended by PBS pre-cooled at 4°C and then centrifuged again. The supernatant was removed carefully. Cells were dispersed in the binding buffer and subjected to the Annexin V-FITC staining manufacturer’s protocol (Keygen Biotech, Nanjing, China). FACS Calibur flow cytometer (BD Biosciences, Mountain View, CA) was used to analyze the samples.
2.9. Cell migration assay
After 48 hours of transfection, cells were detached with trypsin and then suspended with a culture medium free of FBS. Then, 100 μl cell suspension containing 5 × 104 cells was placed in the upper chamber of an 8 μm microporous filter. The lower chamber of the transwell was added with 600 μl of medium containing 10% FBS. The plate was placed in a 37°C incubator for 24 hours. The cells in the upper chamber were wiped off with cotton sticks. The upper and lower chambers were washed twice with PBS. Cells were immobilized by polyformaldehyde for 15 minutes, stained with 0.1% crystal violet for 5 minutes, and finally washed with PBS. The basement membrane was cut off and the migrated cells were counted under a microscope (IX73, Olympus).
2.10. Cell invasion assay
The transwell chambers were loaded with 100 μl matrigel (BD Biosciences, Bedford, MA) in a 24-well plate at 37°C for 4 hours. The subsequent treatment was similar as protocols in migration assay as described above.
2.11. Wound healing assay
Cells were seeded in a 6-well plate and cultured until 90% confluence. Pipette tips were used to would cell monolayers and then washed cell monolayers three times with PBS. A serum-free medium was added to the plate and the cells were cultured for another 24 hours. An inverted microscope (Nikon, TS100, Japan) was used to determine the number of migrated cells at 0 and 24 hours.
2.12. Western blotting
For immunoblotting, cells were collected and treated with RIPA Lysis Buffer (CWBIO, China) supplemented with protease inhibitor cocktail (CWBIO). After heating at 100°C for 10 minutes, the sample was subjected to SDS-PAGE. Briefly, at a constant voltage of 80 V, 20 μg proteins were loaded for electrophoresis for 2 hours. Then, the protein was transferred to the PVDF membrane under the condition of 4°C and 300 mA constant current for 150 min. A polyvinylidene difluoride membrane (Millipore) containing proteins was incubated with primary antibodies against Bax (1:2000; 50,599-2-lg; PTG), E-Cadherin (1:1000; 20,874-1-AP; PTG), N-Cadherin (1:2000; 22,018-1-AP; PTG), SEC61G (1:500; 11,147-2-AP; PTG), GAPDH (1:5000; 60,004-1-Ig; PTG), BCL-2 (1:1000; AF6139; Affinity Biosciences, USA), Cleaved-Caspase 3 (1:1000; AF6311; Affinity), and β-Catenin (1:1000; AF6266; Affinity) and secondary antibody (anti-rabbit or anti-mouse immunoglobulin G conjugated with horseradish peroxidase) (1:5000; S0001 or S0002; Affinity) sequentially. Finally, the Enhanced Chemiluminescence System (Amersham Biosciences, Little Chalfont, UK) was used to detect proteins.
2.13. Statistical analysis
Statistical analysis was conducted by SPSS version 18.0. The data are expressed as the mean ± SD and each in vitro experiment was repeated at least three times. The student’s t-test was used to compare the differences between the si-SEC61G group and the NC group. A p-value of less than 0.05 was considered statistically significant.
3. Results
3.1. SEC61G is overexpressed in HCC and is associated with the different stages of HCC progression
Based on GEPIA information, the expression of SEC61G in HCC tumor tissues was significantly higher than that in non-tumor tissues (Figure 1(a)). According to the median value of SEC61G expression, the survival time of patients with high SEC61G expression was shorter than that of patients with low SEC61G expression (Figure 1(b)). To verify these, we next determined the expression of SEC61G in 85 pairs of tumor and paracancerous tissues from HCC patients by IHC assay. Accordingly, the expression of SEC61G in HCC tumor tissues was significantly higher than that in paracancerous tissues (Figure 1(c)). Taken together, these results hinted that SEC61G might contribute to HCC development.
Figure 1.

SEC61G is overexpressed in liver cancer and is associated with the survival time of patients. (a) The expression of SEC61G in liver cancer was significantly higher than that in paracancerous tissue by analyses from TCGA database. Number of tumor tissues was 369 and that of non-tumor tissues was 160. (b) SEC61G expression was negatively associated with survival time of patients. (c) IHC assay for tissue microarray paraffin blocks of HCC patients (N = 85) showed that SEC61G was also up-regulated in tumor tissues compared to paracancerous tissues. *** P < 0.01 compared with paracancerous tissues
3.2. Knockdown of SEC61G in HCC cell lines
The SEC61G expression was then silenced by the transfection of siRNAs, as result, Q-PCR assay showed that si-SEC61G-4 had the best silencing effect among the four siRNAs against SEC61G in both HepG2 and 7402 cell lines (Figure 2(a)). Next, the protein level of in SEC61G si-SEC61G-4 transfected HepG2 cells and 7402 cells were examined by western blotting. We found that that the protein expression of SEC61G decreased significantly in the two cell lines transfected with si-SEC61G-4 compared to that in the two cell lines transfected with si-NC (Figure 2(b)). Overall, these results demonstrate that si-SEC61G-4 was effective to knock down the expression of SEC61G, thereby SEC61G-4 was selected in subsequent studies.
Figure 2.
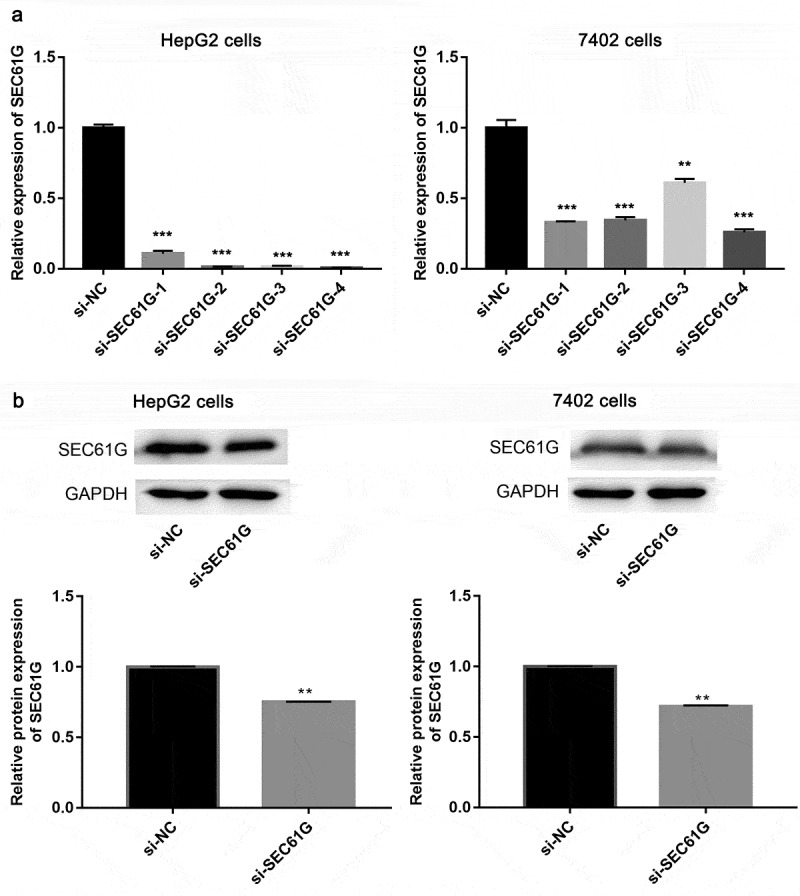
Examination of siRNA effect. (a) Q-PCR showed the mRNA expression level of SEC61G in HepG2 and 7402 cells transfected with si-SEC61G-1, si-SEC61G-2, si-SEC61G-3, si-SEC61G-4 or si-NC. (b) Western blotting showed the protein expression level of cells SEC61G in HepG2 and 7402 transfected with si-SEC61G-4. All experiments were carried out for three times. Data are expressed as mean ± standard deviation (SD). ** P < 0.01 and *** P < 0.001 compared with si-NC group
3.3. Knockdown of SEC61G inhibits the proliferation of HCC cells
To investigate the possible function of SEC61G in regulating the proliferation of HCC cells, CCK8 assay was used to examine the cell viability every 24 hours. We found that the proliferation of HepG2 cells transfected with si-SEC61G was significantly weaker than that of NC group (Figure 3(a)). Accordingly, decreased cell viability was found in si-SEC61G transfected 7402 cells (Figure 3(a)). A colony formation assay was conducted to further verify the role of SEC61G in cell proliferation. The results showed that the number of clones of HepG2 cells and 7402 cells transfected with si-SEC61G was significantly less than that of the NC group (Figure 3(b)), confirming that SEC61G was important for the proliferation of HCC cells.
Figure 3.
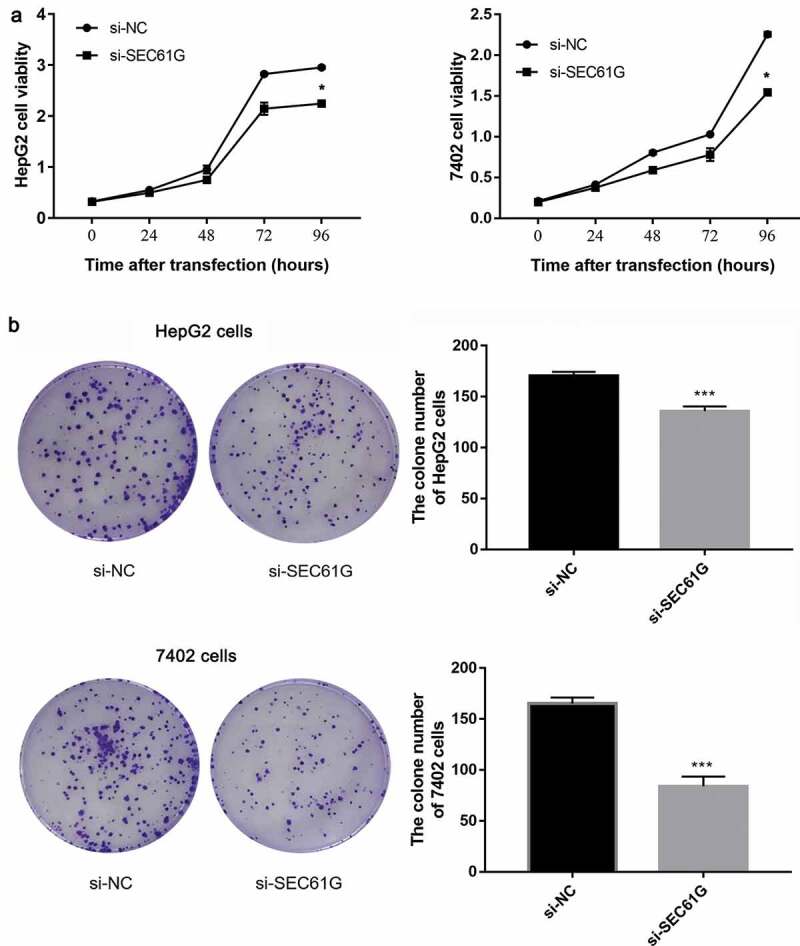
SEC61G was required for cell proliferation of HepG2 and 7402 cells. (a) CCK8 assay demonstrated the proliferation of HepG2 and 7402cells transfected with si-SEC61G was significantly weaker than that of si-NC group. (b) Colony formation assay indicated that the clone number of HepG2 and 7402 cells transfected with si-SEC61G was significantly less than that of si-NC group. All experiments were carried out for three times. Data are expressed as mean ± SD. * P < 0.05 and *** P < 0.001 compared with si-NC group
3.4. SEC61G plays an anti-apoptotic role in HCC cells
To reveal the effect of SEC61G on HCC cell apoptosis, we detected the expression of key proteins involved in cell apoptosis. The expression of Bax and cleaved caspase 3 was found to be significantly increased in si-SEC61G transfected HepG2 and 7402 cells, while the expression of BCL-2 was dramatically decreased in si-SEC61G transfected HepG2 and 7402 cells (Figure 4(a)). Further flow cytometry showed that the number of apoptotic cells was significantly increased in si-SEC61G transfected HepG2 and 7402 cells compared with that in the si-NC group (Figure 4(b)). These results indicated that SEC61G is involved in cell apoptosis.
Figure 4.

SEC61G was involved in cell apoptosis. (a) Western blotting examination of the expression of apoptosis-associated proteins in si-SEC61G transfected HepG2 and 7402 cells, respectively. (b) Flow cytometry showed that the number of apoptotic cells increased significantly in si-SEC61G transfected HepG2 and 7402 cells, respectively. All experiments were carried out for three times. Data are expressed as mean ± SD. *** P < 0.001 compared with si-NC group
3.5. SEC61G is involved in the migration and invasion of HCC cells
Cancer cell migration is important for tumor development [27–30]. So we sought to investigate the role of SEC61G in cell migration by transwell assay. As demonstrated in Figure 5(a), the migration abilities of si-SEC61G transfected HepG2 and7402 cells were all significantly weaker than that of the NC group, indicating that SEC61G suppressed the migration of HepG2 and7402 cells. Moreover, the effect of SEC61G in HCC cell invasion was also detected by transwell assay. We found that cells were able to invade freely through the matrigel in the control group, whereas this ability was hampered in si-SEC61G transfected HepG2 and 7402 cells (Figure 5(b)). The wound-healing assay was used to confirm the role of SEC61G in HCC cell migration. As shown in Figure 6, cells were able to migrate freely across the plate in the control group, whereas this ability was partially hampered in si-SEC61G transfected HepG2 cells and 7402 cells. Taken results described above together, we found that SEC61G played an important role in cell migration and invasion.
Figure 5.
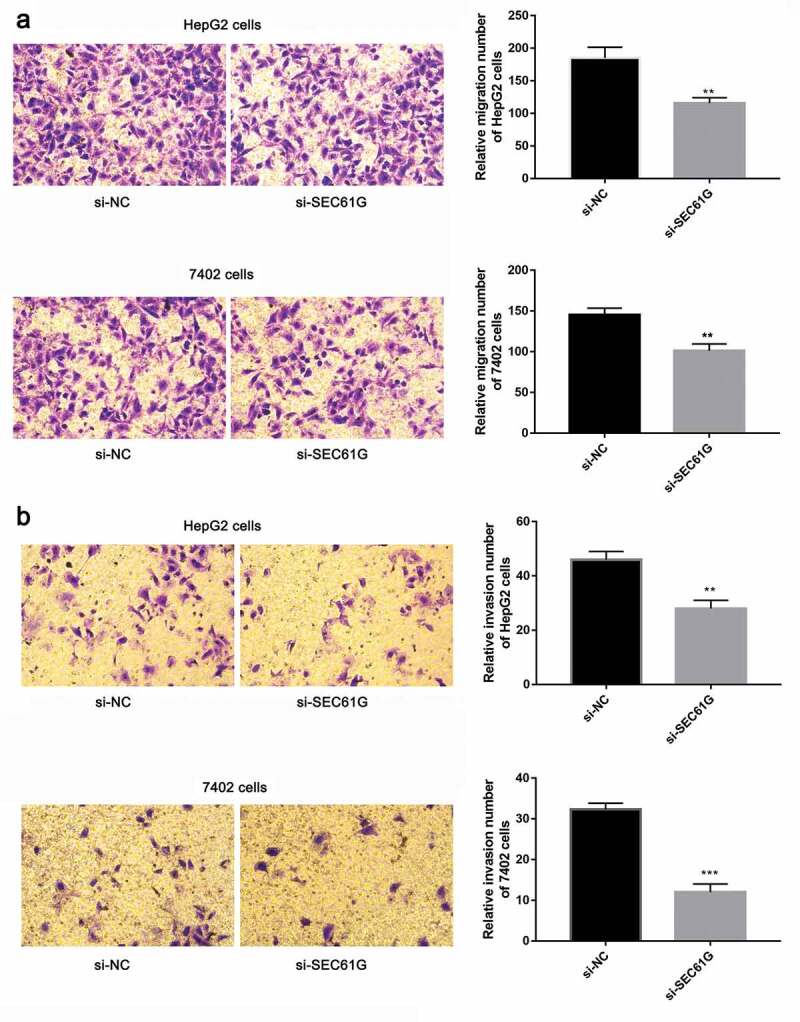
Transwell assay showed the role of SEC61G in HCC cell migration and invasion. (a) SEC61G functions in the migration of HepG2 and 7402 cells. (b) SEC61G functions in the invasion of HepG2 and 7402 cells. All experiments were carried out for three times. Data are expressed as mean ± SD. ** P < 0.01 and *** P < 0.001 compared with si-NC group
Figure 6.
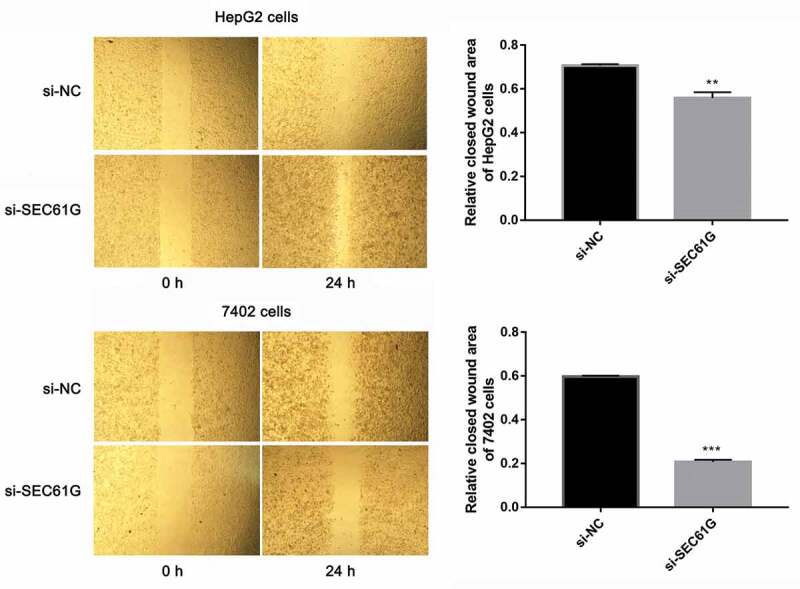
The role of SEC61G in wound healing of HepG2 and 7402 cells. The experiments were carried out for three times. Data are expressed as mean ± SD. ** P < 0.01 and *** P < 0.001 compared with si-NC group
To explore the molecular mechanism of SEC61G participating in cell migration and invasion, we next examined the expression of a panel of proteins involved in these processes. We found that compared to the si-NC group, the expression of N-cadherin and β-catenin was significantly decreased in si-SEC61G transfected HepG2 and 7402 cells, while the expression of E-cadherin, another member of cadherin, was markedly increased in si-SEC61G transfected HepG2 and 7402 cells (Figure 7). These data suggested the complex mechanism of SEC61G in mediating HCC cell migration and invasion.
Figure 7.

The expression of proteins involved in cell migration and invasion in HepG2 and 7402 cells after transfection with si-SEC61G or si-NC. The experiments were carried out for three times. Data are expressed as mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001 compared with si-NC group. 27 Oct
4. Discussion
HCC is one of the most common types of threatening malignancies in humans worldwide. New cases increased steeply every year, rising to 748,300 with 695,900 deaths [31,32]. The patients who undergo tumorectomy are likely to grow new tumors from their remnant liver due to the invasive features of HCC [33,34]. Currently, it is well known that the key tactic to improve the prognosis of HCC is to diagnose the tumor as early as possible, especially when the patient is symptomless and the function of the liver remains [35]. However, due to the lack of an ideal biomarker, it is harder to distinguish to the normal liver with the liver that would turn cancerous if without any interference. HCC patients are often diagnosed in the late stages. Once in the late stages, it is so difficult to cure that the mortality rate is very high. To data, few diagnostic and prognostic biomarkers and targets for HCC have been reported. Therefore, it is urgent to further reveal the molecular mechanism involved in the development of HCC.
Genetic changes of SEC genes have been attributed to multiple terrible diseases, including human cancers, establishing an important basis for devising new therapeutic strategies against some threatening diseases in the last decades [16]. In previous studies, SEC61G was found to be overexpressed in breast and gastric cancers [36,37]. SEC61G expression has been identified as a promising prognostic marker of poor survival, and a predictor of poor outcome to therapies in patients with glioblastoma [38]. Consistent with previous findings [36,37], we also found that SEC61G was overexpressed in HCC tumor tissues. Moreover, knockdown of SEC61G significantly inhibited proliferation, increased apoptosis, and suppressed migration and invasion of HepG2 and 7402 cells in vitro. These data suggest that SEC61G may play an oncogenic role in HCC.
Cell proliferation and apoptosis are of importance in the growth and development of many tumors [39]. It is reported that miR-4677-3p/SEC61G axis is a key mechanism mediating the role of lncRNA LINC02418 in regulating proliferation and apoptosis of non-small cell lung cancer cells [40]. Moreover, the BCL-2 family is the best-characterized group of apoptosis-mediating factors, which is involved in HCC development [41]. BCL-2 inhibitor and Bax activator are revealed as promising approaches for cancer therapy [42,43]. Furthermore, caspases-3 is a key mediator of mitochondrial events of apoptosis [44]. In our study, the expression of Bax and cleaved caspase 3 was found to be significantly increased in si-SEC61G transfected HepG2 and 7402 cells, while the expression of BCL-2 was dramatically decreased. Based on our results, we speculate that SEC61G may promote tumor cell growth in vitro, possibly through an anti-apoptotic mechanism.
Furthermore, the migration and invasiveness of cancer cells are also key steps of cancer development. Epithelial to mesenchymal transition (EMT) is a crucial regulatory mechanism in tumor cell migration, invasion, and metastasis in a variety of cancers [45,46]. It has been revealed that Sirtuin6 promotes the EMT of HCC by stimulating the autophagic degradation of E-cadherin [47]. Zhan et al. demonstrated that reduced expression of N-cadherin was associated with metastatic potential and poor surgical outcomes of HCC [48]. Besides, it is reported that several transcription factors are direct repressors of E-cadherin, including β-catenin, a key component of the canonical Wnt signaling pathway [49]. A very recent study showed that TXNDC12 promotes EMT and metastasis of HCC cells by activation of β-catenin [50]. In our study, knockdown of SEC61G suppressed HCC cell migration and invasion. Moreover, knockdown of SEC61G resulted in the decreased expression of N-cadherin and β-catenin as well as increased expression of E-cadherin in HCC cells. It can be speculated that SEC61G may be involved in HCC cell migration and invasion via regulating the expression of these EMT-related proteins.
This study firstly reported the function of SEC61G in HCC cells. Although we found that high expression of SEC61G was correlated with the short survival time of patients, we did not investigation the association between SEC61G and tumor stage, which limited the clinical application of SEC61G as a target of HCC treatment to a certain degree. Moreover, we only performed a series of in vitro assay to explore the possible mechanism of SEC61G in HCC cells, in vivo experiments were not conducted to confirm the role of SEC61G in HCC. More in vivo experiments may be strong evidence to support our observation. Meanwhile, the heterotrimeric Sec61 complex is revealed to be critical for the transport of newly synthesized and nascent precursor polypeptides into the ER [16]. Exploration of indicators for the detection of ER will help to elucidate the detailed mechanism of SEC61G in HCC.
In conclusion, our results indicated that SEC61G is overexpressed in HCC samples and high expression of SEC61G is relevant to the short survival time of HCC patients. Moreover, SEC61G is involved in multiple biological processes of HCC cells, including proliferation, anti-apoptosis, migration, and invasion. SEC61G may serve as a promising biomarker or target for the diagnosis and treatment of this disease.
Funding Statement
This work was supported by the National Natural Science Foundation of China under Grant number 81602129; and the Natural Science Foundation of Shandong Province under Grant number ZR2016HB13.
Authors’ contributions
HG, JN, and CP made substantial contributions to the conception and design of the study. HG, WN, CG, and ZH were responsible for data acquisition, analysis, and interpretation. HG and CP were involved in drafting the article and critically revising it for important intellectual content. All authors provided final approval of the version to be published. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
All experiments involving human participants were authorized by the Ethics Committee of Shanghai Outdo Biotech Company and Qilu Hospital, Shandong University. All procedures involving human participants were performed in accordance with the ethical standards of the institutional and/or national research committee, and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was provided by each patient.
Disclosure statement
The authors declare that they have no competing interests.
References
- [1].Bi M, Naczki C, Koritzinsky M, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. Embo J. 2005;24(19):3470–3481. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Feldman DE, Chauhan V, Koong AC.. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. [DOI] [PubMed] [Google Scholar]
- [3].Koumenis C, Wouters BG.. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4:423–436. [DOI] [PubMed] [Google Scholar]
- [4].Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Song S, Tan J, Miao Y, et al. Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J Cell Physiol. 2018;233:3867–3874. [DOI] [PubMed] [Google Scholar]
- [6].Lee AS, Hendershot LM. ER stress and cancer. Cancer Biol Ther. 2006;5:721–722. [DOI] [PubMed] [Google Scholar]
- [7].Urra H, Dufey E, Avril T, et al. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer. 2016;2:252–262. [DOI] [PubMed] [Google Scholar]
- [8].Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. [DOI] [PubMed] [Google Scholar]
- [9].Cassim S, Vučetić M, Ždralević M, et al. Warburg and Beyond: the Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers (Basel). 2020;12(5):1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cassim S, Raymond V, Lacoste B, et al. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget. 2018;9:26868–26883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. [DOI] [PubMed] [Google Scholar]
- [12].Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018;69:169–181. [DOI] [PubMed] [Google Scholar]
- [14].Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- [15].Ren B, Liu H, Gao H, et al. Celastrol induces apoptosis in hepatocellular carcinoma cells via targeting ER-stress/UPR. Oncotarget. 2017;8:93039–93050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Linxweiler M, Schick B, Zimmermann R. Let’s talk about Secs: sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct Target Ther. 2017;2:17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zimmermann R, Muller L, Wullich B. Protein transport into the endoplasmic reticulum: mechanisms and pathologies. Trends Mol Med. 2006;12:567–573. [DOI] [PubMed] [Google Scholar]
- [18].Mori Y, Sato F, Selaru FM, et al. Instabilotyping reveals unique mutational spectra in microsatellite-unstable gastric cancers. Cancer Res. 2002;62:3641–3645. [PubMed] [Google Scholar]
- [19].Schulmann K, Brasch FE, Kunstmann E, et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology. 2005;128:590–599. [DOI] [PubMed] [Google Scholar]
- [20].Casper M, Weber SN, Kloor M, et al. Hepatocellular carcinoma as extracolonic manifestation of Lynch syndrome indicates SEC63 as potential target gene in hepatocarcinogenesis. Scand J Gastroenterol. 2013;48:344–351. [DOI] [PubMed] [Google Scholar]
- [21].Lu Z, Zhou L, Killela P, et al. Glioblastoma proto-oncogene SEC61gamma is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. 2009;69:9105–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li WT, Zou AE, Honda CO, et al. Etiology-Specific Analysis of Hepatocellular Carcinoma Transcriptome Reveals Genetic Dysregulation in Pathways Implicated in Immunotherapy Efficacy. Cancers (Basel). 2019;11:1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yeo W, Chan SL, Mo FK, et al. Phase I/II study of temsirolimus for patients with unresectable Hepatocellular Carcinoma (HCC)-a correlative study to explore potential biomarkers for response. BMC Cancer. 2015;15:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Azim HA, Peccatori FA, Brohée S, et al. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015;17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang TL, Diaz LA Jr., Romans K, et al. Q-PCR Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc Natl Acad Sci U S A. 2004;101:3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans. 2017;45:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Paul CD, Mistriotis P, Konstantopoulos K. Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer. 2016;17(2):131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Douglas H, Weinberg RA. Hallmarks of Cancer: the Next Generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- [30].Benoit L, Valérie-Ann R, Shamir C, et al. Highly tumorigenic hepatocellular carcinoma cell line with cancer stem cell-like properties. Plos One. 2017;12:e0171215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brawley OW. Avoidable cancer deaths globally. CA Cancer J Clin. 2011;61:67–68. [DOI] [PubMed] [Google Scholar]
- [32].Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- [33].Roayaie S, Blume IN, Thung SN, et al. A system of classifying microvascular invasion to predict outcome after resection in patients with hepatocellular carcinoma. Gastroenterology. 2009;137:850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Goessling W. Deciphering hepatocellular carcinoma: from bench to bedside and back. Gastroenterology. 2009;137:786–788. [DOI] [PubMed] [Google Scholar]
- [35].Dimitroulis D, Damaskos C, Valsami S, et al. From diagnosis to treatment of hepatocellular carcinoma: an epidemic problem for both developed and developing world. World J Gastroenterol. 2017;23:5282–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Reis-Filho JS, Pinheiro C, Lambros MB, et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J Pathol. 2006;209:445–453. [DOI] [PubMed] [Google Scholar]
- [37].Tsukamoto Y, Uchida T, Karnan S, et al. Genome-wide analysis of DNA copy number alterations and gene expression in gastric cancer. J Pathol. 2008;216:471–482. [DOI] [PubMed] [Google Scholar]
- [38].Liu B, Liu J, Liao Y, et al. Identification of SEC61G as a Novel Prognostic Marker for Predicting Survival and Response to Therapies in Patients with Glioblastoma. Med Sci Monit. 2019;25:3624–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Evan G, Vousden K. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. [DOI] [PubMed] [Google Scholar]
- [40].Han B. LncRNA LINC02418 regulates proliferation and apoptosis of non-small cell lung cancer cells by regulating miR-4677-3p/SEC61G. Eur Rev Med Pharmacol Sci. 2019;23:10354–10362. [DOI] [PubMed] [Google Scholar]
- [41].Guo X-Z, Shao X-D, Liu M-P, et al. Effect of bax, bcl-2 and bcl-xL on regulating apoptosis in tissues of normal liver and hepatocellular carcinoma. World J Gastroenterol. 2002;8:1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xin M, Li R, Xie M, et al. Small-molecule Bax agonists for cancer therapy. Nat Commun. 2014;5:4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. [DOI] [PubMed] [Google Scholar]
- [44].Lakhani SA, Masud A, Kuida K, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. [DOI] [PubMed] [Google Scholar]
- [46].Thompson EW, Newgreen DF. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–5995. [DOI] [PubMed] [Google Scholar]
- [47].Han LL, Jia L, Wu F, et al. Sirtuin6 (SIRT6) promotes the EMT of hepatocellular carcinoma by stimulating autophagic degradation of E-cadherin. Mol Cancer Res. 2019;17:2267–2280. [DOI] [PubMed] [Google Scholar]
- [48].Zhan D, Wei S, Liu C, et al. Reduced N‐cadherin expression is associated with metastatic potential and poor surgical outcomes of hepatocellular carcinoma. J Gastroenterol Hepatol. 2012;27:173–180. [DOI] [PubMed] [Google Scholar]
- [49].Wang Y, Bu F, Royer C, et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat Cell Biol. 2014;16:1092–1104. [DOI] [PubMed] [Google Scholar]
- [50].Yuan K, Xie K, Lan T, et al. TXNDC12 promotes EMT and metastasis of hepatocellular carcinoma cells via activation of β-catenin. Cell Death Differ. 2020;27:1355–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]