

ABSTRACT
We recently made an important discovery that radiation induces myofibroblasts, which play a role in radiation-related carcinogenesis via tumor microenvironment formation. Here, we investigated the threshold dose and the mechanisms of myofibroblast induction to assess adverse radiation effects on normal cells. Single-dose of healthy human fibroblasts in vitro promotes myofibroblast induction at high doses (≥ 5 Gy). In contrast, repeated low dose of fractionated radiation is at least equivalent to high-dose single radiation regarding myofibroblast induction. ROS play a pivotal role in the process of myofibroblast induction in normal tissue injury. Antioxidants, such as epicatechin and ascorbic acid can prevent myofibroblast induction by scavenging ROS. We further investigated the role of DNA damage responses (DDR) on myofibroblast induction. Blocking the DDR using DNA-PK or AKT inhibitors enhanced cellular sensitivity to radiation and facilitated myofibroblast induction, whereas an ATM inhibitor also enhanced radiation sensitivity but abrogated ROS accumulation and myofibroblast induction. In contrast to standard culture conditions, myofibroblasts remained after low or moderate doses of radiation (below 2.5 Gy) under growth-restricted conditions. In conclusion, the recovery of damaged cells from radiation is essential for myofibroblast clearance, which restores stromal cell dormancy and prevents tumor microenvironment formation. However, residual ROS, by way of sustaining myofibroblast presence, can facilitate tumor microenvironment formation. Targeting ROS using antioxidants is effective in the mitigation of radiation-related adverse effects, such as growth retardation and myofibroblast induction, and helps protect normal tissues.
KEYWORDS: myofibroblast, ROS, cell growth recovery, radiation, tumor microenvironment
Introduction
Clinically, radiation is widely used for the treatment and diagnosis of cancer. The advantages of radiotherapy include excellent tumor control, preservation of normal tissues, and minimal systemic influences. However, radiation is recognized as a carcinogen, as it produces carcinogenic changes by altering gene expression.[1] Various epidemiological studies in humans indicate that ionizing radiation increases the risk for leukemia and solid cancers among those who receive over 100 mSv.[2,3] However, radiation risks associated with levels lower than 100 mSv cannot be determined directly from the epidemiological data due to a lack of a large sample size. Therefore, elucidation of key mechanistic insights into radiation-related carcinogenesis is indispensable for estimating radiation-associated cancer risks.
Tumor development is regulated not only by the growth of malignant cancer cells themselves, but also by mutual communication among cancer cells and tumor stromal cells. Fibroblasts are cells in connective tissue that function to synthesize and deposit extracellular matrix (ECM) to control the tissue microenvironment. Fibroblasts can acquire an activated phenotype at which point they are referred to as myofibroblasts, which are related to cancer-associated fibroblasts (CAFs) in the tumor microenvironment (TME).[4–6] Fibroblast-to-myofibroblast differentiation is often characterized by the expression of α- smooth muscle actin (α-SMA).[7] CAFs control the release of paracrine signals that promote tumor growth and cancer cell aggressiveness, such as tumor angiogenesis, ECM remodeling, tumor proliferation, invasion, and metastasis.[8] A previous study reported on the role of stromal fibroblasts in radiation-related carcinogenesis and showed that mitochondrial ROS affects TME formation via fibroblast activation. [9] Importantly, reactive oxygen species (ROS) function in the process of fibroblast activation in normal tissue injury and fibrosis via ECM remodeling and connective tissue turnover. [10–12] We previously reported that repeated low-dose-fractionated radiation (FR) progressively damages fibroblast mitochondria and elevates mitochondrial ROS levels when cells are exposed to FR for over 21 d [13]. Excessive mitochondrial ROS activate transforming growth factor-beta (TGF-β) signaling, thereby inducing differentiation of myofibroblasts from fibroblasts and facilitating tumor growth through TME formation. [9]
Here, we investigated the mechanisms of radiation-induced myofibroblasts induction and their association with single-dose radiation (SR) and FR dosing. We also discussed the properties of radiation-induced myofibroblasts.
Results
Radiation-induced myofibroblasts detected by α-SMA-positive fiber staining
In order to determine the threshold dose for myofibroblast induction, human lung fibroblasts (TIG-3 and MRC-5 cells) were exposed to acute SR at the dose range from 0.1 Gy to 10 Gy and then immunostained for the myofibroblast marker α-SMA.[7] Fibroblasts presented with altered morphology: flatter and larger without their normal spindle-like structure at 24 h after irradiation. These cells displayed a fiber-like staining pattern of α-SMA as shown in Figure 1(A). We manually counted cells that were positive for α-SMA-positive stress fibers under a fluorescence microscope. Compared to nonirradiated cells, we observed a statistically significant increase in the number of α-SMA-fibers in irradiated TIG-3 and MRC-5 cells exposed to over 5 Gy of SR, while radiation below 5 Gy did not affect the α-SMA staining pattern (Figure 1(B)). We next used FR to examine the effects of different radiation exposure methods on myofibroblast induction. A previous report indicated that cells exposed to ≥0.5 Gy per fraction could no longer divide under FR exposure, while cells exposed to 0.01 or 0.05 Gy per fraction continued to grow exponentially for 31 d [14]. FR treatments with 0.01 or 0.05 Gy per fraction over 21 d (total doses 0.3 Gy or 1.5 Gy, respectively) induced α-SMA-fiber-positive myofibroblasts from TIG-3 cells (Figure 1(A,B)). FR for over 21 d also induced α-SMA-positive cells from MRC-5 cells (data not shown). We further quantified the fluorescence intensity values of α-SMA staining using Keyence imaging software. An increase in the number of α-SMA-positive cells was observed after exposure to high-doses of SR or low-doses of FR for over 21 d in TIG-cells (Figure 1(C)) as well as in MRC-5 cells (data not shown).
Figure 1.
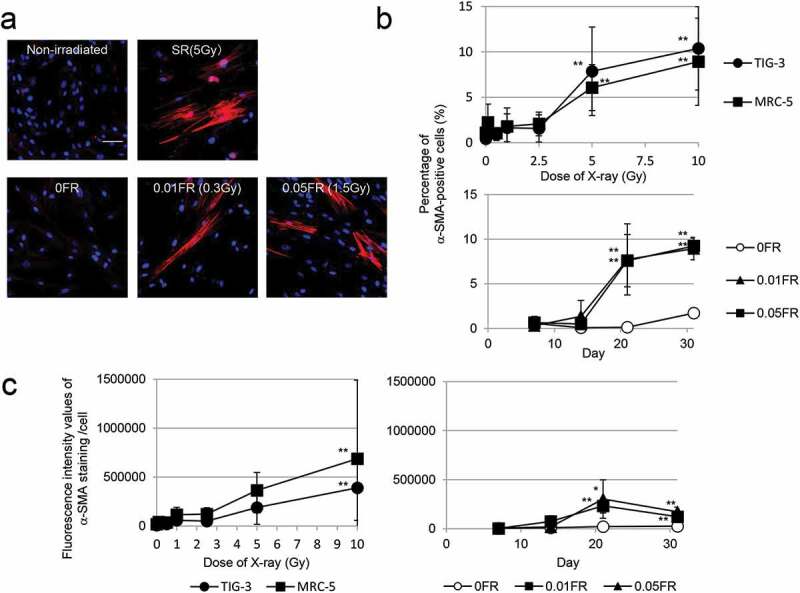
Induction of α-SMA expression
Images of α-SMA staining in the indicated cells. (A) FR was administrated to cells for 21 d. Scale bar = 50 µm. (B) The percentage of TIG-3 and MRC-5 cells with α-SMA staining after each irradiation method is shown in the graph. The asterisk indicates a significant increase in the percentage of α-SMA-positive cells compared with nonirradiated control cells. For FR cells, a significant increase in the percentage of α-SMA-positive cells was compared with nonirradiated FR cells at theindicated day. (C) Fluorescence intensity of α-SMA staining was measured after SR (left) or FR (right). The asterisk indicates a significant increase in fluorescence intensity values of α-SMA staining compared with nonirradiated control cells. For FR cells, a significant increase in fluorescence intensity values of α-SMA staining was compared with nonirradiated FR cells at the indicated day.
Relationship between the DNA damage response (DDR) and myofibroblast induction
We next investigated the influence of radiation sensitivity on myofibroblast induction. Inhibitors such as the ATM inhibitor KU55933[15], the DNA-PK inhibitor NU7026 [16,17], and the AKT inhibitor API-2 [13,18] were used to impede the DDR. Preradiation treatment with all inhibitors enhanced the effect of radiation on the growth retardation of TIG-3 cells (Figure 2(A,B)). ROS levels were measured by staining with 2ʹ,7ʹ-dichlorofluorescin diacetate (DCFDA), a fluorescent probe oxidized by ROS, using FACScan (Becton Dickinson, Mountain View, CA, USA). This staining showed that treatment with KU55933 prevented an increase in ROS levels following radiation exposure. In contrast, ROS levels were significantly high when cells were pretreated with both NU7026 and API-2 and irradiated at 2.5 Gy compared with cells exposed to radiation alone (Figure 2(C)). Under these circumstances, KU55933 also completely eliminated the induction of α-SMA-positive fibers after irradiation, while α-SMA-fiber-positive myofibroblasts appeared at lower doses (below 2.5 Gy) in cells pretreated with NU7026 or API-2 (Figure 2(D)). Thus, both inhibitors lowered the threshold for myofibroblast induction. We further quantified the α-SMA expression levels by western blotting. α-SMA expression was increased in TIG-3 cells after exposure to high doses of radiation (≥ 5 Gy). α-SMA expression did not change after irradiation plus KU55933 treatment. In contrast, preradiation treatment with NU7026 or API-2 enhanced α-SMA expression even after low-dose irradiation (Figure 2(E)).
Figure 2.
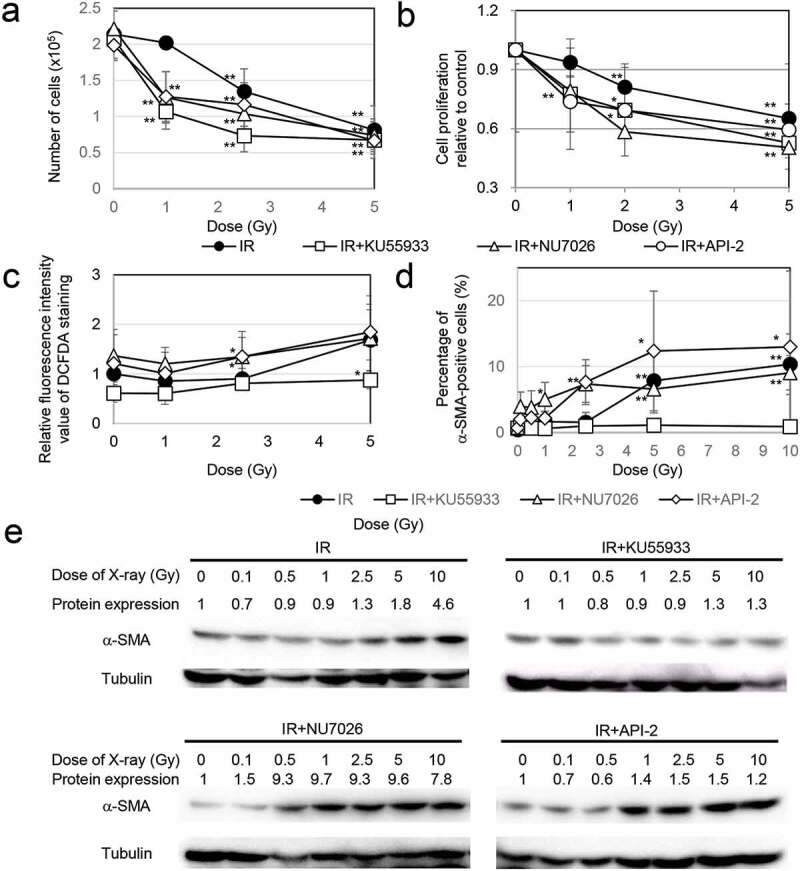
Association of DDR with myofibroblast induction
(A) Effects of inhibitors on cell growth after irradiation. The asterisk indicates a significant decrease in the number of cells compared with nonirradiated control cells. (B) Viability of TIG-3 cells as evaluated by MTT assay. The cell viability of the treatment group was normalized to the value of the nonirradiated group and is shown as the mean value ± SD. The asterisk indicates a significant decrease in cell proliferation rate compared with nonirradiated control cells. (C) The relative fluorescence intensity values of DCFDA staining were normalized to that of the nonirradiated controls. The asterisk indicates a significant increase in fluorescence intensity value of DCFDA staining compared with nonirradiated control cells. (D) The percentage of α-SMA-positive TIG-3 cells in each treatment group is shown in the graph on the right. The asterisk indicates a significant increase in the percentage of α-SMA-positive cells compared with nonirradiated control cells. (E) Western blotting for α-SMA among the indicated treatment groups. The amounts of α-SMA were normalized by the corresponding tubulin level. The values are expressed relative to the untreated control value.
The role of cell growth on myofibroblast induction after irradiation
To clarify the mechanism of action of radiation on myofibroblast clearance, growth inhibition was achieved by removal of FCS (0% FCS) from the culture medium or by treatment with a medium containing a low concentration of FCS (0.5% FCS) 24 h before irradiation (Figure 3(A)). Blocking cell proliferation compared with nonirradiated control cells was also observed at high doses of radiation (≥5 Gy) in standard medium (Figure 3(A)). In contrast to what was seen when cells were cultured in standard medium, α-SMA-fiber-positive myofibroblasts were observed after exposure of TIG-3 cells to low and moderate doses of radiation (0.1 Gy–2.5 Gy) under nongrowth conditions (Figure 3(B)).
Figure 3.
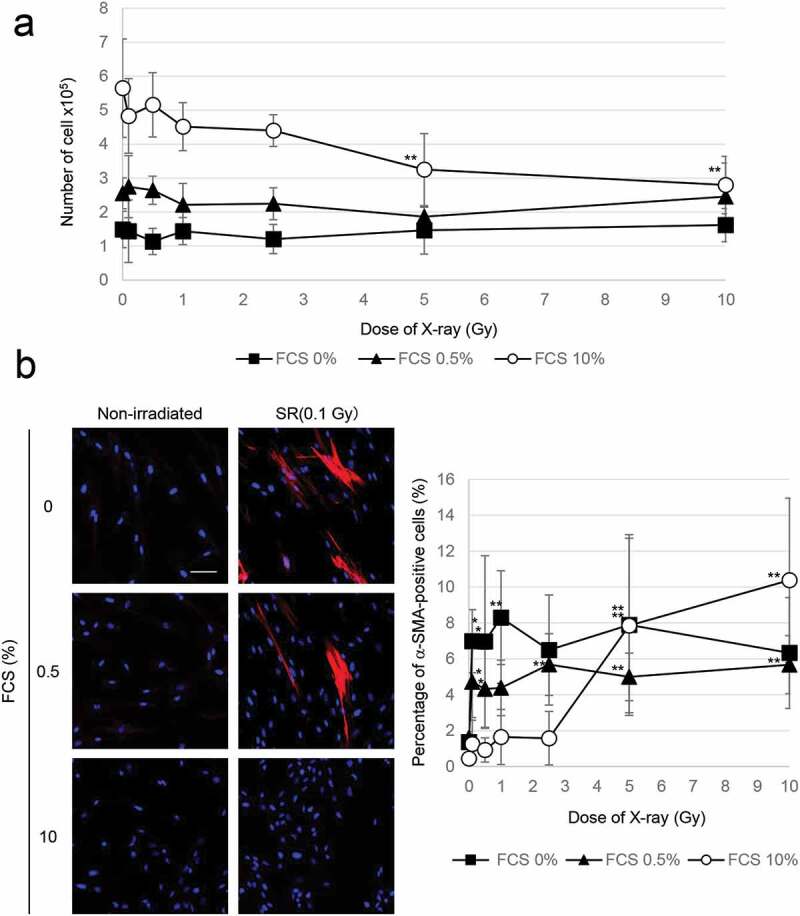
Growth inhibition enhanced myofibroblast induction
(A) Cell growth after irradiation at the indicated doses under different culture conditions. The asterisk indicates a significant decrease in the number of cells compared with nonirradiated control cells. (B) Images of α-SMA staining in the indicated cells. Scale bar = 50 µm. The percentage of α-SMA-positive TIG-3 cells in different culture conditions is shown in the graph on the right. The asterisk indicates a significant increase in the percentage of α-SMA-positive cells compared with nonirradiated control cells.
It was reported that mitochondrial ROS trigger myofibroblast induction via activation of TGF-β signaling and downstream α-SMA expression in irradiated cells. [9] We examined the effect of growth inhibition on the generation of ROS by staining with DCFDA following radiation. Exposure to ≥5 Gy of SR increased ROS levels under growth conditions (Figure 4). In contrast, ROS appeared by exposure to irradiation with any doses even at low doses in the growth-restricted conditions in which cells were cultured in 0.5% or 0% FCS. These results indicated that residual ROS under the growth-restricted conditions is associated with myofibroblast induction in cells irradiated at a low dose.
Figure 4.
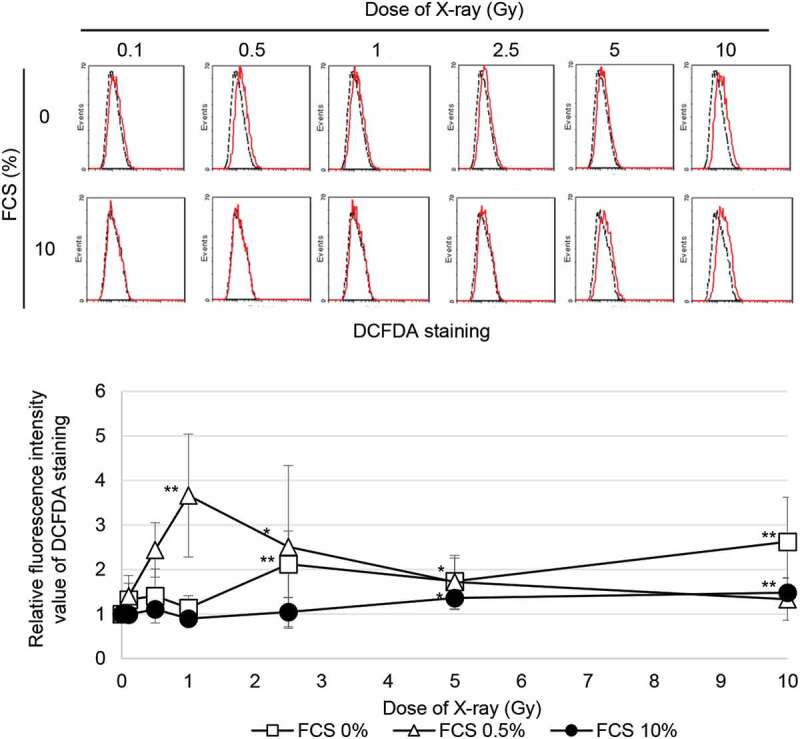
ROS generation FACS results for DCFDA staining 24 h after SR in TIG-3 cells that were either nonirradiated (dotted line) or irradiated (solid line) at the indicated doses. The relative fluorescence intensity values of DCFDA staining were normalized to nonirradiated controls. Asterisk indicates a significant increase in fluorescence intensity value of DCFDA staining compared with nonirradiated control cells
EC and ACA prevent myofibroblast induction by scavenging ROS
A previous report indicated that epicatechin (EC) and ascorbic acid (ACA) are immediately effective in the prevention of mitochondrial ROS-mediated oxidative stress in human fibroblasts and in vivo in mouse platelets after irradiation. [19] Here, we investigated the role of EC and ACA in radiation-induced myofibroblast activation. Preradiation and postradiation treatment with EC or ACA prevented radiation-induced growth retardation in TIG-3 cells (Figure 5(A)). Statistical comparisons revealed that the treatment of irradiated TIG-3 cells with EC or ACA suppressed the persistent accumulation of ROS (Figure 5(B)) and the induction of α-SMA-positive fibers (Figure 5(C)).
Figure 5.
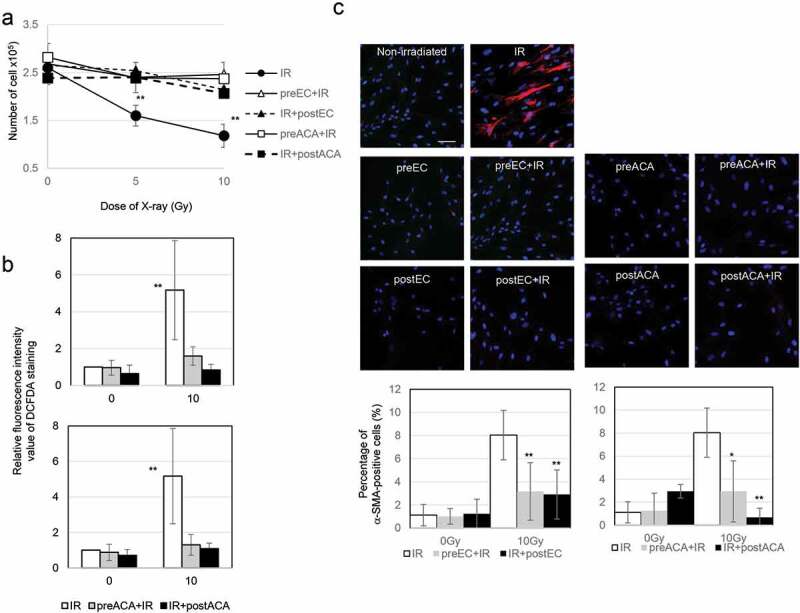
The role of EC and ACA on cell growth, ROS production, and α-SMA staining pattern after irradiation
(A) Preradiation and postradiation treatment with EC and ACA protects against radiation-induced growth retardation. The asterisk indicates a significant decrease in the number of cells compared with nonirradiated control cells. (B) The relative fluorescence intensity values of DCFDA staining were normalized to those of nonirradiated controls. The asterisk indicates a significant increase in fluorescence intensity value of DCFDA staining compared with nonirradiated control cells. (C) Images of α-SMA staining in the indicated cells. Scale bar = 50 µm. The percentage of α-SMA-positive TIG-3 and MRC-5 cells for each irradiation method is shown in the graph. The asterisk indicates a significant increase in the percentage of α-SMA-positive cells compared with nonirradiated control cells.
Discussion
Radiation risks from medical radiation exposure encompass an important issue in terms of the health effects of radiation on humans, especially in children, because a longer post-exposure life expectancy increases the probability of radiation-induced malignancies. [20] Therefore, understanding the mechanistic insights associated with radiation-related carcinogenesis are essential to assess radiation effects in normal tissues. We previously reported that radiation-induced myofibroblasts promote tumor growth through angiogenesis and the release of growth factors. [9] Here, we investigated the mechanism of radiation-induced myofibroblasts which have an abnormal, flat, large morphology with α-SMA-positive fibers. FR is clinically relevant and that this form of irradiation exposure is at least equivalent to high-dose SR regarding α-SMA over time. Myofibroblast differentiation is associated with mitochondrial oxidative stress triggered by radiation exposure. [9] As a byproduct, superoxide molecules leak during mitochondrial oxidative phosphorylation (OXPHOS) and then consume intracellular antioxidant glutathione (GSH). [21]
We previously reported that ROS transiently accumulates in human fibroblasts following SR. [13,15] Upon SR with 1 Gy or 2 Gy, ROS increase from 3 h to 10 h compared with those of nonirradiated controls, and then restored to nonirradiated control levels at 24 h. In contrast, ROS persists at least 24 h after FR[13]. ROS levels were unchanged between nonirradiated cells and FR cells until 14 d, while strong ROS appeared when cells were exposed to FR for over 21 d and were further increased at 31 d. [13] Repeated radiation exposure by FR decreases the GSH levels and leads to the continuous generation of ROS via OXPHOS, which in turn, leads to the accumulation of mitochondrial ROS. [13] ROS accumulation stimulates TGF-β signaling including α-SMA expression, which then stimulates differentiation of myofibroblasts from normal fibroblasts. Therefore, FR showed a lower threshold for the acquisition of myofibroblasts compared with acute SR.
Myofibroblasts are an activated form of fibroblasts that play important roles in tissue repair and regeneration. [22] Figure 6 shows a model of myofibroblast turnover and myofibroblast persistence following irradiation. Myofibroblasts are responsible for excessive ECM production, which leads to fibrosis. [7] Once healing is complete, the number of activated fibroblasts decreases, and the resting phenotype is restored. However, high doses of radiation induce irreversible growth arrest and radiation-induced myofibroblasts remain. Similarly, radiation-induced myofibroblasts appeared in the growth-restricted conditions even at low-doses. These results indicated that blocking cell growth is associated with myofibroblast persistence.
Figure 6.
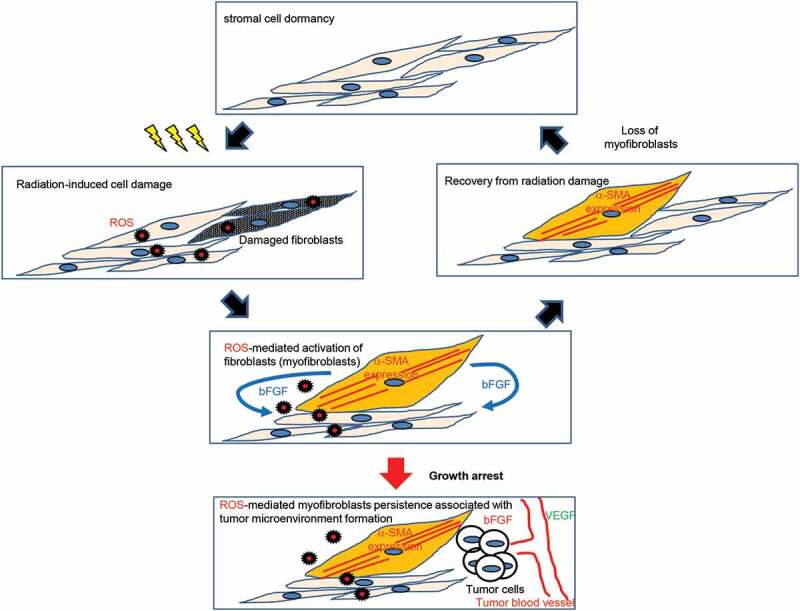
Schematic representation of myofibroblast clearance and persistence following radiation
The ATM inhibitor KU55933 enhanced radiation sensitivity but abrogated the accumulation of ROS and myofibroblast differentiation following radiation. ATM is crucial for crosstalk between nuclear DNA damage and mitochondria in irradiated cells. ATM loss eliminates the effects of radiation on mitochondria, such as ΔΨm elevation, ROS increase, Parkin focus formation, [23] and subsequent mitochondrial ROS-mediated myofibroblast induction. On the contrary, enhanced radiation sensitivity by treatment with NU7026 or API-2 facilitated myofibroblast differentiation. NU7026 or API-2 treatments abrogate DNA repair or cell survival responses in irradiated cells. Damaged DNA sends signals to mitochondria to activate mitochondrial OXPHOS and subsequent generation of ROS facilitates myofibroblast induction.
Alterations in fibroblasts play a key role in radiation-related tumorigenesis. [5,8] Myofibroblasts are likely to be associated with tumor microenvironment formation by releasing tumor growth factor and VEGF for tumor cell growth and angiogenesis [9]. Suppression of myofibroblast induction following radiation may be useful to mitigate the harmful late radiation effects of cancer. We previously reported on the effects of the antioxidant, N-acetyl-cysteine (NAC), a GSH precursor, which can increase intracellular GSH levels[13]. Treatment of long-term FR cells with NAC during FR exposures suppressed the induction of α-SMA in TIG-3 and MRC-5 cells[9]. Here, we demonstrated that preradiation and postradiation treatment with EC and ACA can suppress the generation of ROS and myofibroblast induction. Therefore, these antioxidants are effective in protecting normal cells from radiation by suppressing myofibroblast induction.
In conclusion, myofibroblasts were preferentially induced following FR rather than SR. Growth recovery contributes to myofibroblast clearance. Since ROS causes radiation-induced myofibroblast activation, antioxidants, epicatechin, and ascorbic acid were effective in the suppression of radiation-induced myofibroblast activation by scavenging ROS.
Materials and methods
Cell culture conditions and drugs
Normal human diploid lung fibroblasts (MRC-5 and TIG-3) were purchased from the Health Science Research Resources Bank and were cultured in minimum essential medium (Nacalai Tesque, Kyoto, Japan) supplemented with 10% heat-inactivated fetal calf serum (FCS). In some experiments, cell growth was inhibited by the removal of FCS or by lowering the FCS concentration in the culture media. KU55933 and NU7016 were purchased from Sigma (San Diego, CA, USA). API-2 was purchased from Millipore. These inhibitors were added to the media 2 h before irradiation. Epicatechin (EC) and ascorbic acid (ACA) were purchased from Wako Junyaku KK (Osaka, Japan) and Sigma, respectively. These agents (0.1 mM) were added to the culture medium 2 h before (preradiation treatment) or 2 h after (postradiation treatment) radiation.
Irradiation experiments
Cells were irradiated at the indicated doses at a dose rate of 0.7 Gy/min using a 150-kVp X-ray generator (Model MBR-1505R2, Hitachi) with 0.5-mm Cu and 0.1-mm Al filters. For fractionated radiation (FR), X-ray fractions (0.01 or 0.05 Gy) were administered twice a day for 5 d/wk. Cells were not irradiated on Saturday and Sunday. The total doses delivered over 21 d were 0.3 (=0.01 Gy/fraction × 2 times × 5 d × 3 wk) and 1.5 Gy (=0.05 Gy/fraction × 2 times × 5 d × 3 wk) for cells exposed to FR of 0.01 and 0.05 Gy, respectively. When cells reached 80% confluency, they were subcultured in a new flask.
Immunofluorescence
Immunofluorescence staining was performed as previously described.[14,18] Briefly, the cells were seeded onto 18 × 18-mm cover slips, placed in 35-mm tissue culture dishes, and cultured overnight. The cells on cover slips were fixed in 4% formaldehyde for 10 min and permeabilized with 0.5% Triton X-100 for 5 min. An antibody against α-SMA (A2547, Sigma) and secondary antibodies conjugated to Alexa Fluor 488 (A11034; Molecular Probes, Eugene, OR, USA) or Alexa Fluor 647 (A21236; Molecular Probes, Eugene, OR, USA) were used. Cells were counterstained with Hoechst 33,258 [4 µg/mL in 50% glycerol/PBS(−)] to visualize the DNA. Images were captured using a charge-coupled device camera attached to a fluorescence microscope (Keyence Corporation, Osaka, Japan). For each data point, >50 cells were manually counted from at least three independent samples. Image acquisition and evaluation were conducted with a Keyence BZ-X700 fluorescence microscope and Hybrid Cell Count software (BZ-II Analyzer; Keyence Corporation).
Western blotting
Western blotting was performed as previously described. [24] Cell extracts were collected 24 h after IR. Primary antibodies against α-SMA (A2547, Sigma) and β-tubulin (10,068-1-AP, Proteintech) as well as secondary goat anti-mouse (R&D Systems, Minneapolis, USA) and anti-rabbit antibodies conjugated to horseradish peroxidase (GE Healthcare, Little Chalfont, UK) were used. Protein bands were visualized using Chemi-Lumi One L Western blotting substrate (Nacalai Tesque), and band intensities were measured using Image Lab software (Bio-Rad). α-SMA expression levels were normalized to β-tubulin. The values are expressed relative to the control value of nonirradiated cells.
MTT assay
Cell proliferation was examined using a Cell Counting Kit-8 assay (Dojindo, Kumamoto Japan) according to the manufacturer’s protocol.
ROS detection
Cells were stained with 5 μM DCFDA (Sigma) in a serum-free minimum essential medium for 30 min. DCFDA-stained cells were detected with a FACScan automated flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
Statistical analysis
Error bars represent standard deviations. All experiments were repeated at least three times using independent samples. Following one-way ANOVA, the Dunnett test was used to detect significant differences among the means of three or more independent groups. Double and single asterisks indicate significant differences with p values of <0.01 and <0.05, respectively. Asterisks indicate the significant difference in irradiated cells compared with nonirradiated cells among the indicated treatment groups.
Acknowledgments
We thank Drs. Naoki Kunugita, Kazuyuki Ishi and Kenji Hattori for their support on this study. Sources of support: This research was supported by a grant from the JSPS KAKENHI Grant Number 18H03377, Research on Health effects of radiation organized by the Japanese Ministry of the Environment, Industrial Disease Clinical Research Grants from the Japanese Ministry of Health, Labour, and Welfare and in part by NIFS Collaborative Research Program (NIFS13KOBA028). This work was performed at the Joint Usage/Research Center (Radiation Biology Center), Kyoto University, and the Program of the network-type joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University, and Fukushima Medical University.
Funding Statement
This work was supported by the Japan Society for the Promotion of Science [18H03377].
Disclosure statement
No potential conflicts of interest were disclosed.
References
- [1].Little JB. Radiation carcinogenesis. Carcinogenesis. 2000;21(3):397–404. [DOI] [PubMed] [Google Scholar]
- [2].Hsu WL, Preston DL, Soda M, et al. The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950-2001. Radiat Res. 2013;179:361–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ozasa K, Shimizu Y, Suyama A, et al. Studies of the mortality of atomic bomb survivors, Report 14, 1950-2003: an overview of cancer and noncancer diseases. Radiat Res. 2012;177:229–243. [DOI] [PubMed] [Google Scholar]
- [4].Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–598. [DOI] [PubMed] [Google Scholar]
- [5].Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- [6].Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010;15:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. [DOI] [PubMed] [Google Scholar]
- [8].Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. [DOI] [PubMed] [Google Scholar]
- [9].Shimura T, Sasatani M, Kawai H, et al. Radiation-Induced Myofibroblasts Promote Tumor Growth via Mitochondrial ROS-Activated TGFbeta Signaling. Mol Cancer Res. 2018;16:1676–1686. [DOI] [PubMed] [Google Scholar]
- [10].Samarakoon R, Overstreet JM, Higgins PJ. TGF-beta signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pohlers D, Brenmoehl J, Loffler I, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–756. [DOI] [PubMed] [Google Scholar]
- [12].Anscher MS. Targeting the TGF-beta1 pathway to prevent normal tissue injury after cancer therapy. Oncologist. 2010;15:350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shimura T, Sasatani M, Kamiya K, et al. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget. 2016;7:3559–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shimura K, Miyazawa T, Hanaoka T, et al. Fischer-Tropsch synthesis over alumina supported cobalt catalyst: effect of crystal phase and pore structure of alumina support. J Mol Catal A Chem. 2014;394:22–32. [Google Scholar]
- [15].Shimura T, Sasatani M, Kawai H, et al. ATM-mediated mitochondrial damage response triggered by nuclear DNA damage in normal human lung fibroblasts. Cell Cycle. 2017;16:2345–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shimura T, Martin MM, Torres MJ, et al. DNA-PK is involved in repairing a transient surge of DNA breaks induced by deceleration of DNA replication. J Mol Biol. 2007;367:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shimura T, Ochiai Y, Noma N, et al. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle. 2013;12:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shimura T, Toyoshima M, Adiga SK, et al. Suppression of replication fork progression in low-dose-specic p53-dependent S-phase DNA damage checkpoint. Oncogene. 2006;25:5921–5932. [DOI] [PubMed] [Google Scholar]
- [19].Shimura T, Koyama M, Aono D, et al. Epicatechin as a promising agent to countermeasure radiation exposure by mitigating mitochondrial damage in human fibroblasts and mouse hematopoietic cells. Faseb J. 2019;33:6867–6876. [DOI] [PubMed] [Google Scholar]
- [20].Kutanzi KR, Lumen A, Koturbash I, et al. Pediatric Exposures to Ionizing Radiation: carcinogenic Considerations. Int J Environ Res Public Health. 2016;13:1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shimura T, Kunugita N. Mitochondrial reactive oxygen species-mediated genomic instability in low-dose irradiated human cells through nuclear retention of cyclin D1. Cell Cycle (Georgetown, Tex). 2016;15:1410–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–289. [DOI] [PubMed] [Google Scholar]
- [23].Shimura T, Kobayashi J, Komatsu K, et al. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle (Georgetown, Tex). 2016;15:1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shimura T, Kakuda S, Ochiai Y, et al. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3 beta-mediated cyclin D1 overexpression. Oncogene. 2010;29:4826–4837. [DOI] [PubMed] [Google Scholar]