

Abstract
The bacterial strain PO100/5 was isolated from a skin abscess taken from a pig (Sus scrofa domesticus) in the Alentejo region of southern Portugal. It was identified as Corynebacterium pseudotuberculosis using biochemical tests, multiplex PCR and Pulsed Field Gel Electrophoresis. After genome sequencing and rpoB phylogeny, the strain was classified as C. ulcerans. To better understand the taxonomy of this strain and improve identification methods, we compared strain PO100/5 to other publicly available genomes from C. diphtheriae group. Taxonomic analysis reclassified it and three others strains as the recently described C. silvaticum, which have been isolated from wild boar and roe deer in Germany and Austria. The results showed that PO100/5 is the first sequenced genome of a C. silvaticum strain from livestock and a different geographical region, has the unique sequence type ST709, and could be could produce the diphtheriae toxin, along with strain 05–13. Genomic analysis of PO100/5 showed four prophages, and eight conserved genomic islands in comparison to C. ulcerans. Pangenome analysis of 38 C. silvaticum and 76 C. ulcerans genomes suggested that C. silvaticum is a genetically homogeneous species, with 73.6% of its genes conserved and a pangenome near to be closed (α > 0.952). There are 172 genes that are unique to C. silvaticum in comparison to C. ulcerans. Most of these conserved genes are related to nutrient uptake and metabolism, prophages or immunity against them, and could be genetic markers for species identification. Strains PO100/5 (livestock) and KL0182T (wild boar) were predicted to be potential human pathogens. This information may be useful for identification and surveillance of this pathogen.
Introduction
The genus Corynebacterium from the phylum Actinobacteria has Gram-positive bacteria of biotechnological, veterinary and medical relevance with free, commensal and pathogenic lifestyles. Within pathogenic species, the most prominent species are the nearly exclusively human pathogen C. diphtheriae and the zoonotic C. pseudotuberculosis and C. ulcerans. These three compose the C. diphtheriae group, a clade of species that can be lysogenized by phages harboring the diphtheria toxin (DT) gene (tox) [1]. Within this group, three new species were recently described. C. rouxii [2] and C. belfantii are reclassifications of some of the C. diphtheriae biovar Belfanti strains [3]. C. belfantii is also a synonym of C. diphtheriae subspecies lausannense [2]. C. silvaticum [4] is a reclassification of atypical C. ulcerans strains. Strains of C. silvaticum were previously described as atypical non-toxigenic but tox-gene-bearing (NTTB) strains of C. ulcerans, isolated from wild boars and roe deer in Germany and Austria, which caused caseous lymphadenitis similar to C. pseudotuberculosis infections [5–8]. This variant, examined using genomics and proteomics, was initially named as the “wild boar cluster” (WBC) of C. ulcerans [5–7] and later reclassified as C. silvaticum [4].
The strain PO100/5 was isolated from caseous lymphadenitis lesions in a Black Alentejano pig (Sus scrofa domesticus) from a swine farm in the Alentejo region of Portugal. It was identified as Corynebacterium pseudotuberculosis by both biochemical tests (Api Coryne® kit) and by multiplex PCR and Pulsed Field Gel Electrophoresis [9]. Genome sequencing and rpoB phylogeny showed that this strain was closer to C. ulcerans and the genome was deposited in GenBank as a strain within this species (accession number CP021417.1). Recently the description of C. silvaticum was published and PO100/5 was suggested to be a strain of this species by rpoB phylogeny [4], while a genomic analysis of 28 C. ulcerans strains suggested that PO100/5, W25 and KL1196 could represent a new species [10]. KL1196 had already been classified as C. silvaticum [4].
Pigs and boars are reservoirs of C. silvaticum [4–7] and C. ulcerans [11–13], and are known to transmit pathogens to humans and other domestic animals [11, 12, 14]. Rapid, simple and reliable identification of this new species is essential for diagnosis, treatment and surveillance [15, 16]. To better understand the taxonomy of PO100/5, we performed a comparative analysis of 34 C. silvaticum and 80 C. ulcerans genomes, as well as other publicly available genomes from the C. diphtheriae group in order to explore the genomic diversity of C. silvaticum and to identify molecular markers of this species. We have reclassified PO100/5, established the other three strains recently deposited as C. silvaticum, and found both a unique sequence type and genes that can be useful for species classification.
Materials and methods
Genomes, assembly, and annotation
For the taxonomic, phylogenetic and genome plasticity analyses, a total of 120 genomes (S1 File) were selected, including 80 C. ulcerans, 34 C. silvaticum strains and six type strains from the C. diphtheriae group. Assembled genomes were retrieved from the Pathosystems Resource Integration Center (PATRIC) [17], while genomes available as sequencing reads were assembled in PATRIC using the SPAdes [18] strategy. All genomes were annotated using the Rapid Annotation using Subsystems Technology (RASTtk) pipeline [19] that is available in PATRIC.
Taxonomic analysis
Average Nucleotide Identity (ANI) was estimated using FastANI v1.3 [20]. An automatic genome-based taxonomic analysis was performed using the Type (Strain) Genome Server (TYGS) (https://tygs.dsmz.de) [21]. This pipeline first identifies the closest type strains using MASH [22] for genomic sequences and BLAST [23] for 16S rRNA sequences. It then identifies the 10 closest type strains using Genome Blast Distance Phylogeny (GBDP) [24]. Finally, it clusters species and subspecies using digital DNA:DNA hybridization (dDDH) with a formula that is independent of genome length, being robust against the use of incomplete genomes (formula d4) [24]. It uses a threshold of 70 and 79%, respectively [25]. The difference in G+C content is also evaluated and expected to vary no more than 1% within a species [26]. Those analysis were performed for C. ulcerans strains from GenBank, using either the assembled genomes or sequencing data to check for misidentification of C. silvaticum strains.
Phylogenetic trees of the rpoB and tox genes were built using the Maximum Likelihood method [27] implemented in MEGA v10.1.6 [28]. The tox tree included all sequences from the genomes of C. ulcerans and included outgroups from C. silvaticum and C. pseudotuberculosis. C. rouxii was not included due to all sequenced genomes being tox- [2]. All trees were visualized using iTOL [29].
Multi Locus Sequence Typing (MLST) was performed using MLSTcheck [30], using the scheme for C. diphtheriae and C. ulcerans (genes atpA, dnaE, dnaK, fusA, leuA, odhA and rpoB) [13]. The Minimum Spanning Tree (MST) generated using goeBURST Full MSLT algorithm was built using PHYLOViZ v2.0 [31].
Genome plasticity analysis
Prophages of PO100/5 were predicted using PHASTER [32]. Genomic islands were predicted using GIPSy v1.1.2 [33], with C. ulcerans NCTC7910T and C. pseudotuberculosis ATCC19410T used as references. A circular map was generated using BRIG v0.95 [34]. The presence of niche and virulence factors of Corynebacterium [35, 36] was verified using PATRIC’s Protein Family Sorter and Proteome Comparison tools. Gene neighborhoods were compared with other strains using the Artemis Comparison Tool 17.0.1 [37]. Signal peptide and conserved protein domains were verified using InterProScan [38], while cell wall sorting signal (CWSS) was verified using CW-PRED [39]. Mapping of sequencing reads to sequences of interest was performed using CLC Genomics Workbench v7 [40]. Specific nucleotide sequences in other genomes were identified using BLASTn [41] and GenBank non-redundant (nr) database [42].
The identification of groups of homologous genes (orthogroups) was performed using OrthoFinder v2.3.12 [43]. The output files Orthogroups.tsv and Orthogroups_UnassignedGenes.tsv from OrthoFinder were used as input for pangenome analyses using in-house scripts. The pangenome was represented by all orthogroups and the core genome by orthogroups conserved across all (100%) genomes. The accessory (or dispensable) genome was represented by the genes not conserved across all genomes. Within this subset, singletons were exclusive to a single genome, and shared genome (or dispensable genome minus singletons) are shared between two or more, but not all genomes [44]. To develop molecular markers, we identified subsets of orthogroups conserved and exclusive to a group of genomes (exclusive core). The development of a pangenome was calculated according to Heaps’ law fit formula n = κ*Nγ, in which n is the number of genes, N is the number of genomes, and κ and γ (α = γ -1) are free parameters determined empirically. Heap’s law establishes the pangenome as being closed when α > 1 (γ < 0), which means that there is no significant increase with the addition of new genomes. It also defines a pangenome as open when α ≤ 1 (0 < γ < 1). The development of core genome and singletons were calculated using least-squares fit of the exponential regression decay n = κ*exp[-N/τ] + tg(θ), in which n is the number of genes, N is the number of genomes, and κ, τ, and tg(θ) are free parameters determined empirically [44]. A functional annotation of genes was performed using the eggNOG-mapper v2 [45].
The pathogenicity of C. silvaticum to humans was predicted using PathogenFinder v. 1.1 [46]. The prediction is performed using CD-HIT-2D [47] against a database of protein families associated with human pathogens. Strains PO100/5 and KL0182T were used to represent livestock (domestic pig) and wild boars isolates, respectively.
Results
Taxonomic analysis
ANI results showed that C. ulcerans strains PO100/5, 04–13, 05–13 and W25 had identity values ≥ 99.74% with C. silvaticum KL0182T, and ≤ 91.03% with C. ulcerans NCTC7910T (S2 File). The taxonomic classification using TYGS classified those strains as C. silvaticum due to genome and 16S rRNA GBDP trees, dDDh > 70% and G+C content difference > 1% with C. ulcerans genomes (S3 to S8 Files). In the rpoB phylogeny, the C. ulcerans strains PO100/5, 04–13, 05–13 and W25 clustered with C. silvaticum KL0182T, while other C. ulcerans strains formed two clades (Fig 1). In the phylogenetic tree of tox gene, the same four strains (PO100/5, 04–13, 05–13 and W25) also clustered with C. silvaticum, and were distinct from the C. ulcerans, C. diphtheriae and C. pseudotuberculosis clusters (Fig 2). MLST analysis classified strains 04–13, 05–13 and W25 as ST578 (53-60-121-70-76-66-57) and identified PO100/5 as having a unique and new sequence type, ST709 (53-60-121-70-76-82-57) where it differed from ST578 in the locus odhA. Due to those results those four strains were reclassified for the next analyses, changing the number of C. silvaticum genomes from 34 to 38 and C. ulcerans genomes from 80 to 76. Three new STs were identified, ST710 and ST711 in C. ulcerans lineage 1 and ST712 in lineage 2 (S9 File, Fig 3).
Fig 1. Phylogenetic tree of rpoB gene from Corynebacterium species.

The phylogeny was inferred using the Maximum Likelihood method and the Tamura-Nei (TN93 + G) model implemented in the Mega v10.1.6. The Corynebacterium ulcerans strains PO100/5, 04–13, W25 and 05–13 cluster with the Corynebacterium silvaticum KL0182T (yellow). C. ulcerans strains form lineage 1 (red, collapsed) and lineage 2 (orange).
Fig 2. Phylogenetic tree of the tox gene from Corynebacterium species.

The phylogeny was inferred using the Maximum Likelihood method and the Tamura-Nei (T92 + G) model implemented in Mega v10.1.6. The strains PO100/5, 04–13, W25 and 05–13 cluster with Corynebacterium silvaticum. Clade colors represent rpoB clades of C. silvaticum (yellow) and C. ulcerans (red and orange).
Fig 3. goeBURST diagram for the MLST data set of 38 Corynebacterium silvaticum and 76 C. ulcerans strains generated using PHILOViZ 2.0.
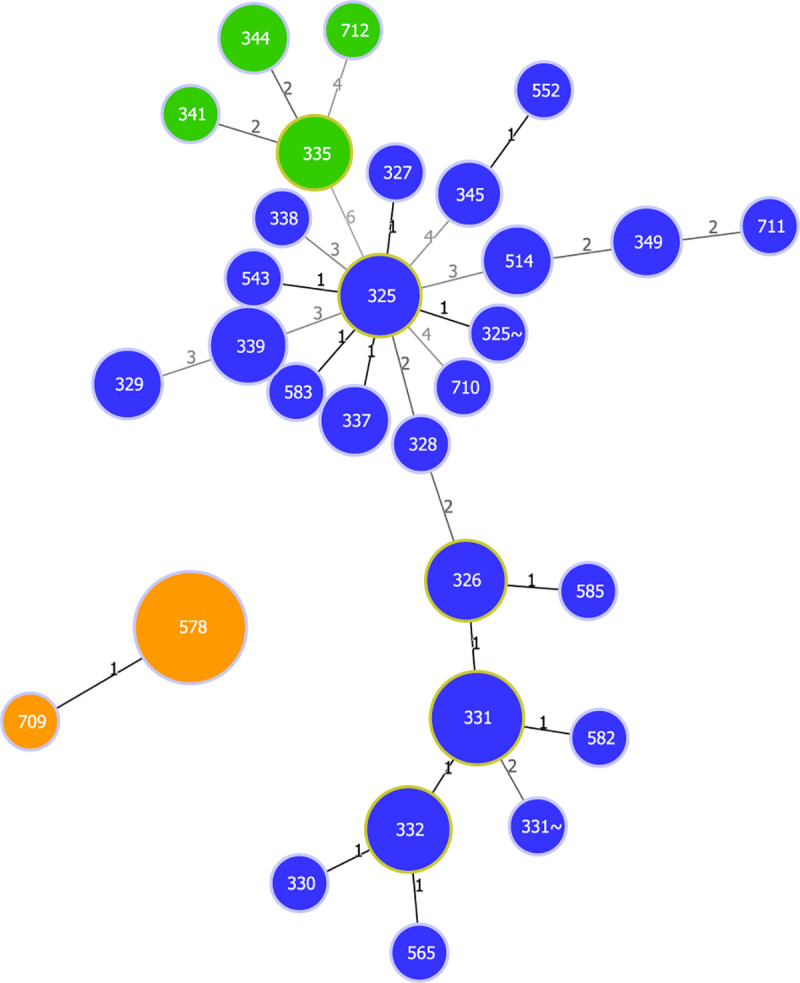
Blue–C. ulcerans lineage 1. Green—C. ulcerans lineage 2. Orange–C. silvaticum. The numbers on the links indicate the number of divergent alleles between STs.
The taxonomic analyses led to additional insights. TYGS classified nine C. ulcerans strains (03–8664, 04–7514, 131002, FRC11, KL0349, LSPQ-04227, LSPQ-04228, NCTC8666 and NCTC12077) as being part of a potential new species. These genomes had dDDH values greater than 70% (99.8–75.9%) within them and less than 70% (63 to 67.2%) with other C. ulcerans genomes, although the difference in the G+C content was less than 1% (S3 File). In ANI analysis, those nine genomes were more similar to each other than to the other genomes. They had values between 95.52 and 96.57% with C. ulcerans NCTC7910T, and ≥ 97.82% when one of them (NCTC12077) was used as reference for the other eight (S2 File). MLST analysis classified them as having the unique sequence types ST335, ST341, ST344 and the new ST ST712 (Fig 3, S9 File). The ANI analysis showed 99.3% identity between C. diphtheriae lausannense strains CHUV2995 and C. belfantii FRC0043T (S2 File). A further analysis using TYGS classified C. diphtheriae lausannense strains CHUV2995T and CMCNS703 as belonging to C. belfantii, and as C. diphtheriae the non-type strains with genomes deposited in GenBank as C. belfantii (https://www.ncbi.nlm.nih.gov/genome/78252/) (S10 File). For this reason, C. belfantii 2937 was renamed to C. diphtheriae 2937 in Fig 2.
Genome plasticity analysis
The presence of genes encoding 16 niche and virulence factors described in the genus Corynebacterium [35, 36] were examined in C. silvaticum (Table 1). The genes rhuM, rpb and tspA were not found, and all the pilus genes, except spaB, were found to be pseudogenized, lacking the signal peptide or CWSS. C. silvaticum has the two pilus gene clusters structured as srtA, spaBC, and srtB, spaD, srtC and spaEF, despite fragmentation of pilin genes. Only eight genomes had the tox gene (04–13, 05–13, KL0182, KL0884, KL0957, KL1196, PO100/5 and W25). PO100/5 and 05–13 do not have a two bases insertion (GG) after position 44, in a homopolymer of four guanines, that introduces a frameshift (S1 Fig). The mapping of PO100/5 sequencing reads to its assembled genome, showed an insertion of one guanine in the beginning of the homopolymer, in 5% of the reads (S2 Fig). The tspA gene was present in all C. ulcerans strains, but rpb was only found in strain 809, and rhuM was found in the 16 strains from Austria, France and Germany that had been isolated from humans, cats and dogs (02–13, FRC58, KL0195, KL0246-cb3, KL0251-cb4, KL0252-cb5, KL0349, KL0387-cb8, KL0475, KL0497, KL0541, KL0547, KL0796, KL0867, KL0880, NCTC12077).
Table 1. Presence of 16 known niche and virulence factors of Corynebacterium in C. silvaticum.
| Gene | Product | Reference locus tag | Reference protein family | C. silvaticum protein family | Manual curation |
|---|---|---|---|---|---|
| endoE | Endoglycosidase E (former corynebacterial protease CP40) | CULC809_01974 | PLF_1716_00006954 | PLF_1716_00006954 | Present |
| cwlH | Cell wall-associated hydrolase | CULC809_01521 | PLF_1716_00062893 | PLF_1716_00062893 | Present |
| nanH | Sialidase (neuraminidase H) | CULC809_00434 | PLF_1716_00002393 | PLF_1716_00002393 | Present |
| pld | Phospholipase D | CULC809_00040 | PLF_1716_00029465 | PLF_1716_00029465 | Present |
| rbp | Shiga-like ribosome-binding protein | CULC809_00177 | PLF_1716_00033486 | - | Absent |
| rhuM | RhuM-like protein | CulFRC58_0285 | PLF_1716_00026137 | - | Absent |
| rpfI | Resuscitation-promoting factor-interacting protein | CULC809_01133 | PLF_1716_00001449 | PLF_1716_00001449 | Present |
| spaB | Surface-anchored protein (minor pilus subunit) | CULC809_01980 | PLF_1716_00010184 | PLF_1716_00010184 | Present |
| spaC | Surface-anchored protein (pilus tip protein) | CULC809_01979 | PLF_1716_00004783 | PLF_1716_00004783 | Pseudogene, no cell wall sorting signal |
| spaD | Surface-anchored protein (major pilus subunit) | CULC809_01952 | PLF_1716_00090862 | PLF_1716_00102654 | Pseudogene, no cell wall sorting signal |
| spaE | Surface-anchored protein (minor pilus subunit) | CULC809_01950 | PLF_1716_00007274 | PLF_1716_00079271 | Pseudogene, no signal peptide |
| spaF | Surface-anchored protein (pilus tip protein) | CULC809_01949 | PLF_1716_00006760 | PLF_1716_00006760 | Pseudogene, no signal peptide |
| tox | Diphtheria toxin | CULC0102_0213 | PLF_1716_00005191 | PLF_1716_00005191 | Present in 8 out of 38 genomes |
| tspA | Trypsin-like serine protease | CULC809_01848 | PLF_1716_00007827 | - | Absent |
| vsp1 | Venom serine protease | CULC809_00509 | PLF_1716_00104602 | PLF_1716_00104343 | 64% identity with C. ulcerans 809 |
| vsp2 | Venom serine protease | CULC809_01964 | PLF_1716_00015799 | PLF_1716_00116381 | 74% identity with C. ulcerans 809 |
| - | C. diphtheriae DIP0733 homolog | CULC22_00609 | PLF_1716_00030114 | PLF_1716_00030114 | Present |
Sixteen and eight genomic islands were predicted by comparing PO100/5 with the reference strains C. pseudotuberculosis ATCC19410T and C. ulcerans NCTC7910T, respectively. No island was detected when it was compared to C. silvaticum KL0182T (Table 2, Fig 4). The genes in the discovered islands are provided in S11 File. They include one complete and three incomplete prophages. Prophage I harbors the tox gene and is similar to Gordonia phage Nyceirae (NC_031004.1) (S11 File, Fig 5). BLASTn of the tox+ prophage sequence using the GenBank nr database identified the best hits as C. ulcerans strains 0102 and 0211, with the same coverage (63%) and identity (92.68%). The best hits with other species were C. diphtheriae lausannense (C. belfantii) CMCNS703 (37 and 85.95%), C. diphtheriae strain B-D-16-78 (41 and 85.86%) and 15 strains of C. pseudotuberculosis (14 and 84.94%). Fig 5 shows the alignment of PO100/5 and C. ulcerans 0102 tox+ prophages.
Table 2. Genomic islands in strain PO100/5 compared to Corynebacterium pseudotuberculosis ATCC19410T and C. ulcerans NCTC7910T.
| n | Position compared to Cp | Size | Position compared to Cul | Size | Type | Prophage content |
|---|---|---|---|---|---|---|
| 1 | 29362–38109 | 8.75 kb | - | - | - | - |
| 2 | 55224–60448 | 5.22 kb | 55224–60448 | 5.22 kb | PA | - |
| 3 | 69256–74863 | 5.6 kb | - | - | PA, RE, SY | - |
| 4 | 97842–103378 | 5.53 kb | - | - | PA, RE | - |
| 5 | 167859–206438 | 38.58 kb | 167859–206209 | 38.35 kb | - | Prophage I |
| 6 | 311533–318567 | 7.03 kb | 311533–318567 | 70.34 kb | - | - |
| 7 | 422293–430839 | 85.46 kb | - | - | RE, SY | - |
| 8 | 694806–746966 | 52.16 kb | 694806–746966 | 52.16 kb | RE | Prophage II |
| 9 | 925047–938225 | 13.18 kb | 925047–938225 | 13.18 kb | - | - |
| 10 | 1235769–1244625 | 8.86 kb | 1235769–1244625 | 8.86 kb | - | - |
| 11 | 1613625–1644953 | 31.33 kb | 1614013–1639302 | 25.29 kb | - | Prophage III |
| 12 | 1793740–1802246 | 8.5 kb | - | - | PA, ME | - |
| 13 | 2029016–2035120 | 6.1 kb | - | - | - | - |
| 14 | 2109299–2139505 | 30.2 kb | 2110317–2139505 | 29.19 kb | - | Prophage IV |
| 15 | 2255307–2261370 | 6.06 kb | - | - | - | - |
| 16 | 2517632–2529863 | 12.23 kb | - | - | ME | - |
Cp–C. pseudotuberculosis, Cul–C. ulcerans, PA–pathogenicity island, RE–resistance island, ME–metabolic island, SY–symbiotic island.
Fig 4. Circular map of Corynebacterium silvaticum genomes generated using BRIG v0.95.

From inner to outer circle: strain PO100/5 (reference); CG content; CG Skew; strains KL0182T, KL0183, KL0259, KL0260, KL0374, KL0382, KL0386, KL0394, KL0395, KL0396, KL0400, KL0401, KL0581, KL0598, KL0615, KL0707, KL0709, KL0773, KL0774, KL0882, KL0883, KL0884, KL0886, KL0887, KL0938, KL0957, KL0968, KL1003, KL1006, KL1007, KL1008, KL1009, KL1010, KL1196, 04–13, 05–13, W25; genomic islands compared to C. pseudotuberculosis ATCC19410T (blue) and C. ulcerans strain NCTC7910T (grey); and prophages (black). Genomic islands (GI) and prophage detection were performed using GIPSy and PHASTER, respectively.
Fig 5. Alignment of tox+ prophages form Corynebacterium silvaticum PO100/5 and C. ulcerans 0102.

The red lines connect sequences with at least 75% identity. Light blue–Phage integrase; Orange–Phage-related transcriptional regulator; Pink–Phage-related coding sequence (CDS); Yellow–Diphtheria toxin CDS; Dark blue–other CDS; Green–tRNA.
For the pangenome analysis, the number of orthogroups in each subset is shown in Table 3 and S12 File for C. silvaticum and C. ulcerans. The core genome represented 73.6% and 40% of orthogroups for C. silvaticum and C. ulcerans, respectively. The pangenome, core genome and singletons development graphs and formulas are shown in Fig 6. Both species had genes conserved in all strains that were absent in the other species, or the exclusive core. In C. silvaticum, 172 orthogroups were detected in this subset. They are represented in strain PO100/5 by 174 proteins, 81 of which are located across genomic islands 1, 2, 5, 6, 8, 9, 10, 11, 12 and 14. C. ulcerans lineage 2 had a hypothetical protein with 37 amino acids (S12 File). A graph comparing the distribution of Cluster of Homologous Groups (COG) categories of the exclusive core genome of both species is shown in Fig 7.
Table 3. Number of genes (orthogroups) in subsets across Corynebacterium silvaticum and C. ulcerans genomes.
| Species | Genomes | Pangenome | Core genome | Accessory genome | Shared genome | Singletons | α |
|---|---|---|---|---|---|---|---|
| C. silvaticum | 38 | 3,002 | 2,209 | 703 | 603 | 190 | 0.9520 |
| C. ulcerans | 76 | 4,351 | 1,747 | 2,604 | 1,706 | 898 | 0.8142 |
| C. silvaticum and C. ulcerans | 114 | 4,916 | 1,618 | 3,298 | 2,349 | 949 | _ |
The pangenome is the entire repertoire of orthogroups, the core genome is the subset conserved across all genomes (100%), the accessory genome is the subset not conserved across all genomes, singletons are exclusive from a genome, and the shared genome are orthogroups shared by two or more, but not all genomes.
Fig 6. Pangenome, core genome and singletons development graphs and formulas for 38 genomes of Corynebacterium silvaticum and 76 C. ulcerans genomes.
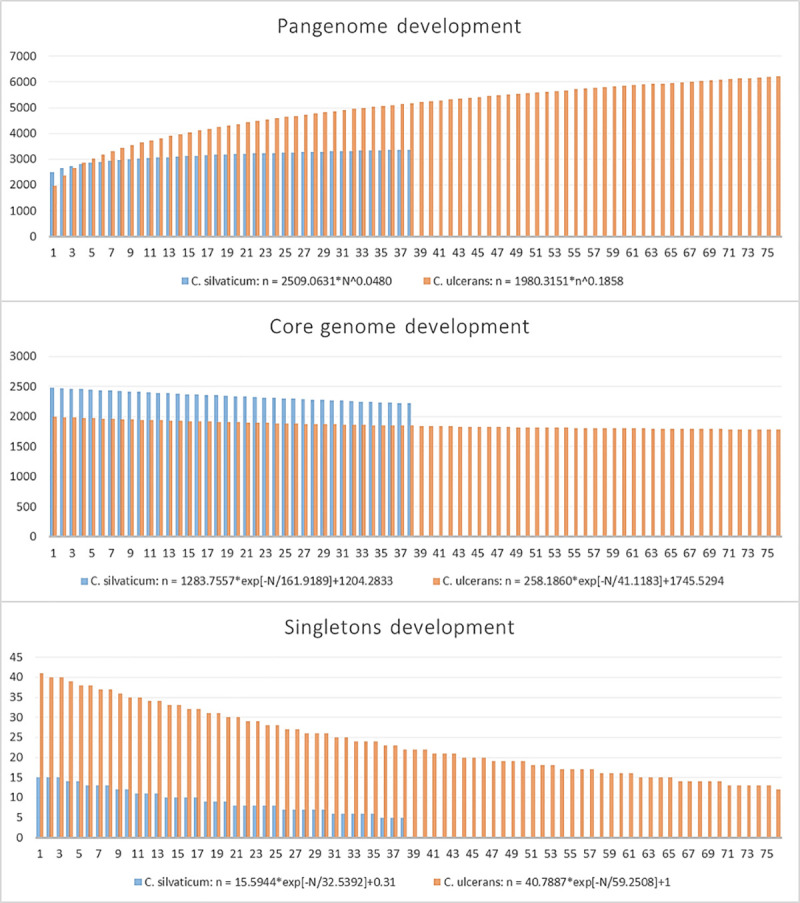
Fig 7. Clusters of orthologous groups (COGs) in the accessory and exclusive core genomes of Corynebacterium silvaticum and C. ulcerans annotated using eggNOG-mapper v2.

COG categories are sorted from most abundant to less abundant in C. silvaticum. In C. silvaticum, 356 out of 793 proteins from the accessory genome and 93 out of 174 proteins of the exclusive core genome had a COG category. In C. ulcerans, 1,065 out of 2,064 proteins from the accessory genome and six out of nine proteins of the exclusive core genome had a COG category.
Finally, PO100/5 (isolated from domestic pig) and KL0182T (wild boar) were predicted to be potential human pathogens by PathogenFinder, with 14 and 13 matches with proteins associated to pathogens, respectively (S13 File).
Discussion
Strain PO100/5 was originally classified as C. pseudotuberculosis. Its resistance profile was tested for 13 antimicrobial compounds (Amoxycillin/Clavulanic acid, Ampicillin, Chloramphenicol, Cephalexin, Gentamicin, Cefotaxime, Enrofloxacin, Nalidixic acid, Penicillin G, Streptomycin, Sulfamethoxazole/Trimethoprim, Tetracycline and Vancomycin) and it was found to be resistant to nalidixic acid and streptomycin [9]. It was suggested as C. silvaticum by a recent rpoB phylogeny [4]. We analyzed the genome diversity of this species, using publicly available genomes from the C. diphtheriae group (S1 File).
Taxonomic analysis showed that PO100/5, W25, 04–13 and 05–13 are strains of the recently described C. silvaticum [4]. This is supported by ANI values above 95% [20] (S2 File), genome and 16S rRNA GBDP clustering [24], dDDH > 70%, G+C content difference > 1% with C. ulcerans genomes [21, 24, 26] (S3–S5 Files), rpoB phylogenetic clustering (Fig 1) and the unique sequence type ST578 from C. silvaticum [4] (Fig 3). Strain PO100/5 has the new ST709 (S9 File, Fig 4). The misclassification of those strains is expected as, prior to the development of methods to identify C. silvaticum [4, 5], the use of biochemistry tests (API Coryne and VITEK2-compact) and the clinical picture would classify these strains as C. pseudotuberculosis [4, 48], while DNA sequence analysis and Fourier-transform Infrared Spectroscopy would classify it as C. ulcerans [6, 7, 48].
Analysis of genome plasticity identified unique characteristics C. silvaticum. The analysis of 16 known niche and virulence factors showed the absence of rpb, rhuM, and tsA. spaB was the only non-fragmented pili gene in C. silvaticum (Table 1). The Shiga-like ribosome-binding protein (rpb) has a ribosome inactivating protein domain that has only been reported in C. ulcerans 809 [35, 49]. The new species also has a RhuM-like protein (rhuM), which has only been seen previously in the C. ulcerans strain KL0387 [50]. A RhuM mutant of Salmonella enterica had a significant decrease in epithelial cell invasion [51]. We identified this protein in 15 other strains from humans, dogs and cats form Austria, France and Germany.
Serine proteases can promote the survival and dissemination of pathogens in the host [52], and we looked for these virulence factors in the genomes we analyzed (Table 1). Venom serine proteases (vsp1 and vsp2) and Trypsin-like serine protease (tspA) are secreted proteases that could have multiple potential pathogenic functions [53]. There is homology between the two serine proteases found in C. ulcerans in this new species (Table 1), but tspA was not found in C. silvaticum. Its absence could be used as a marker to differentiate it from C. ulcerans.
Bacterial pili are adhesion structures required for colonization of host tissues. The Corynebacterium pili are SpaA-type, with a heterotrimeric structure composed by major (pilus shaft), minor and tip pilins, the last two required for adhesion. The pilus is assembled and anchored to the cell wall by the housekeeping sortase SrtA and pili sortases SrtB and SrtC [54]. As seen in C. ulcerans [35], C. silvaticum has the two pili gene clusters spaBC and spaDEF, although only spaB appears to be functional due to the presence of a signal peptide and a CWSS. The SpaB is a minor pilin that in C. diphtheriae has a role in adhesion on pharyngeal epithelial cells and could be functional when linked to the cell wall [55] as shown for the heterodimeric structure SpaB-SpaC in C. diphtheriae [56] and suggested for C. ulcerans [35].
The tox gene was found in only eight out of the 38 C. silvaticum strains (04–13, 05–13, KL0182, KL0884, KL0957, KL1196, PO100/5 and W25), although the strains lacking it were reported to be NTTB [6]. This can be seen in the circular map as a blank space in the tox gene region of the other 30 strains (Fig 4). The absence of the tox gene in the other strains could be the result of an assembly artifact, due to a repetitive region prior to this gene. Additionally, the tox sequences from PO100/5 and 05–13 lack the insertion of two guanines in position 44 (S1 and S2 Figs) that causes pseudogenization, characteristic of other C. silvaticum strains [10]. A recent publication showed that strains 04–13 and 05–13 from Austria produce the tox transcript by reverse transcriptase quantitative PCR (RT-qPCR) [8]. As 04–13 has the frameshift, 05–13 and PO10/5 could be the only known toxigenic C. silvaticum strains. The production of DT has yet to be tested.
In PO100/5, four incomplete prophages were found, one harboring the tox gene. When PO100/5 was compared to C. pseudotuberculosis ATCC 19410T, sixteen genomic islands were identified. When it was compared to C. ulcerans NCTC7910T, only eight islands were found. Four of the islands contained the prophages: GI5, GI8, GI11 and GI13 (S11 File, Figs 4 and 5). No island was found in comparison to C. silvaticum KL0182T. Genomic islands are mobile genetic elements (MGEs) acquired by horizontal gene transfer that can provide adaptive traits [33]. In a previous study, MGEs containing tox in C. diphtheriae were identified as known prophages, while in C. ulcerans they can be different prophages or an alternative pathogenicity island. These mobile elements showed nearly species-specific clades, including the atypical C. ulcerans clade that now represents C. silvaticum. This implies independent events of acquisition of virulence factors in zoonotic species that could influence their pathogenic potential [6].
C. silvaticum was estimated to be more genetically homogeneous than C. ulcerans and to have a pangenome near to being closed, with bigger values of core genome development (Fig 6) and α closer to 1 (Table 3). This result could be influenced by the samples of C. silvaticum being from only two separate countries, Germany (n = 37) and Portugal (n = 1), and from two different species of host (Sus scrofa and Capreolus capreolus). This estimation could change once more genomes are sequenced. A total of 172 and 8 orthogroups were uniquely shared by all C. silvaticum and C. ulcerans, respectively, some in the described genomic islands (S12 File, Fig 7). For C. silvaticum, the most abundant functions are involved in nutrient acquisition such as transport and metabolism of inorganic ions, carbohydrates and amino acids (COG categories E, G and P), or are related to phages or immunity against them (COG category L). For example, two of them are a Type I restriction-modification system [57] in genomic island 11 and an “ABC-type dipeptide oligopeptide nickel transport system”. The function of those genes in the phenotype and infection must be investigated, but they are candidates for genetic markers for a rapid and cost-effective diagnostic using multiplex polymerase chain reaction (PCR) [58–60] and other established methods [4, 5].
In addition to being of veterinary importance, C. silvaticum could have medical relevance, as strains PO100/5 and KL1082 were predicted to be potential human pathogens (S13 File). The known host range of C. silvaticum is limited to wild boars, domestic pigs and roe deer [4, 7, 9]. Wild boars are reservoirs for viruses, bacteria and other parasites that can be transmitted to livestock and humans, during opportunities provided by deforestation and use of lands for agricultural purposes, hunting activities and consumption of wild boar meat [14]. Although they are transmitted additionally by other hosts, pigs and boars are a reservoir of C. ulcerans, which can cause zoonotic transmission to humans [11–13]. By the same route, C. silvaticum could be transmitted to humans and cause infection. In addition, it could be misidentified as C. ulcerans or C. pseudotuberculosis due to limitations in the standard methodology [4, 5].
Additionally, the TYGS results suggest that nine C. ulcerans corresponding to lineage 2 [49] is a potential new species, with dDDH of less than 70% with lineage 1 genomes. (S3–S6 Files). Further investigation is required to verify whether this lineage could be classified as a new species. Recently, C. belfantii and C. diphtheriae lausannense were suggested as synonyms [2]. Our analysis using TYGS corroborated that suggestion. In addition, besides strains FRC0043T, CHUV2995T and CMCNS703, the other nine genomes deposited in GenBank as C. belfantii (https://www.ncbi.nlm.nih.gov/genome/78252/) were classified as C. diphtheriae (S10 File). These results suggest the limitation of using only one cutoff as a parameter for taxonomic classification.
Conclusions
The taxonomic analysis shows PO100/5 and four other genomes deposited as C. ulcerans are from the recently described species C. silvaticum. The comparative genomic analysis showed this species is more genetically homogeneous than C. ulcerans, has SpaB as the only probably functional pilin subunit, and has conserved genomic islands and 172 genes that could be used as molecular markers for PCR identification. In contrast to the other strains from the same species, PO100/5 is the first one to be isolated from livestock and outside Germany and Austria, and to have the unique ST709. A non pseudogenized tox gene in PO100/5 and 05–13 suggest those strains could produce the diphtheria toxin.
Supporting information
The alignment was performed using MUSCLE algorithm implemented in MEGA v10.1.6. C. silvaticum strains PO100/5 and 05–13 do not have a two guanines insertion that lead to a frameshift in other strains from this species.
(TIF)
(TIF)
(XLSX)
Strains highlighted in blue are Corynebacterium ulcerans strains from lineage 2. ANI values under 78.64% are showed as "NA".
(XLSX)
The analysis was performed using Type Strain Genome Server.
(XLSX)
The analysis was performed using Type Strain Genome Server. Strains 04/13 and 05/13 were classified as C. silvaticum, while 03–8664, 04–7514, 131002 were classified as a potential new species.
(PDF)
The analysis was performed using Type Strain Genome Server. Strains PO100/5 and W25 were classified as C. silvaticum, while FRC11, LSPQ-04227, LSPQ-04228, NCTC8666 and NCTC12077 were classified as a potential new species.
(PDF)
The analysis was performed using Type Strain Genome Server. Strain KL0349 was classified as a potential new species.
(PDF)
The analysis was performed using Type Strain Genome Server. All strains were classified as C. ulcerans.
(PDF)
The analysis was performed using Type Strain Genome Server. All strains were classified as C. ulcerans.
(PDF)
The analysis was performed using Type Strain Genome Server. C. diphtheriae lausannense strains CHUV2995T and CMCNS703 were classified as C. belfantii, while C. belfantii strains except the type strain FRC0043T were classified as C. diphtheriae.
(XLSX)
The analysis was performed using MLSTchecker.
(PDF)
(XLSX)
Gene homology groups were predicted using OrthoFinder v2.12.2 and functional annotation was performed using eggNOG-mapper v2.
(XLSX)
PathogenFinder v. 1.1 was used to identify proteins associated to bacterial pathogens in the proteome of C. silvaticum PO100/5 (isolated from domestic pig) and KL0182T (wild boar).
(XLSX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico); CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior); Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG); Universidade Federal de Minas Gerais (UFMG); and Pró-Reitoria de Pesquisa da UFMG (PRPQ-UFMG); A. R. Wattam was supported by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201400027C. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bernard AL, Funke G. Corynebacterium. Bergey’s Manual of Systematic of Archaea and Bacteria (Online). John Wiley & Sons, Bergey’s Manual Trust; 2015. pp. 1–70. 10.1002/9781118960608 [DOI] [Google Scholar]
- 2.Badell E, Hennart M, Rodrigues C, Passet V, Dazas M, Panunzi L, et al. Corynebacterium rouxii sp. nov., a novel member of the diphtheriae species complex. Res Microbiol. 2020;171: 122–127. 10.1016/j.resmic.2020.02.003 [DOI] [PubMed] [Google Scholar]
- 3.Dazas M, Badell E, Carmi-Leroy A, Criscuolo A, Brisse S. Taxonomic status of Corynebacterium diphtheriae biovar Belfanti and proposal of Corynebacterium rouxii sp. nov. Int J Syst Evol Microbiol. 2018;68: 3826–3831. 10.1099/ijsem.0.003069 [DOI] [PubMed] [Google Scholar]
- 4.Dangel A, Berger A, Rau J, Eisenberg T, Kämpfer P, Margos G, et al. Corynebacterium silvaticum sp. nov., a unique group of NTTB corynebacteria in wild boar and roe deer. Int J Syst Evol Microbiol. 2020. 10.1099/ijsem.0.004195 [DOI] [PubMed] [Google Scholar]
- 5.Rau J, Eisenberg T, Peters M, Berger A, Kutzer P, Lassnig H, et al. Reliable differentiation of a non-toxigenic tox gene-bearing Corynebacterium ulcerans variant frequently isolated from game animals using MALDI-TOF MS. Vet Microbiol. 2019;237: 108399 10.1016/j.vetmic.2019.108399 [DOI] [PubMed] [Google Scholar]
- 6.Dangel A, Berger A, Konrad R, Sing A. NGS-based phylogeny of diphtheria-related pathogenicity factors in different Corynebacterium spp. implies species-specific virulence transmission. BMC Microbiol. 2019;19: 1–16. 10.1186/s12866-018-1372-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenberg T, Kutzer P, Peters M, Sing A, Contzen M, Rau J. Nontoxigenic tox-bearing Corynebacterium ulcerans Infection among Game Animals, Germany. Emerg Infect Dis. 2014;20: 448–452. 10.3201/eid2003.130423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaeffer J, Huhulescu S, Stoeger A, Allerberger F, Ruppitsch W. Draft Genome Sequences of Six Corynebacterium ulcerans Strains Isolated from Humans and Animals in Austria, 2013 to 2019. Gill SR, editor. Microbiol Resour Announc. 2020;9: 1–3. 10.1128/MRA.00946-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliveira M, Barroco C, Mottola C, Santos R, Lemsaddek A, Tavares L, et al. First report of Corynebacterium pseudotuberculosis from caseous lymphadenitis lesions in Black Alentejano pig (Sus scrofa domesticus). BMC Vet Res. 2014;10: 218 10.1186/s12917-014-0218-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Möller J, Musella L, Melnikov V, Geißdörfer W, Burkovski A, Sangal V. Phylogenomic characterisation of a novel corynebacterial species pathogenic to animals. Antonie van Leeuwenhoek, Int J Gen Mol Microbiol. 2020;5 10.1007/s10482-020-01430-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuhegger R, Schoerner C, Dlugaiczyk J, Lichtenfeld I, Trouillier A, Zeller-Peronnet V, et al. Pigs as Source for Toxigenic Corynebacterium ulcerans. Emerg Infect Dis. 2009;15: 1314–1315. 10.3201/eid1508.081568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger A, Boschert V, Konrad R, Schmidt-Wieland T, Hörmansdorfer S, Eddicks M, et al. Two Cases of Cutaneous Diphtheria Associated with Occupational Pig Contact in Germany. Zoonoses Public Health. 2013;60: 539–542. 10.1111/zph.12031 [DOI] [PubMed] [Google Scholar]
- 13.Konig C, Meinel DM, Margos G, Konrad R, Sing A. Multilocus Sequence Typing of Corynebacterium ulcerans Provides Evidence for Zoonotic Transmission and for Increased Prevalence of Certain Sequence Types among Toxigenic Strains. J Clin Microbiol. 2014;52: 4318–4324. 10.1128/JCM.02291-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng XJ, Lindsay DS, Sriranganathan N. Wild boars as sources for infectious diseases in livestock and humans. Philos Trans R Soc B Biol Sci. 2009;364: 2697–2707. 10.1098/rstb.2009.0086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seth-Smith HMB, Egli A. Whole genome sequencing for surveillance of diphtheria in low incidence settings. Frontiers in Public Health. 2019. pp. 1–13. 10.3389/fpubh.2019.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger A, Hogardt M, Konrad R, Sing A. Detection Methods for Laboratory Diagnosis of Diphtheria. Corynebacterium diphtheriae and Related Toxigenic Species. Dordrecht: Springer Netherlands; 2014. pp. 171–205. 10.1007/978-94-007-7624-1_9 [DOI] [Google Scholar]
- 17.Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017;45: D535–D542. 10.1093/nar/gkw1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19: 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brettin T, Davis JJ, Disz T, Edwards R A, Gerdes S, Olsen GJ, et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5: 1–6. 10.1038/srep08365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9: 5114 10.1038/s41467-018-07641-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meier-Kolthoff JP, Göker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019;10: 2182 10.1038/s41467-019-10210-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17: 132 10.1186/s13059-016-0997-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10: 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14 10.1186/1471-2105-14-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier-Kolthoff JP, Hahnke RL, Petersen J, Scheuner C, Michael V, Fiebig A, et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 2014;9: 2 10.1186/1944-3277-9-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meier-Kolthoff JP, Klenk HP, Göker M. Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int J Syst Evol Microbiol. 2014;64: 352–356. 10.1099/ijs.0.056994-0 [DOI] [PubMed] [Google Scholar]
- 27.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10: 512–26. 10.1093/oxfordjournals.molbev.a040023 [DOI] [PubMed] [Google Scholar]
- 28.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Battistuzzi FU, editor. Mol Biol Evol. 2018;35: 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47: W256–W259. 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Page AJ, Taylor B, Keane JA. Multilocus sequence typing by blast from de novo assemblies against PubMLST. J Open Source Softw. 2016;1: 118 10.21105/joss.00118 [DOI] [Google Scholar]
- 31.Nascimento M, Sousa A, Ramirez M, Francisco AP, Carriço JA, Vaz C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics. 2017;33: 128–129. 10.1093/bioinformatics/btw582 [DOI] [PubMed] [Google Scholar]
- 32.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44: W16–W21. 10.1093/nar/gkw387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soares SC, Geyik H, Ramos RTJ, de Sá PHCG, Barbosa EGV, Baumbach J, et al. GIPSy: Genomic island prediction software. J Biotechnol. 2016;232: 2–11. 10.1016/j.jbiotec.2015.09.008 [DOI] [PubMed] [Google Scholar]
- 34.Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12: 402 10.1186/1471-2164-12-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trost E, Al-Dilaimi A, Papavasiliou P, Schneider J, Viehoever P, Burkovski A, et al. Comparative analysis of two complete Corynebacterium ulcerans genomes and detection of candidate virulence factors. BMC Genomics. 2011;12: 383 10.1186/1471-2164-12-383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tauch A, Burkovski A. Molecular armory or niche factors: virulence determinants of Corynebacterium species. FEMS Microbiol Lett. 2015;67: fnv185 10.1093/femsle/fnv185 [DOI] [PubMed] [Google Scholar]
- 37.Carver T, Berriman M, Tivey A, Patel C, Böhme U, Barrell BG, et al. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24: 2672–2676. 10.1093/bioinformatics/btn529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30: 1236–1240. 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fimereli DK, Tsirigos KD, Litou ZI, Liakopoulos TD, Bagos PG, Hamodrakas SJ. CW-PRED: A HMM-Based Method for the Classification of Cell Wall-Anchored Proteins of Gram-Positive Bacteria. 1st ed. In: Maglogiannis I, Plagianakos V, Vlahavas I, editors. Artificial Intelligence: Theories and Applications. 1st ed. Springer, Berlin, Heidelberg; 2012. pp. 285–290. 10.1007/978-3-642-30448-4_36 [DOI] [Google Scholar]
- 40.CLC Genomics Workbench. Available: http://www.clcbio.com/products/clc-main-workbench/
- 41.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST plus: architecture and applications. BMC Bioinformatics. 2009;10: 1 Artn 421\nDoi 10.1186/1471-2105-10-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44: D7–D19. 10.1093/nar/gkv1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16: 1–14. 10.1186/s13059-014-0572-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11: 472–477. 10.1016/j.mib.2008.09.006 [DOI] [PubMed] [Google Scholar]
- 45.Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, Von Mering C, et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 2017;34: 2115–2122. 10.1093/molbev/msx148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cosentino S, Voldby Larsen M, Møller Aarestrup F, Lund O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS One. 2013;8: e77302 10.1371/journal.pone.0077302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22: 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 48.Contzen M, Sting R, Blazey B, Rau J. Corynebacterium ulcerans from Diseased Wild Boars. Zoonoses Public Health. 2011;58: 479–488. 10.1111/j.1863-2378.2011.01396.x [DOI] [PubMed] [Google Scholar]
- 49.Subedi R, Kolodkina V, Sutcliffe IC, Simpson-Louredo L, Hirata R, Titov L, et al. Genomic analyses reveal two distinct lineages of Corynebacterium ulcerans strains. New Microbes New Infect. 2018;25: 7–13. 10.1016/j.nmni.2018.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meinel DM, Margos G, Konrad R, Krebs S, Blum H, Sing A. Next generation sequencing analysis of nine Corynebacterium ulcerans isolates reveals zoonotic transmission and a novel putative diphtheria toxin-encoding pathogenicity island. Genome Med. 2014;6: 113 10.1186/s13073-014-0113-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tenor JL, McCormick BA, Ausubel FM, Aballay A. Caenorhabditis elegans-Based Screen Identifies Salmonella Virulence Factors Required for Conserved Host-Pathogen Interactions. Curr Biol. 2004;14: 1018–1024. 10.1016/j.cub.2004.05.050 [DOI] [PubMed] [Google Scholar]
- 52.Liu H, Dang G, Zang X, Cai Z, Cui Z, Song N, et al. Characterization and pathogenicity of extracellular serine protease MAP3292c from Mycobacterium avium subsp. paratuberculosis. Microb Pathog. 2020;142: 104055 10.1016/j.micpath.2020.104055 [DOI] [PubMed] [Google Scholar]
- 53.Ramirez NA, Das A, Ton-That H. New Paradigms of Pilus Assembly Mechanisms in Gram-Positive Actinobacteria. Trends Microbiol. 2020; 1–11. 10.1016/j.tim.2020.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mandlik A, Swierczynski A, Das A, Ton-That H. Pili in Gram-positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol. 2008;16: 33–40. 10.1016/j.tim.2007.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mandlik A, Swierczynski A, Das A, Ton-That H. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol Microbiol. 2007;64: 111–124. 10.1111/j.1365-2958.2007.05630.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang C, Mandlik A, Das A, Ton-That H. Cell surface display of minor pilin adhesins in the form of a simple heterodimeric assembly in Corynebacterium diphtheriae. Mol Microbiol. 2011;79: 1236–1247. 10.1111/j.1365-2958.2010.07515.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loenen WAM, Dryden DTF, Raleigh EA, Wilson GG. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014;42: 20–44. 10.1093/nar/gkt847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pacheco LGC, Pena RR, Castro TLP, Dorella F A., Bahia RC, Carminati R, et al. Multiplex PCR assay for identification of Corynebacterium pseudotuberculosis from pure cultures and for rapid detection of this pathogen in clinical samples. J Med Microbiol. 2007;56: 480–486. 10.1099/jmm.0.46997-0 [DOI] [PubMed] [Google Scholar]
- 59.Almeida S, Dorneles EMS, Diniz C, Abreu V, Sousa C, Alves J, et al. Quadruplex PCR assay for identification of Corynebacterium pseudotuberculosis differentiating biovar Ovis and Equi. BMC Vet Res. 2017;13: 1–8. 10.1186/s12917-016-0931-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badell E, Guillot S, Tulliez M, Pascal M, Panunzi LG, Rose S, et al. Improved quadruplex real-time PCR assay for the diagnosis of diphtheria. J Med Microbiol. 2019;68: 1455–1465. 10.1099/jmm.0.001070 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The alignment was performed using MUSCLE algorithm implemented in MEGA v10.1.6. C. silvaticum strains PO100/5 and 05–13 do not have a two guanines insertion that lead to a frameshift in other strains from this species.
(TIF)
(TIF)
(XLSX)
Strains highlighted in blue are Corynebacterium ulcerans strains from lineage 2. ANI values under 78.64% are showed as "NA".
(XLSX)
The analysis was performed using Type Strain Genome Server.
(XLSX)
The analysis was performed using Type Strain Genome Server. Strains 04/13 and 05/13 were classified as C. silvaticum, while 03–8664, 04–7514, 131002 were classified as a potential new species.
(PDF)
The analysis was performed using Type Strain Genome Server. Strains PO100/5 and W25 were classified as C. silvaticum, while FRC11, LSPQ-04227, LSPQ-04228, NCTC8666 and NCTC12077 were classified as a potential new species.
(PDF)
The analysis was performed using Type Strain Genome Server. Strain KL0349 was classified as a potential new species.
(PDF)
The analysis was performed using Type Strain Genome Server. All strains were classified as C. ulcerans.
(PDF)
The analysis was performed using Type Strain Genome Server. All strains were classified as C. ulcerans.
(PDF)
The analysis was performed using Type Strain Genome Server. C. diphtheriae lausannense strains CHUV2995T and CMCNS703 were classified as C. belfantii, while C. belfantii strains except the type strain FRC0043T were classified as C. diphtheriae.
(XLSX)
The analysis was performed using MLSTchecker.
(PDF)
(XLSX)
Gene homology groups were predicted using OrthoFinder v2.12.2 and functional annotation was performed using eggNOG-mapper v2.
(XLSX)
PathogenFinder v. 1.1 was used to identify proteins associated to bacterial pathogens in the proteome of C. silvaticum PO100/5 (isolated from domestic pig) and KL0182T (wild boar).
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.