

SUMMARY
A surprising complexity of ubiquitin signaling has emerged with identification of different ubiquitin chain topologies. However, mechanisms of how the diverse ubiquitin codes control biological processes remain poorly understood. Here, we use quantitative whole proteome mass spectrometry to identify yeast proteins that are regulated by lysine 11 (K11)-linked ubiquitin chains. The entire Met4 pathway, which links cell proliferation with sulfur amino acid metabolism, was significantly affected by K11 chains and selected for mechanistic studies. Previously we demonstrated that a K48-linked ubiquitin chain represses the transcription factor Met4. Here we show that efficient Met4 activation requires a K11-linked topology. Mechanistically our results propose that the K48 chain binds to a topology-selective tandem ubiquitin binding region in Met4 and competes with binding of the basal transcription machinery to the same region. The change to K11 enriched chain architecture releases this competition and permits binding of the basal transcription complex to activate transcription.
In brief
Li et al. discovered that a chain topology change from K48 to K11 linkages on the transcription factor Met4 relives competition between the K48 chain and the basal transcription complex for binding to the Met4 tandem-UBD. These findings provided insight into the role of ubiquitin chain topologies in ubiquitin signaling.
Graphical Abstract
INTRODUCTION
Modification with ubiquitin has long been known to regulate protein stability, but degradation-independent signaling functions triggered by ubiquitylation are also critical for cell and organismal physiology (Finley et al., 2012; Varshavsky, 2012). The various signaling functions depend on the type of ubiquitylation as well as substrate context. In addition to the distinction between mono, multi, and poly ubiquitylation, ubiquitin chain density and particularly chain topology play major roles in signaling. More recently, posttranslational modifications on ubiquitin, such as phosphorylation, have further expanded complexity of the ubiquitin code (Swatek and Komander, 2016).
Ubiquitin chains are synthesized by covalent attachment of the terminal carboxyl group of an incoming ubiquitin to an amino group of an acceptor ubiquitin. Seven lysine-based amino groups (K6, K11, K27, K29, K33, K48, K63) and the amino terminus are available for chain formation. Chain branching through attachment of two ubiquitin molecules to different lysine residues in one ubiquitin molecule, and mixed chains further diversify the ubiquitin code (Komander and Rape, 2012; Yau and Rape, 2016). All chain topologies, except K63 and linear (amino terminal linked) chains, have been suggested to serve as degradation signals that target modified substrates to the 26S proteasome (Meierhofer et al., 2008; Peng et al., 2003b; Xu et al., 2009b). However, substrate context can significantly alter signaling functions of ubiquitin.
One of the best-studied examples for how substrates can transform signal identity of ubiquitin chains is the yeast transactivator Met4. The cullin-RING ligase SCFMet30 and its two substrates Met4 and Met32 form the core of a metabolic cell cycle checkpoint system that links cell proliferation with methionine metabolism (Ouni et al., 2011; Thomas and Surdin-Kerjan, 1997). The same system coordinates heavy metal stress with cell cycle progression (Barbey et al., 2005; Yen et al., 2012; Yen et al., 2005). The Met4 transactivation potential is repressed by a K48-linked ubiquitin chain attached to lysine 163 in Met4 (Flick et al., 2004). Despite being the canonical degradation signal, the K48 chain on Met4 does not induce degradation, but rather functions as an activity switch and directly represses Met4 transactivation function (Flick et al., 2004; Kaiser et al., 2000). The mechanism of how ubiquitylation represses Met4 activity is unknown, but transformation of the degradation signal of the K48-linked chain to a non-proteolytic regulatory signal has been linked to two ubiquitin binding domains (UBD) in the N-terminal region of Met4. Indeed, this tandem-UBD is transferable and can prevent degradation of heterologous ubiquitylated proteolytic substrates (Flick et al., 2006; Tyrrell et al., 2010). Interestingly, the tandem-UBD is identical with the minimal transactivation domain of Met4 (Pacheco et al., 2018). Physiologically the non-proteolytic regulation of Met4 is important, because, independent of its ubiquitylation status, Met4 serves as a substrate receptor in the context of the expanded SCFMet30/Met4 ubiquitin ligase. SCFMet30/Met4 promotes ubiquitylation and degradation of Met32 and thereby allows recovery from cell cycle arrest when nutrient stress is alleviated (Ouni et al., 2010).
In this study, we use a SILAC label-swap approach that combines high-resolution protein fractionation and an LC–MS/MS platform to examine the proteomes of two yeast strains, a wild-type (WT) strain and a ubiquitin mutant (K11R) strain, wherein the K11 is replaced by arginine. The K11R mutation in ubiquitin prevents the formation of ubiquitin chain topologies that are linked through K11. Our results reveal a profound downregulation of methionine biosynthesis enzymes in the K11R mutant strain, indicating a so far unappreciated role of K11-linked ubiquitin chains in regulating the SCFMet30-Met4 network. We find that a chain topology change from K48 to a K11 linkage enriched chain on Met4 relives competition between K48-ubiquitin chains and the basal transcription complex for binding to the Met4 tandem-UBD, which functions as a transactivation domain. Our study discovered additional complexity in regulation of the methionine controlled metabolic checkpoint system and provides insight into how a K48-linked ubiquitin chain can regulate protein function in a proteolysis-independent manner.
RESULTS
High-coverage Proteomics Aids Pathway Related Functional Study
We used a proteomics approach to identify pathways that are regulated by K11-linked ubiquitin chains. System-wide protein abundance was compared between wild-type yeast and a yeast strain that cannot synthesize K11-linked ubiquitin chains due to a lysine to arginine mutation at position 11 of ubiquitin (K11R). Total cell lysate (TCL) samples were prepared from yeasts cultured in SILAC media (Figure 1A), separated by SDS PAGE to reduce sample complexity, and digested with Lys-C prior to LC–MS/MS analysis (Figure 1B).
Figure 1. SILAC-based proteomic profiling of WT and ubiquitin K11R mutant yeast.

(A) Outline of label-swap experiments for comparing WT and K11R TCL samples.
(B) Sampling the yeast proteome by LC–MS/MS.
(C) Proteome coverage of current study.
(E) Proteome coverage of current study in comparison to previous studies.
(F) MW distribution of missed and identified proteins. The percentage represents the coverage of core proteins for the indicated MW range.
We analyzed the resulting peptide mixtures and identified 4,580 proteins with 99% certainty. Despite using a simpler approach, the protein coverage identified (68.2%) was slightly higher than the 65.5% reported in the landmark study by de Godoy et al. (de Godoy et al., 2008). Of these proteins, 4,267 were identified in forward and 4,171 in reverse experiment. The intersection of these two datasets comprised 3,858 proteins (Figure S1A–S1C) and the average sequence coverage was 33.3% (Figures 1C and S1D).
The Saccharomyces Genome Database (SGD) had 5,155 proteins with annotated functions, of which 4,188 were present in our dataset (81.2% coverage). Therefore, our approach not only used a simplified strategy, but also increased the number of identified proteins and improved the quality of identification (Figures 1C, 1E and Table S1). Of the 4,580 proteins, 88.9% were identified by two or more unique peptides (Figure S1E). The average number of identified peptides per protein was 14.5 (Figure 1C).
Four representative yeast proteomes, published by Ghaemmaghami et al. (tandem affinity purification; “TAP”) (Ghaemmaghami et al., 2003), Huh et al. (green fluorescent protein; “GFP”) (Huh et al., 2003), Deutsch et al. (“PeptideAtlas”) (Deutsch et al., 2008), and de Godoy et al. (“Mann 2008”) (de Godoy et al., 2008), were compared to our dataset. The union of these five datasets comprised 5,314 proteins, of which 86.2% were present in our dataset (Figures 1D and S1F). Of the proteins that were missing in our dataset, the majority had MW ≤ 20 kDa (Figure 1E). These included uncharacterized proteins, as well as products of dubious genes. The coverage for core proteins with MW ≤ 20, 20–90 and 90–190 kDa was 34.1, 77.5 and 90.7%, respectively.
To relate our proteomics datasets with gene expression, in-depth RNA expression analysis was performed using RNA-seq. A total of 5,833 genes were detected using the filter criterion of read counts ≥ 5 (Figure S2A). Our proteome and the RNA-seq dataset had 4,518 gene products in common, occupying 98.6% and 77.5% of identified proteins and sequenced gene transcripts, respectively (Figure S2B). Of the 4,580 identified proteins, 356 of 752 (47.3%) putative and hypothetical proteins were identified with MS evidences in our dataset (Figure S1F). However, confirmation of expression from dubious ORFs could only achieve 4.4% coverage, similar to that of RNA-seq datasets (Figure S2C) and aforementioned yeast proteomes (Figure S1F). Interestingly, of the 26 pseudogenes listed in the SGD, four were present in our dataset (Figure S2D) and the presence of YIL170W was confirmed by the alignment of mass spectra (Figure S2E).
The dynamic range of the protein copy numbers based on intensities (Picotti et al., 2009) was two magnitudes larger than that of transcript abundances (Figure S3A, S3B and Table S1), implying that current proteomics platform had sufficient sensitivity to pursue low-abundance proteins. The coverage for KEGG pathways averaged 84.2% (Figure S3C). One of the most active pathways, mitosis and its associated proteins were catalogued into five subgroups (i.e., midbody, centrosome, kinetochore, telomere and spindle) based on MiCroKiTS 4.0 (Ren et al., 2010). All subgroups had over 76% proteins uniquely identified by two or more peptides (Figure S3D). This allowed for a deeper analysis of protein expression patterns in yeast.
K11-linked Ubiquitin Chains Profoundly Affect Transcript and/or Protein Levels of Enzymes Related to Methionine Metabolism
The homeostasis of the cellular proteome is strictly controlled by the ubiquitin–proteasome system (UPS). Our previous work has revealed that seven ubiquitin linkages are abundant in yeast (Xu et al., 2009b). K11 linkages form the most abundant chain architecture in vivo after the canonical K48-chain topology and the degradation independent K63 chain (Xu et al., 2006; Xu et al., 2009b). However, K11-chains are much less well characterized compared to K48-linked chains. Systematic comparison of TCL-derived ubiquitin conjugates has revealed Ubc6 as a K11 linkage modified protein substrate that mediates protein degradation via the endoplasmic reticulum-associated degradation (ERAD) pathway (Xu et al., 2009b). However, apart from this example, the identification of ubiquitin chain topology specific substrates has been restricted by technological limitations. We surveyed our SILAC-based approach for its ability to classify ubiquitin chain linkage selective substrates.
We compared the log2(K11R/WT) ratio distribution of proteins identified in forward and reverse experiments on a two-dimensional central scatter plot (Figure 2A). We also mapped the differences in transcript abundance onto the central scatter plot. Our results showed that most gene products experienced no significant changes in protein or transcript abundance (blue points). However, the K11 mutation did affect the protein (red points) and transcript abundances (green points) of some genes (Figure 2A and Table S2).
Figure 2. Potential protein substrates of K11-linked ubiquitin chains.

(A) Log2 (K11R/WT) ratio distributions of quantified proteins. Blue, unchanged proteins; red, significantly changed proteins identified through proteomics; green, significantly changed proteins identified through proteomics and transcriptomics. SDE gene stood for the significant different expressed gene.
(B) Gene ontology analysis of potentially K11-linked ubiquitylated substrates, namely the upregulated protein in K11R datasets.
(C) Members of the methionine biosynthesis pathway are colored according to fold change. The protein and mRNA ratios of K11R to WT were determined for each protein-coding gene.
We identified 135 significantly downregulated proteins (lower left quadrant; P-value < 0.01) and 112 significantly upregulated proteins (upper right quadrant) in K11R mutants. We previously showed that protein substrates of K11-linked ubiquitin chains are degraded by the 26S proteasome (Xu et al., 2009b). Such substrates are located in the upper right quadrant of the central scatter plot. Considering the regulation on the level of gene transcription, we identified 107 candidate protein substrates of K11-linked ubiquitin chains, including 68 significantly upregulated proteins with no corresponding changes in the transcriptome, 27 unchanged proteins with significant downregulation in the transcriptome, as well as 12 significantly upregulated proteins with downregulation in the transcriptome (Table S2). Interestingly, Ubc6, a previously confirmed substrate of K11-linked ubiquitin chains, was present among these 107 candidate substrates. Gene Ontology (GO) (Ashburner et al., 2000) classification of these K11-linked ubiquitin chain substrates indicated that the K11 mutation perturbed a broad range of cellular processes (Figure 2B).
Functionally, upregulated proteins were mostly associated with primary metabolite biosynthesis (Figure 2B), whereas downregulated proteins were mostly associated with methionine biosynthesis (Figure 2A). We manually checked the light- and heavy-isotope labeled peptides representing Met genes (Met6 as an example in Figure S4A) and confirmed the decrease of each protein. These results suggested that K11 linkages play a role in regulating methionine metabolism. Indeed, our quantitative proteomics succeeded in profiling the entire methionine biosynthesis pathway (Figure 2C and Table S2).
In contrast to MET proteins, many other proteins showed abundance changes independent of transcription (Figures 2A, S3F and S3G). We therefore used SILAC quantification for global half-life analysis of the proteome of WT and K11R strains as described before (Christiano et al., 2014; Schwanhausser et al., 2011) to identify K11-chain dependent protein degradation. After completely labeling with heavy lysine and arginine, yeast cells were transferred into light SC medium. The cells were harvested at 0, 5, 15, 30, 60, 120, and 240 minutes and then analyzed by LC-MS/MS. We identified 2,783 and 2,762 proteins with 1181 and 1247 quantified proteins with at least two unique peptides in WT and K11R strains, respectively. The ratio (H/L) was used to calculate the decay rates (Kdeg). Proteins with a coefficient of determination R2≥0.9 were included for half-life analysis (Schwanhausser et al., 2011).
The half-lives of a large proportion of proteins were increased in K11R mutant cells (Figure 3A), indicating that proteasome degradation of a significant portion of the yeast proteome is mediated directly or indirectly by K11 ubiquitin chains. For example, the potential substrates Sam4, Rib5, and Ddr48, were more stable in K11R mutant strains (Figures 3B and S4C–S4E), while degradation of proteins expected to be independent of K11-mediated degradation, such as Cdc48, were unchanged (Figure S4F and S4G). Among the 107 potential K11-linked ubiquitin chain modified protein substrates, we quantified 69 proteins in half-life datasets, of which 40 proteins were more stable in K11R mutants as compared to WT cells (Figure 3A, 3B and Table S3), suggesting that these proteins are potential K11-ubiquitin marked proteasome substrates. In contrast, we found that the half-life of proteins in the methionine metabolism pathway, for example Met6 and Met5, were not affected in the K11R mutant strain (Figures 3C, S4F and S4H). These results further suggested that the K11 ubiquitin chain mediated change in methionine biosynthesis enzyme (hereafter called MET proteins) abundances is due to transcriptional regulation of MET genes. Accordingly, we found significantly positive correlations (r = 0.857) between the proteome and transcriptome for members of the methionine biosynthesis pathway (Figure 3D), indicating that K11-linked ubiquitin chains are necessary for efficient MET gene transcription.
Figure 3. Members of the methionine biosynthesis pathway are regulated by K11-linked ubiquitin chains independent of protein stability.

(A) Global half-life analysis of proteomes of WT and ubiquitin K11R strains. Scatterplot comparing the half-life of whole proteome (n = 1,375, grey points) with proteins showing increased abundance in K11R mutants (upper right quadrant Figure 2A; i.e. potential K11 modified proteasome substrates) (n = 69, red points) are displayed.
(B and C) Decay curves for Sam4 (B) and Met6 (C), respectively.
(D) Correlations between the proteome and transcriptome. The Pearson correlation coefficient was 0.86.
The transcriptional activator Met4 is known to be the primary methionine-sensitive regulator of MET gene expression (Lee et al., 2010; Thomas and Surdin-Kerjan, 1997). Recent studies have shown that a K48-linked ubiquitin chain attached to lysine residue K163 in Met4 represses Met4 activity in a non-proteolytic manner (Flick et al., 2004). The ubiquitin chain forms an intramolecular interaction with two tandemly arranged UBDs, which shields Met4 from the 26S proteasome and prevents the assembly of active Met4 transcription complexes through an unknown mechanism (Flick et al., 2004; Flick et al., 2006; Tyrrell et al., 2010). The transcriptional downregulation we observed for the entire methionine biosynthesis pathway in K11R mutants implicates K11-linked ubiquitin chains in Met4 activation. Importantly, protein levels of Met4 and its co-factors were unchanged in the K11R mutant (Figure S4B and Table S2), suggesting that Met4 activity is stimulated by K11-linked ubiquitin chains. K48 and K11-linked ubiquitin chains would therefore have opposing effects on Met4 transactivation.
The Chain Topology of the Met4 Attached Ubiquitin Chain Changes from K48 to K11 Enriched Chains during Met4 Activation.
To survey ubiquitin linkages on Met4, we constructed endogenous 3HA-tagged Met4 in WT and K11R mutant strains (Figure S5A and S5B). We applied a tandem purification approach to further improve the purity of Met4 and covalently attached ubiquitin linkages. Proteins immunopurified by HA antibodies were subjected to 0.5% SDS so that most noncovalently bound contaminants would disassociate from purified Met4 and then diluted tenfold with PBS and purified again with HA antibodies prior to LC–MS/MS analysis. This resulted in the identification of only nine purified proteins (Table S4), including three with a single peptide. The most abundant protein identified was ubiquitin itself, suggesting that enriched proteins were heavily ubiquitylated. Not surprisingly, Met4 was the second most abundant protein. All other identified proteins were at least one order of magnitude lower in abundance (Figure 4A), suggesting that our technique efficiently purified ubiquitylated Met4. More importantly, this purification procedure enabled the measurement of strictly Met4-associated ubiquitin linkages (Figure 4B). The precise identification of ubiquitin linkages was performed with synthetic heavy-labeled peptides as internal standards. Interestingly, as previously reported by Mirzaei et al. (Mirzaei et al., 2010), under repressive growth conditions we detect abundant K48 chains and small amounts of K11 linkages on the purified Met4 (Figure 4B). Manual annotation of K11-linked peptides isolated with Met4 is shown in Figure S5C and S5D. No other ubiquitin linkages were detected, except K63 at very small amounts (0.3%±0.1%), which was close to background levels.
Figure 4. The topology of the Met4 attached ubiquitin chain changes from K48 to K11 during activation.

(A) Intensity distribution of co-IP purified proteins with 3HA-tagged Met4 proteins.
(B) Ubiquitin linkages on 3HA-tagged Met4 proteins identified by LC–MS.
(C) K48-linked chains on 3HA-tagged Met4 proteins during metabolic shift to methionine depleted medium were quantified by LC–MS. The amount was normalized based on the sample from methionine containing medium. Results are shown for mean +/− SEM. Samples were prepared from wild-type cells, K11R mutants, Met4K163R, and met30Δ met32Δ double mutants expressing cells. The met32Δ deletion was required to suppress the lethality of met30Δ mutants.
(D) Same as panel C, but the K11-linkage was quantified.
Previous studies have shown that when methionine levels are high, yeast represses MET genes through SCFMet30-dependent ubiquitylation of Met4 with a K48-linked ubiquitin chain (Flick et al., 2004; Kaiser et al., 2000; Patton et al., 2000). We asked whether a difference in Met4 ubiquitylation could be detected in WT and K11R strains shifted from repressive culture conditions (with methionine) to conditions that activate Met4 (without methionine). We quantified K48- and K11-linked chains on Met4 by LC-MS analysis as described above during an activation time course (Figure 4C and 4D). Remarkably, the ubiquitin-chain topology on Met4 showed a distinct dynamic during the metabolic shift. The repressive K48-linked chains gradually decreased (Figure 4C), whereas K11 linkages on Met4 significantly increased upon methionine limitation (Figure 4D). The small amount of K63 linkages detected in repressive conditions disappeared during activation. These findings support the proteomics results that suggested a role for K11-chains in Met4 activation. The increase of K11 linkages on Met4 during activation was further confirmed by probing immunopurified Met4 with K11-linkage selective antibodies (Matsumoto et al., 2010), which showed a signal specifically on Met4 during methionine depleted growth conditions (Figure S6).
SCFMet30 and the canonical ubiquitin acceptor Lysine 163 in Met4 are required for K11 chain formation
SCFMet30 is the only ubiquitin ligase currently known to ubiquitylate Met4, and lysine 163 (K163) in Met4 is the critical ubiquitin acceptor site (Flick et al., 2004). Using the same mass spectrometric analyses described above, we monitored ubiquitin chains on Met4 in a yeast strain lacking the F-box protein Met30, and in cells expressing Met4K163R (Flick et al., 2004). Both mutations blocked K11 chain formation (Figure 4D), demonstrating that SCFMet30 and K163 in Met4 are required. These results, however, do not necessarily establish that SCFMet30 directly supports synthesis of K11 chains, but may indicate the requirement for a priming ubiquitin attached to K163 by an SCFMet30 mediated reaction, for K11 chain assembly on Met4. Nevertheless, Met4 regulation by a single ubiquitin chain attached to K163 is strongly suggested and Met4 activation appears to be mediated by ubiquitin chain topology changes of a single chain.
K48 but not K11-linked Ubiquitin Chains Bind to the Met4 Tandem UBD.
The observed increase in K11-linked chains attached to Met4 during transcriptional activation, while only correlative, indicates a possible mechanistic involvement of the ubiquitin chain topology change in the activation of the transcription factor Met4. To gain insight into this process we probed communication between K48 and K11 ubiquitin chains with the tandem ubiquitin binding domain in Met4.
Ample evidence supports a non-proteolytic role of the K48 ubiquitin chain attached to K163 in Met4. However, limited mechanistic insight has been achieved into how ubiquitylation can repress Met4 activity, and how activation is accomplished. It is clear that multiple mechanisms are contributing to this phenomenon. Full repression of MET genes has been linked to a Cdc48-dependent active dissociation of Met4 from target promoters, which depends on ubiquitylation (Ndoja et al., 2014). However, this mechanism can only account for a small part of repression, and additional functions of Met4 ubiquitylation in repression of transcription remain to be identified. Furthermore, what triggers the Met4 activation process is unknown.
Because the tandem UBD in the C-terminus of Met4 is an important regulatory element of this system we determined binding characteristics of K48 and K11 chains using biolayer interferometry (BLI). The N-terminal region from residue 76 to 160 of Met4 containing both the UIM and the UIML domains was linked to the sensor dip and K48 as well as K11 tetraubiquitin chains were used as ligands. K48 tetraubiquitin bound to Met4 with a dissociation constant of about 0.40 μM (Figure 5A). Surprisingly, K11 tetraubiquitin did not bind to Met4 with any measurable affinity (Figure 5B). The tandem UBD in Met4 thus strictly distinguishes between the K48 and K11 chain topologies.
Figure 5. The tandem ubiquitin binding domain in Met4 is selective for K48 ubiquitin chains and interacts with the mediator component Med15.

(A) Biolayer interferometry with sensor immobilized recombinant Met4 (residues 76 to 160) and K48-linked tetraubiquitin. Concentrations used were: 1450, 483, 161, 53, 17, and 5.6 nM.
(B) Same as panel A, but K11-linked tetraubiquitin was used as the ligand. No detectable binding was observed.
(C) Same as panel B, but interaction with the mediator complex Med15 (residues Med15 1–651 Δ239–271, Δ373–483) was analyzed.
K48 but not K11-linked Ubiquitin Chains Compete with Recruitment of the Basal Transcription Machinery to the Met4 Tandem UBD
In a recent effort to define the minimal transactivation domain of Met4, transactivation activity was mapped to the tandem UBD (Pacheco et al., 2018). Furthermore, this study demonstrated that the mediator complex, a multi protein co-activator complex, interacts with the region of Met4 that contains the tandem UBD. The Med15 subunit of the mediator was found to form the main contact with Met4 (Pacheco et al., 2018). We therefore measured binding characteristics of the Med15 subunit to the same N-terminal Met4 fragment (residue 76–160) for which we determined tetraubiquitin binding. The dissociation constant for Med15 binding was measured at 0.10 μM using biolayer interferometry (BLI) (Figure 5C). The observed KD is consistent with fluorescence polarization measurements of this interaction (Pacheco et al., 2018).
The linkage selective binding of this Met4 region to ubiquitin chains suggested an intriguing molecular role for the topology change from K48 to K11 chains during Met4 activation. During the repressed condition, the K48 linked chain on Met4 binds to the transactivation/UBD domain and prevents recruitment of the mediator complex. During activation the change to the K11 topology opens access to the transactivation domain to allow mediator binding.
To test this hypothesis, we asked whether K48 tetraubiquitin and the mediator component Med15 compete for the same binding surface on Met4. Indeed, addition of excess recombinant Med15, but not an unrelated protein, to a preformed Met4/K48 tetraubiquitin complex resulted in displacement of ubiquitin and association of Med15 with Met4 (Figures 6A and S7A). Note that for technical reasons both K48 tetraubiquitin affinity to Met4 and competition experiments were conducted under a trans binding model. However, the in vivo mode of K48 ubiquitin binding to Met4 is intramolecular as the chain is linked to K163 in Met4 (Flick et al., 2004). These experiments are therefore likely significantly underestimating the competition between K48 ubiquitin chains and the mediator complex. Indeed, we expect that due to the kinetic advantage of the intramolecular binding mode, presence of a K48 ubiquitin chain on Met4 prevents Med15 binding in vivo. Accordingly, the switch to the K11 topology is predicted to be important during Met4 activation to allow mediator access to the transactivation domain in Met4.
Figure 6. K48 and K11 linked ubiquitin chains on Met4 control expression through modulation of mediator binding.

(A) K48 ubiquitin chains and the mediator component Med15 have overlapping, exclusive binding sites. Immobilized Met4 (76–160) was bound to K48 tetraubiquitin. Elution of K48 tetraubiquitin by addition of 0, 0.25, 0.5, 1, or 3-fold molar excess of Med15 (residues Med15 1–651 Δ239–271, Δ373–483) was analyzed by immuno blotting of the eluted fraction and the bead bound fraction.
(B) Mediator recruitment to MET gene promoters depends on K11 ubiquitin chains. Wild-type, K11R, and Δmet4 mutants were cultured in medium containing repressive amounts of methionine, shifted to methionine depleted growth medium for 30 minutes to activate MET gene transcription before re-repression by addition of methionine for 15 minutes. Mediator binding was monitored by ChIP using the endogenously HA3-tagged mediator subunit Med14. Four Met4-dependent genes (MET3, MET17, CYS4, SAM2) and two Met4-independent genes (CCW12, CDC19) were analyzed. Results are shown for n=3 +/− SD.
(C) Yeast cells lacking the ability to form K11-linked ubiquitin chains (K11R mutants) show a significant defect in activation of Met4 controlled genes involved in methionine biosynthesis. RNA abundance of a panel of MET genes was analyzed on a custom NanoString panel. Three biological replicates are shown. Data were normalized to three housekeeping genes.
We therefore followed recruitment of the mediator complex to Met4 target genes during an activation/repression time course in wild-type and K11R mutants by chromatin immunoprecipitation. Cells were grown in medium containing repressive amounts of methionine, shifted to growth conditions without methionine for 30 minutes to activate transcription of MET genes, and finally transcription was repressed again by adding repressive amounts of methionine for 15 minutes (Figure 6B). As previously shown (Leroy et al., 2006), the mediator complex was readily recruited to MET genes (MET3, MET17, CYS4, and SAM2) during activation, and recruitment was strictly dependent on Met4. Importantly, mediator recruitment to these MET genes was highly dependent on the ability to form K11 ubiquitin chains because recruitment was significantly blunted in K11R mutants (Figure 6B). This defect was specific for Met4-dependent genes because K11 ubiquitin chains were not required for mediator recruitment to Met4-independent genes (CCW12 and CDC19) (Figure 6B).
These in vivo results are consistent with our in vitro data and support the proposed requirement for a K48 to K11 chain topology change to allow mediator binding to Met4. Consistent with mediator recruitment dynamics during activation in nutrient stress conditions, yeast strains expressing K11R ubiquitin mutants were significantly impaired in MET gene activation as compared to wild-type yeast (Figure 6C). Almost all MET genes and proteins affected by the K11R mutation during steady state abundance measurements (Figures 2A and 3D) were also affected during the acute response to nutrient stress (Figure 6C). The two notable exceptions are CYS4 and MUP1, whose transcriptional induction is significantly impaired in K11R mutants during nutrient stress (Figure 6C), but steady state protein abundance is increased (Figure 2A). There are clearly additional gene-specific regulatory mechanisms, such as translational modulation, at work in addition to the general control by the ubiquitin topology change from K48 to K11 enriched chains.
Together these lines of experiments provide mechanistic insight into signaling by different ubiquitin chain topologies, and offer conceptual understanding of the role of UBDs in this process.
DISCUSSION
Progress of High Coverage Proteomics
A longstanding goal in MS-based shotgun proteomics is to identify the entire proteome that is expressed in a cell or tissue type. A major challenge is the wide dynamic range of protein abundance. Advanced sample pretreatment, as well as high sequencing speed and sensitivity are means to increase the sequencing coverage of the proteome. By taking full advantage of high-resolution SDS–PAGE, optimized LC gradients (Xu et al., 2009a) and the faster scan speed of the LTQ Orbitrap Velos mass spectrometer (Li et al., 2012), we identified proteins spanning nearly six orders of magnitude (Figure S3A) and detected 81.2% basally expressed haploid yeast proteins with high accuracy (Figure S1F).
Protein Substrates of K11-linked Ubiquitin Chains Revealed by Quantitative Proteomics
Our previous study showed that K11 linkages are among the most abundant types of ubiquitin linkages (Xu et al., 2009b). By comparing both the entire yeast proteome and ubiquitylated proteins in WT and K11R strains, our current study revealed that a chain topology change from K48 to K11 enriched chains on the transcription factor Met4 is important to coordinate the entire methionine biosynthesis pathway with cell cycle control.
The methionine biosynthesis pathway was missed in our previous study (Xu et al., 2009b), including Met4 itself, as we detected only a fraction of the MET gene network (Figure S3E). However, the significantly higher proteome coverage achieved by our modified approach has helped us uncover all members of the methionine biosynthesis pathway that span nearly five orders of abundance magnitude (Figure 2C and Table S2). To our knowledge, this is the first time global shotgun quantitative proteomics was able to profile an entire metabolic pathway (Figure 2C and Table S2). Our comprehensive analysis on the yeast proteome and transcriptome has provided a resource not only for understanding the dynamic response at both transcriptional and proteome level related to K11-linked ubiquitin chain modifications, but also for identifying potential K11-chain substrates. Half-life analysis of the proteome by SILAC pulse-chase quantification demonstrated that K11-linked ubiquitin chains are important for degradation of many proteins (Figure 3A). Whether the stability of these proteins is affected directly or indirectly by K11-chains is currently unknown and requires further studies using K11-chain selective purification approaches.
Chain Selective Ubiquitin Binding Motifs and Chain Topology Switches Provide a Mechanism for Complex Ubiquitin Signaling
The proteomics discovery that K11 ubiquitin chains are important for homeostasis of most or all proteins required for methionine metabolism was surprising (Figure 2). The proteome changes could be quickly traced to altered transcriptional activity of the transactivator and master regulator of methionine biosynthesis, Met4 (Thomas and Surdin-Kerjan, 1997). However, these findings generated a conundrum. Met4 ubiquitylation has been extensively studied and exclusively linked to Met4 repression (Kaiser et al., 2000; Kuras et al., 2002; Ndoja et al., 2014; Patton et al., 2000), yet, the proteomics results suggest a requirement of K11-linked ubiquitylation in Met4 activation. Clearly, the K48 linked ubiquitin chain on Met4 keeps the transcription factor in a repressed state (Aghajan et al., 2010; Flick et al., 2004; Kaiser et al., 2000; Patton et al., 2000), but here we show that K11-linked ubiquitin chains on Met4 correlate with Met4 activation. These results indicate distinct roles for the different ubiquitin chain topologies. Interestingly, these functions appear to be independent of their canonical roles in promoting protein degradation by the 26S proteasome (Yau and Rape, 2016), although we cannot exclude that a subset of Met4 is targeted for degradation by K11 chains.
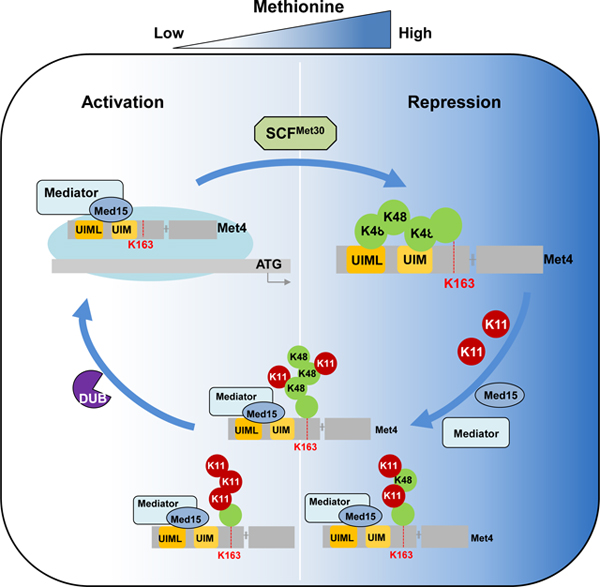
Mechanistically this study provides insight into how a K48-linked ubiquitin chain contributes to transcription factor repression in a proteolysis independent manner, and how the change to the K11 enriched topology promotes transcriptional activation. We show that the tandem UBD, which serves as a transactivating region in Met4 engages with the K48 ubiquitin chain intramolecularly and thereby prevents recruitment of the mediator complex. The switch to K11 enriched ubiquitin chains helps to resolve this repressed state due to the K48 chain selectivity of the Met4 tandem UBD. The mediator complex can now bind to the exposed transactivation/tandem ubiquitin binding domain and initiate transcription (Figure 7).
Figure 7. Model of ubiquitin-controlled mediator recruitment to Met4.

During growth with abundant methionine, MET gene expression is repressed through a Met4-attached K48-linked ubiquitin chain. The K48-ubiquitin chain interacts intramolecularly with the K48-selective tandem UBD in Met4 and prevents mediator recruitment to the tandem UBD/transactivation region. The system is activated when cells experience methionine limitation, which induces a change in topology of the ubiquitin chain attached to K163 in Met4. The new topology could be a homotypic K11 chain (A), a branched K48/K11 chain (B), or a heterotypic K48/K11 chain (C). After initiation of activation through changes in ubiquitin chain topology, Met4 deubiquitylation completes the activation process.
The exact composition of the activating K11 linkage enriched ubiquitin chain is not clear. A significant amount of K48 linkages persists during activation, and it is possible that the activating chain is a heterotypic mixed K11/K48 chain as recently reported for mitotic substrates and misfolded proteins in mammals (Yau and Rape, 2016). The Met4 tandem UBD requires a minimum of two consecutive K48 linkages for binding (Tyrrell et al., 2010), it is thus likely that even a heterotypic K11/K48 chain where K11 linkages dominate will not bind to the transactivation/tandem UBD domain and will thus allow mediator recruitment. However, it is also conceivable that a mixed population of Met4 exists during activation, with homotypic K48 or K11 chains. For example, only chromatin bound Met4 may be activated by K11 chains. This notion may be supported by detection of two pools of Met4 with different deubiquitylation dynamics. During acute inactivation of Met4 ubiquitylation using the temperature sensitive met30–6 allele (Kaiser et al., 2000), one fraction of Met4 was rapidly deubiquitylated (<10 min), whereas another fraction maintained the ubiquitylated state for a longer period (Figure S7B).
The lack of K48 and K11 chains on Met4K163R mutants suggests that Met4 suppression and activation depends on single chains with different topologies. The role of the K11 enriched chain on Met4 is most likely restricted to initiation of activation by terminating competition between ubiquitin and mediator for binding to the tandem UBD/transactivation domain. The K11 chain is not required to maintain activity because Met4K163R is fully active (Flick et al., 2004). Whether the K11 chain on Met4 has additional intrinsic functions such as facilitating recruitment of deubiquitylating enzymes or components of the transcription machinery for rapid activation of the Met4 dependent expression program will be interesting questions for future studies.
The Role of Ubiquitin Binding Domains in Signaling
Ubiquitin binding domains are well known as readers and mediators of ubiquitin signaling (Dikic et al., 2009; Gao et al., 2016; Kirkin and Dikic, 2007; Winget and Mayor, 2010). However, beyond mediating protein interactions with ubiquitylated proteins we have little insight of how UBDs mediate signaling. The recent discovery that the tandem UBD of Met4 overlaps with its minimal transactivation domain provided an opportunity to probe mechanisms of ubiquitin signaling (Pacheco et al., 2018).
Here we show that UBDs can be embedded in regions with additional biological functions (transactivation) and that ubiquitin binding can control these overlapping activities. These findings conceptually expand the role of ubiquitin binding in signaling and provide insight into the complexity of the signaling process.
The regulation of Met4 transactivation activity illustrates that ubiquitin chains (K11 and K48) with seemingly similar signaling functions in protein degradation can have very different effects on protein function. Ubiquitin signaling needs to consider protein context and specificity of the involved UBDs, which function as signal reader and interpreters. Ubiquitin chain selectivity of UBDs is likely a central component of signaling processes and changes in chain topologies are prone to mediate and modulate signaling outcomes.
As with all conceptual advances many new interesting questions arise. We currently do not know what ubiquitin ligase or conjugating enzyme is modifying Met4 with a K11 chain. No other ligase than SCFMet30 and its canonical E2, Cdc34, have so far been linked to Met4 regulation. Surprisingly, deletion of MET30 blocked K11 modification. However, these findings may indicate the requirement for a priming ubiquitin attached by Cdc34/SCFMet30 followed by chain extension with K11 linkages catalyzed by an alternative conjugation system. Ubiquitin chain types are specified by orientation of the acceptor ubiquitin in a way that presents a competent acceptor lysine. Orientation can be achieved through monomeric E2s (Wickliffe et al., 2011) or accessory factors analogous to ubiquitin E2 variants (UEV) (Eddins et al., 2006; VanDemark et al., 2001). Future experiments will need to explore whether alternative E2/E3 components or recruitment of accessory factors to Cdc34/SCFMet30 trigger the switch to K11 modification of Met4.
The improved whole proteome analyses applied here to study the role of K11-linked ubiquitin chains provides insight into molecular concepts of ubiquitin signaling and highlights the importance of ubiquitin binding domains and how they are employed to mediate chain type selective functions. Our results also propose a model where ubiquitin binding domains are embedded within domains of distinct biological activities, such as transactivation in the context of Met4, and thereby form the core for signaling processes controlled by ubiquitin.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ping Xu ( xuping@mail.ncpsb.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and culture
The key resource table shows the yeast strains used in this study. SUB592 had all of its endogenous ubiquitin genes deleted and a synthetic ubiquitin gene expressed under the control of the CUP1 promoter in a 2μ plasmid (Finley et al., 1994). The wild type (WT, JMP024) was generated for the ubiquitin gene, and ubiquitin K11R mutant strain (JMP025) was generated for ubiquitin K11R mutant as described previously for SILAC quantitative analysis (Xu et al., 2009b).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-Met4 | Mike Tyers’s lab | N/A |
| α-K11 polyubiquitin linkage (Clone 2A3/2E6) | Genentech | N/A |
| α-Ub P4D1 | Santa Cruz Biotechnology | sc-8017, RRID:AB_628423 |
| α-HA (Y-11 ) | Santa Cruz Biotechnology | sc-805, RRID:AB_631618 |
| α-streptavidin | Abcam | ab7239, RRID:AB_305786 |
| α-tubulin | Abcam | ab210797 |
| HRP-conjugated goat anti-human IgG, Fcγ | Jackson | 109–005-008, |
| fragment specific-488 | ImmunoResearch | RRID: AB_2337534 |
| HRP-conjugated goat α-mouse | Thermo Fisher | P-21129, RRID:AB_2539816 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lys-C | Meizhiyuan | N/A |
| Trypsin | Meizhiyuan | N/A |
| TRIzol reagent | Invitrogen | 15596026 |
| RNase-free DNase I | Promega | M6101 |
| Dynabeads mRNA DIRECT Kit | Invitrogen | 61011 |
| random hexamer primers | SYBR | N/A |
| DNA polymerase I | SYBR | N/A |
| T4 DNA polymerase | SYBR | N/A |
| Pfu polymerase | NEB | N/A |
| [13C6] L-lysine | Cambridge Isotope Labs | CLM-2247-H |
| [13C6/15N4] L-arginine | Cambridge Isotope Labs | CNLM-539-H |
| K11 diubiquitin | Boston Biochem | UC-40B |
| K48 diubiquitin | Boston Biochem | UC-200B |
| K63 diubiquitin | Boston Biochem | UC-300B |
| K11 tetra-ubiquitin chains | Boston Biochem | UC-45 |
| K48 tetra-ubiquitin chains | Boston Biochem | UC-210B |
| Dynabeads™ Protein G for Immunoprecipitation | Life technologies | 10004D |
| Ni-NTA agarose | Qigen | 30250 |
| Immobilon-NC Transfer Membrane | Millipore | HATF04700 |
| Streptavidin Dip and Read biosensors | ForteBio | 18–5019 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma-Aldrich | P7626 |
| N-ethylmaleimide | Sigma-Aldrich | E3876–25G |
| 0.5 mm glass beads | Biospec Products Inc | 1107900–105 |
| Hepes | Fisher Scientific | BP-310 |
| Deoxycholic Acid | Sigma | D-6750 |
| EDTA | Fisher Scientific | S-311 |
| NaCl | Fisher Scientific | S-640 |
| Triton | Fisher Scientific | BP-151 |
| LiCI | Sigma | L-9650 |
| NP-40 | Boston Bioproducts | P-877 |
| Tris | Invitrogen | 15504 |
| L-Methionine | Sigma | M-5308 |
| Formaldehyde | Sigma | F-8775 |
| Glycogen | Acros Organics | 422950010 |
| RNase A | Sigma | R-5503 |
| Proteinase K | Thermo | 26160 |
| Antifoam Y-30 | Sigma | A-5758 |
| Protein A Sepharose/Salmon Sperm blocked | Millipore | 16–157 |
| Microtube screw cap | Olympus plastics | 21–266 |
| 1.5 mL Bioruptor Microtubes with Caps for | ||
| Bioruptor Pico | Diagenode | C30010016 |
| iTaq Universal SYBR Green SuperMix | Biorad | 1725120 |
| Dynabeads™ MyOne™ Streptavidin C1 | Thermo Fisher Shanghai Leon Chemical | 65002 |
| IISGIGVGGIAVLSPMLISEVAPK | Ltd. Shanghai Leon Chemical | N/A |
| TLSDYNIQK (Ub, unmodified 1, Light & Heavy) | Ltd. | N/A |
| LISEEDLGM*QIFVK(GG)TLTGK (Ub, K6 | Shanghai Leon Chemical | |
| oxidative, Light & Heavy) | Ltd. | N/A |
| LISEEDLGMQIFVK(GG)TLTGK (K6, Light & | Shanghai Leon Chemical | |
| Heavy) | Ltd. | N/A |
| TLTGK(GG)TITLEVESSDTIDNVK (K11, Light & | Shanghai Leon Chemical | |
| Heavy) | Ltd. | N/A |
| TITLEVESSDTIDNVK(GG)SK (K27, Light & | Shanghai Leon Chemical | |
| Heavy) | Ltd. Shanghai Leon Chemical | N/A |
| SK(GG)IQDK (K29, Light & Heavy) | Ltd. Shanghai Leon Chemical | N/A |
| IQDK(GG)EGIPPDQQR (K33, Light & Heavy) | Ltd. Shanghai Leon Chemical | N/A |
| LIFAGK(GG)QLEDGR (K48, Light & Heavy) | Ltd. | N/A |
| TLSDYNIQK(GG)ESTLHLVLR (K63, Light & | Shanghai Leon Chemical | |
| Heavy) | Ltd. | N/A |
| Deposited Data | ||
| Raw data | This study | http://www.proteomexchange.org |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background: BY4741 | ATCC | ATCC 4040002 |
| SUB592 | Spence J, et al., 2000 | N/A |
| JMP024 (WT) | Xu P, et al., 2009 | N/A |
| JMP025 (Ub K11R) | Xu P, et al., 2009 | N/A |
| PY283 | Kaiser P, et al., 2000 | N/A |
| PX001 : JMP024, met4::3HA-MET4 | This study | N/A |
| PX002: JMP025, met4::3HA-MET4 | This study | N/A |
| PX003: JMP024, met4::3HA-MET4 K163R | This study | N/A |
| PX004: JMP024, met4::3HA-MET4, met30:: | ||
| natNT2, met32:: hphNTI | This study | N/A |
| PK2308: JMP024 (WT) | ||
| med14::MED14–3HA::KAN | This study | N/A |
| PK2309: JMP025 (K11R) | ||
| med14::MED14–3HA::KAN | This study | N/A |
| Recombinant DNA | ||
| pET28(a) Avi-6xHis-SUMO-Met4 (amino acids | ||
| 76–160) | This study | N/A |
| pBirAcm plasmid coding for BirA | This study | N/A |
| pAL01 base (6xHis- Med15 1–651 Δ239–271, Δ373–483) | This study | N/A |
| Oligonucleotides | ||
| Primers for MET3, see Table S6 | This study | N/A |
| Primers for MET17, see Table S6 | This study | N/A |
| Primers for CYS4, see Table S6 | This study | N/A |
| Primers for SAM2, see Table S6 | This study | N/A |
| Primers for CCW12, see Table S6 | This study | N/A |
| Primers for CDC19, see Table S6 | This study | N/A |
| Primers for 18SrRNA, see Table S6 | This study | N/A |
| Software and Algorithms | ||
| Trans-Proteomic Pipeline | Pedrioli PG, et al., 2010 | N/A |
| Sequest-Sorcerer algorithm (version 4.0.4 build) | Eng JK, et al., 1994 | N/A |
| SILVER | Chang C, et al., 2014 | N/A |
| MaxQuant (1.5.3.0) | Cox, J., et al., 2008 | N/A |
| Xcalibur v2.0 | Thermo Finnigan | N/A |
| ForteBio Data Analysis 9.0 | Pall ForteBio | N/A |
| Other | ||
| Capillary column 75 μm i.d. × 15 cm; C18, 3 μm | Beijing SpectraPeaks | N/A |
To examine ubiquitin linkages on Met4, we deleted endogenous Met4 from JMP024 and replaced it with a G418-resistant gene from pFA6a kanMX4. PX001 was generated from JMP024 (Figure S5A) by PCR-mediated homologous recombination with the plasmid pRS316–3HA–Met4 (a gift from Dr. Laurent Kuras). In the same way, PX002 was generated from JMP025 (Figure S5B).
In general, the yeast strains were grown at 30 °C i n YPD medium (1% yeast extract, 2% Bacto-peptone, and 2% dextrose) to A600 = 1.0 before harvest unless indicated. The protocol for SILAC was described previously (Xu et al., 2009b). Minimal SD medium (0.17% yeast nitrogen base without ammonium sulfate, 0.5% ammonium sulfate, 2% glucose and supplemented with the appropriate amino acids) was used to analyze the expression of Met4 in yeast. The plates were then cultured at 30 °C and recorded as indicated. To estimate the number of yeast cells, we made the assumption that A600 = 1.0 equates to 10 million CFU mL−1.
METHOD DETAILS
Sample preparation and MS analysis
In order to achieve a higher coverage of the yeast proteome, JMP024 (WT) and JMP025 (K11R) strains were cultured in SILAC medium separately. Yeast cells were harvested at the early exponential phase and lysed in a 1.5 mL centrifuge tube with denaturing lysis buffer (8 M urea, 50 mM NH4HCO3, 5 mM iodoacetamide) and 0.5 mm glass beads (Biospec Products Inc., Bartlesville, OK, USA).
Proteins in TCL samples were separated into 34 (for forward experiments) or 35 gel bands (for reverse experiments) on the basis of MW by SDS–PAGE with Coomassie blue staining (Xu et al., 2009a). Each band was in-gel digested with endoproteinase Lys-C overnight to generate short peptides. After digestion, the peptide mixture was suspended in an extraction buffer (5% FA, 45% ACN) and dried using a vacuum dryer (LABCONCO CentriVap, MO, USA).
Peptides were analyzed using an LC–MS/MS platform comprising a nanoACQUITY ultra-performance liquid chromatography system (Waters, Milford, MA, USA) and an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). The peptide mixtures were loaded onto a 75 μm i.d. × 15 cm capillary column (Beijing SpectraPeaks, Beijing, China) packed with 3 μm C18 reverse-phase fused silica (Michrom Bioresources, Inc., Auburn, CA, USA) and eluted with a 60–140 min nonlinear gradient ramped from 8–40% of mobile phase B (phase B: 0.1% FA in ACN, phase A: 0.1% FA and 2% ACN in water) at a flow rate of 0.3 μL min−1. Eluting peptides were analyzed using the LTQ Orbitrap Velos mass spectrometer. The MS1 precursor was analyzed with a mass range 350–1,800 at a resolution of 30,000 at m/z = 400. The automatic gain control (AGC) was set at 1 × 106 and the maximum injection time (MIT) was set at 150 ms. MS2 spectra were collected in data-dependent mode for the 20 most intense ions, which were subjected to fragmentation via collision induced dissociation (CID) with 35% normalized collision energy in the LTQ. For each scan, the AGC was set at 5,000 and the maximum injection time at 25 ms. The dynamic range was set at 30–60 s to suppress repeated fragmentation of the same peaks.
Proteomics database searching
All raw files were converted into mzXML using Trans-Proteomic Pipeline (version 4.5.2) (Pedrioli, 2010), and searched by the Sequest–Sorcerer algorithm (version 4.0.4 build, Sage-N Research, Inc., San Jose, CA, USA) (Eng et al., 1994) against the combined target-decoy protein FASTA file from the Saccharomyces genome database (version released in 2011.02, 6717 entries, SGD, http://www.yeastgenome.org) along with 112 common contaminants (ftp.thegpm.org/fasta/cRAP). The decoy components were constructed by pseudo-reversed sequences of all target proteins (Elias and Gygi, 2007; Peng et al., 2003a).
For identification, semi-digestion of Lys-C was allowed with a maximum of two missed cleavages. Precursor mass tolerance was 20 ppm. Carbamidomethylation of cysteine was specified as a fixed modification. Oxidation of methionine and the labeling of 13C6 of lysine were added as variable modifications and the labeling efficiency of heavy-labeled lysine reached up to 99% in the yeast. A minimal peptide length of seven amino acids was required. The search results were processed using an in-house software (Xu et al., 2009b) with FDR < 1% on peptide and protein level. As peptides are targets of the shotgun proteomics strategy, it is difficult to guarantee that the proteins with highly homologous amino acid sequences will be unambiguously identified by unique peptides to all of the proteins within such high homology groups. When matching filtered peptides to proteins, we assigned the proteins sharing the same peptide(s) in one group, in which the top protein with the highest number of peptide matches was selected to represent the group. If multiple proteins in the same group had the same sequence coverage, one of them was randomly selected as representative of the identification. Therefore, the other ones were assigned as missing proteins, even though these proteins had identified peptides. After that, the spectra generated from the proteins with only one identified peptide were manually checked to confirm the identification produced via automated processing of our MS data.
After identification, data from our SILAC label-swap experiments were analyzed through our in-house software as described previously (Xu et al., 2009b). P-value was calculated for significantly changed proteins in WT and K11R (Figure 2A) (Cox and Mann, 2008). Only proteins with P-value < 0.01 were considered as significantly changed proteins in the proteome. The other proteins were considered as unchanged proteins. The same rule was applied on the analysis of significantly changed transcripts.
For protein copy number quantification, the area under the extracted ion chromatograms (XICs) for fully digested peptides in label-swap samples was calculated using SILVER (Chang et al., 2014). The intensity of a peptide was first normalized by the median of all peptide intensities in the corresponding sample, then the geometric mean of the intensities from four samples was calculated as the final intensity for each peptide. The mean and standard deviation of the intensity of each unique peptide from each corresponding sample was calculated. The peptides with intensity beyond the mean by at least ± 2 standard deviations were removed as outliers. The sum of the remaining peptides was divided by the protein MW as the final intensity of each protein. With the help of high sequence coverage identification, the influence of the bias of peptide detectability by MS/MS (Mallick et al., 2007) on quantification can be corrected to some extent. The correlation of transcript abundance and protein intensity reached up to 0.65, indicating that the abundance of proteins is mainly affected by the levels of corresponding mRNAs (Marguerat et al., 2012).
Bioinformatics analysis of identified peptides and proteins
Protein information, including gene symbol, chromosome loci, gene model and modifications, was mainly extracted from SGD annotations. Part of the ubiquitination annotations came from the database UUCD 2.0 (http://uucd.biocuckoo.org) (Gao et al., 2013). Four representative proteome datasets, namely tandem affinity purification (TAP) (Ghaemmaghami et al., 2003), green fluorescent protein (GFP) (Huh et al., 2003), PeptideAtlas (Deutsch et al., 2008) and Mann 2008 (de Godoy et al., 2008), were compared with our proteome dataset (Figures 1D and S1F). According to the SGD annotations, all proteins was classified into three catalogs including “Core”, “Hypothetical” and “Dubious”. “Core” proteins refer to verified ORFs or uncharacterized ORFs with essential function. “Hypothetical” proteins refer to putative or hypothetical uncharacterized ORFs. “Dubious” proteins refer to dubious ORFs. Among the 26 proteins annotated as the products of pseudogene, only four were present in our dataset (Figure S2D). Among the remaining 22 pseudogenes, only one was covered by the TAP dataset, and the other 21 were missed in all the published datasets (Figure S2E).
Mitosis annotations (Figure S3D) were extracted from database MiCroKiTS 3.0 (http://microkit.biocuckoo.org) (Ren et al., 2010). A Venn diagram was drawn using the online tool jvenn (http://bioinfo.genotoul.fr/jvenn/example.html) (Bardou et al., 2014).
Synthesized peptides for validation of pseudogenes
Peptides for validation of identified pseudogenes were synthesized by Shanghai Leon Chemical Ltd. (Shanghai, China). The peptides (0.1–1 pmol) were dissolved in a buffer containing 5% ACN and 1% FA and desalted with a homemade StageTip as described previously (Zhai et al., 2013). The peptide samples were analyzed with LC–MS/MS as described above (Figure S2E).
RNA extraction, RNA-seq and data analysis
The same batches of yeast cultures (WT and K11R) as for proteomics analysis were used for RNA extraction. Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and treated with RNase-free DNase I (Promega, Madison, WI, USA). Poly(A) RNA was purified with the Dynabeads mRNA DIRECT Kit (Invitrogen, Carlsbad, CA, USA) from 10 μg of total RNA for each sample according to the manufacturer’s instructions, and then used to synthesize cDNA using random hexamer primers and DNA polymerase I. Double-stranded cDNA was end-repaired with T4 DNA polymerase, after which a single adenosine was added to the 3’ end of cDNA. Illumina adapters were ligated to the repaired cDNA ends, allowing for the subsequent hybridization to the flow cell surface. Fragments of 400–500 bp were separated from 2% agarose gel. Libraries were amplified by PCR with Pfu polymerase (NEB, Beverly, MA, USA). All libraries were sequenced using the Illumina HiSeq 2000. Each sample was sequenced to 5 M reads.
RNA sequencing reads were mapped to Saccharomyces Genome Database (version released in 2011.02, 6717 entries, SGD, http://www.yeastgenome.org/download-data). In each sample, genes with more than 5 mapped reads were deemed as high confident identifications. The RNA-seq data was processed using algorithms built into tophat and cufflinks (Bloom et al., 2009). The number of fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) was calculated to quantify each gene transcript. RNA-seq analysis achieved saturation and identified slightly more protein-coding genes than the MS-based proteome, which proved the high depth of our transcriptomic dataset.
Protein half-life analysis
Protein half-lives were performed mainly described as previously (Christiano et al., 2014; Schwanhausser et al., 2011). The yeast JMP024 and JMP025 strains were heavily labelled in SC medium with 30 mg/L [13C6] L-lysine and 20 mg/L L-arginine [13C6/15N4] (Cambridge Isotope Labs, Tewksbury, MA, USA). After completely SILAC labeling, cells were washed with cold SC medium without lysine and arginine and diluted to optical density OD=0.3 (A600) in 200 mL SC medium containing light lysine and arginine. The cells were harvested at 0, 5, 15, 30, 60, 120, and 240 minutes. Then the samples were lysed in denatured condition (8M urea, 50mM NH4HCO3 and digested in solution with Lys-C for 6 hr and then with trypsin overnight. The peptide sample was not fractionated and analyzed with LC-MS/MS with technical replication. The MS raw files were analyzed with MaxQuant (1.5.3.0) and at least two points were used to calculate decay rates as described (Christiano et al., 2014). The coefficient of determination linear regressions (R2) were calculated and protein with R2≥0.9 were kept for half-life calculation.
Protein extraction and western blot assay
TCL samples were prepared under denaturing condition in a urea buffer (8 M urea, 100 mM Na2HPO4, 10 mM Tris, 5 mM iodoacetamide, 1 mM PMSF, 5 mM N-ethylmaleimide, pH 8.0). Cells were broken with glass beads for 5 × 20 s in a Bertin precellys™ 24 homogenizer (Bertin Technologies, France) with 1 min breaks on ice between runs. Cell debris was removed by centrifugation for 25 min at 100,000 g. Proteins in TCL samples were separated by a 10% SDS–PAGE.
For western blot, separated proteins were transferred to an NC membrane and probed with the monoclonal mouse Met4 antibody from Mike Tyers (University of Montreal)) (1:1000 dilution), followed by HPR-conjugated anti-mouse secondary antibody (1:10,000 dilution). The signal was detected by enhanced chemiluminescence (PerkinElmer life Science, MA, USA).
To confirmed the specificity of anti-K11 polyubiquitin linkage-specific antibody, we took equal amount of K11-linked diubiquitin (UC40B, Boston Biochem, Cambridge, MA, USA), K48-linked diubiquitin (UC200B, Boston Biochem, Cambridge, MA, USA) and K63-linked diubiquitin (UC300B, Boston Biochem, Cambridge, MA, USA) and loaded onto NC membrane as well, probed with the human anti-K11 polyubiquitin linkage-specific antibody (Clone 2A3/2E6, Genentech, South San Francisco, CA, USA) (1:500 dilution), followed by HPR-conjugated anti-human secondary antibody (1:10,000 dilution). The signal was detected by enhanced chemiluminescence (PerkinElmer life Science, MA, USA).
Purification of 3 x HA tagged Met4
Yeast cells were broken by glass beads and total protein was extracted in a native Tris buffer (50 mM Tris, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 5% glycerol, 1 mM PMSF, 5 mM N-ethylmaleimide, pH 7.5) in a Bertin precellys™ 24 homogenizer as described previously. Cell debris was removed by centrifugation at 13,000 g for 10 min. The monoclonal mouse HA antibody (HA.11; Covance, Dedham, MA, USA) was added to the cell extract. Samples were incubated at 4 °C for 2 hr, and washed the beads four times with native Tris buffer after binding to protein G agarose (Life technologies, Rockford, IL, USA) for 2 h at 4 °C. T hen bound 3HA-tagged Met4 was eluted with SDS–PAGE loading buffer (80 °C, 5 min) containing 5 mM DTT to eliminate the link between the light- and heavy-chain in antibodies.
To remove the contaminants from the immunoprecipitated Met4 under native conditions, we performed TAP on Met4 (Burr et al., 2013). Briefly, 3HA-tagged Met4 in yeast TCL samples was bound to the HA antibody and protein G agarose as mentioned previously. After washing four times with native Tris buffer, the beads were re-suspended by eluting buffer (10 mM Tris, 0.5% SDS, pH 7.5), and boiled at 95 °C for 5 min. The supernatant containing enriched Met4 was transferred to a new tube. The solution was then diluted with 10 mM Tris (pH 7.5) to a final concentration of 0.02% SDS. The Met4 was then reacted with the same monoclonal mouse HA antibody again at 4 °C for 2 h. The 3HA-tagged Met4–HA antibody complex was bound to protein G agarose for 2 h at 4 °C. After washing by native tris buffer, the purified Met4 was eluted with SDS–PAGE loading buffer and then separated by SDS–PAGE, digested by trypsin and analyzed by LC–MS/MS. On the other hand, separated proteins were transferred to an NC membrane and probed with the human anti-K11 polyubiquitin linkage-specific antibody (Clone 2A3/2E6, Genentech, South San Francisco, CA, USA) (1:500 dilution), followed by HPR-conjugated anti-human secondary antibody (1:10,000 dilution). The signal was detected by enhanced chemiluminescence (PerkinElmer life Science, MA, USA). After that, washed off all the antibody bound on the NC membrane with stripping buffer for 30min. then probed with monoclonal mouse HA antibody (HA.11; Covance, Dedham, MA, USA) (1:1000 dilution), followed by HPR-conjugated anti-mouse secondary antibody (1:10,000 dilution). The signal was detected by enhanced chemiluminescence (PerkinElmer life Science, MA, USA) as well.
Targeted detection of polyUb linkages on Met4
To measure constitution and relative abundance of ubiquitin chains modified on HA–Met4, seven kinds of heavy-labeled GG-peptide (GG-remnant modified lysine peptides, standing for different ubiquitin linkage modification) were used as standards for precise identification and relative quantification. After TAP, the resulting peptide samples were dissolved in a sample buffer (1% FA, 1% ACN) and analyzed by LC–MS/MS platform mentioned above. Eluted peptides were detected by the LTQ Orbitrap Velos mass spectrometer in a survey scan (300–1600 m/z, resolution 30,000) followed by selective reaction monitoring (SRM) scans for light- and heavy-labeled GG-peptides in the LTQ (Xu et al., 2006; Xu et al., 2009b). The peptides for SRM are provided in the key resource table. The intensities of peptides were analyzed by ion chromatograms using Xcalibur v2.0 software (Thermo Finnigan, San Jose, CA, USA). Three biological repeats were applied in this study.
Purification of truncated Met4 and Med15
The fragment of yeast Met4 (amino acids 76–160) was expressed from a modified pET28(a) vector with an N-terminal Avi-6xHis-SUMO tag. BL21 (DE3) cells were cotransformed with the Met4 expression construct and a pBirAcm plasmid coding for BirA for biotinylation of the Avi tag. Protein expression was induced with 0.5 mM IPTG in the presence of 5 ug/ml biotin at 16 °C overnight. Cell s were harvested, resuspended in lysis buffer (50 mM Tris, 500 mM NaCl, 2 mM PMSF, 5 mM imidazole, and 0.1% triton X, pH8.0), sonicated on ice, and the lysate was centrifuged at 4 °C, 11,000 g for 20 min. The supernatant was incubated with Ni-NTA resin (Qigen, Dusseldorf, Germany) for 1 hr at 4 °C with constant tumbling. The Ni-NTA resin was l oaded on to a column and washed with 50 mM Tris (pH 8.0), 500 mM NaCl, and 20 mM imidazole followed by elution with 4 times 500 μL of 50 mM Tris (pH 8.0), 500 mM NaCl, and 250 mM imidazole. Samples were separated on a 12% SDS-PAGE and then transferred to Immobilon-NC Transfer Membrane (Millipore, MA, USA). Amido black staining was used to determine purification and Western blot, and streptavidin-HRP was used to detect biotin incorporation. Fractions containing biotinylated Met4 (amino acids 76–160) were combined and concentrated on an Amicon Ultra Centrifugal Filter.
Yeast Med15 construct, pAL01(6xHis- Med15 1–651 Δ239–271, Δ373–483) in BL21 (DE3) RIL, was provided to us by Derek Pacheco as described (Pacheco et al., 2018; Tuttle et al., 2018). Cells were grown in YT media with ampicillin at 37 °C to an OD600 of 0.8 and the temperature was reduced to 16 °C. T he protein expression was induced with 0.5 mM IPTG at 16 °C overnight. Cells were harvested, resuspended in lysis buffer (50 mM HEPES, 500 mM NaCl, 5 mM imidazole, 10% glycerol, 1 mM DTT, and 1 mM PMSF, pH 7.0), sonicated on ice, and the lysate was centrifuged at 4 °C 12000 rpm for 30 mins. The supernatant was incubated with Ni-NTA resin (GE Healthcare, PA, USA) for 1 h at 4 °C with constant tumbling. The Ni-NTA resin was loaded on to a column and washed with 50 mM HEPES (pH 7.0), 500 mM NaCl, 25 mM imidazole, 10% glycerol, and 1 mM DTT followed by elution with 5 times 500 μL of 50 mM HEPES (pH 7.0), 500 mM NaCl, 250 mM imidazole, 10% glycerol, and 1 mM DTT. Samples were separated on a 10% SDS-PAGE and then transferred to Immobilon-NC Transfer Membrane (Millipore, MA, USA). Amido black staining was used to determine purification. Pure fractions were combined and concentrated on an Amicon Ultra Centrifugal Filter.
Expression analyses using NanoString
For Nanostring analysis, RNA was isolated from frozen pellets with RNeasy™Plus Mini Kit (Qiagen, Düsseldorf, Germany) as per manufacturer’s protocol. 100 ng of RNA of three biological replicates were processed on a NanoString nCounter (NanoString Technologies, Seattle, WA, USA) with a costume code set (Table S5) according to the manufacturer’s protocol. The code set contains 21 Met4 target genes (Lee et al., 2010) as well as three housekeeping genes of different abundance for normalization. Detailed information about the code set can be obtained on request. The results were analyzed using nSolver (4.0 software).
Biolayer Interferometry
The binding affinity of tetra-ubiquitin chains (K11 and K48, Boston Biochem, Cambridge, MA, USA) and mediator component Med15 to Met4(76–160) was determined by using ForteBio Octet RED96 (Pall ForteBio LLC, Fremont, CA, USA). All proteins were diluted in assay buffer (50 mM Tris, 150 mM NaCl, 0.01 mg/mL BSA, and 0.02% tween 20, pH7.5) as follows: Met4(76–160) final concentration 20 μg/mL, the quenching solution (biocytin) was diluted to 5 μg/mL, and the tetra-ubiquitin chains were diluted to a concentration range between 1.45 μM – 0.0028 μM. Streptavidin Dip and Read biosensors (ForteBio, CA, USA) were hydrated with assay buffer for 10 min prior to the experiment. The monitoring conditions were as follows: Initial baseline for 60 s, loading for 120 s, quenching for 120 s, baseline for 30 s, association for 30 s, dissociation for 60 s; shake speed 1,000 rpm, and plate temperature 30° C. To de termine binding parameters the data were analyzed using ForteBio Data Analysis 9.0. The following concentrations of proteins were used: K48 tetraubiquitin [nM]: 1450, 483, 161, 53, 17, and 5.6; Med15 [nM]: 1000, 500, 250, and 125.
Tetraubiquitin and Med15 binding to Met4
Purified Met4 (residues 76 to 160) was immobilized on magnetic streptavidin beads (Dynabeads™ Streptavidin C1, Thermo Fisher, San Jose, CA, USA). K48 tetraubiquitin (Tetra-Ubiquitin/Ub4 WT Chains, Boston Biochem, Cambridge, MA, USA) was bound to immobilized Met4 in binding buffer (20mM Tris, 100 mM NaCl, 0,05% NP40, pH 7.5) for 4 hr at room temperature. Beads were washed 3 times with 150 μL binding buffer, resuspended in binding buffer and distributed to 5 different tubes. The Met4/K48 tetraubiquitin complexes were then incubated for 1 h in a total volume of 10 μL containing either no Med15, or the following molar ratios compared to Met4 (Met4/Med15: 0.25, 0.5, 1, 3). The supernatant was analyzed for replaced K48 by immunoblotting. Proteins bound to Met4 were analyzed after 2 washes of the bead bound complexes with 150 μL binding buffer and elution in SDS loading buffer by immuno blotting.
ChIP analysis of recruitment to MET gene promoters
The targeted genes primers and materials used for ChIP was listed in the key resource table and Table S6. Wild-type, K11R, and Δmet4 mutants were used for analysis and the mediator binding was monitored by ChIP using the endogenously HA3-tagged mediator subunit Med14. Four Met4-dependent genes (MET3, MET17, CYS4, SAM2) and two Met4-independent genes (CCW12, CDC19) were analyzed.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical data are from three independent experiments. Results are shown as mean ± SEM unless mentioned otherwise. Statistical analysis was performed by the Student t test for forward and reverse SILAC-based quantification. MS data were analyzed with Xcalibur 2.2 (Thermo Software).
DATA AND SOFTWARE AVAILABILITY
The proteomics data supporting this study is available from ProteomeXchange (http://www.proteomexchange.org) (Vizcaino et al., 2014) with ID PXD001928.
Supplementary Material
Annotations for proteins and protein-coding genes
List of proteins quantified in this experiment
Global protein half life analysis
List of 3HA-tagged Met4 proteins
Nanostring analysis RNAs of Met4 target genes
Primers used for ChIP
Highlights.
Lack of K11 ubiquitin chains affects transcription of methionine pathway enzymes
A K48 ubiquitin chain on the transcription factor Met4 prevents mediator binding
Met4 activation is initiated by a change from K48 to K11 linkages on Met4
K11 linkages do not compete with mediator binding and allow transcription
ACKNOWLEDGMENTS
We are indebted to Dr. Junmin Peng for support in the early stage of this project. We thank Steve Hahn, Simin He, Matthew J Higgins, Jin-Kwang Kim, Don Kirkpatrick, Laurent Kuras, Lanlan Li, Hui Jiang, Feng Qiao, Hengliang Wang, Qi Xie, T. Yao, Zhihu Zhao, Na Su and Li Zhu for gracious gifts of their reagents, help with experiments, and discussion. We thank Henning Hermjakob, Siqi Liu, and Jun Qin for critical reading and editing. This work was funded by the MOST (2017YFC0906600, 2017YFA0505002, 2017YFA0505100 & 2016YFA0501300), the National Natural Science Foundation of China (91839302, 31670834, 31700723 & 31870824), Innovation Foundation of Medicine (16CXZ027, BWS17J032 & BWS14J052), National Megaprojects for Key Infectious Diseases (2018ZX10302302001), the Foundation of State Key Lab of Proteomics (SKLP-Y201501 & SKLP-K201705), Guangzhou science and technology innovation & development project (201802020016), the Hitachi-Nomura Award to L. L., and the National Institute of Health (R01 GM-066164 to P.K. and T32 CA09054 to MA.V.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and six tables can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aghajan M, Jonai N, Flick K, Fu F, Luo M, Cai X, Ouni I, Pierce N, Tang X, Lomenick B, et al. (2010). Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotechnol 28, 738–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, and Eppig JT (2000). Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbey R, Baudouin-Cornu P, Lee TA, Rouillon A, Zarzov P, Tyers M, and Thomas D (2005). Inducible dissociation of SCF(Met30) ubiquitin ligase mediates a rapid transcriptional response to cadmium. EMBO J 24, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardou P, Mariette J, Escudie F, Djemiel C, and Klopp C (2014). jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JS, Khan Z, Kruglyak L, Singh M, and Caudy AA (2009). Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays. BMC Genomics 10, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr ML, van den Boomen DJ, Bye H, Antrobus R, Wiertz EJ, and Lehner PJ (2013). MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proceedings of the National Academy of Sciences 110, 14290–14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Zhang J, Han M, Ma J, Zhang W, Wu S, Liu K, Xie H, He F, and Zhu Y (2014). SILVER: an efficient tool for stable isotope labeling LC-MS data quantitative analysis with quality control methods. Bioinformatics 30, 586–587. [DOI] [PubMed] [Google Scholar]
- Christiano R, Nagaraj N, Frohlich F, and Walther TC (2014). Global proteome turnover analyses of the Yeasts S. cerevisiae and S. pombe. Cell Rep 9, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- de Godoy LM, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, and Mann M (2008). Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254. [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Lam H, and Aebersold R (2008). PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO reports 9, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S, and Walters KJ (2009). Ubiquitin-binding domains - from structures to functions. Nature reviews Molecular cell biology 10, 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins MJ, Carlile CM, Gomez KM, Pickart CM, and Wolberger C (2006). Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nature structural & molecular biology 13, 915–920. [DOI] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Finley D, Sadis S, Monia BP, Boucher P, Ecker DJ, Crooke ST, and Chau V (1994). Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol 14, 5501–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T, and Kaiser P (2012). The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick K, Ouni I, Wohlschlegel JA, Capati C, McDonald WH, Yates JR, and Kaiser P (2004). Proteolysis-independent regulation of the transcription factor Met4 by a single Lys 48-linked ubiquitin chain. Nat Cell Biol 6, 634–641. [DOI] [PubMed] [Google Scholar]
- Flick K, Raasi S, Zhang H, Yen JL, and Kaiser P (2006). A ubiquitin-interacting motif protects polyubiquitinated Met4 from degradation by the 26S proteasome. Nat Cell Biol 8, 509–515. [DOI] [PubMed] [Google Scholar]
- Gao T, Liu Z, Wang Y, Cheng H, Yang Q, Guo A, Ren J, and Xue Y (2013). UUCD: a family-based database of ubiquitin and ubiquitin-like conjugation. Nucleic Acids Res 41, D445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Li Y, Zhang C, Zhao M, Deng C, Lan Q, Liu Z, Su N, Wang J, Xu F, et al. (2016). Enhanced Purification of Ubiquitinated Proteins by Engineered Tandem Hybrid Ubiquitin-binding Domains (ThUBDs). Mol Cell Proteomics 15, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, and Weissman JS (2003). Global analysis of protein expression in yeast. Nature 425, 737–741. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, and O’Shea EK (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. [DOI] [PubMed] [Google Scholar]
- Kaiser P, Flick K, Wittenberg C, and Reed SI (2000). Regulation of transcription by ubiquitination without proteolysis: Cdc34/SCF(Met30)-mediated inactivation of the transcription factor Met4. Cell 102, 303–314. [DOI] [PubMed] [Google Scholar]
- Kirkin V, and Dikic I (2007). Role of ubiquitin- and Ubl-binding proteins in cell signaling. Current opinion in cell biology 19, 199–205. [DOI] [PubMed] [Google Scholar]
- Komander D, and Rape M (2012). The ubiquitin code. Annu Rev Biochem 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Kuras L, Rouillon A, Lee T, Barbey R, Tyers M, and Thomas D (2002). Dual regulation of the met4 transcription factor by ubiquitin-dependent degradation and inhibition of promoter recruitment. Mol Cell 10, 69–80. [DOI] [PubMed] [Google Scholar]
- Lee TA, Jorgensen P, Bognar AL, Peyraud C, Thomas D, and Tyers M (2010). Dissection of combinatorial control by the Met4 transcriptional complex. Mol Biol Cell 21, 456–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy C, Cormier L, and Kuras L (2006). Independent recruitment of mediator and SAGA by the activator Met4. Mol Cell Biol 26, 3149–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Adams RM, Chourey K, Hurst GB, Hettich RL, and Pan C (2012). Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J Proteome Res 11, 1582–1590. [DOI] [PubMed] [Google Scholar]
- Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, et al. (2007). Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol 25, 125–131. [DOI] [PubMed] [Google Scholar]
- Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, and Bahler J (2012). Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M, et al. (2010). K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell 39, 477–484. [DOI] [PubMed] [Google Scholar]
- Meierhofer D, Wang X, Huang L, and Kaiser P (2008). Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J Proteome Res 7, 4566–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei H, Rogers RS, Grimes B, Eng J, Aderem A, and Aebersold R (2010). Characterizing the connectivity of poly-ubiquitin chains by selected reaction monitoring mass spectrometry. Mol Biosyst 6, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndoja A, Cohen RE, and Yao T (2014). Ubiquitin signals proteolysis-independent stripping of transcription factors. Mol Cell 53, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouni I, Flick K, and Kaiser P (2010). A transcriptional activator is part of an SCF ubiquitin ligase to control degradation of its cofactors. Mol Cell 40, 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouni I, Flick K, and Kaiser P (2011). Ubiquitin and transcription: The SCF/Met4 pathway, a (protein-) complex issue. Transcription 2, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco D, Warfield L, Brajcich M, Robbins H, Luo J, Ranish J, and Hahn S (2018). Transcription Activation Domains of the Yeast Factors Met4 and Ino2: Tandem Activation Domains with Properties Similar to the Yeast Gcn4 Activator. Mol Cell Biol 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Peyraud C, Rouillon A, Surdin‐Kerjan Y, Tyers M, and Thomas D (2000). SCFMet30‐mediated control of the transcriptional activator Met4 is required for the G1–S transition. The EMBO Journal 19, 1613–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrioli PG (2010). Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods in molecular biology (Clifton, NJ) 604, 213–238. [DOI] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, and Gygi SP (2003a). Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2, 43–50. [DOI] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, and Gygi SP (2003b). A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21, 921–926. [DOI] [PubMed] [Google Scholar]
- Picotti P, Bodenmiller B, Mueller LN, Domon B, and Aebersold R (2009). Full Dynamic Range Proteome Analysis of S. cerevisiae by Targeted Proteomics. Cell 138, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu Z, Gao X, Jin C, Ye M, Zou H, Wen L, Zhang Z, Xue Y, and Yao X (2010). MiCroKit 3.0: an integrated database of midbody, centrosome and kinetochore. Nucleic Acids Res 38, D155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Swatek KN, and Komander D (2016). Ubiquitin modifications. Cell research 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, and Surdin-Kerjan Y (1997). Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiology and molecular biology reviews : MMBR 61, 503–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle LM, Pacheco D, Warfield L, Luo J, Ranish J, Hahn S, and Klevit RE (2018). Gcn4-Mediator Specificity Is Mediated by a Large and Dynamic Fuzzy Protein-Protein Complex. Cell Rep 22, 3251–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell A, Flick K, Kleiger G, Zhang H, Deshaies RJ, and Kaiser P (2010). Physiologically relevant and portable tandem ubiquitin-binding domain stabilizes polyubiquitylated proteins. Proc Natl Acad Sci U S A 107, 19796–19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDemark AP, Hofmann RM, Tsui C, Pickart CM, and Wolberger C (2001). Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer. Cell 105, 711–720. [DOI] [PubMed] [Google Scholar]
- Varshavsky A (2012). The ubiquitin system, an immense realm. Annu Rev Biochem 81, 167–176. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, and Rape M (2011). The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell 144, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winget JM, and Mayor T (2010). The diversity of ubiquitin recognition: hot spots and varied specificity. Mol Cell 38, 627–635. [DOI] [PubMed] [Google Scholar]
- Xu P, Cheng D, Duong DM, Rush J, Roelofs J, Finley D, and Peng J (2006). A proteomic strategy for quantifying polyubiquitin chain topologies. Israel Journal of Chemistry 46, 171–182. [Google Scholar]
- Xu P, Duong DM, and Peng J (2009a). Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res 8, 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, and Peng J (2009b). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R, and Rape M (2016). The increasing complexity of the ubiquitin code. Nat Cell Biol 18, 579–586. [DOI] [PubMed] [Google Scholar]
- Yen JL, Flick K, Papagiannis CV, Mathur R, Tyrrell A, Ouni I, Kaake RM, Huang L, and Kaiser P (2012). Signal-induced disassembly of the SCF ubiquitin ligase complex by Cdc48/p97. Mol Cell 48, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen JL, Su NY, and Kaiser P (2005). The yeast ubiquitin ligase SCFMet30 regulates heavy metal response. Mol Biol Cell 16, 1872–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L, Chang C, Li N, Duong DM, Chen H, Deng Z, Yang J, Hong X, Zhu Y, and Xu P (2013). Systematic research on the pretreatment of peptides for quantitative proteomics using a C(1)(8) microcolumn. Proteomics 13, 2229–2237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotations for proteins and protein-coding genes
List of proteins quantified in this experiment
Global protein half life analysis
List of 3HA-tagged Met4 proteins
Nanostring analysis RNAs of Met4 target genes
Primers used for ChIP
Data Availability Statement
The proteomics data supporting this study is available from ProteomeXchange (http://www.proteomexchange.org) (Vizcaino et al., 2014) with ID PXD001928.