

Abstract
RNA interference (RNAi) plays key roles in post-transcriptional and chromatin modification levels as well as regulates various eukaryotic gene expressions which are involved in stress responses, development and maintenance of genome integrity during developmental stages. The whole mechanism of RNAi pathway is directly involved with the gene-silencing process by the interaction of Dicer-Like (DCL), Argonaute (AGO) and RNA-dependent RNA polymerase (RDR) gene families and their regulatory elements. However, these RNAi gene families and their sub-cellular locations, functional pathways and regulatory components were not extensively investigated in the case of economically and nutritionally important fruit plant sweet orange (Citrus sinensis L.). Therefore, in silico characterization, gene diversity and regulatory factor analysis of RNA silencing genes in C. sinensis were conducted by using the integrated bioinformatics approaches. Genome-wide comparison analysis based on phylogenetic tree approach detected 4 CsDCL, 8 CsAGO and 4 CsRDR as RNAi candidate genes in C. sinensis corresponding to the RNAi genes of model plant Arabidopsis thaliana. The domain and motif composition and gene structure analyses for all three gene families exhibited almost homogeneity within the same group members. The Gene Ontology enrichment analysis clearly indicated that the predicted genes have direct involvement into the gene-silencing and other important pathways. The key regulatory transcription factors (TFs) MYB, Dof, ERF, NAC, MIKC_MADS, WRKY and bZIP were identified by their interaction network analysis with the predicted genes. The cis-acting regulatory elements associated with the predicted genes were detected as responsive to light, stress and hormone functions. Furthermore, the expressed sequence tag (EST) analysis showed that these RNAi candidate genes were highly expressed in fruit and leaves indicating their organ specific functions. Our genome-wide comparison and integrated bioinformatics analyses provided some necessary information about sweet orange RNA silencing components that would pave a ground for further investigation of functional mechanism of the predicted genes and their regulatory factors.
Introduction
In multicellular eukaryotes, wide range of biological functions including genome rearrangement, antiviral defense, heterochromatin formation and development patterning and timing are fine-tuned by generally two types of small RNA (sRNA; including 21–24 nucleotides), named microRNA (miRNA) and short interfering RNA (siRNA) [1–3]. These sRNA molecules are involved in both transcriptional and post-transcriptional gene silencing as well as natural immunity system [2, 4–7]. In plants, the sRNA biogenesis process is significantly regulated by the proteins encoded by respective Dicer-like (DCL), Argonate (AGO) and RNA-dependent RNA polymerases (RDR) gene families. In plants, RDRs are inevitable gene silencing members that synthesize dsRNA by using RNA template and actually intensify the gene silencing signals [8–13]. The DCLs are responsible for the cleavage of dsRNAs into 21–24 nucleotide long small RNAs (i.e. siRNA or miRNA). The specification to the endonuclease-containing, RNA-induced silencing complex (RISC) is provided by these sRNAs which facilitate the AGO proteins with RNaseH-type activities to degrade the target homologous RNAs with the sequence complementary to the small RNAs [14, 15]. These are also involved in the transcriptional gene silencing by the implementation of chromatin reformation [16, 17].
DCL proteins, which mainly process the small mature RNAs from the long double-stranded RNAs [18–22] are a major component of RNA interference (RNAi) pathway (also known as small RNA (sRNA) biogenesis process). The DCL proteins have the functional domains, named DEAD/ResIII, Helicase_C, Dicer_Dimer, PAZ, RNase III and DSRM [23] which play an important role for the proteins to be functional. The PAZ domain acts to bind the siRNA as well as the dsRNA which is cleaved by the two catalytic RNaseIII domains. The main components of RNAi are the AGO proteins which play the core role of gene silencing [24]. All the AGO proteins include the Argo-N/Argo-L, PAZ, MID and PIWI significant functional domains [14]. A significant specific binding pocket is contained in the PAZ domain. Additionally, to anchor the sRNA onto the AGO proteins, the specific pocket of MID domain binds the 5' phosphate of the small RNAs [25]. The siRNA 5' end is bonded to the target RNA by the PIWI domain [26]. Among the three groups of AGO proteins i.e. Ago -like, PIWI-like and C. elegens-specific group 3 AGO proteins [27, 28], the AGO-like proteins are presented and expressed in plants, animals, fungi and bacteria, while PIWI-like proteins have only been found in animals [29]. Some important catalytic residues are missed by C. elegens -specific group 3 AGO proteins [27] while the other AGOs conserved them and the expression of PIWI-like group is restricted in human germ-cell and in rat and some mammals [29]. The third major RNAi associated protein is RDR which has not been identified in insects or vertebrates [30] but is present in fungi, nematodes and plants. The only special conserved catalytic RNA-dependent RNA polymerase (RdRP) domain is shared by the RDR which makes the RDR proteins a consistent member of RNAi gene family [31–33]. Among the three types of RDR: RDRα, RDRβ and RDRγ, the RDRβ group has not been found in plants [31, 32].
In case of plants, the DCL, AGO and RDR gene families related to distinct RNAi pathways [34–36] vary from 20 genes in Arabidopsis [37] to 51 genes in Brassica [38] species. The member of these RNAi associated gene families has been identified in many plants species such as 32 genes in rice [14], 28 genes in maize [39] and tomato [33], 38 genes in foxtail millet [40], 22 genes in grapevine [41] and pepper [42] and 20 genes in cucumber [43]. Recently 23 genes in Barley (Hordeum vulgare L.) [44], 36 genes in sugarcane (Saccharum spontaneum) [45] and 25 genes in sweet orange (Citrus sinensis) [46] belonging to DCL, AGO and RDR genes have been identified and characterized. Besides, their expression pattern was also investigated under various conditions.
In A. thaliana, AtDCL1, AtDCL3 and AtAGO4 influenced the RNA-directed DNA methylation of the FWA transgene linkage to the histone H3 lysine 9 (H3K9) methylation [47, 48]. AtDCL2 is associated with the virus defence and siRNA production while the AtDCL4 is related to the regulation of vegetative phase change [23, 49]. AtDCL1 and AtDCL3 function for A. thaliana flowering [50]. Moreover, in rice if the OsDCL1 is knocked down then it fails to perform siRNA metabolism which causes pleiotropic phenotype in rice [51]. Besides, AGO proteins related to various forms of RNA silencing, such as AtAGO1 is associated with the transgene-silencing pathways [52] and AtAGO4 with the epigenetic silencing [47]. AtAGO7 and AtAGO10 influence the plant growth [53] and meristem maintenance [54]. Additionally, other AGOs also have a significant role in RNAi pathways. On the other hand, previous studies reported that the RDR genes are responsible for different gene silencing including co-suppression, virus defence, chromatin silencing and PTGS in plants such as in A. thaliana and maize [11, 35, 55–57]. Also the RDRα type enzyme was recognized playing a vital role in endogenous gene regulation [15, 58] antiviral silencing [8, 59, 60], arrangement of heterochromatin and genome resistance [61, 62].
The sweet orange (C. sinensis) is considered a great natural source of vitamin C, antioxidants and high nutrition important for the human body [46, 63–65]. It is considered the second highest amount of fruit producing plant all over the world (FAO Statistics 2006) and around USD 9 billion estimated price value was reported for the total production of sweet orange in 2012 [46, 66]. It not only has the market value, but also contains about 170 phytonutrients and over 60 flavonoids which work as antioxidant, anti-inflammation, anti-cancer and anti-arteriosclerosis compounds. It also protects us from many chronic diseases like arthritis, obesity and coronary heart diseases [67–70]. In spite of extensive studies of RNAi-related genes in many other plant species, very little information is available in the literature about these gene families for sweet orange. Until now, 13 AGO, 5 DCL and 7 RDR genes have been identified and investigated regarding their roles in fruit abscission process in C. sinensis [46]. However, sub-cellular location, functional pathways and associated regulatory factors (transcription and cis-acting) of these gene families in C. sinensis are not yet widely investigated. Therefore, in this study, an attempt is made to accomplish a comprehensive in silico analyses for genome-wide identification and characterization of AGO, DCL and RDR gene families and their associated regulatory elements in C. sinensis. Our results provide first insights into the genome-wide composition study, predicted function and factors influencing regulatory process of RNAi pathway genes in C. sinensis.
Materials and methods
Data source of DCL, AGO and RDR genes
For genome-wide identification of DCL, AGO and RDR genes in sweet orange (C. sinensis), protein sequences were downloaded from the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html) by taking advantage of completed C. sinensis genome sequence [64]. The previously identified sRNA biogenesis protein sequences of the model plant A. thaliana (AtDCLs, AtAGOs and AtRDRs) were collected from The Arabidopsis Information Resource (TAIR) (http://www.arabidopsis.org) and used to search the protein sequence of C. sinensis. The Basic Local Alignment Search Tool (BLASTP) program was used against C. sinensis genome in the Phytozome database (Fig 1).
Fig 1. The working flowchart of the integrated bioinformatics analyses approaches to select the best candidates for DCL, AGO and RDR genes and their associated regulatory elements in C. sinensis.
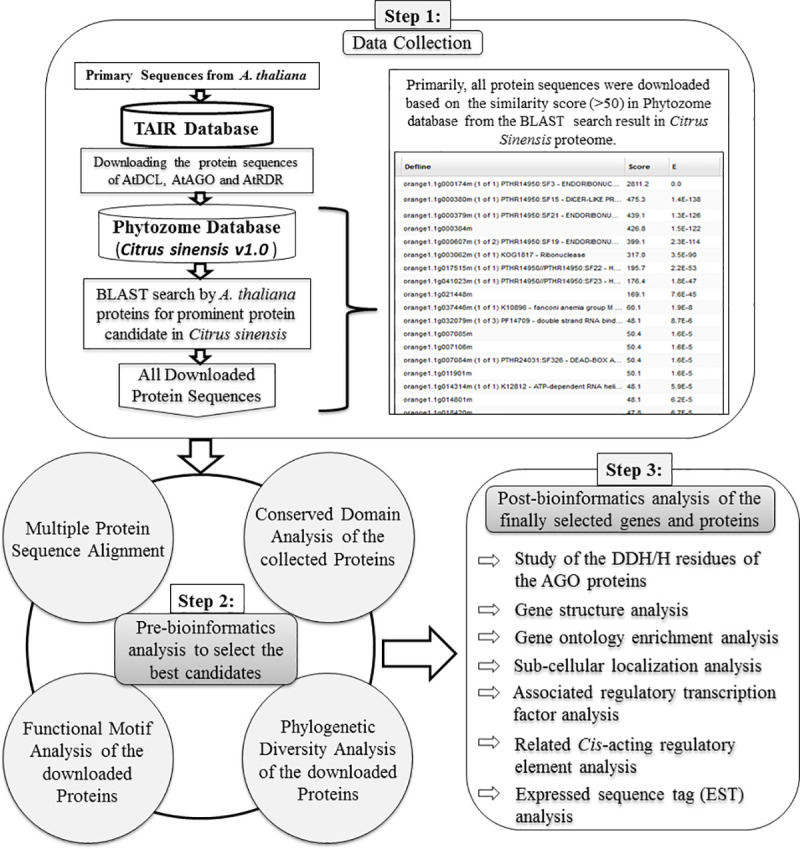
The derived paralog protein sequences from C. sinensis were downloaded with the significant score (≥50) and the significant E-values. For avoiding the redundancy of sequences, only the primary transcripts were considered in this analysis. The conserved domains of all retrieved sequences were searched and predicted by using the Pfam (http://pfam.sanger.ac.uk/) and the NCBI-CD database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and the SMART analysis. By this time, the different genomic information including the primary transcript name, genomic length and the chromosomal location of genes, ORF length, encoded protein length was downloaded from the C. sinensis genome in Phytozome database. In this study, the computationally identified new CsDCLs, CsAGOs and CsRDRs genes in C. sinensis genome were named according to the nomenclature based on phylogenetic relatedness of the similar family-members of the A. thaliana genes as named previously. The molecular weight of the selected protein sequences was predicted by using the ExPASyComputepI/Mwtool (http://au.expasy.org/tools/pitool.html).
Integrated bioinformatics analyses approaches
The integrated bioinformatics analyses approaches which included the sequence alignment and phylogenetic tree construction, prediction of the functional domain and motif structure of the proteins, the exon-intron structure of the RNAi candidate genes, gene ontology (GO) analysis, prediction of subcellular location, regulatory network among the gene transcription factors and C. sinensis RNAi candidate genes, cis-acting regulatory element (CARE) analysis, express sequence tag (EST) analysis, were carried out for comprehensive genome-wide identification, characterization, diversification analysis and to retrieve regulatory transcription components of C. sinensis RNA silencing machinery genes (Fig 1). These approaches are described in the following sub-sections.
Sequence alignment and phylogenetic analysis
In this in silico identification, the multiple sequence alignments of the encoded protein sequences of the predicted genes were conducted by using the Clustal-W method [71] with the MEGA5 program [72]. Finally, the phylogenetic tree analysis was carried out using the Neighbor-joining method [73] implemented on the aligned sequenced and the 1,000 bootstrap-replicates [74] were used to check this evolutionary relationship. The evolutionary distances were computed using the Equal Input method [75].
Conserved domain and motif analysis
To investigate the functional domains of the predicted genes the NCBI-CDD, Pfam database and SMART analysis were utilized to retrieve the conserved domains. The reported RNAi related proteins in C. sinensis containing the maximum number of significant functional domains similar to the Arabidopsis proteins AtDCLs, AtAGOs and AtRDRs were selected. In motif investigation, the most significant conserved metal-chelating catalytic triad residues in the PIWI domain of AGO proteins, i.e. aspartate, aspartate and histidine (DDH) [14] as well as histidine at 798 positions (H798) were investigated for reported CsAGO proteins (Fig 2). The conserved motif divergences among all the predicted RNAi related proteins were conducted by a complete online program for protein sequence analysis i.e. Multiple Expectation Maximization for Motif Elicitation (MEME-Suite) [76]. For this purpose, the following parameters were specified: (i) optimum motif width as ≥6 and ≤50; (ii) maximum 20 motifs.
Fig 2. The multiple sequence alignment profile of PIWI domain of the amino acids sequences of C. sinensis and Arabidopsis AGO proteins by Clustal-W program in MEGA5.

The downward yellow arrows indicate the position of conserved DDH triad of PIWI domain and the conserved H798 positions are surrounded by red box.
Gene structure and genomic location analysis
The gene structure of the predicted genes was constructed using the online Gene Structure Display Server (GSDS 2.0, http://gsds.cbi.pku.edu.cn/index.php) [77]. The structures of the selected genes were compared with the gene structure of A. thaliana to compare the exon-intron composition of the predicted genes in C. sinensis. The genomic location of the reported genes were represented using online tool MapGene2Chromosome V2 (http://mg2c.iask.in/mg2c_v2.0/).
Gene ontology and sub-cellular localization analysis
To check the engagement of our predicted RNAi associated genes with the cluster of different biological processes and molecular functional pathways, the Gene Ontology (GO) analysis was conducted using online tool implemented in PlantTFDB [78]. Here, the respective p-values were determined by Fisher’s exact test and Benjamini-Hochberg’s corrections. We considered the p-value < 0.05 as statistically significant for the GO enrichment results corresponding to the predicted genes. For the reported gene products, the sub-cellular location was investigated into the cell considering the different organelles. Web-based integrative subcellular location predictor tool called plant subcellular localization integrative predictor (PSI) [79] was used to predict the subcellular location of the identified genes.
Regulatory relationship and network analyses between TFs and C. sinensis RNAi related genes
In this study, the analysis of associated TFs family with the predicted RNAi related genes in C. sinensis was conducted from the widely used plant transcription factor database, PlantTFDB (http://planttfdb.cbi.pku.edu.cn//). After identification of the related regulatory TFs of the C. sinensis RNAi associated genes, the regulatory network and sub-network were constructed and visualized using Cytoscape 3.7.1 [80] to find out the hub proteins and the related important hub TF through the interaction network. The key hub factors were selected based on the highest degree of connectivity into the interaction network. The networks were constructed to investigate the key regulatory relationship between the TFs and reported RNAi related genes.
Cis-regulatory element analysis
To investigate cis-elements in the promoter sequences of three RNAi-related (CsDCL, CsAGO and CsRDR) gene families, 1.5 kb sequences upstream of the initiation codon (ATG) were collected and subjected to stress response-related cis-acting element online prediction analysis with Signal Scan search program in the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) [81]. The collected cis-regulatory element was classified into five categories: light responsive (LR), stress responsive (SR), hormone responsive (HR), other activities (OT) and unknown function. The known and established cis-elements of CsDCLs, CsAGOs and CsRDR are represented separately.
In silico Expressed Sequence Tag (EST) analysis
For the important and valuable information about the gene expression, the in silico expressed sequence tag (EST) data analysis was conducted according to Mirzaei et al., 2014 [82] for the reported genes. The PlantGDB database (http://www.plantgdb.org/cgi-bin/blast/PlantGDB/) was used for EST mining against the proposed RNAi related genes in C. sinensis. The default parameter with e-value = 1e-10 was considered for BLASTN search for the EST mining in PlantGDB database. The PlantGDB is a regularly updated online platform where the EST data from NCBI-dbEST and GeneBank are accessible [83]. The further heatmap was constructed to represent the specific RNAi associated gene expression into different tissue and organ in this fruit plant.
Results and discussion
Identification and characterization of CsDCLs, CsAGOs and CsRDRs genes
To identify the best candidates of RNAi related pathway in C. sinensis similar to the A. thaliana, all the previously downloaded sequences were gone through various kinds of analysis (Fig 1). Finally, we have identified 4 DCL, 8 AGO and 4 RDR genes encoding CsDCLs, CsAGOs and CsRDRs proteins, respectively, in the C. sinensis genome. On the basis of HMMER analysis with regards of all six types of conserved domains DEAD, Helicase_C, Dicer_dimar, PAZ, RNase III and DSRM; four DCL loci were identified in sweet orange genome. The genome length of predicted CsDCL genes varied from 10603 bp to 12728 bp corresponding to CsDCL1 and CsDCL2 with the coding potentiality of 1931 and 1396 amino acids (Table 1) when the ORF varied from 4191 bp (CsDCL2, orange1.1g000607m) to 5796 bp (CsDCL1, orange1.1g000174m). This findings are similar with Sabbione et al., 2019 except for the CsDCL2b (orange1.1g003062) protein which was additionally reported [46]. In this analysis the identified CsDCLs genes did not have any paralogs within the four subgroups in C. sinensis. The isoelectric point (pI) values of the CsDCLs proteins indicated that the proteins are more likely to be acidic where only the CsDCL3 have the highest pI value 8.01.
Table 1. Basic information about C. sinensis Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families.
| Serial Number | Gene Name | Accession Number | Chromosomal location | ORF length (bp) | Gene Length (bp) | Protein | |||
|---|---|---|---|---|---|---|---|---|---|
| No. of Intron | Molecular Weight (Da) | Protein Length (aa) | pI | ||||||
| CsDCLs | |||||||||
| 1 | CsDCL1 | orange1.1g000174m | scaffold00001:3480331..3490933 | 5796 | 10603 | 18 | 216424.20 | 1931 | 5.96 |
| 2 | CsDCL2 | orange1.1g000607m | scaffold00367:74912..87639 | 4191 | 12728 | 21 | 158487.91 | 1396 | 7.65 |
| 3 | CsDCL3 | orange1.1g000379m | scaffold00013:998934..1009643 | 4815 | 10710 | 24 | 178949.30 | 1604 | 8.01 |
| 4 | CsDCL4 | orange1.1g000380m | scaffold00068:219006..230745 | 4806 | 11740 | 25 | 179592.47 | 1601 | 6.46 |
| CsAGOs | |||||||||
| 1 | CsAGO1 | orange1.1g001466m | scaffold00674:51962..59926 | 3222 | 7965 | 20 | 118338.09 | 1073 | 9.38 |
| 2 | CsAGO4 | orange1.1g002449m | scaffold00028:907375..916121 | 2763 | 8747 | 21 | 102997.53 | 920 | 8.98 |
| 3 | CsAGO5a | orange1.1g002204m | scaffold00595:85931..92382 | 2865 | 6452 | 21 | 106772.65 | 954 | 9.27 |
| 4 | CsAGO5b | orange1.1g037086m | scaffold00595:96826..99593 | 1278 | 2768 | 10 | 48003.28 | 426 | 9.05 |
| 5 | CsAGO5c | orange1.1g003630m | scaffold03700:12..6517 | 2421 | 6506 | 21 | 91163.31 | 806 | 9.03 |
| 6 | CsAGO6 | orange1.1g002661m | scaffold00067:338566..346634 | 2688 | 8069 | 21 | 100499.54 | 895 | 9.41 |
| 7 | CsAGO7 | orange1.1g001684m | scaffold00003:3309646..3313553 | 3093 | 3908 | 2 | 117035.78 | 1030 | 9.24 |
| 8 | CsAGO10 | orange1.1g001954m | scaffold00011:54251..63917 | 2979 | 9667 | 20 | 111515.83 | 992 | 9.34 |
| CsRDRs | |||||||||
| 1 | CsRDR1 | orange1.1g002586m | scaffold00058:231382..236081 | 2715 | 4700 | 2 | 103382.03 | 904 | 6.51 |
| 2 | CsRDR2 | orange1.1g001183m | scaffold00020:1255446..1250927 | 3396 | 4520 | 3 | 128958.15 | 1131 | 6.59 |
| 3 | CsRDR3 | orange1.1g001771m | scaffold00027:638984..650509 | 3048 | 11526 | 18 | 115670.83 | 1015 | 6.80 |
| 4 | CsRDR6 | orange1.1g041430m | scaffold d00051:398116..402488 | 3594 | 4373 | 1 | 136437.20 | 1197 | 6.13 |
[Information containing the gene names, accession number, chromosomal location, ORF length, genome length, protein length were collected from Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html) and the molecular weight and isoelectric point (pI) values were predicted by the ExPASy ComputepI/Mwtool (http://au.expasy.org/tools/pi_tool.html). Molecular weights are in Da (Daltons) and “aa” means amino acid.]
Based on the conserved domain PAZ and PIWI from the putative polypeptide sequences by HMM and HMMER analysis, we have isolated a total of 8 AGO genes in the C. sinensis genome. Conserved domain analysis by the Pfam database, NCBI databases and SMART analysis reported that all the selected AGO proteins (CsAGO1-8) shared an N-terminus PAZ domain and a C-terminus PIWI super family domain that are the core properties of plant AGO proteins.
From the previous study, it is observed that the PIWI domain demonstrating expansive homology to RNase H binds the siRNA 5' end to the target RNA [26] and cracks the target RNAs that represent the complementary sequences to small RNAs [84]. Interestingly, the catalytic trait, three conserved metal-chelating residues (D = aspartate, D = aspartate and H = histidine) in PIWI domain, are related to the previous event and this trait was firstly shown in the model plant A. thaliana on AGO1 [14].
Moreover, another critical conserved histidine residue in AGO1 for in vitro endonuclease activity [85] was found. The genome length of the selected CsAGO genes varied from 2768 bp to 9667 bp produced by the CsAGO5b (orange1.1g037086m) and CsAGO10 (orange1.1g001954m), respectively, with the coding potentiality of 426 and 992 amino acids. The genes having the ORF ranging from 1278 to 3222 bp (CsAGO5b and CsAGO1) encode the reported CsAGO proteins homologous. In this study, the multiple sequence alignment of the PIWI domains of all CsAGO proteins with the orthologs AtAGOs from A. thaliana using the CLUSTAL-W method was utilized (Fig 2).
This alignment revealed that the five CsAGO proteins represented the conserved DDH triad residues like A. thaliana AGO1. We also investigated the important DDH/H motif among the reported CsAGO proteins. The DDH/H motif was found in CsAGO1, CsAGO7 and CsAGO10 proteins where the DDH/P motif and the DDH/S motif were identified in the CsAGO4 and CsAGO6 protein. The DDY/H motif and the DDY/P motif were found in CsAGO5a and CsAGO5c protein, respectively.
Among the CsAGO proteins, the CsAGO5b represented two missing PIWI domain catalytic residue(s) in the second aspartate at the 845th position (D845) and third histidine at the 986th position (H986) (Table 2). But the other two CsAGO proteins, CsAGO5a and CsAGO5c had the catalytic trait but with a replacement in the third histidine residue at the 986th position by tyrosine (Y) residue. Therefore, the DDH catalytic trait structure does not become conserved among the CsAGOs proteins in C. sinensis.
Table 2. Comparison of the Argonaute proteins with missing catalytic residue(s) in PIWI domains between C. sinensis and A. thaliana.
| Serial No. | Citrus sinensis | Arabidopsis thalianab | ||
|---|---|---|---|---|
| Argonaute | Motifsa | Argonaute | Motifsa | |
| 1 | CsAGO1 | DDH/H | AtAGO1 | DDH/H |
| 2 | CsAGO4 | DDH/P | AtAGO4 | DDH/S |
| 3 | CsAGO5a | DDY/H | AtAGO5 | DDH/H |
| 4 | CsAGO5b | D_ _ /R | AtAGO6 | DDH/P |
| 5 | CsAGO5c | DDY/P | AtAGO7 | DDH/H |
| 6 | CsAGO6 | DDH/S | AtAGO10 | DDH/H |
| 7 | CsAGO7 | DDH/H | ||
| 8 | CsAGO10 | DDH/H | ||
aComparison of conserved motif corresponds to D760, D845, H986/H798 of Arabidopsis AGO1; D: aspartate, H: histidine, P: proline, R: arginine, S: serine, Y: tyrosine and “_” represents the missing catalytic residue(s).
bFrom Kapoor et al. (2008)
Surprisingly, the histidine residue at the 786th position was replaced by proline (P) in CsAGO4 and CsAGO5c; by arginine (R) in CsAGO5b and in CsAGO6, H786 residue was replaced by serine (S) residue (Table 2). Due to the replacement of the conserved DDH/H motif residues in the reported CsAGO proteins, it can be assumed that the newly identified amino acid residues in the metal-chelating catalytic triad positions (DDH/H) may appear for genetic diversification or natural mutation. These changes indicate that the correspondent CsAGO proteins may fail to perform the endonuclease cleavage activities or the newly introduced residues may reflect new significant biological function in C. sinensis that can be explored through the expression analysis of the reported genes. Therefore, more expression analysis is required to investigate the functionality of the PIWI domain with the new catalytic residues in C. sinensis. Besides, two catalytic residues are missed in CsAGO5b protein but not in CsAGO5a although they are paralogous and the chromosomal location are in the same scaffold. The pI values of the CsAGOs indicated that the proteins have the basic properties as the pI values are greater than 7 and above 9.
The newly identified 4 CsRDR proteins that shared a common domain RdRP which consist of a sequence motif corresponds to the catalytic β' subunit of DNA-dependent RNA polymerases [86]. The CsRDRs have the genome length varying from 4373 bp to 11526 bp corresponding coding potentiality of 1157 and 1015 amino acids for CsRDR6 (orange1.1g041430m) and CSRDR3 (orange1.1g001771m) protein, respectively. In C. sinensis, no CsRDR4 and CsRDR5 candidates were identified in this analysis in comparison with the A. thaliana. The gene encoding ORF length was varied from 2715 to 3594 bp corresponding to CsRDR1 and CsRDR6, respectively. The previous study reported that more RDR proteins (CsRDR1b/c; CsRDR6b) were found as subgroup members of CsRDR1(a/b/c) and the CsRDR6(a/b) while CsRDR4/5 types were not found [46]. The identified CsRDRs genes were close orthologues of the AtRDRs in structures, evaluation and characteristics found in C. sinensis.
Phylogenetic analysis of DCL, AGO and RDR proteins in C. sinensis and A. thaliana
To investigate the phylogenetic relationship of C. sinensis RNAi related genes, phylogenetic tree was constructed for CsDCL, CsAGO and CsRDR proteins along with the candidate proteins of A. thaliana. Phylogenetic tree was generated from the full-length aligned protein sequences (S1 Data) of the 4 CsDCLs and 4 AtDCLs from C. sinensis and A. thaliana (Fig 3A). The four CsDCL proteins (CsDCL1-4) were divided into four subgroups along with the corresponding DCLs in A. thaliana (AtDCL1-4) with well-supported bootstrap values. The CsDCL proteins showed high sequence conservation with their corresponding counterpart in A. thaliana. Every DCL subfamily comprised a single CsDCL protein.
Fig 3.

Phylogenetic tree for the (A) Dicer-like (DCL) proteins (B) Argonaute (AGO) proteins and (C) RDR proteins from C. sinensis and A. thaliana. All the phylogenetic trees were constructed by neighbour-joining method considering significant bootstrap values. The accession number and the abbreviations of proteins from A. thaliana are given below while others are tabulated in (Table 1): (A) Four DCL proteins, AtDCL1 (At1g01040), AtDCL2 (At3g03300), AtDCL3 (At3g43920) and AtDCL4 (At5g20320) were used in DCL analysis. (B) AtAGO1 (At1g48410), AtAGO4 (At2g27040), AtAGO5 (At2g27880), AtAGO6 (At2g32940), AtAGO7 (At1g69440) and AtAGO10 (At5g43810) were considered for AGO analysis. (C) Among six AtRDR proteins, the phylogenetic tree exhibited only four major classes with CsRDR proteins. The RDR proteins from A. thaliana were AtRDR1 (At1g14790), AtRDR2 (At4g11130), AtRDR3 (At2g19910) and AtRDR6 (At3g49500). The three different gene families from A. thaliana are indicated by different colours in the constructed tree adjacent to the designation.
To construct the phylogenetic tree for CsAGO proteins, the full-length protein sequence of the 8 CsAGOs and 6 AtAGOs were considered (S2 Data). The tree exhibited six subfamilies, AGO1, AGO4, AGO5, AGO6, AGO7 and AGO10 with the AtAGOs (Fig 3B). The AGO1 subfamily consists only a single C. sinensis protein named CsAGO1 with the A. thaliana AGO protein AtAGO1. Among the other AGO subfamilies, each showed a group containing a single C. sinensis AGO protein with a single A. thaliana AGO protein, except AGO5 cluster.
The AGO5 subfamily included three C. sinensis proteins with a single A. thaliana protein AtAGO5, which were named CsAGO5a, CsAGO5b and CsAGO5c on the basis of higher sequence similarity to AtAGO5. In CsAGO5 group, three paralogs were identified in C. sinensis when the CsAGO5a/b were located in similar scaffold location having unique genomic structures. The AGO1, AGO4, AGO6, AGO7 and AGO10 groups exhibited each separated cluster with a single AGO protein from C. sinensis and a single protein from A. thaliana.
Four main classes of RDR genes in C. sinensis were revealed by the phylogenetic analysis of the full-length protein sequences (S3 Data) of RDR proteins of C. sinensis and A. thaliana (Fig 3C). The CsRDR proteins were designated as CsRDR1, CsRDR2, CsRDR3 and CsRDR6 corresponding to the RDR proteins of Arabidopsis AtRDR1, AtRDR2, AtRDR3 and AtRDR6, respectively, for the increased sequence similarity. The predicted CsRDR proteins were clustered according to their high sequence conservation with their reflection part in A. thaliana RDR proteins.
The number of DCL, AGO and RDR proteins are conserved in different species that may or may not be similar. For instance, 4 DCLs for Arabidopsis, 8 for rice, millet, and B. napus, 5 for Barley, 4 for sugarcane and many more were reported for different monocots and dicot species [14, 40–42, 44, 45]. The number of identified AGO proteins exhibited a wide range of clades across the plant lineage. The maximum number of AGO proteins and their clades were observed in the flowering plants, for example, 22 AGO proteins in soybean (Glycine max, a paleopolyploid) [87], 21 in sugarcane [45], 19 in rice [14], and 17 in maize [40]. Although the genomic diversity exists among the AGO proteins in flowering plant, the three common clades can be observed through phylogenetic analysis: the AGO1/5/10, AGO2/3/7, and AGO4/6/8/9 clades [88]. The reported CsAGO proteins also contained at least one member for each of the significant clades. On the other hand, the number of RDR gene family members varied from minimum 5 [14, 40, 41, 89] to maximum 16 [38]. In our analysis, we have identified four CsRDR genes, while CsRDR4/5 were found to be absent in C. sinensis genome indicating their evolutionary structure and functional effectiveness may be substituted or changed in C. sinensis. The DCL4 and AGO1 are two common and effective RNAi related genes found in all monocots and dicots [45] that have also been identified in our analysis.
Conserved domain and motifs analysis of predicted proteins
The functional conserved domains of the predicted CsDCLs, CsAGOs and CsRDRs were retrieved by conserved domain searching databases Pfam, NCBI-CDD and Simple Modular Architecture Research Tool (SMART) analysis. The summary results are tabulated in Table 3.
Table 3. Domain analysis of the DCLs, AGOs and RDRs proteins of the predicted gene mapping on C. sinensis with Pfam, SMART and NCBI-CDD.
| Serial Number | Gene Name | Accession Number | Domains | ||
|---|---|---|---|---|---|
| Pfam | SMART | NCBI-CDD | |||
| CsDCLs | |||||
| 1 | CsDCL1 | orange1.1g000174m | DEAD, Helicase_C, Dicer_dimer, PAZ, Ribonuclease_3, Ribonuclease_3, dsrm, DND1_DSRM | DEXDc, Helicase_C, Dicer_dimer, PAZ, RIBOc, RIBOc, DSRM, DSRM | PAZ super family, helicase_C, Rnc, RIBOc, Dicer_dimer, DSRM |
| 2 | CsDCL2 | orange1.1g000607m | DEAD, Helicase_C, Dicer_dimer, PAZ, Ribonuclease_3, Ribonuclease_3 | DEXDc, Helicase_C, Dicer_dimer, PAZ, RIBOc, RIBOc | helicase_C, RIBOc, PAZ super family, Dicer_dimer, RIBOc |
| 3 | CsDCL3 | orange1.1g000379m | ResIII, Helicase_C, Dicer_dimer, PAZ, Ribonuclease_3 | DEXDc, Helicase_C, Dicer_dimer, Low complexity PAZ, RIBOc | helicase_C, PAZ super family, RIBOc, Dicer_dimer, Rnc super family |
| 4 | CsDCL4 | orange1.1g000380m | DEAD, Helicase_C, Dicer_dimer, PAZ, Ribonuclease_3, Ribonuclease_3, DND1_DSRM | DEXDc, Helicase_C, Dicer_dimer, PAZ, RIBOc, RIBOc, DSRM, DSRM | helicase_C, RIBOc, Dicer_dimer, RIBOc, PAZ super family, DSRM |
| CsAGOs | |||||
| 1 | CsAGO1 | orange1.1g001466m | Gly-rich_Ago1, ArgoN, ArgoL1, PAZ, ArgoL2, ArgoMid, Piwi | Gly-rich_Ago1, ArgoN, DUF1785, ArgoL1, PAZ, ArgoL2, ArgoMid, Piwi | Piwi-like super family, Gly-rich_Ago1 |
| 2 | CsAGO4 | orange1.1g002449m | ArgoN, ArgoL1, PAZ, ArgoL2, ArgoMid, Piwi | ArgoN, DUF1785, PAZ, ArgoL2, ArgoMid, Piwi | Piwi-like super family |
| 3 | CsAGO5a | orange1.1g002204m | ArgoN, ArgoL1, PAZ, ArgoL2, ArgoMid, Piwi | ArgoN, DUF1785, PAZ, ArgoL2, ArgoMid, Piwi | Piwi-like super family |
| 4 | CsAGO5b | orange1.1g037086m | PAZ, ArgoL2, ArgoMid, Piwi | PAZ, ArgoL2, ArgoMid, Piwi | Piwi-like super family, PAZ |
| 5 | CsAGO5c | orange1.1g003630m | ArgoN, ArgoL1, PAZ, ArgoL2, ArgoMid, Piwi | ArgoN, DUF1785, PAZ, ArgoMid, Piwi | Piwi-like super family |
| 6 | CsAGO6 | orange1.1g002661m | ArgoN, ArgoL1, PAZ, ArgoL2, Piwi | ArgoN, DUF1785, PAZ, ArgoL2, Piwi | Piwi-like super family |
| 7 | CsAGO7 | orange1.1g001684m | ArgoN, ArgoL1, PAZ, Piwi | ArgoN, DUF1785, PAZ, ArgoMid, Piwi | Piwi_ago-like, PAZ, ArgoL1, ArgoN |
| 8 | CsAGO10 | orange1.1g001954m | ArgoN, ArgoL1, PAZ, ArgoL2, ArgoMid, Piwi | ArgoN, DUF1785, PAZ, ArgoL2, ArgoMid, Piwi | Piwi-like super family |
| CsRDRs | |||||
| 1 | CsRDR1 | orange1.1g002586m | RdRP | RdRP | RdRP |
| 2 | CsRDR2 | orange1.1g001183m | RdRP | RdRP | RdRP, RRM_SF super family |
| 3 | CsRDR3 | orange1.1g001771m | RdRP | RdRP | RdRP |
| 4 | CsRDR6 | orange1.1g041430m | RdRP | RdRP | RdRP |
The CsDCLs proteins showed all the conserved domains through the SMART analysis and also exhibited some unknown regions and low complexity regions besides the expected domains (Fig 4).
Fig 4. The conserved domains of the predicted proteins were drawn by using Pfam database.

From the conserved domain search results from the Pfam database, NCBI database and the SMART analysis, all proteins reflected that half of the predicted DCL proteins (CsDCL1 and CsDCL4) were conserved with the DEAD/ResIII, Helicase_C, Dicer-dimer, PAZ, RNase III and dsRM domains, which were preserved by all the plant DCL proteins from the DCL genes family (class 3 RNase III family) [90, 91]. On the other hand, CsDCL2 and CsDCL3 have missed a single dsRM domain while others are contained with a second dsRM domain. The non-plant DCL proteins lacked the dsRM domain completely [23]. Compared to others DCL proteins, the CsDCL1 had the N-terminal DEAD domain which might consist of three adjacent segments of amino acid sequence within the full domain length (152 amino acids), resulted by the analysis of Pfam databases and SMART. The CsDCL3 also revealed the ResIII domain instead of the DEAD domain. Previous study revealed that the joint activities of the DCL2 and DCL3 are very important in disease response whereas DCL4 is also responsive in viral disease defense [92]. Therefore, the expression analysis of the reported DCL genes is necessary to explore their extensive biological role in C. sinensis.
The AGO proteins are the main elements for processing the double stand RNA in single stand and trigger the whole target RNA cleavage process. The findings showed that the PAZ and PIWI domain are the key functional domain for constructing RNA Induced Silencing Complex (RISC) in all species [14, 25, 26]. All the reported CsAGO proteins also preserved the other conserved domains like the Arabidopsis i.e. ArgoN, ArgoL1, DUF1785, ArgoL2. Moreover, the conserved domain ArgoMid was present in all the CsAGO proteins except the CsAGO6 (orange1.1g002661m) which does not contain any MID domain. The six of the 8 CsAGO proteins started with the ArgoN domain while only the CsAGO1 (orange1.1g001466m) started with the Gly-rich domain and the CsAGO5b (orange1.1g037086m) started with the PAZ domain. Although the CsAGO5b posed the PAZ, ArgoMid and the PIWI domain, it did not contain the common DUF1785 domain. This seems that the CsAGO5b is found as a novel member of the RNAi related gene family in C. sinensis. The identified CsDCL and CsAGO proteins also contain the opulent number of functional domains including the main functional domain as in A. thaliana and some additional regions (Table 3). The RNA dependent RNA polymerase (RdRp) is one of the most versatile enzymes of RNA viruses that is indispensable for replicating the genome as well as for carrying out transcription. All the reported CsRDR consistently posed the RdRp domain while the CsRDR2 showed the RRM_SF super family region, found from NCBI-CDD analysis. The putatively functional RDR1 and RDR2 genes have a significant impact on siRNA biogenesis and accelerate the RNAi process [93, 94].
By MEME-suite analysis, the DCLs proteins had 19 (in CsDCL2 and CsDCL3) and 20 (in CsDCL1 and CsDCL4) motifs among the 20 motifs as mentioned before. The predicted motifs were well distributed among the DCL domains for all CsDCLs proteins. The MEME analysis of CsAGOs proteins identified 16 common conserved motifs among all the AGO proteins from C. sinensis and A. thaliana, except the CsAGO5b having 9 conserved motifs. The predicted conserved motifs were distributed among the AGO domains in C. sinensis AGO proteins.
Among the CsAGO proteins one had 16 different motifs (CsAGO7), three proteins (CsAGO4, CsAGO5c and CsAGO6) reflected 17 motifs and others three proteins (CsAGO1, CsAGO5a and CsAGO10) contained 20 conserved motifs (Fig 5). Although from the analysis it is observed that the conservation within the AGO proteins of C. sinensis and A. thaliana is balanced there still has some variability of motif distribution between the different subfamilies of AGO proteins in C. sinensis. Moreover, this analysis suggested that the conserved predicted motifs might play important roles in these AGO proteins.
Fig 5. Conserved motifs of the proteins of different gene families are drawn by MEME-suite (maximum 20 motifs are displayed).

Different colour represented various motifs distributed in the domains of the proteins.
In RDR protein family, the MEME analysis exhibited that the least 6 conserved motifs in CsRDR3 coincided with the AtRDR3. Among other CsRDRs proteins, the CsRDR2 and CsRDR6 presented 20 out of 20 conserved motifs which are well distributed on the RdRP domain and the CsRDR1 had 18 out of 20 conserved motifs (Fig 5). Although the predicted motifs were well conserved in the major part of the RDR domain, the motif schemes of different RDR subfamilies did not follow the same distributional pattern. The RDR proteins also reflected some additional motifs besides the RdRP domain having unknown functional role. However, the MEME-suite analysis reflected that the CsDCLs, CsAGOs and CsRDRs proteins were enriched with balanced conservation and distribution of the motifs throughout the subfamilies. This analysis suggested that the multiple functional domains and predicted motifs might play vital roles in the functional importance of these genes in C. sinensis in different developmental stages which can be investigated through expression analysis.
Gene structure and genomic location of CsDCLs, CsAGOs and CsRDRs
To observe the gene structure of the predicted CsDCLs, CsAGOs and CsRDRs gene, their exon-intron configuration was explored by using GSDS with the respective genes family of A. thaliana. The exon-intron configuration of the predicted genes represented higher conservation as expected for that of DCLs, AGOs and RDRs genes in the model plant A. thaliana (Fig 6). The gene structure of CsDCLs exhibited having the number of intron 18–25 (Table 1), bearing higher similarity with AtDCLs.
Fig 6. Gene structure of the predicted CsDCLs, CsAGOs and CsRDRs genes in C. sinensis with A. thaliana using Gene Structure Display Server (GSDS 2.0, http://gsds.cbi.pku.edu.cn/index.php) [77].

On the other hand, out of eight CsAGOs, six genes displayed 20 or 21 introns in the gene structure except the CsAGO5b and CsAGO7 having the number of introns 10 and 2, respectively, (Fig 6). This structure indicated that CsAGOs genes are highly similar to the AtAGOs. The CsRDRs showed up with the equal number of introns with their orthologs from A. thaliana, except CsRDR3, which, having 18 introns, is just one short of that in AtRDR3.
The genomic location of the predicted RNAi pathway related genes in C. sinensis was conducted by observing the position of the genes in different scaffold location. The predicted CsDCLs, CsAGOs and CsRDRs genes were distributed among the 15 different scaffolds through the entire genome (S1 Table). All genes had a unique scaffold position while the two CsAGO genes (CsAGO5a and CsAGO5b) were placed in the scafold000595 in different location. Furthermore, the chromosomal location was constructed and analyzed to check the genomic distribution of the reported genes (Fig 7). The identified RNAi related genes were scattered among the chromosomes of the C. sinensis when none of them were located in the chromosome 1, 3 and 8. The CsDCL3 and CsDCL4 were distributed on the chromosome 4 when the chromosome 6 and the unknown chromosome contained the CsDCL2 and CsDCL1 gene only. Among the CsAGO genes, 5 genes were found in the chromosome 2 (CsAGO4/5) and chromosome 7 (CsAGO5a/5c/7).
Fig 7. The genomic location of the reported CsDCL, CsAGO and CsRDR genes.

The chromosomal length indicating scale is provided on the left. The ChrUn means the unknown chromosome.
The CsAGO1 and CsAGO10 appeared in the chromosome 5 and 10 separately (Fig 7). In the chromosome 7, two paralogous of the CSAGO5 (CsAGO5a/c) were neighboring in very close genomic location indicating a higher possibility of tandem duplication. Also, the CsAGO7 and CsRDR6 were located closely in chromosome 7. Therefore, it can be pre-assumed that these genes will perform a diverse expression pattern due to their appearance that can be studied further under various condition and stresses. The four CsRDR genes are scattered in chromosome 2, 4, 5, and 7 (Fig 7).
Gene ontology enrichment analyses
The Gene Ontology (GO) analysis predicts the location or functional similarity of genes within the cells that are over- or under-expressed where the information are gathered from literature, database and computational evidences [95, 96]. The different GO terms describes the engagement with the various functional pathways linked to the reported genes. In order to better understand the biological roles of the predicted RNAi pathway related genes and characterize them, GO enrichment analysis was performed (Fig 8 and S4 Data) from the PlantTFDB. From the analysis result it was observed that of the reported genes 12 were involved in post-transcriptional gene silencing (PTGS) pathway (GO: 0016441; p-value: 3.60e-27), 10 were related to RNA interference (GO: 0016246; p-value: 8.50e-25) and 12 genes were associated with gene silencing (GO: 0016458; p-value: 7.40e-24). The RNAi is closely related to the phenomenon named post-transcriptional gene silencing (PTGS) in plants [97]. As most of the predicted genes significantly showed the GO terms those are associated with the RNAi, it clearly indicates that these genes have a great involvement with mRNA degradation process in the C. sinensis.
Fig 8.

The heatmap for the predicted GO terms corresponding to the reported RNAi genes are presented for (A) biological process (B) cellular components (C) molecular function whether the genes are related (Present) or not (Absent). The p-value corresponding to the GO terms are showed in histogram adjacent to the heatmap, using -log10 (p-value). The Ven diagrams are drawn to observe the shared GO terms by three gene families considering the (D) biological process (E) cellular components (F) molecular functions.
The GO enrichment analysis showed that five predicted RNAi pathway genes (among 16) were related to the endonuclease activity (GO: 0004519; p-value = 4.20e-09) which (S4 Data) indicates a positive linkage with the RNA-induced silencing complex (RISC) mediated cleavage activities into the cell. This multimeric protein complex (i.e. RISC) guides protein degradation. Among the RNAi proteins, Argonautes work for the cleavage called endonucleolytic activities, which result in the final PTGS for specific mRNA substrate [98]. The CsAGOs may also have the various biological activities that can be revealed by their expression analysis against any biological question. There were 13 genes related to nucleic acid binding (GO: 0003676; p-value = 1.10e-08), 7 genes to RNA binding (GO: 0003723; p-value = 2.10e-07) and 12 genes related to protein binding (GO: 0005515; p-value = 0.00022) activities (S4 Data) which indicate the RNAi protein’s participation to the RISC as well as interference processes conducted. The predicted CsAGO proteins contained the special domains called PAZ and PIWI domain that play the key role in making a complex with RNA or DNA. The PAZ domain has a nucleic acid-binding fold that promotes the domain to bind to the specific position of the nucleic acids [99, 100] by binding with the target mRNA for degradation. The GO enrichment analysis also showed the attachment of the predicted genes to the numerous biological processes. Significantly, most of the reported genes were engaged with the regulation of biological process (GO: 0050789; p-value = 3.90e-08), negative regulation of gene expression (GO: 0010629; p-value: 2.30e-20) and dsRNA fragmentation (GO: 0031050; p-value: 4.10e-23) (S4 Data) which are also parts of the greater RNAi process.
The C. sinensis RNAi candidate genes were also involved in virus response (GO: 0009615; p-value: 6.70e-28), immune response (GO: 0006955; p-value: 4.10e-14) as these were reported for AtDCL and AtRDR [11, 23, 35, 49, 55–57]. These GO enrichment analysis for biological processes (S1A Fig), molecular functions (S1B Fig) and cellular component (S1C Fig) undoubtedly indicated that the predicted genes are deeply interrelated with the RNAi pathway in C. sinensis. In addition, the predicted genes act with the hydrolase activity, acting on ester bond that was predicted from the GO analysis (S4 Data).
The Ven Diagram of the GO terms for three clusters of the RNAi associated genes was drawn (Fig 8). It was observed that the CsDCL, CsAGO and CsRDR genes had significant number of GO pathways in common. In biological process, there were 89 GO enrichment pathways (Fig 8) were shared by the reported proteins, which indicate the involvement of the RNAi gene members in numerous biological processes together in C. sinensis. Also, in molecular function and cellular component, the predicted genes exhibited a group of mutual GO pathways. So, this GO analysis provides a significant indication of the predicted RNAi member genes in this study.
Sub-cellular localization of the reported genes and proteins
The subcellular localization studies of the predicted proteins were observed to uncover their cellular appearance. By the sub-cellular localization annotation, it has been shown that all the predicted proteins reported in this study appear into the cytosol (Fig 9). As PTGS occurs into the cytoplasmic region [101], this result implies that the reported RNAi proteins may directly involve to the PTGS process. On the other hand, four CsAGO and one CsRDR proteins exhibited their appearance into the nucleus whereas no CsDCLs were located there. These bring a significant importance whether the CsDCLs are not found in nucleus. Further expression pattern analysis will provide deeper insight about the CsDCLs. Some of the identified RNAi proteins were also distributed into the cell membrane, plastid and mitochondria (Fig 9).
Fig 9.

Sub-cellular localization analysis for (A) CsDCL (B) CsRDR and (C) CsAGO proteins. (D) The percentage of protein appears in different cellular components. Here cytosol (cytos), endoplasmic reticulum (ER), extracellular (extra), golgi apparatus (golgi), membrane (membr), mitochondria (mito), nuclear (nucl), peroxisome (pero), plastid (plast) and vacuole (vacu). Overall report is tabulated in (S2 Table).
Previous studies reported that the RNAi genes are not only highly related with PTGS but also with transcriptional gene silencing (TGS) [101]. In protein transcriptional process, RNA polymerase type II complexes are directly involved [102]. For PTGS, the RNAi proteins have greater participation in RNA-induced silencing complex (RISC) mediated cleavage activities by the help of DCL, AGO and RDR proteins with other molecules [98]. The PTGS happens into the cytoplasmic region for targeted mRNA protein degradation [102].
Regulatory relationship between TFs and RNAi genes
Transcription factors (TFs) play the central roles as drivers of gene expression in all living organisms since they control the rate of genetic transcription and coordinate the action of any genetic network [103, 104]. The studies evidence that the various family of TFs are associated with the growth and development of the aboveground and underground parts of plants, abiotic stress responses, response to pathogens and many more [103, 105–109]. Thus, identification of the regulatory TFs of the reported RNAi genes can help to improve our understanding of gene silencing process in C. sinensis. In this analysis a total of 235 TFs those regulate the predicted RNAi genes (S5 Data) were identified. The identified TFs were distributed into 27 groups based on the TF families. The TFs MYB, Dof, ERF, NAC, MIKC_MADS, WRKY and bZIP families may play significant role in regulating RNAi genes. Particularly those of ERF, NAC, WRKY and bZIP families, which are the top four families, contained 29, 20, 20 and 10 TFs respectively, and accounted 57.66% of the total identified TFs (S3 Table). This finding indicates that those TFs can be important in regulating RNAi genes.
From the resultant network it was observed that different groups of TFs exhibited distinct structure. For example, TFs belonging to ERF family mainly linked to the gene CsAGO5a (Fig 10B and S2 Fig). However, some RNAi genes such as CsAGO5c, CsDCL4 and others were also regulated by ERF family; all of them were also linked to CsAGO5a (Fig 11A). Very similar results were also observed for the hub TFs NAC, WRKY and bZIP (Fig 11B–11D).
Fig 10.

(A) The regulatory network among the TFs and the predicted RNAi genes. The nodes of the network were coloured based on RNAi genes and TFs. DCL, AGO and RDR genes were represented by blue, red and green node colour, respectively, and the TFs were represented by yellow node colour. Different node symbols were used for different families of TFs. Magenta node level was used for the hub TFs. (B) The map representing the associated number of TFs with the CsRNAi genes.
Fig 11.

RNAi gene mediated sub-network for (A) ERF, (B) NAC, (C) bZIP, and (D) WRKY TF family. (E) Sub-network among the hub TFs those regulate more than five RNAi genes.
Moreover, six hub TFs were identified on the basis of node degree which had more than five interacting partners with the RNAi related genes (Fig 11E). All of the hubs TFs were connected to eight RNAi genes. Out of eight RNAi genes corresponding to hub TFs, five are AGO, two are DCL and only one is RDR. Three RNAi genes (2 AGOs: CsAGO10, CsAGO7; 1 DCL: CsDCL1) were predicted to be regulated by the entire six hub TFs. Among them 3 belonged to Dof TFs family and other three were in MIKC_MSDS, C2H2 and bZIP (Fig 11E). The Dof TFs family is directly involved with the DNA binding activities by the N- and C-terminal region and causes the regulation of gene activation or repression of the target genes which is the main theme of RNAi. The Dof TFs family also works for the biosynthesis of flavonoids and glucosinolates, stress tolerance, seed germination and controlling the photoperiodic flowering [110–114]. The MYB TFs are available in both plants and animals which contain MYB domain (a 52 amino acid motif) [115]. The MYB TF plays a significant role in biotic and abiotic response in Arabidopsis [116, 117]. The expression patterns of the WRKY family of TFs are associated with the defence response against biotrophic pathogens, necrotrophic pathogens and also works as anti-microbial defence [118–120]. Moreover, the expression of the RDR gene families may be influenced by the MYB, NAC and the WRKY TFs during various stress conditions and they have a direct or indirect involvement in plant development and stress response in plant [115, 120–123]. The expression of the defensive genes is regulated through the interaction activities of Calmodulin (CaM) with the specific TFs MYB, NAC and the WRKY [124, 125]. Our study indicates that the further gene expression study needs to clarify whether the calcium/CaM related pathways are playing a vital role in RNAi-related pathways in C. sinensis or not. From the network analysis it is observed that the MIKC_MADS (orange1.1g027691m) TF regulates maximum seven RNAi related genes and the rest of the TFs regulate five RNAi genes (Fig 11E). This MIKC_MADS TF family also involves with the transcription of OsRDR1 genes to augment the tolerance power against the Rice stripe virus (RSV) in rice (Oryza sativa) [126]. The regulatory network clearly exposes that these predicted genes and the associated TFs of RNAi process in C. sinensis will exhibit a wide ranges expression pattern that can be retrieved by deeper investigation of these genes in future.
Cis-acting regulatory element analysis
The cis-acting regulatory elements (CAREs) are short motifs (5±20 bp) where the TFs can bind to the specific targeted genes to initiate the transcription and control gene regulation process [127]. The cis-elements are also involved in plant defence response, stress responsiveness [127, 128]. The wet lab experimental exploration of these inevitable regulatory component is technically challenging and expensive whether their computational identification is being used through various enriched databases [127]. The cis-acting regulatory element analyses were conducted to find out the functional diversity of the motifs related to the promoter region of the proposed RNAi genes into C. sinenesis. The PlantCARE database provided the information about the motifs and their functionality with the genes. The analysis revealed that most of the motifs were light responsive (LR) (Fig 12), widely present in the entire RNAi gene’s promoter. Supporting the EST analysis later, the light responsiveness is associated with the photosynthesis which occurs in leaves. Among the light responsive motifs, the ATCT-motif, ATC-motif, Box-4, AE-box, G-box, I-box, GAT-motif, GT1-motif were shared by the most of the RNAi related genes in C. sinensis (Fig 12) [127, 129–132]. The TC-rich repeats (cis-acting element involved in defense and stress responsiveness) [133], MBS (MYB binding site involved in drought-inducibility) [134] and LTR elements (cis-acting element involved in low-temperature responsiveness) were commonly found as stress responsive motif among the CsDCL/AOG/RDR genes in C. sinensis.
Fig 12. The cis-regulatory element in the promoter region of the identified C. sinenis DCLs, AGOs and RDRs genes, respectively.

The deep color represents the presence of that element with the corresponding genes.
It is known that the plant hormones are essential for plant growth and development. The significant plant hormone responsive (HR) cis-acting elements were identified in this analysis. The ABRE (cis-acting element involved in the abscisic acid responsiveness) [135, 136], AuxRR-core (cis-acting regulatory element involved in auxin responsiveness) [137, 138], GC-motif (enhancer-like element involved in anoxic specific inducibility) [139–141], GARE-motif (gibberellin-responsive element), O2-site (cis-acting regulatory element involved in zein metabolism regulation) [137, 138], P-box (gibberellin-responsive element), TATC-box (cis-acting element involved in gibberellin-responsiveness) [137, 142], TCA-element (cis-acting element involved in salicylic acid responsiveness) and TGA-element (auxin-responsive element) [138, 142, 143] were the hormone responsive cis-elements shared by the CsDCLs, CsAGOs and CsRDRs as phytohormones responsive element (Fig 12). There were some others significant elements identified and represented as others activities. The AT-rich element (binding site of AT-rich DNA binding protein (ATBP-1)), AT-rich sequence (element for maximal elicitor-mediated activation), CAAT-box (common cis-acting element in promoter and enhancer regions), CAT-box (cis-acting regulatory element related to meristem expression), CCAAT-box (MYBHv1 binding site), GCN4_motif (cis-regulatory element involved in endosperm expression), TATA-box (core promoter element around -30 of transcription start), circadian (cis-acting regulatory element involved in circadian control), silencer (GT-1 factor binding site) and TGACG-motif (cis-acting regulatory element involved in the MeJA-responsiveness) [129, 138, 142–144] were recognized as other cis-acting regulatory elements shared by RNAi related genes in C. sinensis (Fig 12). Some unknown cis-regulatory elements were also detected along with the reported cis-elements (S6 Data). In general, the cis-regulatory elements carried out significant evidences about the proposed RNAi genes that will be helpful for further investigation about their role in plant disease response, growth and development.
In silico Expressed Sequence Tag (EST) analysis
The EST mining results obtained from the PlantGDB database indicated that the sweet orange RNAi associated genes are expressed in multiple important tissues and organs. The search results provided 154 unique EST contigs records against the reported RNAi related genes of sweet orange. The GeneBank accession ID of the obtained EST contigs has been supplied in supplementary data file (S7 Data). However, evidence of expression of RNAi pathway related genes in several plant species showed their expression in leaf, root, flower, seeds and other organs [14, 33, 39–43, 82, 145, 146]. A recent study identified and characterized the expression of sweet orange RNAi related genes in several tissues and organs using RNA-seq data [46]. In general, most of the genes were expressed in leaf and fruit indicating their direct involvement in the photosynthesis and fruit developmental stages in C. sinensis (Fig 13).
Fig 13. The in silico Expressed Sequence Tag (EST) analysis of the identified RNAi genes in C. sinensis plant.

The green color represents the existence of expression and off color stands for absent of expression.
Among the identified EST contigs, the expression of CsDCLs were detected in leaves (CsDCL1/3/4), flowers (CsDCL2/4), fruit (CsDCL1/2/4), ovule (CsDCL1/3/4) and bark (CsDCL3), and no expression were found in root. The entire CsAGOs exhibited diverse expression pattern in all the organs (roots, leaves, flowers, ovule, fruit, fruit rind and seeds) of sweet orange (Fig 13). Among the CsAGO genes, EST contigs of CsAGO1 and CsAGO4 were detected in most of the organs in C. sinensis while only the CsAGO1/4 provided the confirmation of expression in seeds. Similarly, almost all the CsRDRs expressed in leaf, flower and fruit when the CsRDR6 showed expression in ovule and bark. Although for the proposed C. sinensis RNAi related genes showed that all the genes have their expression at least in one organ or tissue, no evidence of expression were found for the CsRDR3 in this in silico EST analysis (Fig 13). Collectively, the EST analyses indicated that the proposed RNAi related genes have vast contribution in ovule fertilization, fruit development process, plant photosynthesis which can be validated by tissue specific expression and functional study.
Conclusion
The sweet orange is considered as the second highest produced fruits all over the world. The C. sinensis plant is the major source of sweet orange which is one of the most favourite and nutritious fruits. In silico characterization, diversity analyses and regulatory process of the RNAi-related gene families were essential, since these families play a vital role for silencing of other targeted genes in plant. Our study attempted to identify the RNAi pathway genes, keeping their key transcriptional factors and regulatory elements in focus, in C. sinensis along with the genomic and physicochemical information of the predicted genes and their corresponding proteins. With the phylogenetic analysis, the subgroups of the three gene families were exhibited, the domains and motifs configuration and the gene structures revealed the maximum homogeneity with the respective gene family of A. thaliana. Moreover, the GO enrichment and subcellular localization analysis provided the final confirmation about the reported genes and protein which are the key factor of RNAi process in C. sinensis. In this analysis, we explored regulatory relationship network between TFs and proposed RNAi genes. Potential TFs and cis-acting regulatory elements involved in plant growth and development as well as controlling the gene expression or suppression related to RNAi process were identified. The expressed sequence tag (EST) analysis indicates that the reported RNAi-related genes have diverse involvement into the orange plant growth, development and flowering processes. Thus, the reported genes in this study may exhibit significant expression pattern under different stress conditions in various developmental stages of sweet orange. Therefore, our findings may provide a basis for further functional analysis of RNAi pathway genes in C. sinensis to clarify their roles in growth, development, disease resistance and improve production and quality of sweet orange.
Supporting information
(PDF)
(PDF)
(PDF)
(TXT)
(TXT)
(TXT)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
GO enrichment analysis of the predicted RNAi genes (A) biological process, (B) molecular function and (C) cellular process. In the directed acyclic graph (DAG) the downstream term corresponds to a subset of the upstream term. The significant (p-value < 0.05, FDR < 0.05) GO terms are in colored boxes (the degree of color saturation is positively correlated to the enrichment level of the GO term), and non-significant terms are in white boxes.
(PDF)
Rows of the figure represent the predicted RNAi genes and the columns represent the families of the TFs. The number indicates the TF families regulate the RNAi genes.
(PDF)
Acknowledgments
The authors acknowledge and appreciate the reviewers and the members of the editorial panel for their important comments and suggestions for improving the quality of this manuscript.
Abbreviations
- AGO
Argonaute
- BLAST
Basic Local Alignment Search Tool
- bp
base pair
- DCL
Dicer-Like
- GO
Gene Ontology
- GSDS
Gene Structure Display Server
- HMM
Hidden Markov Model
- MEME
Multiple Em for Motif Elicitation
- miRNA
microRNA
- PTGS
post-transcriptional gene silencing
- RDR
RNA, dependent RNA polymerase
- RNAi
RNA interference
- RISC
RNA-induced silencing complex
- sRNA
small RNA
- siRNA
short interfering RNA
- TF
transcription factor
- TGS
transcriptional gene silencing
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
The author(s) received no specific funding for this work.
References
- 1.Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003. pp. 336–338. 10.1126/science.1085242 [DOI] [PubMed] [Google Scholar]
- 2.Finnegan EJ, Matzke MA. The small RNA world. J Cell Sci. 2003;116: 4689–4693. 10.1242/jcs.00838 [DOI] [PubMed] [Google Scholar]
- 3.Lai EC. microRNAs: Runts of the Genome Assert Themselves Current Biology. Cell Press; 2003. 10.1016/j.cub.2003.11.017 [DOI] [PubMed] [Google Scholar]
- 4.Origin Voinnet O., Biogenesis, and Activity of Plant MicroRNAs. Cell. 2009. pp. 669–687. 10.1016/j.cell.2009.01.046 [DOI] [PubMed] [Google Scholar]
- 5.Wilson RC, Doudna JA. Molecular Mechanisms of RNA Interference. Annu Rev Biophys. 2013;42: 217–239. 10.1146/annurev-biophys-083012-130404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castel SE, Martienssen RA. RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond Nature Reviews Genetics. Nature Publishing Group; 2013. pp. 100–112. 10.1038/nrg3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shabalina SA, Koonin E V. Origins and evolution of eukaryotic RNA interference Trends in Ecology and Evolution. Elsevier; 2008. pp. 578–587. 10.1016/j.tree.2008.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Use Voinnet O., tolerance and avoidance of amplified RNA silencing by plants. Trends in Plant Science. 2008. 10.1016/j.tplants.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 9.Hunter LJR, Brockington SF, Murphy AM, Pate AE, Gruden K, Macfarlane SA, et al. RNA-dependent RNA polymerase 1 in potato (Solanum tuberosum) and its relationship to other plant RNA-dependent RNA polymerases. Sci Rep. 2016;6 10.1038/srep23082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devert A, Fabre N, Floris M, Canard B, Robaglia C, Crété P. Primer-Dependent and Primer-Independent Initiation of Double Stranded RNA Synthesis by Purified Arabidopsis RNA-Dependent RNA Polymerases RDR2 and RDR6. Pooggin MM, editor. PLoS One. 2015;10: e0120100 10.1371/journal.pone.0120100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mourrain P, Béclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, et al. Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell. 2000;101 10.1016/s0092-8674(00)80863-6 [DOI] [PubMed] [Google Scholar]
- 12.Prakash V, Devendran R, Chakraborty S. Overview of plant RNA dependent RNA polymerases in antiviral defense and gene silencing Indian Journal of Plant Physiology. Springer Verlag; 2017. pp. 493–505. 10.1007/s40502-017-0339-3 [DOI] [Google Scholar]
- 13.Vaucheret H. Post-transcriptional small RNA pathways in plants: Mechanisms and regulations. Genes and Development. 2006. pp. 759–771. 10.1101/gad.1410506 [DOI] [PubMed] [Google Scholar]
- 14.Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, et al. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics. 2008;9 10.1186/1471-2164-9-451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fei Q, Xia R, Meyers BC. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell. 2013. 10.1105/tpc.113.114652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wassenegger M, Heimes S, Riedel L, Sänger HL. RNA-directed de novo methylation of genomic sequences in plants. Cell. 1994;76 10.1016/0092-8674(94)90119-8 [DOI] [PubMed] [Google Scholar]
- 17.Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, et al. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;2 10.1371/journal.pbio.0020104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Großhans H, Filipowicz W. Molecular biology: The expanding world of small RNAs. Nature. 2008. 10.1038/451414a [DOI] [PubMed] [Google Scholar]
- 19.Chapman EJ, Carrington JC. Specialization and evolution of endogenous small RNA pathways. Nature Reviews Genetics. 2007. 10.1038/nrg2179 [DOI] [PubMed] [Google Scholar]
- 20.Millar AA, Waterhouse PM. Plant and animal microRNAs: Similarities and differences. Functional and Integrative Genomics. 2005. 10.1007/s10142-005-0145-2 [DOI] [PubMed] [Google Scholar]
- 21.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409 10.1038/35053110 [DOI] [PubMed] [Google Scholar]
- 22.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science (80-). 2001;293 10.1126/science.1064023 [DOI] [PubMed] [Google Scholar]
- 23.Margis R, Fusaro AF, Smith NA, Curtin SJ, Watson JM, Finnegan EJ, et al. The evolution and diversification of Dicers in plants. FEBS Lett. 2006;580 10.1016/j.febslet.2006.03.072 [DOI] [PubMed] [Google Scholar]
- 24.Moazed D. Small RNAs in transcriptional gene silencing and genome defence. Nature. 2009. 10.1038/nature07756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters L, Meister G. Argonaute Proteins: Mediators of RNA Silencing. Molecular Cell. 2007. 10.1016/j.molcel.2007.05.001 [DOI] [PubMed] [Google Scholar]
- 26.Höck J, Meister G. The Argonaute protein family. Genome Biology. 2008. 10.1186/gb-2008-9-2-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yigit E, Batista PJ, Bei Y, Pang KM, Chen CCG, Tolia NH, et al. Analysis of the C. elegans Argonaute Family Reveals that Distinct Argonautes Act Sequentially during RNAi. Cell. 2006;127 10.1016/j.cell.2006.09.033 [DOI] [PubMed] [Google Scholar]
- 28.Hutvagner G, Simard MJ. Argonaute proteins: Key players in RNA silencing. Nature Reviews Molecular Cell Biology. 2008. 10.1038/nrm2321 [DOI] [PubMed] [Google Scholar]
- 29.Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442 10.1038/nature04917 [DOI] [PubMed] [Google Scholar]
- 30.Djupedal I, Ekwall K. Epigenetics: Heterochromatin meets RNAi. Cell Research. 2009. 10.1038/cr.2009.13 [DOI] [PubMed] [Google Scholar]
- 31.Wassenegger M, Krczal G. Nomenclature and functions of RNA-directed RNA polymerases. Trends in Plant Science. 2006. 10.1016/j.tplants.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 32.Willmann MR, Endres MW, Cook RT, Gregory BD. The Functions of RNA-Dependent RNA Polymerases in Arabidopsis. Arab B. 2011;9 10.1199/tab.0146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai M, Yang GS, Chen WT, Mao ZC, Kang HX, Chen GH, et al. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene. 2012;501 10.1016/j.gene.2012.02.009 [DOI] [PubMed] [Google Scholar]
- 34.Cogoni C, Macino G. Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature. 1999;399 10.1038/20215 [DOI] [PubMed] [Google Scholar]
- 35.Dalmay T, Hamilton A, Rudd S, Angell S, Baulcombe DC. An RNA-dependent RNA polymerase gene in arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell. 2000;101 10.1016/S0092-8674(00)80864-8 [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Li C, Wang E. Potential and flux landscapes quantify the stability and robustness of budding yeast cell cycle network. Proc Natl Acad Sci U S A. 2010;107 10.1073/pnas.0910331107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vazquez F. Arabidopsis endogenous small RNAs: highways and byways. Trends in Plant Science. 2006. 10.1016/j.tplants.2006.07.006 [DOI] [PubMed] [Google Scholar]
- 38.Cao JY, Xu YP, Li W, Li SS, Rahman H, Cai XZ. Genome-wide identification of dicer-like, argonaute, and RNA-dependent RNA polymerase gene families in brassica species and functional analyses of their arabidopsis homologs in resistance to sclerotinia sclerotiorum. Front Plant Sci. 2016;7 10.3389/fpls.2016.01614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian Y, Cheng Y, Cheng X, Jiang H, Zhu S, Cheng B. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 2011;30 10.1007/s00299-011-1046-6 [DOI] [PubMed] [Google Scholar]
- 40.Yadav CB, Muthamilarasan M, Pandey G, Prasad M. Identification, characterization and expression profiling of dicer-like, argonaute and rna-dependent RNA polymerase gene families in foxtail millet. Plant Mol Biol Report. 2015;33 10.1007/s11105-014-0736-y [DOI] [Google Scholar]
- 41.Zhao H, Zhao K, Wang J, Chen X, Chen Z, Cai R, et al. Comprehensive Analysis of Dicer-Like, Argonaute, and RNA-dependent RNA Polymerase Gene Families in Grapevine (Vitis Vinifera). J Plant Growth Regul. 2015;34 10.1007/s00344-014-9448-7 [DOI] [Google Scholar]
- 42.Qin L, Mo N, Muhammad T, Liang Y. Genome-wide analysis of DCL, AGO, and RDR gene families in pepper (Capsicum Annuum L.). Int J Mol Sci. 2018;19 10.3390/ijms19041038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gan D, Zhan M, Yang F, Zhang Q, Hu K, Xu W, et al. Expression analysis of argonaute, Dicer-like, and RNA-dependent RNA polymerase genes in cucumber (Cucumis sativus L.) in response to abiotic stress. J Genet. 2017;96 10.1007/s12041-017-0758-y [DOI] [PubMed] [Google Scholar]
- 44.Hamar E, Szaker HM, Kis A, Dalmadi A, Miloro F, Szittya G, et al. Genome-wide identification of RNA silencing-related genes and their expressional analysis in response to heat stress in barley (Hordeum vulgare l.). Biomolecules. 2020;10 10.3390/biom10060929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui DL, Meng JY, Ren XY, Yue JJ, Fu HY, Huang MT, et al. Genome-wide identification and characterization of DCL, AGO and RDR gene families in Saccharum spontaneum. Sci Rep. 2020;10 10.1038/s41598-020-70061-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabbione A, Daurelio L, Vegetti A, Talón M, Tadeo F, Dotto M. Genome-wide analysis of AGO, DCL and RDR gene families reveals RNA-directed DNA methylation is involved in fruit abscission in Citrus sinensis. BMC Plant Biol. 2019;19 10.1186/s12870-019-1998-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zilberman D, Cao X, Jacobsen SE. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science (80-). 2003;299 10.1126/science.1079695 [DOI] [PubMed] [Google Scholar]
- 48.Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet. 2006;38 10.1038/ng1804 [DOI] [PubMed] [Google Scholar]
- 49.Deleris A, Gallago-Bartolome J, Bao J, Kasschau KD, Carrington JC, Voinnet O. Hierarchical action and inhibition of plant dicer-like proteins in antiviral defense. Science (80-). 2006;313 10.1126/science.1128214 [DOI] [PubMed] [Google Scholar]
- 50.Schmitz RJ, Hong L, Fitzpatrick KE, Amasino RM. DICER-LIKE 1 and DICER-LIKE 3 redundantly act to promote flowering via repression of FLOWERING LOCUS C in Arabidopsis thaliana. Genetics. 2007;176 10.1534/genetics.107.070649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, Li PC, Li X, Liu CY, Cao SY, Chu CC, et al. Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiology. 2005. 10.1104/pp.105.063420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fagard M, Boutet S, Morel JB, Bellini C, Vaucheret H. AGO1, QDE-2, and RDE-1 are related proteins required for post-transcriptional gene silencing in plants, quelling in fungi, and RNA interference in animals. Proc Natl Acad Sci U S A. 2000;97 10.1073/pnas.200217597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hunter C, Sun H, Poethig RS. The Arabidopsis Heterochronic Gene ZIPPY Is an ARGONAUTE Family Member. Curr Biol. 2003;13 10.1016/j.cub.2003.09.004 [DOI] [PubMed] [Google Scholar]
- 54.Moussian B, Schoof H, Haecker A, Jürgens G, Laux T. Role of the ZWILLE gene in the regulation of central shoot meristem cell fate during Arabidopsis embryogenesis. EMBO J. 1998;17 10.1093/emboj/17.6.1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jovel J, Walker M, Sanfaçon H. Recovery of Nicotiana benthamiana Plants from a Necrotic Response Induced by a Nepovirus Is Associated with RNA Silencing but Not with Reduced Virus Titer. J Virol. 2007;81 10.1128/jvi.01192-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matzke M, Kanno T, Huettel B, Daxinger L, Matzke AJM. Targets of RNA-directed DNA methylation. Current Opinion in Plant Biology. 2007. 10.1016/j.pbi.2007.06.007 [DOI] [PubMed] [Google Scholar]
- 57.Dorweiler JE, Carey CC, Kubo KM, Hollick JB, Kermicle JL, Chandler VL. Mediator of paramutation 1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell. 2000;12 10.1105/tpc.12.11.2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia R, Chen C, Pokhrel S, Ma W, Huang K, Patel P, et al. 24-nt reproductive phasiRNAs are broadly present in angiosperms. Nat Commun. 2019;10 10.1038/s41467-019-08543-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Csorba T, Kontra L, Burgyán J. Viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virology. 2015. 10.1016/j.virol.2015.02.028 [DOI] [PubMed] [Google Scholar]
- 60.Ding SW. RNA-based antiviral immunity. Nature Reviews Immunology. 2010. 10.1038/nri2824 [DOI] [PubMed] [Google Scholar]
- 61.Borges F, Martienssen RA. The expanding world of small RNAs in plants. Nature Reviews Molecular Cell Biology. 2015. 10.1038/nrm4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nature Reviews Genetics. 2015. 10.1038/nrg3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Etebu E, Nwauzoma AB. A review on sweet orange (citrus sinensis)_ health, diseases and management. Am J Res Commun. 2014;2. [Google Scholar]
- 64.Xu Q, Chen LL, Ruan X, Chen D, Zhu A, Chen C, et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet. 2013;45 10.1038/ng.2472 [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Chen D, Lei Y, Chang JW, Hao BH, Xing F, et al. Citrus sinensis Annotation Project (CAP): A comprehensive database for sweet orange genome. PLoS One. 2014;9 10.1371/journal.pone.0087723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu GA, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol. 2014;32: 656–662. 10.1038/nbt.2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hertog MGL, Feskens EJM, Kromhout D, Hertog MGL, Hollman PCH, Hertog MGL, et al. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet. 1993;342 10.1016/0140-6736(93)92876-u [DOI] [PubMed] [Google Scholar]
- 68.Crowell PL. Prevention and therapy of cancer by dietary monoterpenes. Journal of Nutrition. 1999. 10.1093/jn/129.3.775S [DOI] [PubMed] [Google Scholar]
- 69.Tripoli E, Guardia M La, Giammanco S, Majo D Di, Giammanco M. Citrus flavonoids: Molecular structure, biological activity and nutritional properties: A review. Food Chem. 2007;104 10.1016/j.foodchem.2006.11.054 [DOI] [Google Scholar]
- 70.Di Majo D, Giammanco M, La Guardia M, Tripoli E, Giammanco S, Finotti E. Flavanones in Citrus fruit: Structure-antioxidant activity relationships. Food Res Int. 2005;38 10.1016/j.foodres.2005.05.001 [DOI] [Google Scholar]
- 71.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nei M, Saitou N. The neighbor-joining method: a new method for reco… [Mol Biol Evol. 1987]—PubMed result. Mol Biol Evol. 1987. [DOI] [PubMed] [Google Scholar]
- 74.Felsenstein J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution (N Y). 1985;39 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 75.Tajima F; Nei M. Estimation of evolutionary distance between nucleotide sequences. Mol Biol Evol. 1984;1: 269–85. 10.1093/oxfordjournals.molbev.a040317 [DOI] [PubMed] [Google Scholar]
- 76.Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol. 1995;3 [PubMed] [Google Scholar]
- 77.Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics. 2015;31 10.1093/bioinformatics/btu817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, et al. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45 10.1093/nar/gkw982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu L, Zhang Z, Mei Q, Chen M. PSI: A Comprehensive and Integrative Approach for Accurate Plant Subcellular Localization Prediction. PLoS One. 2013;8 10.1371/journal.pone.0075826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van De Peer Y, et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30: 325–327. 10.1093/nar/30.1.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mirzaei K, Bahramnejad B, Shamsifard MH, Zamani W. In silico identification, phylogenetic and bioinformatic analysis of argonaute genes in plants. Int J Genomics. 2014;2014 10.1155/2014/967461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dong Q, Lawrence CJ, Schlueter SD, Wilkerson MD, Kurtz S, Lushbough C, et al. Comparative plant genomics resources at PlantGDB. Plant Physiol. 2005;139 10.1104/pp.104.059212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rivas F V., Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, et al. Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol. 2005;12 10.1038/nsmb918 [DOI] [PubMed] [Google Scholar]
- 85.Baumberger N, Baulcombe DC. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci U S A. 2005;102 10.1073/pnas.0505461102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iyer LM, Koonin E V., Aravind L. Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct Biol. 2003;3 10.1186/1472-6807-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu X, Lu T, Dou Y, Yu B, Zhang C. Identification of RNA silencing components in soybean and sorghum. BMC Bioinformatics. 2014;15 10.1186/1471-2105-15-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang H, Xia R, Meyers BC, Walbot V. Evolution, functions, and mysteries of plant ARGONAUTE proteins. Current Opinion in Plant Biology. 2015. pp. 84–90. 10.1016/j.pbi.2015.06.011 [DOI] [PubMed] [Google Scholar]
- 89.Garg V, Agarwal G, Pazhamala LT, Nayak SN, Kudapa H, Khan AW, et al. Genome-wide identification, characterization, and expression analysis of small RNA biogenesis purveyors reveal their role in regulation of biotic stress responses in three legume crops. Front Plant Sci. 2017;8 10.3389/fpls.2017.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MacRae IJ, Doudna JA. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Current Opinion in Structural Biology. 2007. 10.1016/j.sbi.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 91.NICHOLSON AW. The ribonuclease III family: forms and functions in RNA maturation, decay, and gene silencing. RNAi a Guid to gene Silenc. 2003; 149–174. Available: http://ci.nii.ac.jp/naid/10028258270/en/ [Google Scholar]
- 92.Katsarou K, Mavrothalassiti E, Dermauw W, Van Leeuwen T, Kalantidis K. Combined Activity of DCL2 and DCL3 Is Crucial in the Defense against Potato Spindle Tuber Viroid. PLoS Pathog. 2016;12 10.1371/journal.ppat.1005936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Venkataraman S, Prasad BVLS, Selvarajan R. RNA dependent RNA polymerases: Insights from structure, function and evolution. Viruses. 2018. 10.3390/v10020076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marker S, Le Mouël A, Meyer E, Simon M. Distinct RNA-dependent RNA polymerases are required for RNAi triggered by double-stranded RNA versus truncated transgenes in Paramecium tetraurelia. Nucleic Acids Res. 2010;38 10.1093/nar/gkq131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.du Plessis L, Škunca N, Dessimoz C. The what, where, how and why of gene ontology-A primer for bioinformaticians. Brief Bioinform. 2011;12 10.1093/bib/bbr002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arnaud MB, Costanzo MC, Shah P, Skrzypek MS, Sherlock G. Gene Ontology and the annotation of pathogen genomes: the case of Candida albicans. Trends in Microbiology. 2009. 10.1016/j.tim.2009.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature. 1998;391 10.1038/35888 [DOI] [PubMed] [Google Scholar]
- 98.Lingel A, Izaurralde E. RNAi: Finding the elusive endonuclease. RNA. 2004. 10.1261/rna.7175704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lingel A, Simon B, Izaurralde E, Sattler M. Structure and nucleic-acid binding of the Drosophila Argonaute 2 PAZ domain. Nature. 2003;426 10.1038/nature02123 [DOI] [PubMed] [Google Scholar]
- 100.Ma JB, Ye K, Patel DJ. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature. 2004;429 10.1038/nature02519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Agrawal N, Dasaradhi PVN, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK. RNA Interference: Biology, Mechanism, and Applications. Microbiol Mol Biol Rev. 2003;67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chery J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016;4 10.14304/surya.jpr.v4n7.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lutova LA, Dodueva IE, Lebedeva MA, Tvorogova VE. Transcription factors in developmental genetics and the evolution of higher plants. Russian Journal of Genetics. 2015. 10.1134/S1022795415030084 [DOI] [PubMed] [Google Scholar]
- 104.Latchman DS. Transcription factors: An overview. Int J Biochem Cell Biol. 1997;29 10.1016/s1357-2725(97)00085-x [DOI] [PubMed] [Google Scholar]
- 105.Khan SA, Li MZ, Wang SM, Yin HJ. Revisiting the role of plant transcription factors in the battle against abiotic stress. International Journal of Molecular Sciences. 2018. 10.3390/ijms19061634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sasaki K. Utilization of transcription factors for controlling floral morphogenesis in horticultural plants. Breeding Science. 2018. 10.1270/jsbbs.17114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cheng W, Jiang Y, Peng J, Guo J, Lin M, Jin C, et al. The transcriptional reprograming and functional identification of WRKY family members in pepper’s response to Phytophthora capsici infection. BMC Plant Biol. 2020;20 10.1186/s12870-020-02464-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perotti MF, Ribone PA, Chan RL. Plant transcription factors from the homeodomain-leucine zipper family I. Role in development and stress responses. IUBMB Life. 2017. 10.1002/iub.1619 [DOI] [PubMed] [Google Scholar]
- 109.Shu Y, Liu Y, Zhang J, Song L, Guo C. Genome-wide analysis of the AP2/ERF superfamily genes and their responses to abiotic stress in Medicago truncatula. Front Plant Sci. 2016;6 10.3389/fpls.2015.01247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wen CL, Cheng Q, Zhao L, Mao A, Yang J, Yu S, et al. Identification and characterisation of Dof transcription factors in the cucumber genome. Sci Rep. 2016;6 10.1038/srep23072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Venkatesh J, Park SW. Genome-wide analysis and expression profiling of DNA-binding with one zinc finger (Dof) transcription factor family in potato. Plant Physiol Biochem. 2015;94 10.1016/j.plaphy.2015.05.010 [DOI] [PubMed] [Google Scholar]
- 112.Skirycz A, Reichelt M, Burow M, Birkemeyer C, Rolcik J, Kopka J, et al. DOF transcription factor AtDof1.1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. Plant J. 2006;47 10.1111/j.1365-313X.2006.02767.x [DOI] [PubMed] [Google Scholar]
- 113.Skirycz A, Jozefczuk S, Stobiecki M, Muth D, Zanor MI, Witt I, et al. Transcription factor AtDOF4;2 affects phenylpropanoid metabolism in Arabidopsis thaliana. New Phytol. 2007;175 10.1111/j.1469-8137.2007.02129.x [DOI] [PubMed] [Google Scholar]
- 114.Noguero M, Atif RM, Ochatt S, Thompson RD. The role of the DNA-binding One Zinc Finger (DOF) transcription factor family in plants. Plant Science. 2013. 10.1016/j.plantsci.2013.03.016 [DOI] [PubMed] [Google Scholar]
- 115.Prakash V, Chakraborty S. Identification of transcription factor binding sites on promoter of RNA dependent RNA polymerases (RDRs) and interacting partners of RDR proteins through in silico analysis. Physiol Mol Biol Plants. 2019;25 10.1007/s12298-019-00660-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seo PJ, Park CM. MYB96-mediated abscisic acid signals induce pathogen resistance response by promoting salicylic acid biosynthesis in Arabidopsis. New Phytol. 2010;186 10.1111/j.1469-8137.2010.03183.x [DOI] [PubMed] [Google Scholar]
- 117.Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends in Plant Science. 2010. 10.1016/j.tplants.2010.06.005 [DOI] [PubMed] [Google Scholar]
- 118.Shim JS, Jung C, Lee S, Min K, Lee YW, Choi Y, et al. AtMYB44 regulates WRKY70 expression and modulates antagonistic interaction between salicylic acid and jasmonic acid signaling. Plant J. 2013;73 10.1111/tpj.12051 [DOI] [PubMed] [Google Scholar]
- 119.Mzid R, Marchive C, Blancard D, Deluc L, Barrieu F, Corio-Costet MF, et al. Overexpression of VvWRKY2 in tobacco enhances broad resistance to necrotrophic fungal pathogens. Physiol Plant. 2007;131 10.1111/j.1399-3054.2007.00975.x [DOI] [PubMed] [Google Scholar]
- 120.Oh SK, Baek KH, Park JM, Yi SY, Yu SH, Kamoun S, et al. Capsicum annuum WRKY protein CaWRKY1 is a negative regulator of pathogen defense. New Phytol. 2008;177 10.1111/j.1469-8137.2007.02310.x [DOI] [PubMed] [Google Scholar]
- 121.Guo H, Wang Y, Wang L, Hu P, Wang Y, Jia Y, et al. Expression of the MYB transcription factor gene BplMYB46 affects abiotic stress tolerance and secondary cell wall deposition in Betula platyphylla. Plant Biotechnol J. 2017;15 10.1111/pbi.12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mao X, Zhang H, Qian X, Li A, Zhao G, Jing R. TaNAC2, a NAC-type wheat transcription factor conferring enhanced multiple abiotic stress tolerances in Arabidopsis. J Exp Bot. 2012;63 10.1093/jxb/err462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marè C, Mazzucotelli E, Crosatti C, Francia E, Stanca AM, Cattivelli L. Hv-WRKY38: A new transcription factor involved in cold- and drought-response in barley. Plant Mol Biol. 2004;55 10.1007/s11103-004-0906-7 [DOI] [PubMed] [Google Scholar]
- 124.Ranty B, Aldon D, Galaud J-P. Plant Calmodulins and Calmodulin-Related Proteins. Plant Signal Behav. 2006;1 10.4161/psb.1.3.2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reddy ASN, Ali GS, Celesnik H, Day IS. Coping with stresses: Roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell. 2011. 10.1105/tpc.111.084988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang H, Jiao X, Kong X, Hamera S, Wu Y, Chen X, et al. A signaling cascade from miR444 to RDR1 in rice antiviral RNA silencing pathway. Plant Physiol. 2016;170 10.1104/pp.15.01283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kaur A, Pati PK, Pati AM, Nagpal AK. In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. Mehrotra R, editor. PLoS One. 2017;12: e0184523 10.1371/journal.pone.0184523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wittkopp PJ, Kalay G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nature Reviews Genetics. 2012. 10.1038/nrg3095 [DOI] [PubMed] [Google Scholar]
- 129.Le Gourrierec J, Li YF, Zhou DX. Transcriptional activation by Arabidopsis GT-1 may be through interaction with TFIIA-TBP-TATA complex. Plant J. 1999;18: 663–668. 10.1046/j.1365-313x.1999.00482.x [DOI] [PubMed] [Google Scholar]
- 130.Giuliano G, Pichersky E, Malik VS, Timko MP, Scolnik PA, Cashmore AR. An evolutionarily conserved protein binding sequence upstream of a plant light-regulated gene. Proc Natl Acad Sci U S A. 1988;85 10.1073/pnas.85.19.7089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Menkens AE, Schindler U, Cashmore AR. The G-box: a ubiquitous regulatory DNA element in plants bound by the GBF family of bZIP proteins. Trends in Biochemical Sciences. 1995. 10.1016/s0968-0004(00)89118-5 [DOI] [PubMed] [Google Scholar]
- 132.Ishige F, Takaichi M, Foster R, Chua NH, Oeda K. A G-box motif (GCCACGTGCC) tetramer confers high-level constitutive expression in dicot and monocot plants. Plant J. 1999;18 10.1046/j.1365-313X.1999.00456.x [DOI] [Google Scholar]
- 133.Arias JA, Dixon RA, Lamb CJ. Dissection of the functional architecture of a plant defense gene promoter using a homologous in vitro transcription initiation system. Plant Cell. 1993;5: 485–496. 10.1105/tpc.5.4.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chon W, Provart NJ, Glazebrook J, Katagiri F, Chang HS, Eulgem T, et al. Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell. 2002;14: 559–574. 10.1105/tpc.010410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maruyama K, Todaka D, Mizoi J, Yoshida T, Kidokoro S, Matsukura S, et al. Identification of cis-acting promoter elements in cold-and dehydration-induced transcriptional pathways in arabidopsis, rice, and soybean. DNA Res. 2012;19: 37–49. 10.1093/dnares/dsr040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ezcurra I, Wycliffe P, Nehlin L, Ellerström M, Rask L. Transactivation of the Brassica napus napin promoter by ABI3 requires interaction of the conserved B2 and B3 domains of ABI3 with different cis-elements: B2 mediates activation through an ABRE, whereas B3 interacts with an RY/G-box. Plant J. 2000;24 10.1046/j.1365-313x.2000.00857.x [DOI] [PubMed] [Google Scholar]
- 137.Kaur A, Pati PK, Pati AM, Nagpal AK. In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. PLoS One. 2017;12 10.1371/journal.pone.0184523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou Y, Hu L, Wu H, Jiang L, Liu S. Genome-Wide Identification and Transcriptional Expression Analysis of Cucumber Superoxide Dismutase (SOD) Family in Response to Various Abiotic Stresses. Int J Genomics. 2017;2017 10.1155/2017/7243973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nishida J, Yoshlda M, Aral K ichi, Yokota T. Definition of a GC-rich motif as regulatory sequence of the human IL-3 gene: Coordinate regulation of the IL-3 gene by CLE2/GC box of the GM-CSF gene in T cellactivation. Int Immunol. 1991;3 10.1093/intimm/3.3.245 [DOI] [PubMed] [Google Scholar]
- 140.Lundin M, Nehlin JO, Ronne H. Importance of a flanking AT-rich region in target site recognition by the GC box-binding zinc finger protein MIG1. Mol Cell Biol. 1994;14: 1979–1985. 10.1128/mcb.14.3.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Martin-Malpartida P, Batet M, Kaczmarska Z, Freier R, Gomes T, Aragón E, et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat Commun. 2017;8: 1–15. 10.1038/s41467-016-0009-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shariatipour N, Heidari B. Investigation of Drought and Salinity Tolerance Related Genes and their Regulatory Mechanisms in Arabidopsis (Arabidopsis thaliana). Open Bioinforma J. 2018;11 10.2174/1875036201811010012 [DOI] [Google Scholar]
- 143.Kim SR, Kim Y, An G. Identification of methyl jasmonate and salicylic acid response elements from the nopaline synthase (nos) promoter. Plant Physiol. 1993;103 10.1104/pp.103.1.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu J, Wang F, Yu G, Zhang X, Jia C, Qin J, et al. Functional Analysis of the Maize C-Repeat/DRE Motif-Binding Transcription Factor CBF3 Promoter in Response to Abiotic Stress. Int J Mol Sci. 2015;16: 12131–12146. 10.3390/ijms160612131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vaucheret H. Plant ARGONAUTES. Trends in Plant Science. 2008. 10.1016/j.tplants.2008.04.007 [DOI] [PubMed] [Google Scholar]
- 146.Nakasugi K, Crowhurst RN, Bally J, Wood CC, Hellens RP, Waterhouse PM. De Novo Transcriptome Sequence Assembly and Analysis of RNA Silencing Genes of Nicotiana benthamiana. PLoS One. 2013;8 10.1371/journal.pone.0059534 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(TXT)
(TXT)
(TXT)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
GO enrichment analysis of the predicted RNAi genes (A) biological process, (B) molecular function and (C) cellular process. In the directed acyclic graph (DAG) the downstream term corresponds to a subset of the upstream term. The significant (p-value < 0.05, FDR < 0.05) GO terms are in colored boxes (the degree of color saturation is positively correlated to the enrichment level of the GO term), and non-significant terms are in white boxes.
(PDF)
Rows of the figure represent the predicted RNAi genes and the columns represent the families of the TFs. The number indicates the TF families regulate the RNAi genes.
(PDF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.