

Graphical abstract

Abbreviations: 2019-nCoV, 2019 novel coronavirus; ACE2, Angiotensin-converting enzyme 2; Covid-19, coronavirus disease 2019; DPP4, dipeptidyl peptidase 4; LARMD, ligand and receptor molecular dynamics; MERS-CoV, Middle East respiratory syndrome coronavirus; MM-GBSA, molecular mechanics-generalized born surface area; MM-PBSA, molecular mechanics-Poisson–Boltzmann surface area; PCA, principal component analysis; PDB, Protein Data Bank; PPIs, protein–protein interactions; Q, fraction of native contacts; RBD, receptor-binding domain; Rg, radius of gyration; RMSD, root mean square deviation; ROF, rule of five; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2
Keywords: Receptor-binding domain, RBD, Angiotensin-converting enzyme 2, ACE2, Spike protein, Protein–protein interface, COVID-19, SARS-CoV-2, Molecular docking, Molecular dynamics simulation
Abstract
Coronavirus disease 2019 (COVID-19) is a fatal infectious disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). The virus infection is initiated upon recognition and binding of the spike (S) protein receptor-binding domain (RBD) to the host cell surface receptor, angiotensin-converting enzyme 2 (ACE2). Blocking the interaction between S protein and ACE2 receptor is a novel approach to prevent the viral entry into the host cell. The present study is aimed at the identification of small molecules which can disrupt the interaction between SARS-CoV-2 S protein and human ACE2 receptor by binding to the interface region. A chemical library consisting of 1,36,191 molecules were screened for drug-like compounds based on Lipinski’s rule of five, Verber’s rule and in silico toxicity parameters. The filtered drug-like molecules were next subjected to molecular docking in the interface region of RBD. The best three hits viz; ZINC64023823, ZINC33039472 and ZINC00991597 were further taken for molecular dynamics (MD) simulation studies and binding free energy evaluations using Molecular mechanics-Poisson–Boltzmann surface area (MM-PBSA) and Molecular mechanics-Generalized Born surface area (MM-GBSA). The protein-ligand complexes showed stable trajectories throughout the simulation time. ZINC33039472 exhibited binding free energy value lower as compared to the control (emodin) with a higher contribution by gas-phase energy and van der Waals energy to the total binding free energy. Thus, ZINC33039472 is identified to be a promising interfacial binding molecule which can inhibit the interaction between the viral S protein and human ACE2 receptor which would consequently help in the management of the disease.
Introduction
Since its first emergence in December 2019 in Wuhan city, Hubei province of China, the highly pathogenic coronavirus known initially as a 2019 novel coronavirus (2019-nCoV) and later described as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has rapidly spread to more than eighty other countries leading to serious global health emergency [1]. The virus has caused a new fatal disease known as coronavirus disease 2019 (COVID-19) with patients reporting a wide range of symptoms such as fever, dry cough, dyspnea, headache, and pneumonia [2,3]. Two other highly pathogenic human coronaviruses have been previously reported viz; the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2003 and Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012. Compared to the previous outbreaks from these two viruses, SARS-CoV-2 has a low mortality rate which ranges between 3 to 5% [1]. Phylogenetic studies have shown that SARS-CoV-2 is a new member within betacoronavirus genus which includes MERS-CoV, SARS-CoV, bat SARS-related coronaviruses (SARSr-CoV) etc. [4]. The closest relative of the SARS-CoV-2 is most likely to be Bat coronavirus RaTG13 which shows a similarity of 93.1% sequence identity in the spike (S) gene [5]. At present, there are no effective prophylactics or therapeutics available for the treatment of COVID-19 and as of May 5, 2020, there has been a total of 3 489 053 confirmed cases and 241 559 confirmed death reports [6].
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a single positive-strand RNA enveloped virus with a genome size of 29.9 kb [7]. There are four structural proteins present in coronavirus viz; spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins [3]. Out of these, S protein, a type I transmembrane glycoprotein mediates key functions such as viral attachment, fusion and entry [8]. The first step in the viral infection cycle is the viral entry into the host cell which is mediated by binding of the S protein receptor-binding domain (RBD) to the host cell surface receptor which is angiotensin-converting enzyme 2 (ACE2) receptor in SARS-CoV and dipeptidyl peptidase 4 (DPP4) in case of MERS-CoV [9,10]. Similarly, ACE2 also serves as a receptor for SARS-CoV-2 S protein. The RBD (residues 318–510) in the spike S1 subunit (residues 17–680) mediates the binding of the virus to the host cell surface receptor and the transmembrane S2 subunit (residues 681–1195) allows the fusion between the virus and the host cell membrane [11,12]. Because of its significant role in viral entry into the host cell, S protein has emerged as an important therapeutic target for the development of antibodies, vaccines and entry inhibitors [6].
Protein–protein interactions (PPIs) play an important role in regulating various cellular processes as well as drive the pathogenesis in many human diseases such as neurodegenerative diseases, cervical cancer, bacterial infection and leukaemia etc. [13]. Computational study of protein–protein interactions plays a significant role in the drug discovery process which can provide useful insights into the biochemical nature of the protein–protein interface, mutations affecting the binding interface and designing small molecules which can regulate the pathological cellular processes [14]. This rational approach has been widely used in designing of many PPI modulators which can be categorized into three different types- a) orthosteric inhibitor b) allosteric inhibitor and c) interfacial inhibitor [15]. The orthosteric inhibitors bind to the protein target preferentially at regions which are normally used by other interacting protein partners for binding and therefore, disrupt the formation of functional biological complexes. A typical example of orthosteric inhibitor is MI-2-2 which binds to menin with nanomolar binding affinity and disrupts the protein–protein interaction between menin and MLL in mixed lineage leukemia (MLL) leukaemia cells [16]. Whereas allosteric inhibitors bind to sites distinct from the protein–protein interface region and cause a conformational change in the target protein and thereby destabilizing the protein–protein complex. BRaf inhibitor, PLX4032 is an example of an allosteric inhibitor that inhibits BRaf-CRaf heterodimerization and activation [17]. The interfacial inhibitors are small molecules which bind to a pocket at the protein–protein interface to form a ternary complex and render the protein ineffective by locking the complex into a non-functional state. An example in this category include the interfacial binding inhibitor BFA binds at the protein–protein interface of ARF1–GDP-Sec7 complex and inhibits conformational changes in ARF1 necessary for Sec7 to displace the GDP molecule [18]. Protein–protein interaction study between human ACE2 and SARS-CoV-2 S protein can be used to design interface binding inhibitors which can disrupt the protein–protein interaction and inhibit the attachment and entry of the virus into the host cell. In this study, we have screened a chemical compound library to screen novel drug-like molecules which can effectively bind to the interface region between RBD of SARS-CoV-2 S protein and human ACE2 which will help in blocking the viral attachment and entry into the host cell.
Materials and methods
Retrieval of three-dimensional crystal structures of target proteins
The three dimensional X-ray crystal structure of SARS-CoV-2 spike receptor-binding domain (RBD) bound with human ACE2 (PDB ID: 6M0J) and the crystal structure of SARS-CoV spike RBD complexed with human ACE2 (PDB ID: 2AJF) were retrieved from Protein Data Bank (PDB) (http://www.rcsb.org/) at a resolution of 2.45 Å and 2.90 Å respectively.
Characterization of protein–protein interface region
The protein–protein interface statistics including interface area, interface residues and residue interactions across the interface (salt bridges, disulphide bonds, hydrogen bonds and non-bonded contacts) were determined using PDBsum program [19]. The hotspot residues in protein–protein interfaces were calculated using alanine scanning mutagenesis program of DrugScorePPI webserver [20].
Sequence alignment and structural superposition
The pairwise sequence alignment between SARS-CoV-2 spike RBD and SARS-CoV spike RBD was carried out using the CLUSTAL W program [21]. The root mean square deviation (RMSD) between structures of SARS-CoV-2 spike RBD and SARS-CoV spike RBD was calculated using UCSF Chimera [22].
Retrieval of three-dimensional structures of ligands
A random set of 1,36,191 chemical compounds were retrieved from the ZINC database [23]. The chemical structures were downloaded and saved in SDF format for the virtual screening process.
Virtual screening of small drug-like molecules
The chemical library was subjected to virtual screening procedure based on selected drug-like filters such as Veber’s rule [24], Lipinski's rule of five (ROF) [25] and in silico toxicity parameters such as mutagenicity, tumorigenicity, irritancy and reproductive effects. The physicochemical properties of the compounds were calculated using the OSIRIS DataWarrior program version 5.0 [26].
Molecular docking studies
Virtual screening of potential hits binding to SARS-CoV-2 spike RBD was performed using ArgusLab docking program version 4.0 [27]. A grid box of size 40 × 40 × 40 Å with a grid resolution of 0.4 was placed at the centre around the interface residues. The other parameters include AScore as scoring function and the DockEngine was set to exhaustive search. The top 100 hits were docked within the interface region using AutoDock Vina program which utilizes sophisticated gradient optimization method [28]. The target protein was prepared by removing the heteroatoms such as ions, water molecules and cocrystal ligands and the addition of polar hydrogen atoms and Kollman charges. Similarly, the chemical compounds were prepared by adding hydrogen atoms, Gasteiger charges and optimally defining the torsions. A grid box encompassing the interface region was centred at XYZ coordinates of −36.223, 27.8467 and 11.8760 with dimensions of X: 27.5694 Å, Y: 45.4190 Å and Z: 30.0170 Å. The binding conformations were clustered and ranked according to their binding affinities. The molecular interactions (hydrogen bonds and hydrophobic interactions) between the target protein and compounds were evaluated using LigPlot + program version 1.4.5 [29].
Molecular dynamics simulation
The molecular dynamics of the spike-ligand docked complexes were performed using AMBER16 program available on Ligand and Receptor Molecular Dynamics (LARMD) (http://chemyang.ccnu.edu.cn/ccb/server/LARMD/). The interactional binding mode (Int_mod) was chosen for 4 ns MD simulation in an explicit water model [30]. The steps involved in the MD simulations consist of (a) assignment of bcc charges to the ligand atoms using the antechamber module of the AMBER16 program [31] (b) generation of the coordinate and topology files of the complex using Tleap module of the AMBER16 program (c)The force fields selected for amino acid residues and ligands include the AMBER ff14SB force field [32] and General AMBER Force Field (GAFF) [33,34] respectively (d) Addition of counter ions (Na+ and Cl−) to the system (e) the systems were explicitly solvated in an octahedral box of TIP3P waters [35] (f) energy minimization with 2000 steps steepest descent method and 3000 steps conjugated gradient method using sander module in the AMBER16 program [36] (g) the systems were gradually heated by raising the temperature from 10 to 300 K for 30 ps. All the atoms were relaxed using periodic boundary conditions at a temperature of 300 K and pressure of 1 atm.
Analysis of trajectories
The geometric properties of the systems such as root mean square deviation (RMSD), radius of gyration (Rg) and fraction of native contacts (Q) values were analysed using the Cpptraj module in the AMBER16 program module [37]. R package was used to analyse the RMSD distribution of protein and ligand in a histogram representation. The non-native contacts were calculated using MDTraj was and Bio3d was used to study the principal component analysis PCA and residue cross-correlation [38].
Binding free energy analysis
The binding free energy () was calculated using the following Eq. (1)
| (1) |
where is the binding energy, is the solvation entropy and is the conformational entropy. While the enthalpy was calculated using the Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) or molecular mechanics generalized Born surface area (MM-GBSA) method [39], entropy was derived using an empirical method [40,41].
Results and discussion
Characterization of the interface region between ACE2 and spike protein
The human angiotensin-converting enzyme 2 (ACE2) and SARS-CoV-2 spike receptor-binding domain (RBD) and human ACE2 and SARS-CoV spike RBD were analysed for their interface regions and key residues contributing to the binding recognition between the interacting protein partners. The interface region between human ACE2 and SARS-CoV spike RBD complex was investigated and there are nineteen interface residues (Gln24, Thr27, Phe28, Lys31, His34, Glu37, Asp38, Tyr41, Gln42, Leu45, Met82, Tyr83, Gln325, Glu329, Asn330, Lys353, Gly354, Asp355 and Arg357) of ACE2 and fifteen residues of spike RBD (Arg426, Tyr436, Tyr440, Tyr442, Leu472, Asn473, Tyr475, Asn479, Gly482, Tyr484, Thr486, Thr487, Gly488, Ile489 and Tyr491) forming an interface area of 832 Å2 and 872 Å2 respectively (Fig. 1 A). The complex is stabilised by 1 salt bridge, 9 hydrogen bonds and 94 non-bonded contacts. Further, alanine scanning mutagenesis of the interface residues was carried out to determine residues (hotspots) contributing more to the binding between the two interacting proteins. Hot spots are residues which contributes a significant increase in the binding free energy of at least 2.0 kcal/mol when replaced with alanine [42]. Both ACE2 receptor and SARS-CoV spike RBD possess two hotspot residues in their interface regions which are Asp38 and Tyr41 in ACE2 and Tyr475 and Tyr491 in spike RBD (Suppl. Fig. S1A). The interface statistics of ACE2 and SARS-CoV-2 spike RBD complex show that there are twenty residues (Gln24, Thr27, Phe28, Asp30, Lys31, His34, Glu35, Glu37, Asp38, Tyr41, Gln42, Leu79, Met82, Tyr83, Asn330, Asp335, Lys353, Gly354, Arg357 and Arg393) belonging to human ACE2 and seventeen residues of RBD (Lys417, Gln443, Gly446, Tyr449, Tyr453, Leu455, Phe456, Ala475, Phe486, Asn487, Tyr489, Gly496, Gln498, Thr500, Asn501, Gly502 and Tyr505) contributing to interface area of 825 Å2 and 863 Å2 respectively (Fig. 1B). The protein–protein complex between ACE2 and SARS-CoV-2 spike RBD were stabilized by 1 salt bridge, 9 hydrogen bonds and 101 non-bonded contacts. Three hotspot residues (Asp38, Tyr41 and Tyr83) were identified in ACE2 and two hot spot residues (Tyr489 and Tyr505) in SARS-CoV-2 spike RBD (Suppl. Fig. S1B). The interface statistics differences in terms of the number of interface residues and non-bonded contacts between the two protein–protein complexes may plausibly explain the differential nature of binding of SARS-CoV-2 and SARS-CoV spike protein to ACE2 receptor. It has been revealed from an earlier study that ACE2 binds to SARS-CoV-2 ectodomain with approximately 15 nM affinity which is around 10–20 fold higher than its binding to SARS-CoV [43].
Fig. 1.

The interface statistics for (A) SARS-CoV spike RBD and human ACE2 receptor complex (B) SARS-CoV-2 spike RBD and human ACE2 receptor complex depicting the interface area, residues and molecular interactions. The interface residues have been coloured according to the physicochemical properties such as positive residues (His, Lys and Arg) in blue, negative residues (Asp, Glu) in red, neutral residues (Ser, Thr, Asn, Gln) in green, aliphatic residues (Ala, Val, Leu, Ile and Met) in grey, aromatic residues (Phe, Tyr and Trp) in purple and other residues (Pro and Gly) in orange.
Sequence alignment and structural superposition
The primary sequence alignment of SARS-CoV-2 and SARS-CoV spike RBD domains show a sequence similarity of 72.38% and out of the five critical residues (Tyr442, Leu472, Asn479, Thr487 and Tyr491) at the SARS-CoV spike RBD and ACE2 interface, only Tyr491 is conserved (Fig. 2 A). The results corroborates with the previous studies by Xie and Chen [44]. It is worth mentioning here that Asn479 and Asn487 are two important residues in RBD which determine SARS progression and tropism, and their mutations may increase animal-to-human or human-to-human transmission [12]. Further, their structural superposition exhibits a root mean square deviation (RMSD) of 0.674 Å (Fig. 2B) which means that the structure of the RBD domain has undergone significant change during viral co-evolution within the host cell.
Fig. 2.

(A) Pairwise sequence alignment between SARS-CoV-2 spike RBD and SARS-CoV spike RBD. The blue encircled residues indicate the five critical interface residues (Tyr442, Leu472, Asn479, Thr487 and Tyr491) where blue asterisk show the conserved interface residue (Tyr491) and the red arrow indicates the amino acid positions (Asn479 and Thr487) prone to mutations which determine animal-to-human or human-to-human transmission. (B) Structural superposition between SARS-CoV-2 spike RBD (cyan) and SARS-CoV spike RBD (tan).
Virtual screening of drug-like molecules
A total of 1,36,191 chemical compounds from the ZINC database were subjected to virtual screening procedure and the molecules were filtered based on their physicochemical properties and toxicity profile. A final set of 42,889 molecules qualified the rule of five, Veber’s rule and found to have favourable drug-like properties such as being non-mutagenic, non-tumorigenic, non-irritant and no adverse effects on the reproductive health.
Molecular docking studies
An initial virtual screening was performed using ArgusLab docking program considering interface region of SARS-CoV-2 spike RBD as the binding pocket for a set of 42,889 drug-like molecules. Out of which a total of 100 high ranked hits were obtained which possess higher docking scores (docking scores ranging between −11.9152 kcal/mol and −13.7791 kcal/mol) to the interface region of spike RBD as compared to the control Emodin (−8.2187 kcal/mol) (Suppl. Table S1). Emodin is a naturally occurring anthraquinone derivative extracted from Rheum officinale and Polygonum multiflorum which has been demonstrated to have anti-SARS activity by blocking the binding of Spike (S) protein to ACE2 and reducing the infectivity of S protein-pseudotyped retrovirus to Vero E6 cells [45]. The 100 high ranked hits were further taken for the second round of virtual screening with Autodock vina program for rescoring and reranking the hits (Suppl. Table S2). The top 3 hits obtained were ZINC64023823 (−8.2 kcal/mol), ZINC33039472 (−8.0 kcal/mol) and ZINC00991597 (−7.9 kcal/mol) which possessed higher binding affinity to SARS-CoV-2 spike RBD as compared to emodin (−6.1 kcal/mol) (Table 1 ). The drug-like properties of the top 3 compounds along with the control are enumerated in Table 2 The interaction between ZINC64023823 and spike protein is strengthened by three hydrogen bonds with residues Asn437, Asn440 and Gln506 and hydrophobic interactions with eight residues including Leu441, Arg509, Ala344, Asn343, Phe374, Ser373, Trp436 and Val503 (Fig. 3 A). Whereas, the second lead molecule ZINC33039472 forms one hydrogen bond with Ty453 within the binding cleft of spike protein and the interaction is further reinforced through hydrophobic interactions with seven residues including Arg403, Tyr495, Tyr505, Asn501, Gly502, Gly496 and Gln498 (Fig. 3B). The third lead molecule ZINC00991597 interacts with the spike protein by establishing one hydrogen bond with Gly496 and hydrophobic interactions contributed by seven residues including Tyr453, Ser494, Asn501, Gly502, Tyr505, Arg403 and Tyr495 (Fig. 3C). Interestingly, the control emodin establishes four hydrogen bonds with residues-Arg346, Tyr351, Asn354 and Asn450 and hydrophobic interactions via five residues such as Ala348, Phe347, Ala352, Ser349 and Leu452 (Fig. 3D).
Table 1.
The binding energies of top 3 ranked molecules along with the control (Emodin) obtained through virtual screening using AutoDock Vina docking program.
| Molecules | Structure | IUPAC name | Binding energy (kcal/mol) |
|---|---|---|---|
| ZINC64023823 |  |
3-(2H-indol-3-yl)-N-[2-[(2R)-2-phenyl-3,5-dihydro-2H-1,4-benzoxazepin-4-yl]ethyl]propanamide | −8.2 |
| ZINC33039472 | 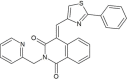 |
(4E)-4-[(2-phenylthiazol-4-yl)methylene]-2-(2-pyridylmethyl)isoquinoline-1,3-dione | −8.0 |
| ZINC00991597 | 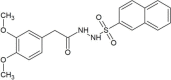 |
2-(3,4-dimethoxyphenyl)-N'-(2-naphthylsulfonyl)acetohydrazide | −7.9 |
| Emodin (CID3220) |  |
1,3,8-trihydroxy-6-methylanthracene-9,10-dione | −6.1 |
Table 2.
Drug-like physicochemical profile of top 3 lead molecules along with the control.
| Molecule | MWa | cLogPb | cLogSc | HBAd | HBDe | TPSAf (Å2) | RBg | Druglikeness | Mutagenic | Tumorigenic | Reproductive effective | Irritant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ZINC64023823 | 441.573 | 1.1227 | −3.549 | 5 | 3 | 56.74 | 7 | 1.5967 | None | None | None | None |
| ZINC33039472 | 423.495 | 3.6596 | −3.855 | 5 | 0 | 91.4 | 4 | 5.92 | None | None | None | None |
| ZINC00991597 | 400.454 | 1.7456 | −3.112 | 7 | 2 | 102.11 | 6 | 3.8539 | None | None | None | None |
| Emodin (CID3220) | 270.239 | 2.3402 | −4.186 | 5 | 3 | 94.83 | 0 | −1.1275 | High | High | High | High |
a: Molecular weight; b: Partition coefficient between n-octanol and water; c: Aqueous solubility at 25° and pH = 7.5; d: Hydrogen bond acceptor; e: Hydrogen bond donor; f: Polar surface area; g: Rotatable bonds.
Fig. 3.

Binding conformations and molecular interactions between SARS-CoV-2 spike RBD and (A) ZINC64023823 (B) ZINC33039472 (C) ZINC00991597 (D) Emodin (CID3220). The binding conformations on the left side panel show the molecules in stick representations surrounded by interacting residues in wireframe with hydrogen bonds depicted in cyan. The LigPlot + results show the molecular interactions in two-dimensional representation with hydrogen bonds depicted in green and residues contributing to hydrophobic interactions are rendered with red arcs with protruding eyelashes.
MD simulation analysis
The binding modes of the best three lead molecules- ZINC64023823, ZINC33039472 and ZINC00991597 along with the control (emodin) were further explored using molecular dynamics simulation study in an aqueous environment for a simulation time of 4 ns. The geometric properties of the protein-ligand complexes such as root mean square deviation (RMSD), radius of gyration (Rg) and fraction of native contacts (Q) values were evaluated to check the stability of the system. The RMSD gives the measurement of root mean square deviation of atomic positions which is used to calculate the average distance between the atoms of superimposed structures of protein and ligand over a period of time [46]. The average RMSD of Cα atoms of spike protein and heavy atoms of ZINC64023823 in spike(RBD)_ZINC64023823 complex was found to be 1.37 ± 0.1644 Å and 1.734 ± 0.3857 Å respectively with respect to the starting structures (Fig. 4 A). In the case of spike(RBD)_ZINC33039472 complex, the average RMSD of Cα atoms of spike protein and heavy atoms of ZINC33039472 were 1.414 ± 0.2028 Å and 1.78 ± 0.5061 Å respectively (Fig. 4B). The average RMSD of Cα atoms of spike protein and heavy atoms of ZINC00991597 in spike(RBD)_ ZINC00991597 complex was found to be 1.308 ± 0.1676 Å and 2.374 ± 0.4118 Å (Fig. 4C). Whereas, spike(RBD)_emodin complex has an average RMSD of 1.25 ± 0.1744 Å for Cα atoms of spike protein and 0.3439 ± 0.1491 Å for heavy atoms of emodin (Fig. 4D). Rg can be explained as the root mean square distance from each atom of the system to its centre of mass [47]. The Rg values for protein-ligand complexes: spike(RBD)_ZINC64023823 and spike(RBD)_ZINC33039472 show stable fluctuation between 18.1 to 18.5 Å whereas spike(RBD)_ ZINC00991597 and spike(RBD)_emodin complexes stably fluctuated between 18.0 to 18.4 Å (Suppl. Fig. S2). The Q gives an idea about conformational dynamics and the transition states of a protein with a folding free energy barrier [48]. The Q values for ZINC64023823, ZINC33039472 and ZINC00991597 along with the control (emodin) were found to be 0.976819 ± 0.006382, 0.965613 ± 0.013479, 0.983011 ± 0.008702 and 0.957289 ± 0.012163 respectively (Suppl. Fig. S3). Principal component analysis (PCA) or essential dynamics (ED) is considered as a powerful method for clustering the conformations of a protein and extracting the large concerted modes of fluctuations from trajectories of MD simulations. The contribution of eigenvector 1 (PC1) towards the total mean square fluctuations were found to be 20.916 Å2 (17.158%), 18.749 Å2 (14.518%), 19.777 Å2 (16.486%) and 16.677 Å2 (14.476%) for spike(RBD)_ZINC64023823, spike(RBD)_ZINC33039472, spike(RBD)_ZINC00991597 and spike(RBD)_emodin respectively (Fig. 5 ). Eigenvector 2 contributions to the total mean square fluctuations for spike(RBD)_ZINC64023823, spike(RBD)_ZINC33039472, spike(RBD)_ZINC00991597 and spike(RBD)_emodin were calculated to be 13.421 Å2 (11.009%), 14.789 Å2 (11.451%), 15.030 Å2 (12.529%) and 12.549 Å2 (10.891%) respectively. Whereas eigenvector 3 (PC3) also shares significant contributions to the total mean square fluctuations in spike(RBD)_ZINC64023823, spike(RBD)_ZINC33039472, spike(RBD)_ZINC00991597 and spike(RBD)_emodin complexes with their corresponding eigenvalues as 11.192 Å2 (9.182%), 9.341 Å2 (7.233%), 7.448 Å2 (6.209%) and 10.663 Å2 (9.255%).
Fig. 4.

Plot of root mean square deviation (RMSD) versus simulation time (ps) for (A) spike(RBD)_ZINC64023823 (B) spike(RBD)_ZINC33039472 (C) spike(RBD)_ZINC00991597 (D) spike(RBD)_Emodin. The black line indicates the RMSD curve of backbone atoms of protein whereas the red line corresponds to RMSD curve of heavy atoms of ligand in each plot.
Fig. 5.

Principal component analysis (PCA) for conformational changes with trajectory frames coloured from blue to white to red in order of MD simulation time for (A) spike(RBD)_ZINC64023823 (B) spike(RBD)_ZINC33039472 (C) spike(RBD)_ZINC00991597 (D) spike(RBD)_Emodin. The first twenty eigenvectors out of 582 eigenvectors (3 N, where N = 194 Cα atoms) capture at least 70% of the total variance and first three eigenvectors (PC1, PC2 and PC3) contributes over 50% to the total variance.
Binding free energy analysis
The binding free energies of ZINC64023823 (ΔPB = −2.91 kcal/mol, ΔGB = −3.00 kcal/mol), ZINC33039472 (ΔPB = −9.08 kcal/mol, ΔGB = −11.73 kcal/mol), ZINC00991597 (ΔPB = −2.53 kcal/mol, ΔGB = −3.67 kcal/mol) and the control (emodin) (ΔPB = −6.02 kcal/mol, ΔGB = −6.41 kcal/mol) were determined using Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) and molecular mechanics generalized Born surface area (MM-GBSA) methods. (Table 3 ). Out of the three lead compounds, ZINC33039472 has a binding free energy value higher than the control with major contribution by Gas phase energy (−33.89 ± 3.56 kcal/mol) and van der Waals energy (−30.20 ± 3.89 kcal/mol). The top ten residues contributing towards the binding interaction between ZINC33039472 and spike protein include Arg403 (−2.43 kcal/mol), Gln493 (−1.12 kcal/mol), Tyr505 (−1.07 kcal/mol), Phe497 (−0.69 kcal/mol), Leu455 (−0.66 kcal/mol), Gly496 (−0.61 kcal/mol), Tyr495 (−0.6 kcal/mol), Tyr453 (−0.31 kcal/mol), Ser494 (−0.27 kcal/mol) and Glu406 (−0.22 kcal/mol) (Fig. 6 A). In case of spike(RBD)_emodin complex, residues such as Tyr449 (−2.29 kcal/mol), Phe490 (−1.77 kcal/mol), Leu452 (−0.62 kcal/mol), Leu492 (−0.27 kcal/mol), Gln493 (−0.19 kcal/mol), Pro491 (−0.08 kcal/mol), Ile472 (−0.07 kcal/mol), Ser494 (−0.06 kcal/mol), Thr470 (−0.06 kcal/mol) and Tyr451 (−0.04 kcal/mol) contribute significantly to the total binding energy (Fig. 6B). In both the cases, the top ten residues have more contribution of gas phase energy and van der Waals energy to the total binding energy as compared to the electrostatic energy contribution.
Table 3.
MM-PB(GB)SA binding free energy calculations for top 3 lead molecules along with the control.
| Molecules | ELEa (kcal/mol) (1) | VDWb (kcal/mol) (2) | GASc (kcal/mol) (3) | PBSOLd (kcal/mol) (4) | PBTOTe (kcal/mol) (5 = 1+2 + 4) | GBSOLd (kcal/mol) (6) | GBTOTe (kcal/mol) (7 = 1+2 + 6) | TSf (kcal/mol) (8) | ΔPBg (kcal/mol) (9 = 5+8) | ΔGBh (kcal/mol) (10 = 7+8) |
|---|---|---|---|---|---|---|---|---|---|---|
| ZINC64023823 | 6.84 ± 4.41 | −26.11 ± 3.4 | −19.26 ± 5.9 | 3.78 ± 4.75 | −15.49 ± 2.94 | 3.68 ± 4.09 | −15.58 ± 3.29 | 12.58 ± 1.75 | −2.91 | −3.00 |
| ZINC33039472 | −3.69 ± 1.78 | −30.20 ± 3.89 | −33.89 ± 3.56 | 12.34 ± 1.94 | −21.55 ± 3.39 | 9.69 ± 1.18 | −24.20 ± 3.05 | 12.47 ± 1.78 | −9.08 | −11.73 |
| ZINC00991597 | −8.89 ± 3.91 | −24.84 ± 2.71 | −33.73 ± 5.51 | 18.04 ± 3.86 | −15.69 ± 4.42 | 16.90 ± 3.2 | −16.83 ± 3.43 | 13.16 ± 2.42 | −2.53 | −3.67 |
| Emodin (CID3220) | −1.80 ± 2.09 | −15.06 ± 3.61 | −16.86 ± 4.77 | 5.35 ± 3.18 | −11.52 ± 2.98 | 4.95 ± 2.07 | −11.91 ± 3.32 | 5.50 ± 3.00 | −6.02 | −6.41 |
a: Electrostatic energy; b: van der Waals energy; c: Gas phase energy; d: Non-polar and polar solvation energy; e: Summation of electrostatic energy, van der Waals energy and non-polar and polar solvation energy; f: Entropic energy; g, h: Final estimated binding free energy.
Fig. 6.

Per-residue binding energy analysis showing the top ten residues contributing significantly to the total binding free energy of two systems-(A) spike(RBD)_ZINC33039472 (B) spike(RBD)_Emodin.
Discussion
The high mortality rates of COVID-19 and lack of effective approved therapeutics remains a global concern. COVID-19 is caused by a positive-stranded RNA virus known as severe acute respiratory syndrome coronavirus-2. The virus makes use of the spike protein receptor-binding domain (RBD) to gain entry into the host cell by recognizing and binding to the host angiotensin-converting enzyme 2 (ACE2) receptor. Blocking the protein–protein interaction between ACE2 and spike protein receptor can be envisioned as a novel approach to prevent the virus from entering the host cells and thus effectively control the pathogenesis of the disease. Hence, herein we have characterized the protein–protein interface of the SARS-CoV-2 RBD and ACE2 protein and used the information to target the spike protein interface region. The SARS-CoV-2 spike protein binding to ACE2 occurs through an interface area of 825 Å2 consisting of twenty residues of RBD with high energetic contributions (hot spot residues) by Tyr489 and Tyr505. SARS-CoV-2 RBD protein sequence shows a similarity of 72.38% with that of SARS-CoV and out of five critical residues (Tyr442, Leu472, Asn479, Thr487 and Tyr491) at the spike receptor-ACE2 complex, we observed the conservation of the only Tyr491. Further, the three-dimensional structural difference between SARS-CoV-2 spike and SARS-CoV spike is subtle with an RMSD of 0.674 Å. This subtle differences in the sequence, structure and key binding residues perhaps explain the higher binding affinity of SARS-CoV-2 S protein to ACE2 as compared to SARS-CoV S protein. A freely accessible database such as ZINC database offers a large repertoire of chemical entities which have been used in the present virtual screening procedure. Out of a random set of 1,36,191 compounds, a total of 42, 889 molecules cleared the ROF, Verber’s rule and in silico toxicity filters and were found to be orally bioactive. The filtered drug-like molecules were docked into the binding pocket within the interface region of RBD and the docking scores were compared with the control (emodin). Emodin is a natural anthraquinone derivative which is previously reported to block the SARS-CoV Spike (S) protein binding to Vero E6 cells and exhibit percent inhibition of 94.12 ± 5.90 % at 50 μM concentration against S protein-pseudotyped retrovirus infection [45]. Finally, three hits were shortlisted based on the docking scores and molecular interaction profile. The protein-ligand complexes of the three hits were further subjected to molecular dynamics simulation studies in an aqueous environment and various geometric and thermodynamic properties of the docked complexes were evaluated. MD simulation studies confirmed the stable trajectories of the protein-ligand complexes as inferred from RMSD, Rg and Q graphs. The principal component analysis highlights significant contributions of the first three eigenvectors (PCA 1–3) to the total mean square fluctuations. Further, the binding free energy calculation using MM-PBSA and MM-GBSA methods reveal that all the three hits exhibited favourable negative free energy values and the hit molecule ZINC33039472 showed higher binding affinity to spike receptor as compared to the control. Further, the energy decomposition analysis of spike(RBD)_ZINC33039472 complex reveals a major contribution of gas-phase energy and van der Waal energy and the top ten residues which contributed majorly to the binding free energy include Arg403, Gln493, Tyr505, Phe497, Leu455, Tyr495, Tyr453, Ser494 and Glu406. The present work has a few limitations such as short duration of molecular dynamics simulation. Hence, it would be quite interesting to explore the dynamics of the protein-ligand complexes at a much longer time scales which would further provide better insights into the binding modes of the lead molecules at the atomic level.
Conclusion
Using protein–protein interface targeted drug design approach, we have successfully identified ZINC33039472 as the best lead molecule which shows stronger interaction with the spike receptor-binding domain (RBD) as compared to emodin and thus the molecule could be a promising lead which can effectively block the interaction between SARS-CoV-2 spike RBD and human ACE2 receptor thereby hindering the pathogenesis of COVID-19. Further, the identified lead could be taken for various preclinical studies for developing into anti-coronavirus drug candidate molecules.
Funding
No funding sources.
Competing interests
None declared.
Ethical approval
Not required.
Acknowledgements
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding of the research through the research group project #RG-1438-015.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jiph.2020.12.014.
Appendix A. Supplementary data
The following is Supplementary data to this article:
References
- 1.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 2.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang N., Shang J., Jiang S., Du L. Subunit vaccines against emerging pathogenic human coronaviruses. Front Microbiol. 2020;11:298. doi: 10.3389/fmicb.2020.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020:1–4. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17:613–620. doi: 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang D., Comish P., Kang R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1008536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raj V.S., Mou H., Smits S.L., Dekkers D.H.W., Müller M.A., Dijkman R. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du L., Kao R.Y., Zhou Y., He Y., Zhao G., Wong C. Cleavage of spike protein of SARS coronavirus by protease factor Xa is associated with viral infectivity. Biochem Biophys Res Commun. 2007;359:174–179. doi: 10.1016/j.bbrc.2007.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science (80-) 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 13.Ryan D.P., Matthews J.M. Protein–protein interactions in human disease. Curr Opin Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez M.W., Kann M.G. Protein interactions and disease. PLoS Comput Biol. 2012;8 doi: 10.1371/journal.pcbi.1002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurung A.B., Bhattacharjee A., Ali M.A., Al-Hemaid F., Lee J. Binding of small molecules at interface of protein–protein complex—A newer approach to rational drug design. Saudi J Biol Sci. 2017;24:379–388. doi: 10.1016/j.sjbs.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi A., Murai M.J., He S., Lund G., Hartley T., Purohit T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood J Am Soc Hematol. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatzivassiliou G., Song K., Yen I., Brandhuber B.J., Anderson D.J., Alvarado R. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 18.Mossessova E., Corpina R.A., Goldberg J. Crystal structure of ARF1• Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol Cell. 2003;12:1403–1411. doi: 10.1016/s1097-2765(03)00475-1. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski R.A., Jabłońska J., Pravda L., Vařeková R.S., Thornton J.M. PDBsum: structural summaries of PDB entries. Protein Sci. 2018;27:129–134. doi: 10.1002/pro.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krüger D.M., Gohlke H. DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein–protein interactions. Nucleic Acids Res. 2010;38:W480–W486. doi: 10.1093/nar/gkq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Irwin J.J., Shoichet B.K. ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 25.Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Sander T., Freyss J., von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 27.Thompson M.A. Molecular docking using ArgusLab, an efficient shape-based search algorithm and the AScore scoring function Philadelphia. ACS Meet. 2004;vol. 172 p. 42. [Google Scholar]
- 28.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 30.Yang J.-F., Wang F., Chen Y.-Z., Hao G.-F., Yang G.-F. LARMD: integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Brief Bioinform. 2020;21:2206–2218. doi: 10.1093/bib/bbz141. [DOI] [PubMed] [Google Scholar]
- 31.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 32.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B., Merz K.M. A fast QM/MM (quantum mechanical/molecular mechanical) approach to calculate nuclear magnetic resonance chemical shifts for macromolecules. J Chem Theory Comput. 2006;2:209–215. doi: 10.1021/ct050212s. [DOI] [PubMed] [Google Scholar]
- 34.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 35.Price D.J., Brooks C.L., III A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121:10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 36.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R., Merz K.M., Jr. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 38.Grant B.J., Rodrigues A.P.C., ElSawy K.M., McCammon J.A., Caves L.S.D. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 39.Hou T., Wang J., Li Y., Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao G.-F., Zhu X.-L., Ji F.-Q., Zhang L., Yang G.-F., Zhan C.-G. Understanding the mechanism of drug resistance due to a codon deletion in protoporphyrinogen oxidase through computational modeling. J Phys Chem B. 2009;113:4865–4875. doi: 10.1021/jp807442n. [DOI] [PubMed] [Google Scholar]
- 41.Pan Y., Gao D., Zhan C.-G. Modeling the catalysis of anti-cocaine catalytic antibody: competing reaction pathways and free energy barriers. J Am Chem Soc. 2008;130:5140–5149. doi: 10.1021/ja077972s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thorn K.S., Bogan A.A. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. doi: 10.1093/bioinformatics/17.3.284. [DOI] [PubMed] [Google Scholar]
- 43.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80-) 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie M., Chen Q. Insight into 2019 novel coronavirus—an updated intrim review and lessons from SARS-CoV and MERS-CoV. Int J Infect Dis. 2020;94:119–124. doi: 10.1016/j.ijid.2020.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho T.-Y., Wu S.-L., Chen J.-C., Li C.-C., Hsiang C.-Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 2007;74:92–101. doi: 10.1016/j.antiviral.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maiorov V.N., Crippen G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J Mol Biol. 1994;235:625–634. doi: 10.1006/jmbi.1994.1017. [DOI] [PubMed] [Google Scholar]
- 47.Lobanov M.Y., Bogatyreva N.S., Galzitskaya O.V. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42:623–628. [PubMed] [Google Scholar]
- 48.Best R.B., Hummer G., Eaton W.A. Native contacts determine protein folding mechanisms in atomistic simulations. Proc Natl Acad Sci. 2013;110:17874–17879. doi: 10.1073/pnas.1311599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.