

Abstract
Since the early days of the genome era, the scientific community has relied on a single ‘reference’ genome for each species, which is used as the basis for a wide range of genetic analyses, including studies of variation within and across species. As sequencing costs have dropped, thousands of new genomes have been sequenced, and scientists have come to realize that a single reference genome is inadequate for many purposes. By sampling a diverse set of individuals, one can begin to assemble a pan-genome: a collection of all the DNA sequences that occur in a species. Here we review efforts to create pan-genomes for a range of species, from bacteria to humans, and we further consider the computational methods that have been proposed in order to capture, interpret and compare pan-genome data. As scientists continue to survey and catalogue the genomic variation across human populations and begin to assemble a human pan-genome, these efforts will increase our power to connect variation to human diversity, disease and beyond.
Much of the field of genomics revolves around the existence of reference genomes, which are roadmaps for a ‘typical’ individual of each species. The creation of each reference was, and still remains, a major focus of the genomics community, with 13 years and US$2.7 billion1 having been spent on the creation of the human reference genome alone. The ability to compare a newly sequenced individual with a reference and find differences has enabled myriad discoveries and innovations, and in human genomics this ability has formed the basis of thousands of studies seeking the genetic origins of disease. However, as the number and scope of sequencing experiments have grown dramatically, scientists have begun to realize the many limitations that a single reference genome imposes upon the community. To better capture the variation missed by using one reference, we can create and utilize a ‘pan-genome’, a collection of all the DNA sequences that occur in a species.
Cataloguing the DNA from all individuals in a species is a daunting task. The first pan-genomes were developed for small, easy-to-sequence bacteria, but, even in that context, pan-genomes provided novel scientific insights. The consideration of genetic diversity within bacterial species has contributed to our understanding of underlying differences in pathogenicity, virulence and drug resistance and can even help predict how pathogenic a new strain will be2–11. Pan-genome studies of plants and animals remained elusive at first, due to the large genome sizes and vast amounts of intergenic sequence in these species. However, in recent years, thanks to dramatic improvements in the efficiency of sequencing technology, the scientific community has been able to sequence dozens, hundreds or even thousands of individuals of a single plant or animal species12. Additionally, new long-read sequencing technologies now allow us to better assemble repetitive regions of large genomes, including centromeric regions, that are difficult to characterize with short reads13,14.
Human sequencing, too, has accelerated. Over the past few years, a flurry of publications have described large collections of newly sequenced human genomes, including population-specific cohorts from Iceland15,16, Denmark17, Sweden18, Papua New Guinea19, Mongolia20 and Africa21–23, as well as large-scale surveys of the entire world24–27. These studies have demonstrated, among other things, that large amounts of sequence in these populations — by some estimates, up to 10% of the total genome size — are missing from the reference genome28. As these genome collections have accumulated, computational scientists have been working to develop new methods to detect, represent and analyse large-scale structural variants, which had previously been sidelined while most genetic studies focused on single-nucleotide polymorphisms (SNPs). New representations must be able not only to capture the variation from large collections of genomes but also to enable efficient means of searching these genomes. Regardless of what methods are chosen, it is now clear that the community must move beyond reliance on a single reference genome (Box 1). While the use of a single reference has advanced genetics immensely, it has not, as some had hoped, allowed us to find the cause of all genetic disease, a shortcoming that has prompted some commentators to call the Human Genome Project a failure29,30. Although we now know that many diseases are caused by complex mixtures of multiple genetic variants, if we are to attempt to uncover the genetic causes of many still-unexplained diseases, one of the many factors we must consider is the vast genetic diversity present in the pan-genome.
Box 1 |. The human reference genome.
An initial draft of the human reference genome was first published in 2001 (REFS108,109). The genome consisted of sequence from approximately 20 individuals who answered an advertisement for volunteers in the Buffalo News, a newspaper in Buffalo, New York, USA. To sequence these individuals, DNA was extracted from a blood sample and was sheared into ~150–200-kb pieces, which were inserted into bacterial artificial chromosomes (BACs) to be sequenced. This approach meant that each ~150-kb segment could be sequenced and assembled separately, reducing errors caused by ubiquitous repeats that occur throughout the genome. Furthermore, a physical map of the genome was created to determine the relative locations of the BAC clones along the chromosomes. Thus, the human reference genome was assembled as a mosaic of these sequenced individuals, where one BAC-length segment might come from one individual, the next segment from a different individual and so on. The individuals who provided the DNA were anonymous.
The original version of the human reference genome contained 2.69 Gb and nearly 150,000 gaps. The genome has undergone many major updates since 2001 to produce the current version, GRCh38.p13, which contains 2.95 Gb of sequence and only 349 gaps110. These updates have included filling in gaps where no sequence was present, replacing rare alleles in the genome with the more common variants and adding alternative sequences representing divergent variants of some portion of the reference genome, although these alternative sequences are often not considered by analysis pipelines. However, the underlying genetic background of the reference remains the same as in the initial version — a mosaic of sequences from a small number of anonymous individuals.
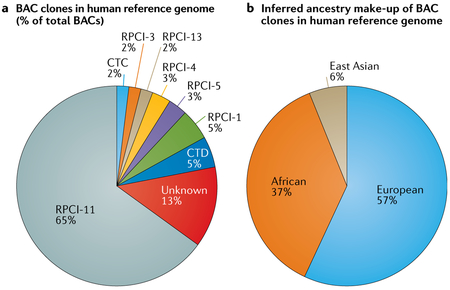
In 2010, a study describing the Neanderthal genome additionally performed an analysis on the human reference (version GRCh37)111. That analysis used the original BAC information to trace which anonymous donor was the source for each segment of the genome and then used population-specific single-nucleotide polymorphisms (SNPs) to determine the ancestry of each donor. This process revealed that approximately two-thirds of the reference genome sequence was composed of DNA from one male donor with the anonymous identifier RPCI-11, and that RPCI-11 was almost certainly 50% African and 50% European. In the figure, we illustrate the full make-up of the BAC clones (part a) and the inferred ancestral make-up of GRCh37 (part b), as described in the earlier study111 (supplemental material, p.146 of REF.111).
Because scientists continue to use the human reference genome as a baseline for nearly all human genetics studies, it is important to acknowledge that it does not represent the whole population. Rather, it is a mixture of ethnicities, predominantly sequence from a European/African admixed individual. Furthermore, as a mosaic of many individuals, it may not represent variant combinations that exist in any individual.
This Review discusses the recent history of work on pan-genomes, first proposed in 2005 for bacterial species4, and more recently applied to plants and animals, including humans. We begin by reviewing the definitions of a pan-genome, core genome and dispensable genome, as well as the considerations and decisions that go into creating them. We then review the efforts to date to build pan-genomes, focusing on the most recent work on the human pan-genome. We provide an overview of the computational challenges associated with both creating and utilizing pan-genomes and of ongoing efforts to develop methods to store and analyse them. Finally, we describe how a human pan-genome promises to solve at least some of the problems faced by ongoing efforts to sequence millions of individual humans and how shifting to pan-genomic approaches promises to lead us into an era marked by new biological discoveries.
Defining a pan-genome
Bacterial pan-genomics.
The concept of a pan-genome was first described by Tettelin et al.4 in 2005, in the context of bacteria. They described a pan-genome as a “core genome containing genes present in all strains and a dispensable genome composed of genes absent from one or more strains and genes that are unique to each strain”; under this definition, the pan-genome captures the whole of the genic content of a species. The dispensable genome is often further subdivided into genes unique to one strain (termed ‘unique genes’) and genes shared between some but not all strains (termed ‘accessory genes’) (FIG. 1a). Defining the pan-genome in terms of genes rather than DNA sequence is sensible for prokaryotes. Not only do genes comprise most (typically 90% or more) of the sequence content in these species, but gene content varies widely; in some bacterial species, unique genes have been found to make up anywhere from 20% to 40% of the pan-genome31. These differences in what genes are present often contribute to pathogenicity, drug resistance and other phenotypes of interest in human health; thus, analysing the dispensable versus core genomes can help explain these phenotypes.
Fig. 1 |. Core and dispensable genomes.
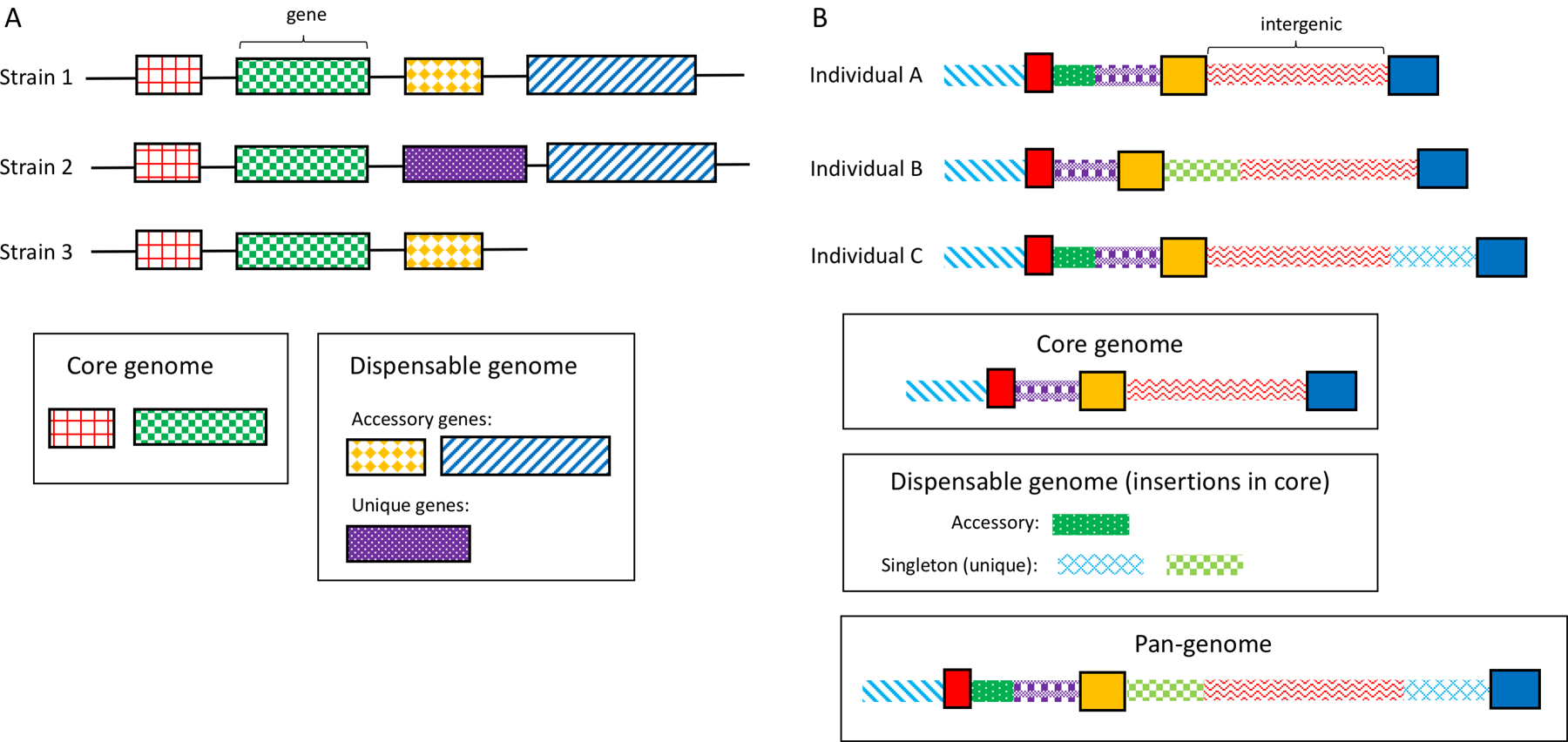
a | Bacterial and other prokaryotic genomes consist predominantly of genes, with little intergenic sequence. The core genome of a species consists of genes shared by all strains. The dispensable genome is made up of genes shared by some but not all strains (accessory genes) and genes present in only one strain (unique genes). Together, the core and dispensable genomes make up the pan-genome. b | Eukaryotic genomes are not highly variable in their genic content. Pan-genomes consider intergenic sequence as well as genes, resulting in an ordered pan-genome of all sequence present in at least one individual.
Eukaryotic pan-genomics.
Restricting the pan-genome to gene content makes less sense in eukaryotes, particularly those with large genomes (>500 Mb), where more than 50% of the genome may be intergenic, and where the gene sequences themselves are dominated by long introns32. In addition, eukaryotes do not exchange DNA as freely as bacteria do, making their gene content much more stable. For a species such as humans, in which exons occupy only ~2% of the genome33, a pan-genome composed only of exonic sequences would yield little information about within-species differences. Thus, a eukaryotic pan-genome is commonly defined to include all the DNA sequence in a collection of genomes, not just the genes. Although eukaryotic pan-genome studies sometimes borrow the terms ‘core’ and ‘dispensable’ genomes, in eukaryotes these descriptors refer additionally to intergenic sequences, rather than sets of genes, with a unique sequence being referred to as a ‘singleton’ (FIG. 1b). In this Review, for eukaryotic genomes we will use the term ‘genic pan-genome’ if intergenic sequences are not considered.
One other type of pan-analysis has been proposed, as well: the ‘pan-transcriptome’. This term describes the collection of all RNA sequences transcribed from the genome of a species, which can be captured using RNA sequencing (RNA-seq) technology. Pan-genomes built from RNA-seq data focus on transcript-level differences between individuals, generally ignoring intergenic sequences and introns but including alternative splice variants and other alternative isoforms that may derive from a single locus on the genome.
Biological considerations.
Pan-genome studies that consider protein-coding genes can use protein sequence conservation in addition to DNA sequence to determine whether genes are homologous. However, with intergenic sequences, defining what is shared versus unique becomes more challenging, particularly in organisms in which divergent repeats both between and within genomes are common. In deciding how to represent a pan-genome, one must consider the following criteria. First is whether to represent genes alone versus all DNA sequences. Related to this decision are whether to represent introns as well as exons (if relevant) and whether to represent differences in gene splicing (if relevant). Second, a decision must be made regarding what constitutes divergent versus shared sequence, and, finally, it must be determined whether to represent location in the genome or unordered presence/absence.
Which of these factors are most critical may depend on the application, but, ultimately, each time we create a pan-genome we must make calculated decisions about what features are important and how we define sequence sharing. The pan-genome efforts reviewed in the next section are based on a variety of choices to this effect.
Beyond bacteria: eukaryotic pan-genomes
Plant pan-genomes.
Pan-genomes have been created for multiple food crop species, including rice34–36, tomato37, soybean38, Brassica oleracea (whose cultivars include cauliflower, cabbage, broccoli, kale etc.)39 and sunflower40. Often the goal of creating a crop’s pan-genome is to determine which variations are linked to phenotypes that ultimately affect agricultural production. Although crops have been selectively bred since their domestication, the genes underlying the selected phenotypes often remain unknown and are sometimes linked to genes with undesirable phenotypes; for example, a cultivar that produces larger fruit might be lacking in disease-resistance genes. Discovering these phenotype-causing genes can help both to breed and to genetically modify plants so as to create crops that are more disease-resistant, are more productive, have a longer shelf life or taste better, without sacrificing desired phenotypes. Genic pan-genomic approaches in plants have already uncovered numerous associations between agronomic phenotypes and the presence or absence of specific genes41.
Rice and tomato are two of the world’s top food crops, with an annual production of more than 482 million tons for rice42, and over 180 million tons for tomatoes37. The rice and tomato pan-genome projects are exploring the boundaries of what can be found with ever-increasing amounts of sequencing data. One rice pan-genome effort has sequenced 3,010 rice genomes, many at low coverage36, whereas another has utilized deep sequencing of 67 varieties to create a pan-genome34. A recent tomato project has sequenced 725 genomes37, and another active project is sequencing 100 tomatoes with long-read sequencing technology43. Small genomic variants that have agricultural benefit have been studied in rice and tomato since their respective reference genome publications in 2012 (REFS44,45), but varieties differ greatly. A genic pan-genome for tomato described 4,873 genes present in one or more varieties that were missing from the reference genome37, whereas the rice projects report that there is great varietal diversity even in well-studied genes, such as those controlling flowering time or hull colour34. The variation captured in the pan-genome will differ according to the approach; for example, utilizing many low-coverage genomes can be helpful in examining the spectrum of gene presence/absence, whereas examining fewer genomes with long reads will have more sensitivity to detect complex structural variation, particularly in repetitive regions.
Pan-transcriptome analyses have been more common than pan-genome development in plants, possibly due to the large genome sizes and the high proportion of mobile elements46 in some species. Pan-transcriptomes have been described for many major crops, including maize47,48 and barley49, as well as for the model organism Arabidopsis thaliana, which has both a pan-genome and a pan-transcriptome50,51. Pan-transcriptomes are a more cost-efficient and targeted way to survey the genic landscape of these plants. The examination of RNA-seq data has aided in discovering, for example, that over 8,500 transcripts detectable in RNA-seq reads have no alignment to the maize reference genome47 and that wild barley varieties have many more disease resistance genes than do cultivated varieties49.
The beginnings of human pan-genomics.
In the past several years, large-scale human sequencing projects have become increasingly common. No project to date has produced a comprehensive, analysable human pan-genome that surveys a wide variety of human populations, captures both genic and intergenic variation, and incorporates this variation into a single utilizable pan-genome. Efforts are under way, however, to create population-specific pan-genomes, as well as to discover as many human SNPs and structural variants as possible, and a recent National Human Genome Research Institute-funded initiative has been launched to build a human pan-genome reference from 350 diverse individuals52. With continued development of computational methods capable of handling larger and larger datasets, these variant catalogues may ultimately provide the data needed to perform pan-genomic analyses in humans.
Human variant catalogues.
Scientists have been cataloguing human variants since well before the completion of the Human Genome Project. However, with the completion of a full reference genome came the ability to catalogue variation genome-wide, leading to the creation of large databases, including dbSNP53 and ClinVar54, as well as continued updates to pre-existing databases such as Online Mendelian Inheritance in Man (OMIM)55. ClinVar and OMIM track variants of clinical interest or with known phenotypic associations, although nearly all the variants tracked to date have been SNPs and small insertions or deletions (indels), relative to the reference genome. Although these variants can be incorporated into genome analyses using SNP-aware aligners such as HISAT2 (REF.56), mrsFAST-Ultra57 and SNPwise58, we now know that any given individual is likely to contain on the order of 20,000 structural variants (of >50 bp) relative to the reference genome59–62. More recent databases, such as dbVar, DGVa63 and DGV64, aim to catalogue these larger variants, although they cannot yet be easily incorporated into most standard alignment and subsequent analysis pipelines. Several projects have attempted to survey the landscape of human structural variation across the globe, including the 1000 Genomes Project (1KGP)27, Trans-Omics Precision Medicine (TOPMed)65 and the Simons Genome Diversity Project24.
The 1KGP was the first attempt at a large-scale global project for human genome sequencing. The 1KGP was performed in three phases, initially collecting SNP array data and later generating low-coverage (mean 7.4×) whole-genome sequence (WGS) data for 2,504 samples from 26 populations. In 2019 an updated re-sequencing of these 2,504 genomes was released in order to improve data quality and consistency. However, to date, no studies have been published analysing this new data release. An analysis of structural variants in the WGS data reported over 40,000 deletions, 6,000 duplications, nearly 3,000 copy number variants and nearly 17,000 mobile element insertions, by comparison with the human reference genome27. In all, 60% of the variants detected were novel relative to the pre-existing Database of Genomic Variants, a database consisting of variants reported from 55 studies at the time of its publication in 2013 (REF.64). In addition to reporting novel variation, one major finding of the 1KGP was the detection of homozygous deletions of large portions of 240 human genes27. The discovery that these genes are missing or severely altered in many individuals studied indicates that these genes are part of the dispensable genic pan-genome, a concept infrequently considered in human genomics. This dispensable gene set was enriched for two classes of proteins, glycoproteins and immunoglobulins, and nearly all the deletions were found in multiple populations. Other deleted regions in their set of over 40,000 deletions are likely to represent dispensable non-genic regions. Although these findings from the 1KGP are an important step in understanding the dispensable and core components of the human pan-genome, deletion discovery is only one step. The reference genome is missing dispensable sequences as well, which would appear as insertions in the 1KGP samples, but low-coverage WGS data are ill-suited to discovering novel insertions, and this was not attempted.
Other global projects have since examined novel sequence content. The Simons Genome Diversity Project generated deep coverage (30–40×) in short-read sequencing of 300 individuals from 142 diverse populations24. The project assembled sequences that failed to align to the reference genome and discovered 5.8 Mb of novel, non-repeat sequences in the collection. They also catalogued 34.4 million SNPs, 2.1 million small indels and 1.6 million short tandem repeats. Many of these variants — up to 11% of the heterozygous SNP variants in one population — were missing from the 1KGP variant calls, despite the 1KGP dataset containing more individuals, highlighting the need to continue collecting additional samples from diverse populations24. A more recent project, TOPMed, has examined short-read WGS data from 53,831 individuals, and using a similar method of assembling unaligned reads revealed 2.2 Mb of novel sequence65. Although the TOPMed data contained many more genomes than the Simons Genome Diversity Project, the investigators discarded any sequence without a good match to one of five hominid genomes, perhaps explaining why they reported a smaller amount of novel sequence.
All these global projects have limitations. Each of them utilized short-read sequencing data (usually 100-bp reads) that they aligned to the human reference genome, so, although some variation can be uncovered, the data necessary to build a pan-genome — that is, the union of all sequences in all humans — remain elusive, in part because reference-based genome assembly methods will entirely miss large insertions in other genomes. Furthermore, none of these large projects has had a primary goal of creating a human pan-genome, and in each case the analysis of novel sequences was secondary to their main findings. Each study has contributed snippets of what is needed, such as dispensable sequences deleted or inserted in many individuals, as well as other detectable variation, small and large, but this variation has not been aggregated in any way into a pan-genome. Although we now have a comprehensive gene set of core and dispensable genes for many bacteria and some plants, even this limited, genic pan-genome view does not yet exist for human populations, in part because we still lack a standardized comprehensive human gene set66,67. The pan-genome with intergenic variation included thus remains even more elusive.
In addition to the global projects that have touched on discovering novel sequence insertions, several recent efforts have focused solely on discovering these novel non-reference sequences within and across populations, utilizing both short-read and long-read technologies. Many of these efforts have stated the explicit goal of building a pan-genome, although to date no project has characterized the indel landscape completely enough to generate a full pan-genome, even for a single homogeneous population. Many of the efforts to do so are ongoing, but they remain complicated by the difficulty of determining which repeat sequences are truly novel, and without telomere-to-telomere assemblies of all human chromosomes, it is difficult to tell where repeat copies fall within each individual genome. Thus, definitions of novel sequence vary widely between projects and, as a result, so do the amounts of novel sequence discovered. Estimates of novel sequence in human populations vary from 0.33 Mb, in 15,219 Icelandic individuals, to 296.5 Mb, in 910 African-ancestry individuals15,17,24,25,28,65,68–74 (TABLE 1).
Table 1 |.
Reported novel sequences from efforts to sequence and analyse structural variation in large cohorts of human individuals
| Population and consortium (if applicable) | Number of individuals | Data type | Total novel sequence reported | Average per individual | Additional requirements | Publication year |
Refs |
|---|---|---|---|---|---|---|---|
| Swedish, SweGen | 1,000 (subset of 2) | Short read (long read) | 46 Mb (17.3 Mb) | 0.6 Mb (12.1 Mb) | Over 300 bp (over 100 bp) | 2019 (2018) | 18,79 |
| Han Chinese | 275 | Short read | 29.5 Mb | ~5 Mb fully unaligned + ~6 Mb partially unaligned to reference | Over 500 bp | 2019 | 69 |
| Mixed, TOPMed | 53,831 | Short read | 2.2 Mb | 0.2–0.5 Mb | Must align to a hominid genome | 2019 | 65 |
| Mixed | 154 | BioNano maps, linked reads (10X Genomics) | 60 Mb | 14.2 Mb | >2 kb | 2019 | 71 |
| Mixed | 15 | Long read | 21.3 Mb | 6.4 Mb | Not in peri-centromeric regions, over 50 bp | 2019 | 68 |
| African ancestry, Consortium on Allergy in African-Ancestry Populations | 910 | Short read | 296.5 Mb | 2.5 Mb | >1 kb | 2019 | 28 |
| Mixed | 17 | Linked reads (10X Genomics) | 2.1 Mb | 0.71 Mb | Breakpoint resolved, over 50 bp of non-repetitive content per sequence | 2018 | 73 |
| Icelandic | 15,219 | Short read | 0.33 Mb | 0.16 Mb | Non-repetitive, breakpoint resolved | 2017 | 15 |
| Danish, Danish Genome Project | 150 | Short read | >15,000 insertionsa,b | Not reported | >50 bp | 2017 | 17 |
| Dutch, Genome of the Netherlands | 769 | Short read | 4.3 Mb | Not reported | >150 bp | 2016 | 70 |
| Mixed | 10,545 | Short read | 3.26 Mb | 0.7 Mb | Non-repetitive, >200 bp | 2016 | 25 |
| Mixed, data from 1KGP | 45 | Short read | 61.6 Mb | 17,700–20,500 insertionsa,c | No size or other restrictions reported | 2016 | 74 |
| Mixed, The Simon’s Genome Diversity Project | 300 | Short read | 5.8 Mb (13 Mb with repetitive elements) | Not reported | Non-repetitive, >500 bp | 2016 | 24 |
| Japanese, Tohoku Medical Megabank Organization | 1,070 | Short read | 9,354 insertionsa | 45 insertionsa | >1 kb | 2015 | 72 |
1KGP, 1000 Genomes Project; TOPMed, Trans-Omics Precision Medicine.
Did not report number of bases.
Estimates separated into the average number of contiguous sequences per population with at least a partial match. The 61.6 Mb reported was based on 30,879 insertions.
Human pan-genome efforts.
Population-specific efforts to capture novel sequence variants have varied widely in their findings, in large part due to disagreement about the definition of novel sequences. However, any novel sequences produced from these efforts are sequences that can be incorporated into a pan-genome, provided they verifiably represent sequences present in at least some humans. Efforts to summarize the novel sequence content from studies of large cohorts have shown that human individuals contain anywhere from 0.16 Mb to 14.2 Mb of novel sequence. These study results are collected in TABLE 1, and several of these efforts are highlighted in this section.
There has been an ongoing national Icelandic effort to sequence its population, resulting in a WGS dataset of 15,219 Icelanders, making this, to our knowledge, the largest single-population WGS effort to date. The Icelandic effort stands out as reporting relatively little novel sequence compared with other studies, with more individuals. One reason for the low yield in novel sequence may be that the study focused on non-repetitive, non-reference (NRNR) sequences and excluded sequences with repeat elements and sequences similar to a constructed Icelandic reference genome created by incorporating Icelandic SNP data into the current version of the human reference sequence, GRCh38 (see BOX 1 for further information on GRCh38). They additionally required that sequences could be unambiguously placed into the reference genome. Despite its strict parameters, this project reported the discovery of 3,791 NRNR sequences, totalling 326.6 kb in length. Genotyping estimates indicated that the average Icelander would carry at least 157.8 kb of this sequence15, demonstrating that even when using the strictest of definitions of novel sequence within a single, homogeneous population, the reference genome is still missing many kilobases of unique sequence for a given individual.
Other studies have taken a less conservative approach to defining novel sequences in the populations they study. The Genome of the Netherlands project70,75,76 detected 4.3 Mb of novel sequence and nearly 20,000 deletions over 100 bp in length, from WGS data of 769 individuals from 250 Dutch families. These researchers noted that each haplotype in their cohort had, on average, 4.8 Mb of sequence affected by non-SNP variants (including previously reported variants), relative to the GRCh38 reference genome. Many of these variants were mobile element insertions (MEIs), which are known sequences inserted in new locations. Although MEIs are generally not considered novel sequence unless they have diverged considerably, two MEIs found in the Dutch cohort, one in a promoter and one in an exon, had a significant impact on gene expression70, indicating that sequence placement in addition to novelty should be considered in the construction of a pan-genome, although the prevalence of such variants is currently unknown.
The attempt to collect insertions in the African pan-genome from 910 individuals of African ancestry produced the largest reported amount of novel sequence: 296.5 Mb, calling into question early estimates that a full pan-genome might contain between 19 and 40 Mb (REF.77) of novel sequence. Although this study required insertions to be longer than 1 kb, many of its novel sequences were divergent copies of known repeats. As such, in addition to novel non-repetitive sequence, this study reported a large collection of novel repeats, predominantly HSAT II and III repeats, which diverge from each other and from their representations in the reference genome28,78. This study reported sequences with less than 90% identity as being divergent enough to be distinct novel sequences, but as efforts to build a human pan-genome progress, decisions must be made regarding how divergent sequences at differing levels of sequence identity should be represented and considered in analyses.
Other studies have utilized BioNano mapping71, linked-read sequencing71,73 and long-read sequencing68,79–81 to draw similar conclusions regarding the amount of novel human sequence. The BioNano mapping estimate (which did not use sequence data, but instead used restriction fragment lengths to create maps) produced the largest estimate of novel sequence per person (14.2 Mb) of any of the studies, possibly due to their inclusion of maps that spanned large peri-centromeric and acrocentric regions that are not well represented in the reference genome. A landmark study that generated deep coverage from 15 individuals using Pacific Biosciences (PacBio) long-read sequencing reported 6.4 Mb of novel sequence per individual68. Although this study included only a small number of individuals, it is unique as the largest long-read human WGS dataset to date. Because long reads can span difficult-to-align regions, variants can be called ‘in-place’ after reference alignment. This is unlike short-read approaches, in which the detection of long insertions requires either full de novo assembly or else alignment followed by assembly of unmapped reads74. While long-read alignment is still reference-biased and may miss variants, long reads are better able to detect variation within difficult-to-assemble repeats than are short reads. These higher estimates, of 6.4 Mb excluding peri-centromeric regions and 14.2 Mb including them, are closer to the long-read assembly-based estimates that revealed 12.8 Mb (REF.81) of novel sequence in one Chinese individual, over 10 Mb per person in two Swedish individuals79 and 16 Mb in a pseudo-diploid genome from two haploid hydatidiform moles80.
It remains unclear how much of this non-reference sequence is shared across individuals, and thus it is unknown how many individuals must be sequenced before the human pan-genome can be considered complete. We expect, however, that far fewer individuals will be needed if only non-repetitive sequence is being considered, as nearly 25 times as much sequence has been found, on average, in studies that have considered repeats than in studies that have not (TABLE 1). While non-repetitive sequences may be simpler to analyse, repeat elements can have substantial biological effects on gene expression70 and disease-related phenotypes78,82 and thus should not be ignored in the creation of a comprehensive human pan-genome.
Pan-genome representations
Although databases of SNPs and structural variants (for example, dbSNP and dbVar) provide a valuable resource for genetic analysis, a comprehensive pan-genome, in whatever form it is stored, is likely to present considerable new challenges for scientists who wish to use it. The amount of sequence data alone will likely be extremely large, especially if the pan-genome includes all variants of repeat sequences. In addition, the number and variety of rearrangements are both large and difficult to capture in a form that is easy to use with current bioinformatics tools. To date, no computational approach has been practically scalable enough to represent and analyse a full human pan-genome created from thousands or millions of individuals.
Inclusion of alternate sequences.
Currently the most commonly used approach to include divergent human sequences in a genetic analysis is simply to include these extra sequences when performing read alignment to the reference genome. The reference genome already contains several hundred of these alternative or ‘alt’ sequences, although their inclusion does not represent any systematic attempt to capture human variation. This strategy poses a number of problems. First, although the Genome Reference Consortium provides locations for the alt sequences and alignments to the main chromosomes of the reference genome83,84, most sequence alignment programs were not designed to handle variant information provided in this manner. As a result, most aligners simply treat the alt sequences as additional sequence tacked onto the genome and, as a result, the variants are treated as repeats. Some aligners, such as bwa85, have created ‘alt-aware’ modes to account for this, but, even with these fixes, this approach is not sustainable. As we continue to add divergent sequences, storing and searching the reference genome becomes increasingly space- and time-intensive. Furthermore, including these additional sequences separately does not accurately represent the underlying biology. Although in some cases we know where these sequences belong in the genome, what sequences they are alternatives to or what populations they are prevalent in, that information is lost by simply including them as additional sequences and continuing to use current algorithms for alignment.
Graphical representations.
A more principled alternative, which considers where the sequences belong and what reference sequence they are alternatives to is to represent the genome plus its variants as a graph. Aligners such as HISAT2 (REF.56) already align to graphical representations of the genome plus small known variants and choose the best path through the graph for each short read (FIG. 2a). A number of recently published methods, including vg86, SevenBridges87, PaSGAL88 and GraphAligner89, include algorithms and data structures for representing more complex structural variants in a graph. One benefit of these graphical representations is their ability to represent nested variation — for example, SNPs within insertions, or multiple variants of an insertion that share some regions but not others (FIG. 2b). This allows complex variants, hyper-variable regions and divergent repeat copies all to be represented much more compactly than would be required by representing every variant separately. Reads can then be aligned to these graphs made by incorporating known structural variation into the reference; for example, vg is able to construct a graph representation with 72,485 structural variants in approximately 14 wall-clock hours and under 50 Gb of RAM, and, once the graph is constructed, reads can be aligned to that graph faster than bwa-mem can perform linear alignments to the standard reference. However, graph genomes introduce a new complication: paths through the graph, particularly in complex regions, may join together variants that never appear together in any individual. Thus, a graph genome implicitly contains an exponentially large number of alleles that do not exist in the population.
Fig. 2 |. Graphical representations of pan-genomes.
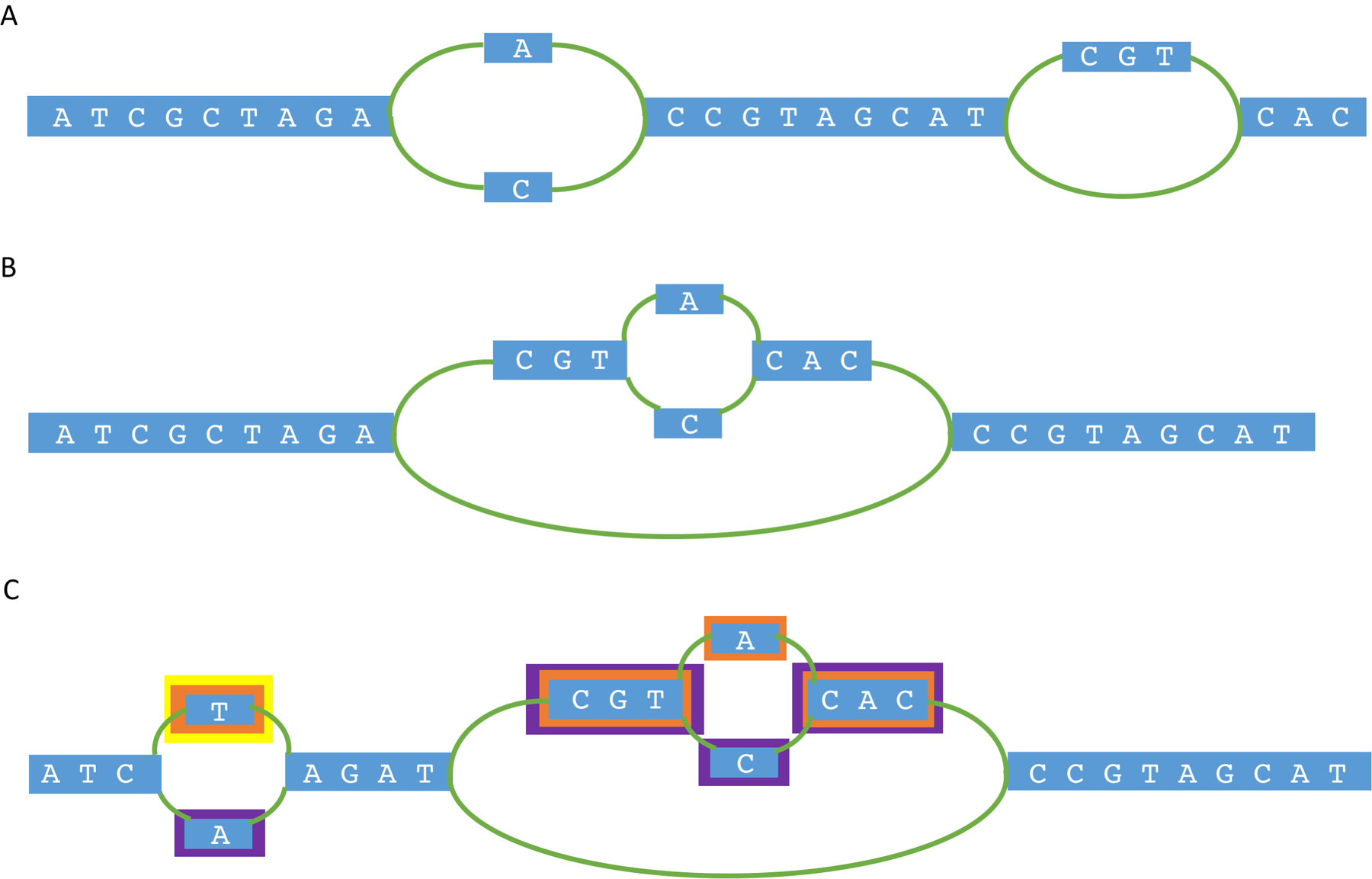
a | A single-nucleotide polymorphism (SNP) or insertion or deletion (indel) can be represented as two diverging paths (black lines) through the genome. Graph aligners can determine, for a read, which path is the best alignment. b | Nested variation can be represented in a graph. Here, both reads that contain the insertion and reads that do not can be aligned to the graph with no mismatches. For reads with the insertion sequence, they can be aligned to one of two paths within the insertion based on the A/C SNP they contain, again resulting in fewer mismatches in the alignments. c | To avoid a read alignment through the graph that does not represent any individual, colours can be tracked to indicate the population or individual of origin (yellow, orange and purple). Segments with no colour are equivalent to all colours, as they must be traversed in all paths. In this graph, a path containing the base A at the first SNP position, the insertion, and A as the within-insertion SNP would be a disallowed path for a read, because it is not colour-consistent: the first A SNP is only purple, and the second is only orange.
Additional information can be encoded in the graph to avoid this exponential blow-up; for example, ‘coloured’ graph approaches such as Cortex90, VARI91 and BFT92 can assign a different colour to each genome or population and then assign colours to each node in the graph according to where that node’s sequence came from. The alignment algorithm can then require that any path through the graph must be colour-consistent, as a path that switches colours may not exist in any individual genome (FIG. 2c). All allowed paths can also be stored more directly, as is the case with vg93, with the same end result. However, this additional information requires each node in the graph to store its ‘colours’, which may require as many colours as there are individuals in the population, and, in the path-based approach, a distinct path must be stored for each genome. Because a pan-genome may be produced from thousands or millions of individuals, retaining this information becomes increasingly computationally intensive as more individuals with new patterns of variation are included in the graph. Recent extensions to vg94 have incorporated text-indexing strategies for graphs95,96, developing efficient implementations to compactly represent biologically accurate variant paths. This work has demonstrated that the creation of such a graph from the 2,504 1KGP individuals, given per-sample variant information, is possible in under a day94. Furthermore, by extrapolating from experiments on human chr17, the study estimated that index construction for a full graph of the 54,035 TOPMed individuals would take 13–14 days, 76,000 CPU hours and 320 Gb of RAM to create an 80–90-Gb index94. A more comprehensive discussion of graph-based pan-genomics algorithms can be found in recent reviews by The Computational Pan-Genomics Consortium97 and Paten et al.98.
While graphical representations provide compact representations for a pan-genome, the goal is not only to store but also to analyse pan-genomic data. Given that short-read sequencing (with read lengths in the range 100–250 bp) is the current standard, this means that researchers must be able to align short-read data to the pan-genome representation. Short reads are difficult to align accurately in repetitive regions, even when aligning to a linear reference. Reads may be misaligned if, for example, a SNP or a sequencing error causes them to be identical to a different copy of a repeat elsewhere in the genome. By adding in large numbers of variants, we increase the number of places a repetitive read might align and increase the chances that a read might be aligned to an incorrect location (FIG. 3). The study describing the FORGe variant prioritization tool demonstrated that when 8–12% of known SNPs are included, graph aligners such as HISAT2 have the fewest number of incorrectly mapped reads. However, when the number of variants included is increased beyond that, accuracy declines99. Although only SNPs and small indels were examined in that analysis, the logic extends to structural variants as well, particularly when the variants belong to a high-copy-number repeat class, such as the HSAT II and III centromeric repeats. Another study demonstrated that whereas graph-based mapping with vg and SevenBridges yields higher accuracy than linear alignment on reads that contain known variants, linear genome alignment is superior when the reads do not contain variants100.
Fig. 3 |. Addition of variants increases alignment ambiguity.
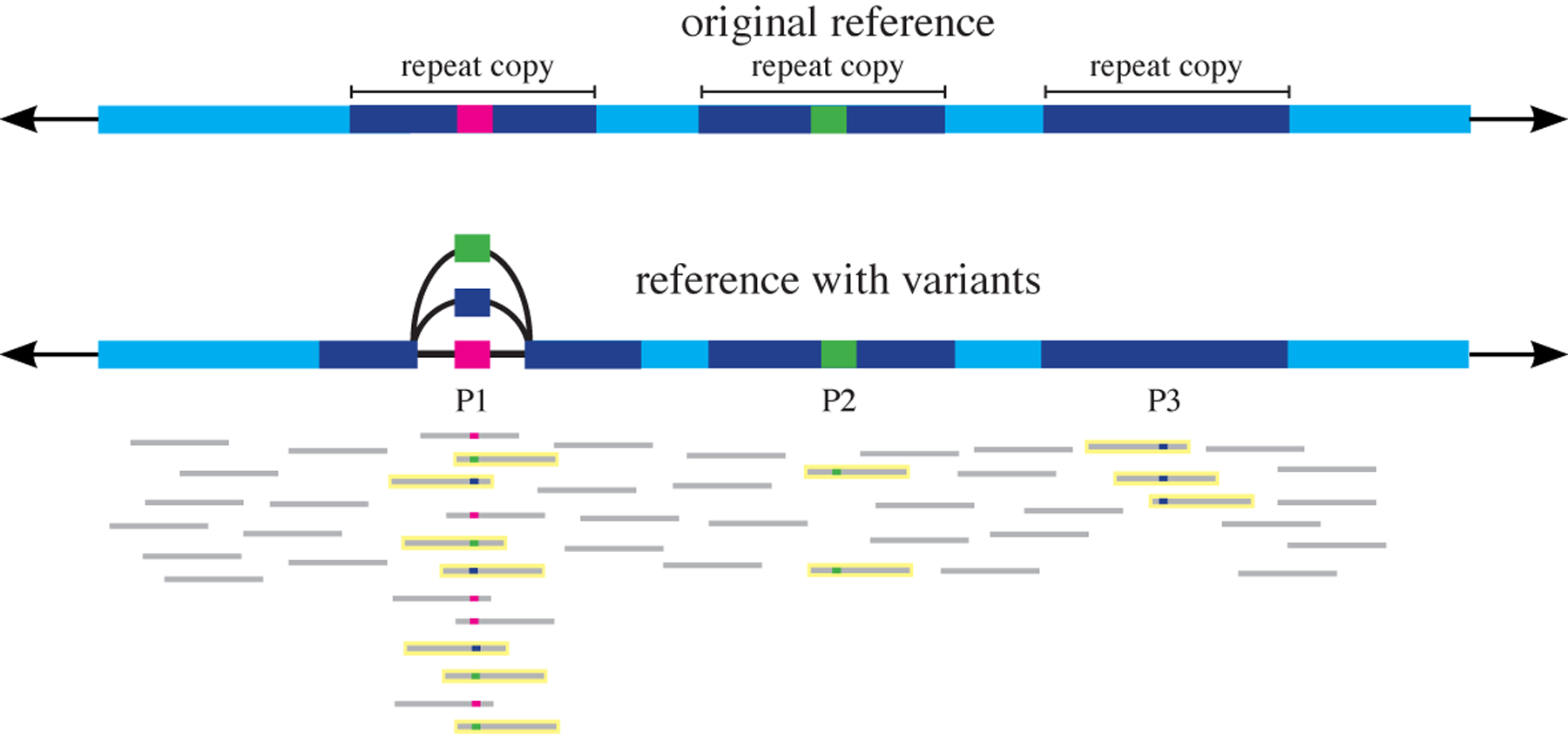
A graph-based representation includes alternate variants (blue, green) at position P1, whereas the reference contains only the pink reference allele. These variants are within a repeat (dark blue). The addition of each alternate variant increases alignment ambiguity. The six reads with the blue variant allele align perfectly only to P3 in the original reference, and now align to P1 and P3 equally well. Likewise, the six reads with the green variant allele now align to P1 or P2 perfectly, not just P2. Ambiguous reads are highlighted with yellow outlines.
Linear references and hybrid approaches.
Another strategy for handling human-scale pan-genomic data is to combine both graph-based and linear alignments. The two-step Graph Mapper100 program first aligns reads using a graph-based method such as vg, then extracts the best predicted linear path through the graph from these mappings and realigns the reads to the linear path (FIG. 4). This approach improved overall mapping accuracy compared with vg and SevenBridges, although its accuracy was slightly lower than vg for reads with variants. Like FORGe, this hybrid approach aims to reduce the size of the variant graph rather than to utilize a full pan-genome, although it does this with an online algorithm, whereas FORGe uses a sample-independent pre-processing step. Although it appears that decreasing the possible locations for reads to align can improve alignment accuracy, it is unclear whether these approaches will work well with graph genomes that contain structural variants, a capability that has not yet been tested with these methods.
Fig. 4 |. Two-step alignment method.
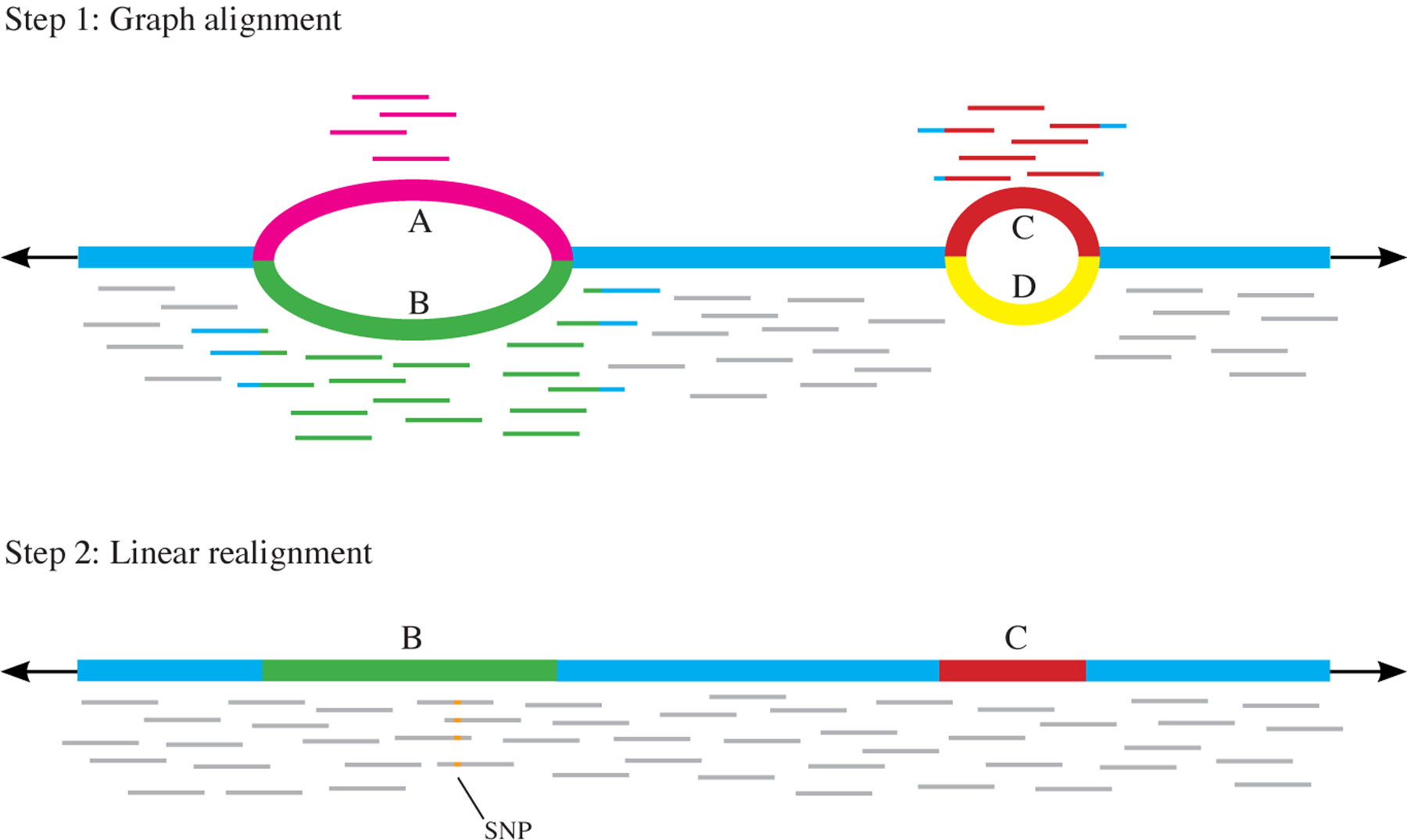
First, alignment to a graph is performed. Reads can align to either variant A or B at the first variant locus, and to C or D at the second. The path through the graph with the most reads aligned to it is then extracted — in this case, the path containing B and then C. In the second step, reads are realigned to the extracted linear genome. This allows for reads that may have been misaligned in the initial step (due to the introduction of variants) to be realigned only to the alleles they are most likely to have originated from. Here, the four reads that aligned to variant A now align to variant B, allowing a single-nucleotide polymorphism (SNP) to be detected that was undetectable from the graph alignment alone, as the reads with the SNP were misaligned.
Another option for capturing the human pan-genome is to build a collection of high-quality linear reference genomes and to align new sequences to the collection. An individual human genome consists of two linear genomes, one for each haplotype; thus, a linear representation with two copies of each chromosome is true to the underlying biology. If we had an abundance of reference genomes, the most closely related genome (or genomes) could be used for the analysis of any new genome, without a need to redesign aligners or other downstream tools. A two-step method might employ an initial pass to determine which reference genome should be used (or which of multiple references for an admixed individual), followed by the more computationally intensive alignment step of aligning the subject’s DNA to the reference genome (or genomes). Recent work has indicated that at least 1,000 genomes can be stored, indexed and searched very efficiently, with only a modest increase in time and space requirements over those required for a single genome101. In support of this strategy, the creation of additional reference-quality genomes is under way102,103.
Applications of pan-genome analyses
Pan-genome analyses have led to novel insights across a wide range of species. The use of bacterial pan-genomes has led to the identification of pathogenic genes in Escherichia coli8,9, Helicobacter pylori5 and other bacterial species present in the human microbiome that occur in both commensal and pathogenic strains. For plants, analysis of the tomato pan-genome has produced insights into agriculturally relevant traits that affect flowering time, fruit yield, domestication and flavour. For example, in a study of a tomato pan-genome that included members of a wild progenitor species (SP) and two varieties of a derived domesticated species, the earlier-derived SLC and later-derived heirloom SLL, an ~4-kb substitution in the promoter region of a flavour-related gene, TomLoxC, was found to be present in more than 90% of SP accessions, 15% of SLC accessions and 2% of heirloom SLL accessions37. This allele, which is associated with increased fruit flavour and is not present in the modern domesticated reference tomato species, was determined to be under negative selection in the domestication process, causing modern varieties of tomato to be less flavourful and aromatic than their wild progenitors. Without the hundreds of plants from each species included in the pan-genome analysis, no such trend from wild to domesticated species could have been determined, as domesticated species can contain the wild allele, and wild species can contain the modern allele, albeit at lower frequencies. Isolation of the alleles behind these desirable traits through this pan-genomic analysis may aid breeders in recovering desired traits, either through engineered genetic modifications or through introgression from wild species, a strategy that has successfully increased the frequency of the TomLoxC non-reference substitution in a more modern SLL variety37.
In humans, a clear advantage of pan-genome analysis is that scientists can discover variants that are missing from a single reference genome and then link those variants to phenotypes, which might include both beneficial and harmful traits. For example, any sequence longer than a few hundred nucleotides that is missing from GRCh38 is essentially invisible to most downstream analysis tools, regardless of how many individuals are sequenced, because any reads containing that sequence will simply fail to align. If such a sequence contains a disease-causing or disease-preventing variant, that variant will be undiscoverable unless this sequence is included in the analysis (FIG. 5). An illustration of this point arose in the Icelandic human-sequencing project, in which their examination of 15,219 Icelandic individuals showed a 766-bp insertion at high allele frequency (65%), the presence of which was found to significantly correlate with a decreased risk of myocardial infarction15. Another recent study discovered a repeat expansion causing neuronal intranuclear inclusion disease, a fatal neurodegenerative disease that causes symptoms ranging from deterioration of motor function to dementia104. That study utilized long reads to discover the repeat expansion and then demonstrated that the repeat expansion could be genotyped in other individuals using short reads alone. Although our ability to efficiently sequence and analyse large collections of human genomes is still limited, these recent examples are a demonstration of the potential to detect new and important variants as we become increasingly able to analyse human pan-genomes.
Fig. 5 |. Variant discovery from a pan-genome reference.
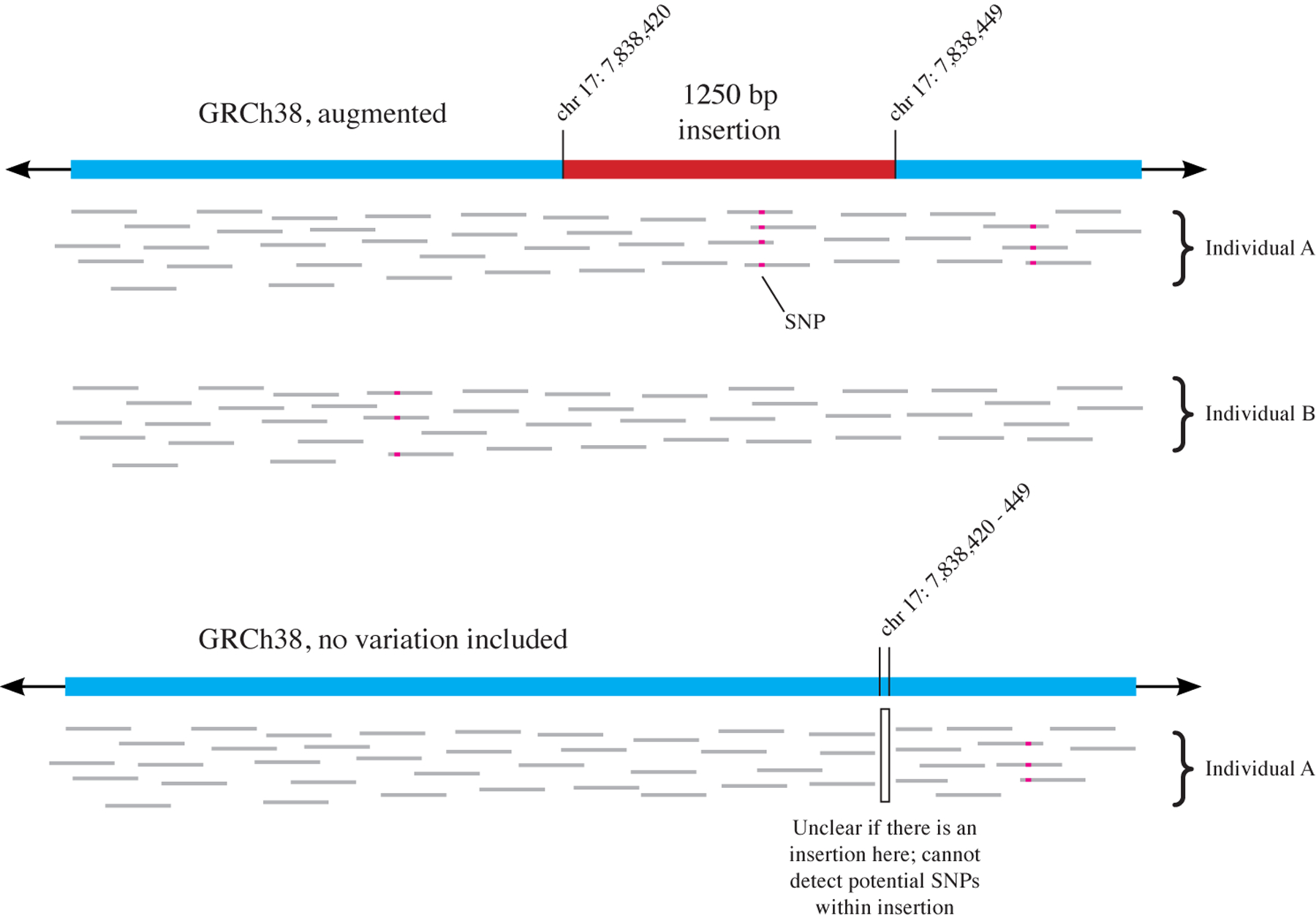
When the reference genome sequence is augmented with a known insertion, reads will align to this region for individuals containing this insertion. The 1,250-bp insertion included on chromosome 17 (chr 17) is within the gene KDM6B and has been reported in numerous studies15,28,65,68, including at a frequency of 1 in the Trans-Omics Precision Medicine (TOPMed) dataset of over 53,000 individuals65, and thus appears to be present in all or most individuals. With the insertion included in a pan-genome reference, reads from sequenced individuals will align to the region, allowing for the detection of single-nucleotide polymorphisms (SNPs). Here a SNP can be detected that is present in individual A but not individual B. However, when no pan-genomic variation is included in the reference, neither the insertion sequence nor the SNP in individual A can be detected. The depicted coordinates and the length of the KDM6B insertion were taken from Sherman et al. (2019)28, although they are nearly identical in all reports.
Conclusions and future perspectives
For many years scientists have understood that considering more than just a single representative genome can help identify genes and phenotypically consequential variants in bacterial and plant species. However, in human studies nearly all analyses still begin by aligning sequence data from a subject or a set of subjects to the human reference genome, discarding sequences that do not align. Considering more than just a single reference genome is necessary if we are to link more phenotypes of interest to their causal variants105. We now know that populations across the globe contain thousands of DNA sequences of various lengths that are not present in the human reference genome and thus that are not examined in standard analyses. Although we are amassing a wealth of pan-genomic data in both global and population-specific studies, what to do with these data remains an open question. The creation of a single, global human pan-genome holds conceptual appeal, and cataloguing all human variation is a noble goal. However, to date, no computational method is capable of aligning human sequences to a pan-genome of all human variation while enforcing that alignments be biologically plausible, although research that is actively under way on the efficient indexing, storage and traversal of graphical representations might solve this problem94,101,106. Population-specific pan-genomes may prove more feasible, and multiple such projects, such as the Icelandic and Danish efforts, are under way. Developing additional linear reference genomes, particularly if they are haplotype-resolved, has the benefit of representing a real individual, and a linear representation does not introduce variants that are never seen together. Furthermore, our ability to accurately assemble these genomes is rapidly improving: recently, the first telomere-to-telomere assembly of a human chromosome107 demonstrated that newly produced genomes have the potential to be of higher contiguity than the current reference. With a plethora of human reference genomes, individuals could be analysed by comparing them to their closest matching population or populations, even if this information is not known a priori. Regardless of what representations are ultimately used to capture these genomes, it seems inevitable that we will soon move beyond our reliance on a single human reference genome. Pan-genomic approaches that capture the vast amounts of variation in the population will be a critical tool in helping us understand and analyse the genetic instructions that make us human.
Acknowledgements
This work was supported in part by the National Institutes of Health under grants R01-HL129239, R01-HG006677 and R35-GM130151.
Glossary
- Reference genomes
A reference genome is a genome sequence that is used as the representative for the species — typically, the most polished and complete sequence available for the species.
- Long-read sequencing
Sequencing reads on the order of 5–10 kb (Pacific Biosciences) or longer, in some cases up to 1–2 Mb in length (Oxford Nanopore Technologies). Long reads are more expensive to generate and have higher error than short reads (100–250 bp in length).
- Core genome
The genes or sequence shared between all individuals of a species (or other grouping).
- Dispensable genome
The genes or sequence not shared between all individuals of a species (or other grouping). Everything that is not a part of the core genome is part of the dispensable genome, and vice versa.
- Singleton
A sequence found only in a single individual in the study population or group.
- Transcriptome
The sequences of only the exon regions, typically inferred by sequencing RNA transcripts rather than DNA directly.
- Alignment
The process of computationally lining up sequencing reads to a genome (typically a reference) in order to determine where they are likely to have originated from in the genome.
- Assembly
The process of overlapping sequencing reads from many copies of a genome in order to piece together short sequences into longer sequences. Assembly is often performed for a whole genome, particularly when no reference is available for alignment, but it can be performed locally, as well as on regions or subsets of reads.
- Haplotype
A sequence on one of the two homologous chromosomes of an organism’s diploid genome. In humans, haplotypes are considered in contrast to using a single sequence to represent that sequence on both homologous copies of a chromosome.
- Admixed
An individual with genetic ancestry from multiple distinct populations.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.National Human Genome Reserach Institute. Human Genome Project FAQ. NIH https://www.genome.gov/human-genome-project/Completion-FAQ (2019). [Google Scholar]
- 2.Rouli L, Merhej V, Fournier PE & Raoult D The bacterial pangenome as a new tool for analyzing pathogenic bacteria. New Microbes New Infect. 7, 72–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pallen MJ & Wren BW Bacterial pathogenomics. Nature 449, 835–842 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Tettelin H et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘pan-genome’. Proc. Natl Acad. Sci. USA 102, 13950–13955 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first work on pan-genomes in bacteria, this paper coined the term ‘pan-genome’ and the associated concepts of the ‘core’ and ‘dispensable’ genomes.
- 5.Ali A et al. Pan-genome analysis of human gastric pathogen H. pylori: comparative genomics and pathogenomics approaches to identify regions associated with pathogenicity and prediction of potential core therapeutic targets. Biomed. Res. Int 2015, 139580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali A et al. Campylobacter fetus subspecies: comparative genomics and prediction of potential virulence targets. Gene 508, 145–156 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Imperi F et al. The genomics of Acinetobacter baumannii: insights into genome plasticity, antimicrobial resistance and pathogenicity. IUBMB Life 63, 1068–1074 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Rasko DA et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol 190, 6881–6893 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salipante SJ et al. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res. 25, 119–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trost E et al. Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia. J. Bacteriol 194, 3199–3215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medini D, Donati C, Tettelin H, Masignani V & Rappuoli R The microbial pan-genome. Curr. Opin. Genet. Dev 15, 589–594 (2005). [DOI] [PubMed] [Google Scholar]
- 12.1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sedlazeck FJ, Lee H, Darby CA & Schatz MC Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Miga KH et al. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 24, 697–707 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kehr B et al. Diversity in non-repetitive human sequences not found in the reference genome. Nat. Genet 49, 588–593 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Jonsson H et al. Whole genome characterization of sequence diversity of 15,220 Icelanders. Sci. Data 4, 170115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maretty L et al. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 548, 87–91 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Eisfeldt J, Martensson G, Ameur A, Nilsson D & Lindstrand A Discovery of novel sequences in 1,000 Swedish genomes. Mol. Biol. Evol 10.1093/molbev/msz176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs GS et al. Multiple deeply divergent Denisovan ancestries in Papuans. Cell 177, 1010–1021.e1032 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Bai H et al. Whole-genome sequencing of 175 Mongolians uncovers population-specific genetic architecture and gene flow throughout North and East Asia. Nat. Genet 50, 1696–1704 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Choudhury A et al. Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans. Nat. Commun 8, 2062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurdasani D et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 517, 327–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathias RA et al. A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nat. Commun 7, 12522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mallick S et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Telenti A et al. Deep sequencing of 10,000 human genomes. Proc. Natl Acad. Sci. USA 113, 11901–11906 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherman RM et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet 51, 30–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports over 300 Mb of novel sequence detected from the examination of African-ancestry individuals, demonstrating that a considerable amount of sequence is missing from the human reference genome.
- 29.Hall SS Revolution postponed. Sci. Am 303, 60–67 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Wade N A decade later, genetic map yields few new cures. N. Y. Times 12 (12 June 2010). [Google Scholar]
- 31.Zou Y et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol 37, 179–185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francis WR & Worheide G Similar ratios of introns to intergenic sequence across animal genomes. Genome Biol. Evol 9, 1582–1598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piovesan A et al. Human protein-coding genes and gene feature statistics in 2019. BMC Res. Notes 12, 315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Q et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet 50, 278–284 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Schatz MC et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 15, 506 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun C et al. RPAN: rice pan-genome browser for approximately 3000 rice genomes. Nucleic Acids Res. 45, 597–605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao L et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet 51, 1044–1051 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Li YH et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol 32, 1045–1052 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Golicz AA et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun 7, 13390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hubner S et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 5, 54–62 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Tao Y, Zhao X, Mace E, Henry R & Jordan D Exploring and exploiting pan-genomics for crop improvement. Mol. Plant 12, 156–169 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Shahbandeh M Rice — statistics & facts. Statistica https://www.statista.com/topics/1443/rice/ (2017). [Google Scholar]
- 43.Alonge M et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomato Genome Consortium. The tomato genomesequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang X et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgante M, De Paoli E & Radovic S Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol 10, 149–155 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Hirsch CN et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 26, 121–135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansey CN et al. Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PLoS One 7, e33071 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma Y, Liu M, Stiller J & Liu C A pan-transcriptome analysis shows that disease resistance genes have undergone more selection pressure during barley domestication. BMC Genomics 20, 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gan X et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao J et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet 43, 956–963 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Ganguly P NHGRI funds centers for advancing the reference sequence of the human genome. NIH https://www.genome.gov/news/news-release/NIH-funds-centers-for-advancing-sequence-of-human-genome-reference (2019). [Google Scholar]
- 53.Sherry ST et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landrum MJ et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hamosh A et al. Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 30, 52–55 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim D, Paggi JM, Park C, Bennett C & Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hach F et al. mrsFAST-Ultra: a compact, SNP-aware mapper for high performance sequencing applications. Nucleic Acids Res. 42, W494–W500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tithi SS, Heath LS & Zhang L in 7th International Conference on Bioinformatics and Computational Biology (BICoB) (eds Saeed F & Haspel N) 187–192 (International Society for Computers and Their Applications, 2015). [Google Scholar]
- 59.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pendleton M et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaisson MJ et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lappalainen I et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 41, D936–D941 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacDonald JR, Ziman R, Yuen RK, Feuk L & Scherer SW The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Preprint at bioRxiv 10.1101/563866 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salzberg SL Next-generation genome annotation: we still struggle to get it right. Genome Biol. 20, 92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pertea M et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 19, 208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Audano PA et al. Characterizing the major structural variant alleles of the human genome. Cell 176, 663–675.e619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors examined 15 PacBio-sequenced genomes to produce the largest long-read structural variant callset to date, and so discovered over 6 Mb of sequence per individual, on average, that was absent from the reference.
- 69.Duan Z et al. HUPAN: a pan-genome analysis pipeline for human genomes. Genome Biol. 20, 149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents a pan-genome for a collection of Chinese individuals, as well as a proposed method to examine collections of human pan-genome data, provided that de novo assemblies can be performed on each individual genome.
- 70.Hehir-Kwa JY et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat. Commun 7, 12989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levy-Sakin M et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun 10, 1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nagasaki M et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun 6, 8018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong KHY, Levy-Sakin M & Kwok PY De novo human genome assemblies reveal spectrum of alternative haplotypes in diverse populations. Nat. Commun 9, 3040 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faber-Hammond JJ & Brown KH Anchored pseudo-de novo assembly of human genomes identifies extensive sequence variation from unmapped sequence reads. Hum. Genet 135, 727–740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boomsma DI et al. The genome of the Netherlands: design, and project goals. Eur. J. Hum. Genet 22, 221–227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet 46, 818–825 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Li R et al. Building the sequence map of the human pan-genome. Nat. Biotechnol 28, 57–63 (2010). [DOI] [PubMed] [Google Scholar]; This study produces some of the first full assemblies of the human genomes of diverse populations. Asian and African genome assemblies are produced, and, based on the assemblies, the researchers estimate that a full human pan-genome might contain between 19 and 40 Mb of DNA missing from the reference.
- 78.Miga KH Centromeric satellite DNAs: hidden sequence variation in the human population Genes 10, 352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ameur A et al. De novo assembly of two Swedish genomes reveals missing segments from the human GRCh38 reference and improves variant calling of population-scale sequencing data. Genes 9, 486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huddleston J et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shi L et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun 7, 12065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barra V & Fachinetti D The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun 9, 4340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Church DM et al. Modernizing reference genome assemblies. PLOS Biol. 9, e1001091 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Church DM et al. Extending reference assembly models. Genome Biol. 16, 13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garrison E et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol 36, 875–879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Vg is one of the leading methods to build and map reads to a variation graph, able to store a human pan-genome graph with ~180 Mb of variant sequences in under 4 Gb, with an index of ~63 Gb. Read alignment from a human genome to the variant graph can be performed in under an hour, although index and graph building are more time-consuming.
- 87.Rakocevic G et al. Fast and accurate genomic analyses using genome graphs. Nat. Genet 51, 354–362 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Jain C, Dilthey A, Misra S, Zhang H & Aluru S Accelerating sequence alignment to graphs. Preprint at bioRxiv 10.1101/651638 (2019). [DOI] [Google Scholar]
- 89.Rautiainen M, Mäkinen V & Marschall T Bit-parallel sequence-to-graph alignment. Bioinformatics 35, 3599–3607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Iqbal Z, Caccamo M, Turner I, Flicek P & McVean G De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet 44, 226–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Muggli MD et al. Succinct colored de Bruijn graphs. Bioinformatics 33, 3181–3187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Holley G, Wittler R & Stoye J Bloom filter trie: an alignment-free and reference-free data structure for pan-genome storage. Algorithms Mol. Biol 11, 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hickey G et al. Genotyping structural variants in pangenome graphs using the vg toolkit. Preprint at bioRxiv 10.1101/654566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Siren J, Garrison E, Novak AM, Paten B & Durbin R Haplotype-aware graph indexes. Bioinformatics 10.1093/bioinformatics/btz575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Durbin R Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 30, 1266–1272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Novak AM, Garrison E & Paten B A graph extension of the positional Burrows-Wheeler transform and its applications. Algorithms Mol. Biol 12, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Computational Pan-Genomics Consortium. Computational pan-genomics: status, promises and challenges. Brief. Bioinform 19, 118–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paten B, Novak AM, Eizenga JM & Garrison E Genome graphs and the evolution of genome inference. Genome Res. 27, 665–676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pritt J, Chen NC & Langmead B FORGe: prioritizing variants for graph genomes. Genome Biol. 19, 220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grytten I, Rand KD, Nederbragt AJ & Sandve GK Assessing graph-based read mappers against a novel baseline approach highlights strengths and weaknesses of the current generation of methods. Preprint at bioRxiv 10.1101/538066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kuhnle A et al. in Research in Computational Molecular Biology Vol. 11467 (ed. Cowen LJ) 158–173 (Springer, 2019). [Google Scholar]
- 102.Liu Q, Shi L & Wang K Ethnicity-specific reference genome assembly by long-read sequencing. J. Mol. Genet. Med 12, 1–3 (2018). [Google Scholar]
- 103.Graves-Lindsay T Reference genome improvement. National Human Genome Research Institute https://www.genome.wustl.edu/items/reference-genome-improvement/ (2018). [Google Scholar]
- 104.Sone J et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet 51, 1215–1221 (2019). [DOI] [PubMed] [Google Scholar]; This research discovers a repeat expansion associated with disease by using long-read sequencing of affected families; the result highlights the limitations of approaches based on short reads to reference alignment and demonstrates that consideration of harder-to-detect variants can lead to clinically relevant discoveries.
- 105.Ballouz S, Dobin A & Gillis JA Is it time to change the reference genome? Genome Biol. 20, 159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gagie T, Navarro G & Prezza N in Proceedings of the Twenty-Ninth Annual ACM-SIAM Symposium on Discrete Algorithms (ed. Czumaj A) 1459–1477 (Society for Industrial and Applied Mathematics, 2018). [Google Scholar]
- 107.Miga KH et al. Telomere-to-telomere assembly a complete human X chromosome. Preprint at bioRxiv 10.1101/735928 (2019). [DOI] [Google Scholar]
- 108.The International Human Genome Sequencing Consortium et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 109.Venter JC et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 110.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Green RE et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]