

Supplemental Digital Content is available in the text.
Keywords: heart failure, right; humans; macrophages; models, animal; pulmonary hypertension
Objective:
To determine whether global reduction of CD68 (cluster of differentiation) macrophages impacts the development of experimental pulmonary arterial hypertension (PAH) and whether this reduction affects the balance of pro- and anti-inflammatory macrophages within the lung. Additionally, to determine whether there is evidence of an altered macrophage polarization in patients with PAH.
Approach and Results:
Macrophage reduction was induced in mice via doxycycline-induced CD68-driven cytotoxic diphtheria toxin A chain expression (macrophage low [MacLow] mice). Chimeric mice were generated using bone marrow transplant. Mice were phenotyped for PAH by echocardiography and closed chest cardiac catheterization. Murine macrophage phenotyping was performed on lungs, bone marrow–derived macrophages, and alveolar macrophages using immunohistochemical and flow cytometry. Monocyte-derived macrophages were isolated from PAH patients and healthy volunteers and polarization capacity assessed morphologically and by flow cytometry. After 6 weeks of macrophage depletion, male but not female MacLow mice developed PAH. Chimeric mice demonstrated a requirement for both MacLow bone marrow and MacLow recipient mice to cause PAH. Immunohistochemical analysis of lung sections demonstrated imbalance in M1/M2 ratio in male MacLow mice only, suggesting that this imbalance may drive the PAH phenotype. M1/M2 imbalance was also seen in male MacLow bone marrow–derived macrophages and PAH patient monocyte-derived macrophages following stimulation with doxycycline and IL (interleukin)-4, respectively. Furthermore, MacLow-derived alveolar macrophages showed characteristic differences in terms of their polarization and expression of diphtheria toxin A chain following stimulation with doxycycline.
Conclusions:
These data further highlight a sex imbalance in PAH and further implicate immune cells into this paradigm. Targeting imbalance of macrophage population may offer a future therapeutic option.
Highlights.
The macrophage low model provides useful models to further study their contribution to pulmonary arterial hypertension (PAH).
Male macrophage low mice develop PAH.
Altered M1/M2 ratio in the lung of macrophage low mice with PAH observed in blood-derived macrophages from patients with PAH.
Macrophage populations may be a future therapeutic target for PAH.
Pulmonary arterial hypertension (PAH) is a complex disease characterized by increased pulmonary vascular resistance due to progressive pulmonary vascular remodeling that leads to right ventricular (RV) failure and ultimately death.1 Excessive infiltration of inflammatory cells has been detected in vascular lesions of both clinical and experimental PAH2–6 and correlated with the development of pulmonary vascular remodeling.7,8 Macrophages are among the inflammatory cells associated with PAH. Increased numbers of CD68+ (cluster of differentiation) macrophages are reported within diseased lungs of patients with and in animal models of PAH.5,9 However, the exact role for macrophages and whether their presence or absence is required for the vascular remodeling seen in PAH remains unclear.
Tissue macrophages are a remarkably versatile and heterogeneous population of immune cells. Their heterogeneity and plasticity is crucial to allow them to perform tissue-specific functions by undergoing polarized activation toward different macrophage subsets.10 Despite the diversity in the nomenclature of macrophage subsets following activation, it is widely accepted that macrophage classification could be viewed as a linear spectrum on which M1 macrophages (classically activated) represent one extreme and the M2 macrophages (alternatively activated) represent the other.11 M1 macrophages produce proinflammatory cytokines and mediate defensive mechanisms against invading pathogens such as bacteria and viruses.10,12 Macrophages, generated in response to different stimuli such as ILs (interleukins), are designated under the M2 subtype. Hence, M2 macrophages are anti-inflammatory and participate in debris scavenging, regulate wound healing, and have immune regulatory functions.12–15 The imbalance in M1 versus M2 phenotypic change has been implicated in the development of different pathological conditions such as tumors, obesity, and osteoarthritis.16–18
Anatomically, 2 main populations of tissue macrophages reside in the lung; the alveolar macrophages (AMs) found within the alveolar compartment and the interstitial macrophages found within the lung parenchymal tissue. In the case of inflammation, both tissue macrophage subtypes can expand by local proliferation of resident macrophages and by the recruitment of monocytes, which then differentiate into macrophages.19,20 These different lung macrophages have different gene expression profiles following exposure to stimuli such as hypoxia.9 Furthermore, specifically in a mouse model of pulmonary hypertension (PH), interstitial macrophages have been described to switch to an anti-inflammatory phenotype in later disease, whereas AMs conserve their proinflammatory phenotype.9
To investigate the role of macrophages in the development of PH, previous studies have used clodronate-containing liposomes to deplete cells of monocyte/macrophage lineage. In rodent models of hypoxia-induced PH, this strategy attenuated pulmonary vascular remodeling3 and reduced pulmonary artery pressure.21 Similarly, treatment targeting stromal-derived factors mediated recruitment of inflammatory cells, reduced numbers of CD68+ macrophages in rats with monocrotaline-induced PH, and was associated with improvements in pulmonary hemodynamic measurements and pulmonary arterial muscularization.5 However, these models are limited by the relative lack of macrophage specificity with a transient and almost complete loss of macrophages. In this current study, we utilized the macrophage low (MacLow) transgenic mouse model of inducible CD68+ macrophage depletion,15 which utilizes the Tetracycline-On system to switch on the expression of the diphtheria toxin A chain (DTA) in CD68-positive cells, leading to cell death. Using this model, up to a 50% reduction of tissue macrophages has been reported following 2 weeks of doxycycline treatment within the liver, spleen, and bone15,22 (increasing exposure did not lead to further reduction).
We now demonstrate that macrophage depletion in MacLow mice leads to the sex-dependant development of PAH in male mice. Utilizing bone marrow (BM) transplant to generate chimeric mice, we show loss of macrophages in both tissue and the circulation is required to induce PAH. PAH phenotype was associated with altered M1/M2 ratio in male MacLow mice, and an altered polarization capacity was also seen in patient-derived macrophages following stimulation in vitro. Conditioned media from MacLow macrophages cultured in vitro contained IL-6 and induced proliferation of pulmonary artery smooth muscle cells (PASMCs). These data collectively further highlight an important modifying role of macrophage in the pathogenicity of PAH and suggest that targeting this imbalance in macrophage population may be a future therapeutic target.
Materials and Methods
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
All animal procedures were approved by the University of Sheffield Ethics Committee and the UK Home Office (project license number 40/3517 and 70/8910) and conducted in accordance with the Animal (Scientific Procedures) Act 1986. Double transgenic CD68-rtTA-EGFP/tetDTA (MacLow) mice were generated by crossing the heterozygous single transgenic mouse CD68-rtTA-EGFP line with a homozygous tetDTA transgenic line, on an FVB background, as described previously.15 To induce macrophage depletion in MacLow mice, animals were treated with a doxycycline-containing diet (625 mg/kg; ENVIGO Laboratories, Inc, Madison, WI) at a dose of 80 mg/kg body weight.
Hypoxia-Induced PH
For the hypoxia-induced PH model, MacLow mice 12 to 13 weeks of age were fed either doxycycline-containing diet or normal laboratory diet (Teklad Global; ENVIGO) for 2 weeks and then either left in normal air or placed in a hypoxic chamber (10% of oxygen) for 2 weeks to induce PH.23–25 Doxycycline-containing diet was continued during this period.
MacLow Model
For the MacLow-induced PAH model, male and female groups of MacLow mice aged 12 to 13 weeks were randomized and fed doxycycline-containing diet for 6 weeks and housed in normal air. Single transgenic littermates (tetDTA) were also fed doxycycline-containing diet for 6 weeks and used as controls.
BM Transplant
To generate chimeric mice, recipient control and MacLow male mice were given a sublethal single dose of whole-body irradiation equaling 10 Gy (1000 rad). Donor BM cells (5–6×106) from male control or MacLow mice aged 5 to 7 weeks were administered via tail vein injection.23–26 After 4 to 8 weeks of recovery on regular normal laboratory diet, all mice were assigned to doxycycline diet for a further 6 weeks to induce PAH. As per the previous experiments, the single transgenic littermates (tetDTA) were used as controls.
PAH Phenotyping
Operators were blinded to genotype and treatment at the time of phenotyping. To assess cardiac size and function, echocardiography was performed using the Vevo 770 system (Visual Sonics, Toronto, Canada) and the RMV 707B scan head. Mice were anaesthetized using 5% v/v isoflurane, 2 L/min oxygen, and then placed on a heated platform. Anesthesia was maintained using 0.5% to 2% v/v isoflurane through oxygen. Measurements were taken from the long axis and the short axis as described previously.23–25 Following echocardiography, left ventricular and RV catheterization was performed using a closed chest method. Left ventricles and RVs were reached through the right internal carotid artery and the right external jugular vein, respectively, and hemodynamic parameters obtained using Millar 1F catheters (SPR-1030 [right], SPR-1045 [left]; Millar Instruments, Inc, Houston, TX) as described previously.23–25 Isoflurane-induced anesthesia was maintained throughout the procedure (0.5%–2% v/v isoflurane). Data were recorded using the Lab Chart Pro software (version 7.0; ADInstruments, Oxfordshire, United Kingdom). RV hypertrophy index was calculated by dividing the weight of the RV by the weight of left ventricular free wall plus septum.
Collection and Culturing of Mouse Primary Cells
To culture BM cells, tibia and femur bones were harvested from male mice and flushed with DMEM. Cells were homogenized, centrifuged, and resuspended in culture medium containing 100 IU/mL penicillin/streptomycin antibiotics, 10% v/v fetal bovine serum, and 20 ng/mL M-CSF (macrophage colony-stimulating factor). At day 7, mature BM-derived macrophages (BMDMs) were obtained. Formation of the mature BMDM was evaluated using flow cytometric analysis and fluorophore-conjugated antibodies to detect cells expressing F4/80 and CD68. Morphology was assessed by EVOS XL Core (Life Technology) hemocytometer, with ×20 magnification. To isolate AMs, mice were culled by anesthetic overdose, and bronchoalveolar lavage fluid was obtained through intratracheal instillation of 4×1 mL cold normal saline. The pellets were centrifuged and resuspended in RPMI 1640 (Roswell Park Memorial Institute) media (containing 10% fetal bovine serum v/v, 100 IU/mL penicillin/streptomycin) and passed through a 35-µm cell strainer and then allowed to adhere in the incubator for 2 hours. Isolation of AMs was confirmed using flow cytometric analysis as described above.
Isolation of Human Peripheral Blood Mononuclear Cells and Culture of Monocyte-Derived Macrophages
Peripheral blood mononuclear cells were isolated using Ficoll density gradient separation method from peripheral blood of healthy individuals and patients with PAH following informed consent according to the Declaration of Helsinki, with local research ethics committee approval and informed written consent from all subjects from the Sheffield Teaching Hospitals Observational study into Pulmonary Hypertension, Cardiovascular and Other Respiratory Diseases (STH-Obs, UK REC 18/YH/0441). Twelve males and 12 females patients with PAH and 4 male and 5 female healthy volunteers (HV) were included in the study (Table). Peripheral blood mononuclear cells were cultured in macrophage media containing 10% newborn calf serum for 3 hours before being washed and refreshed with macrophage differentiation media containing 10% heat-inactivated fetal bovine serum. After 14 days in culture, the resulted monocyte-derived macrophages (MDMs) were assessed using flow cytometry. Morphology was assessed by EVOS XL Core hemocytometer and by scanning electron microscope.
Table.
Demographics and Clinical Characteristics of Patients With PAH and HV
Immunohistochemistry
Formalin-fixed paraffin-embedded lung sections were stained with Alcian blue Elastin Van Gieson and immunostained for αSMA (α-smooth muscle actin; 1:150; Dako, M0851), von Willebrand factor (1:300; Dako, A0082), NF-κB (nuclear factor kappa-B; 1:400; Cell Signaling, D14E12), F4/80 (1:100; Abcam, ab111101), iNOS (inducible NO synthase; 1:100; Abcam, ab15323), and CD206 (1:100; R&D Systems, AF2535). As isotype control, IgG and IgG2a were used. Formalin-fixed paraffin-embedded liver sections were stained with anti-F4/80 to confirm macrophage ablation. For immunohistochemical and immunofluorescent staining, a standard protocol was followed. Histological images were visualized using a Zeiss multislide scanning microscope (Imager.Z2; Carl Zeiss, Ltd) with an Axiocam 506 color camera (Zeiss) for immunohistochemical images and MRm camera (Zeiss) for immunofluorescent images. Slides were scanned sequentially using ×20 magnification objective lens, and the analysis was performed in Zen2 blue edition (Zeiss). For all tissue macrophage quantification, positively stained cells were counted in 6 fields of view at ×200 magnification and normalized to the control group.
Assessment of Pulmonary Vascular Remodeling
The degree of remodeling was assessed in small pulmonary arteries and arteriole <50 µm in diameter by calculating the ratio of the media to the cross-sectional area of the whole vessel in sections stained with αSMA and also by assessing the degree of muscularization in sections stained with Alcian blue Elastin Van Gieson. Vessels were classified as muscularized (double elastic lamina) or nonmuscularized 1 thin elastic lamina layer) and then expressed as a percentage of muscularized vessels to total pulmonary vessels.23–25
Proliferation Assay
Human PASMCs (658401; Lonza, Basel, Switzerland) were subcultured in SmBM containing SmGM-2 SingleQuot Kit supplements and growth factors (Lonza) containing penicillin and streptomycin at 37 °C (5% CO2). All experiments were conducted with cells between passages 4 and 7. PASMCs were seeded into 96-well plates (0.5×104 cells/well) and allowed to adhere for 24 hours. Cells were then synchronized with growth arrest media (DMEM, 0.2% fetal bovine serum, penicillin, and streptomycin) for 48 hours before stimulation. PASMCs were then stimulated with BMDM conditioned media (from MacLow and control mice) or MDM conditioned media (from PAH patients and HV) diluted 2-fold with arrest media. PASMCs stimulated with either PDGF (platelet-derived growth factor; 25 ng/mL) or arrest media were used as positive and negative controls, respectively. Proliferation was assessed after 72 hours using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Southampton, United Kingdom).
TaqMan Polymerase Chain Reaction
Total RNA was extracted from frozen mouse lungs using the Maxwell miRNA tissue kit (Promega). Following reverse transcription using TaqMan Universal Master Mix II (Applied Biosystems), gene expression was measured by performing TaqMan polymerase chain reaction using Gene Expression MasterMix (Applied Biosystems) for TNFα (tumor necrosis factor alpha; Mm00443258_m1), CXCL1 (Mm04207460_m1), IL-6 (Mm00446190_m1), IL-1β (Mm01336189_m1), and IL-10 (Mn01288386_m1) on the 7900HT fast real-time polymerase chain reaction system (Applied Biosystems). Gene expression was calculated using the ΔΔCT comparative quantification method with 18S rRNA as an endogenous control.
ELISA
Macrophage culture media from mouse BMDM were run using assay DY406 (mouse IL-6) as per manufacturer’s instructions (R&D Systems).
Dot Blot Technique for Detection of DTA Protein Expression
For the dot blot experiment, a narrow-mouth pipette tip was used to spot 2 µL of cell lysate (normalized for protein concentration, Pierce 660nm Protein Assay) onto a nitrocellulose membrane (Amersham Protran 0.45 µm NC). The membrane was left to dry for few minutes and then soaked in blocking buffer (30% Odyssey blocking buffer, diluted in PBS; LI-COR) before incubation with anti-DTA antibody (1:2000; Abcam, ab8308). Liver tissue harvested from MacLow mouse that had been treated with doxycycline for 2 weeks was used as a positive control. The membrane was visualized by the Odyssey SA imaging system (LI-COR Biosciences).
Statistical Analysis
No data were excluded from the analysis. All statistics were performed using Prism (version 8.4.2 for Mac; GraphPad Software, San Diego, CA). Statistical comparison between the different groups was established by the use of 1-way ANOVA. However, due to sample size, the normality and variance were not tested to determine whether the applied parametric tests were appropriate. Post hoc analysis was performed using Benjamini, Krieger, and Yekuteli 2-stage linear step-up correction where >4 comparisons were made or by Bonferroni where <4. Values are presented as mean±SEM, and differences of P<0.05 values were accepted as significant.
Results
Reduction of CD68 Cells Has No Effect on Hypoxia-Induced PH
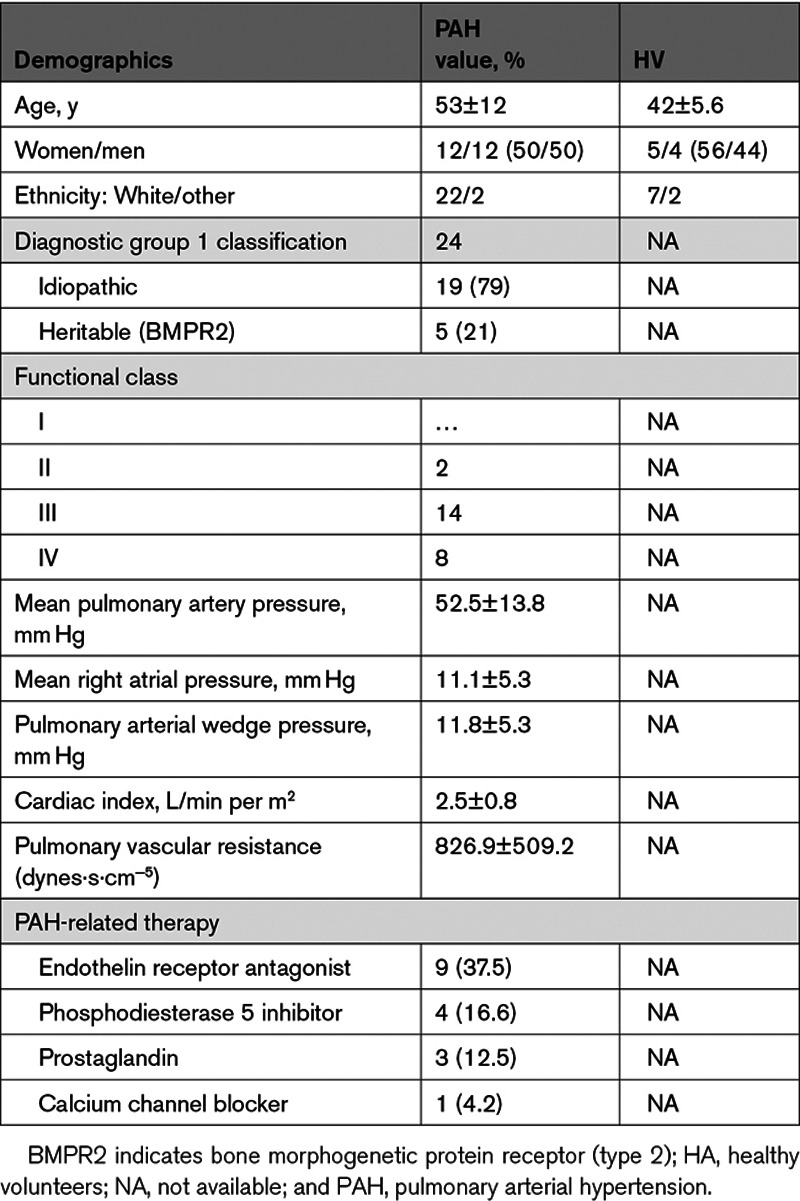
To determine whether macrophages are required for pulmonary vascular remodeling associated with hypoxia-induced PH, double transgenic CD68-rtTA-EGFP×tetDTA, MacLow mice were fed either doxycycline-containing diet (to induce macrophage depletion) or normal laboratory diet (chow) for 2 weeks. PH was then induced by hypoxia (10% oxygen) for further 2 weeks with continuous administration of doxycycline diet (parallel groups were left in room air as controls; Figure 1A). In both normoxia and hypoxia, doxycycline treatment reduced the total number of F4/80+ macrophages within the liver to about 50% compared with untreated control livers (Figure 1B and 1I). However, this had no effect on the hypoxia-induced increase in RV end-systolic pressure (RVESP; Figure 1C), RV hypertrophy (Figure 1D), or pulmonary vascular remodeling (Figure 1E and 1I). Additionally, we observed that MacLow mice treated with doxycycline but not exposed to hypoxia showed a trend toward higher RVESP (Figure 1C) and increased pulmonary vascular remodeling (Figure 1E). To determine the ablation of lung macrophages following the 4-week doxycycline treatment, anti-F4/80, anti-iNOS, and anti-CD206 were used to quantify M total, M1, and M2, respectively. Within the lung tissue, there was a significant increase in the total number of F4/80+ macrophages following exposure to hypoxia of nondoxycycline-treated mice compared with the normoxia group (Figure 1F and 1I). Doxycycline treatment reduced the number of F4/80+ macrophages in both the normoxia and hypoxia groups to a similar and significant level. Quantification of macrophage subtype showed no significant reduction in iNOS+ macrophages (M1; Figure 1G) or in CD206+ macrophages (M2; Figure 1H) in doxycycline-treated groups compared with correspondent controls.
Figure 1.
Loss of CD68+ (cluster of differentiation) macrophages does not protect against hypoxia-induced PH: macrophage low (MacLow) mice were fed either doxycycline-containing diet or normal laboratory diet for 2 wk to induce macrophage ablation, and then PH was induced by hypoxia (10% oxygen) for another 2 wk with continuous doxycycline diet. Parallel groups were left in normal air as controls. A, Experimental timeline. B, Quantification of F4/80+ macrophages remaining within the liver after 4-wk doxycycline treatment. C–E, Assessment of PH phenotype with right ventricular (RV) end-systolic pressure (RVESP; C), RV hypertrophy (RVH) index (D), and medial wall thickness as a ratio of total vessel size (media/cross-sectional area [CSA]; E). F–H, Quantification of macrophages remaining within the lung after 4-wk doxycycline treatment using anti-F4/80 antibody for total macrophages (F), anti-iNOS (inducible NO synthase) antibody for M1 macrophages (G), and anti-CD206 antibody for M2 macrophages (H). I, Representative photomicrographs of liver sections immunostained for F4/80 and lung sections immunostained for F4/80 and αSMA (α-smooth muscle actin) or stained with Alcian blue Elastic van Gieson (ABEVG). n=6 to 10/group. Bars show mean±SEM. Hypoxia, 10% oxygen; normoxia, 21% oxygen. ♂, male mice; ♀, female mice. Scale bar=50 µm. LV&S indicates left ventricular free wall plus septum. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
It was unexpected to report that MacLow mice treated with doxycycline only for 4 weeks showed a trend toward higher pressure, and by further examination, it seems that this trend was driven by the male (♂) mice, suggesting a potential link between sex, macrophage loss, and the development of PH. To explore this further, we examined whether a longer period (6 weeks) of macrophage depletion would result in a more pronounced spontaneous PH phenotype and whether this remained sex dependent.
Reduced CD68 Macrophage Number Causes Pulmonary Vascular Remodeling and PAH in Male but Not Female MacLow Mice
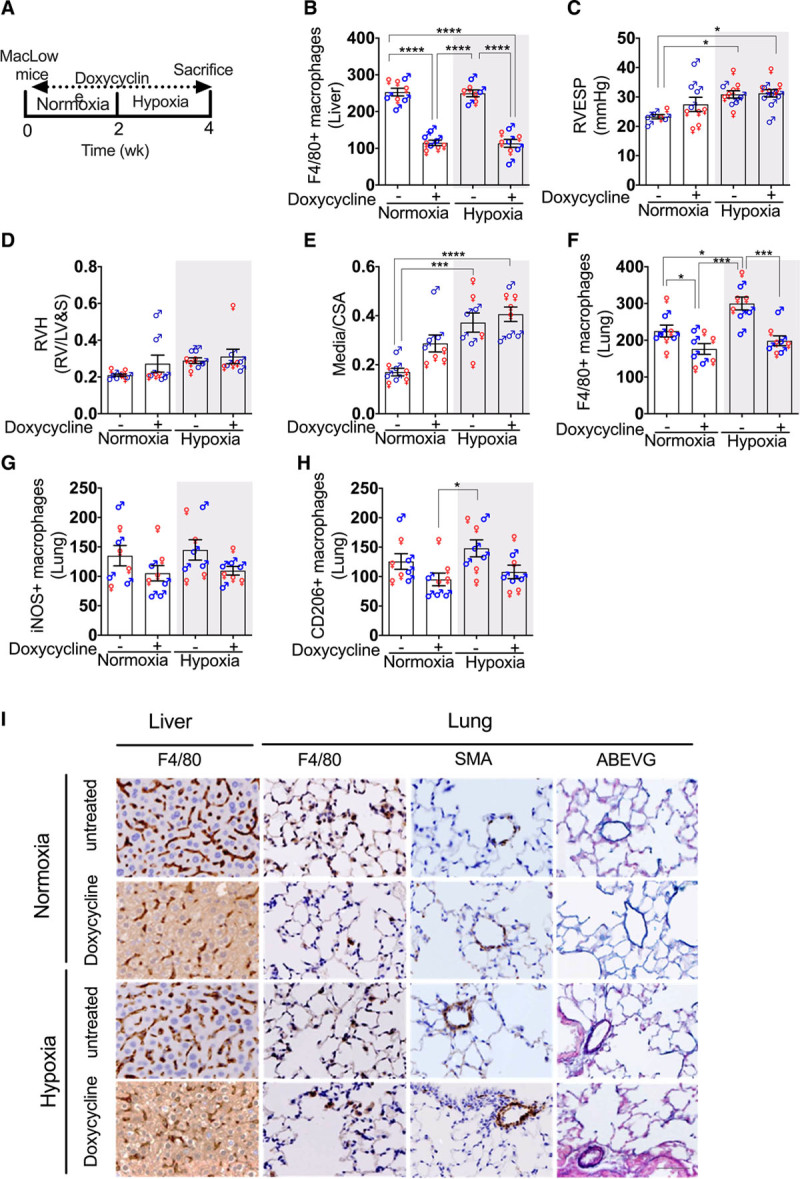
Male and female MacLow and control (single transgenic) mice were treated with doxycycline for 6 weeks in normoxic conditions with no other insult (Figure 2A). As with the previous experiment, a significant reduction of F4/80+ macrophages was confirmed in the liver (Figure 2B). Interestingly, only male MacLow mice developed a significant increase in RVESP (Figure 2C) compared with male controls. This increase in RVESP was also associated with an increase in RV hypertrophy (Figure 2D). There was no significant effect on either cardiac index (Figure 2E) or left ventricular end-systolic pressure (Figure 2F), but there was evidence of significant pulmonary vascular remodeling (Figure 2G through 2I). Immunohistochemical analysis of NF-κB transcription factor demonstrated expression within the remodeled small pulmonary arterial lesions in MacLow lung sections (Figure 2I). Collectively, we showed here that increasing doxycycline treatment period from 4 to 6 weeks caused a more pronounced sex-specific spontaneous PAH phenotype, which suggests that reduction in CD68+ macrophages can lead to spontaneous PAH phenotype in male MacLow mice.
Figure 2.
Loss of CD68+ (cluster of differentiation) macrophages causes PH in male macrophage low (MacLow) mice: male and female groups of MacLow or control mice were fed doxycycline-containing diet for 6 wk. A, Experimental timeline. B, Quantification of F4/80+ macrophages remaining within the liver following 6-wk doxycycline treatment. Assessment of PH phenotype was shown by (C) right ventricular (RV) end-systolic pressure (RVESP), (D) RV hypertrophy (RVH) index, (E) cardiac index, (F) left ventricular end-systolic pressure (LVESP), (G) medial wall thickness as a ratio of total vessel size (media/cross-sectional area [CSA]), and (H) pulmonary vascular remodeling as percentage muscularized vessels. I, Representative photomicrographs of lung sections stained with Alcian blue Elastic van Gieson (ABEVG) or immunostained for VWF (von Willebrand factor), αSMA (α-smooth muscle actin), and NF-κB (nuclear factor kappa-B) from male and female groups. Bars show mean±SEM. ♂, males; ♀, females. Scale bar=50 µm. LV&S indicates left ventricular free wall plus septum. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Imbalance in M1/M2 Ratio Is Associated With the Development of PAH in Male MacLow Mice
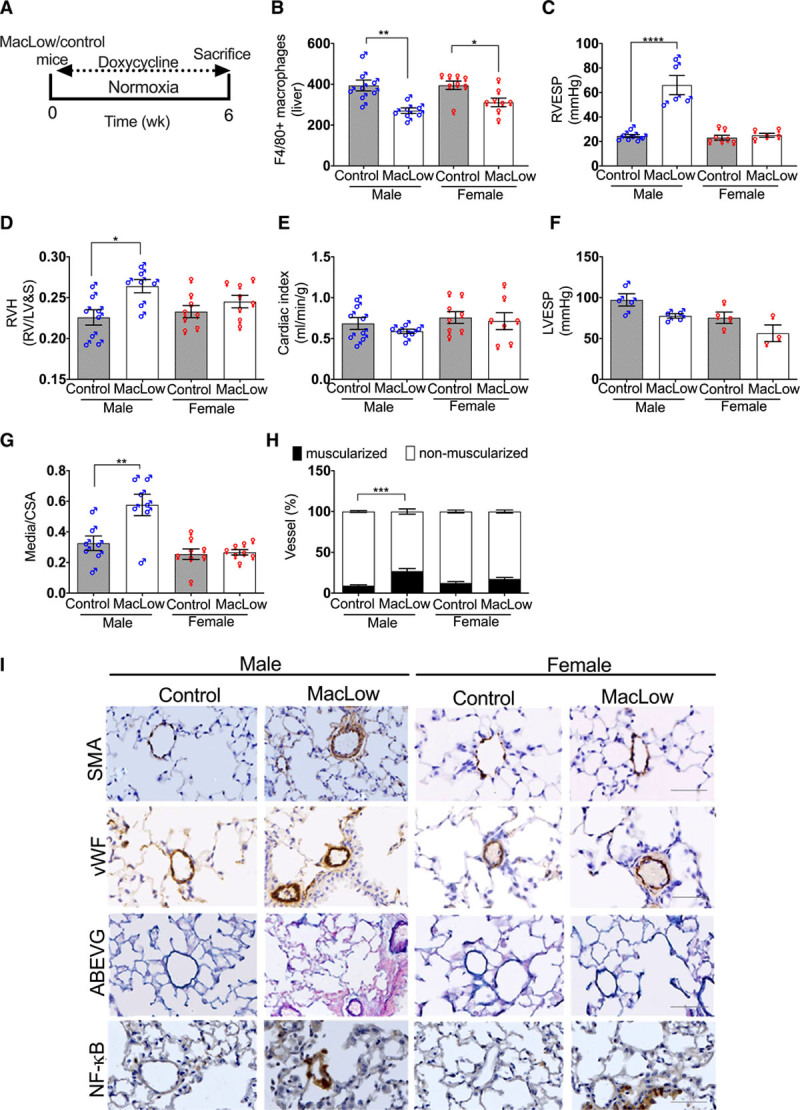
To assess the level of ablation of lung macrophages, immunohistochemical analysis of F4/80, iNOS, and CD206 was performed to quantify total, classically activated (M1) macrophage and alternatively activated (M2) macrophage number, respectively. Significant reduction in total lung macrophages was observed in both male and female MacLow mice undergoing doxycycline treatment compared with controls (Figure 3A and 3E), with no significant difference between male and female mice. Further characterization of macrophage subtype showed a significant reduction in iNOS+ macrophages (M1) in the lung of both male and female MacLow mice (Figure 3B and 3E). However, CD206+ (alternatively activated M2) macrophages were significantly reduced in the lungs of only female MacLow mice (Figure 3C and 3E). Comparison of the M1/M2 ratio (Figure 3D and 3E) highlighted an imbalance between the M1/M2 ratio in only the male MacLow mice suggesting that this is critical to driving the PAH phenotype. Despite reduced numbers, F4/80+ macrophages were observed accumulating within the thickened media of distal pulmonary arterioles of male MacLow mice with PAH (Figure 3F). To investigate whether the altered macrophage accumulation in the pulmonary arterioles was also associated with altered cytokine expression within the lung tissue, we measured the gene expression of TNFα, CXCL1, IL-6, IL-1β, and IL-10, although there was no significant increase in the expression of either cytokine (Figure I in the Data Supplement).
Figure 3.
Imbalance in M1/M2 ratio is associated with the development of pulmonary arterial hypertension in male macrophage low (MacLow) mice: male and female groups of MacLow or control mice were fed doxycycline-containing diet. Quantification of the number of macrophages remaining within the lung after 6 wk was performed using (A) anti-F4/80 antibody for total macrophage count, (B) anti-iNOS (inducible NO synthase) antibody for M1 macrophages, and (C) anti-CD206 (cluster of differentiation) antibody for M2 macrophages. D, M1-to-M2 ratio. E, Lung sections stained with antibodies against F4/80, iNOS, and CD206 and counterstained with hematoxylin in the MacLow-induced PH model. F, Representative immunofluorescence images of paraffin-embedded lung sections stained with anti-F4/80 (red) and SMA (smooth muscle actin; yellow) with cell nuclei labeled with DAPI (4′,6-diamidino-2-phenylindole; blue). Images were captured using a Zeiss multislide scanning microscope at ×20 magnification. Bars show mean±SEM. ♂, males; ♀, females. Scale bar, 50 µm. *P<0.05.
MacLow BM Cells Alone Are Not Sufficient to Induce PAH
Next, we investigated whether depletion of the tissue or BMDM population was sufficient to induce PAH in male mice. BM transplant of male MacLow and control (single transgenic) mice was performed to generate chimeric mice (Figure 4A). Following engraftment (4–9 weeks) and 6 weeks of doxycycline treatment, macrophage depletion was confirmed in the livers of MacLow recipient mice (Figure 4B). Assessment of PAH phenotype identified that only MacLow recipient mice with MacLow BM developed PAH as defined by an increased RVESP (Figure 4C) and pulmonary vascular remodeling (media/cross-sectional area; Figure 4D and 4J). There was no significant increase in RV hypertrophy (Figure 4E). These data imply that depletion of both BMDMs and lung-resident macrophages is required for the development of PAH in this model. Analysis of lung macrophage populations demonstrated a similar reduction to those previously observed in total F4/80 macrophages (Figure 4F) and iNOS+ macrophages (Figure 4G) in MacLow recipients, but as previously observed, the CD206+ macrophage numbers were preserved (Figure 4H). As with previous experiments, there was a significant reduction in the M1/M2 ratio in the MacLow recipient mice with MacLow BM that developed PAH when compared with controls (Figure 4I). Remodeled pulmonary arterioles demonstrating increased smooth muscle cell content also demonstrated NF-κB expression (Figure 4J). Although there was no significant increase in inflammatory cytokine expression, there was a trend for increased expression of IL-6 in MacLow to MacLow group that developed PAH (Figure II in the Data Supplement).
Figure 4.
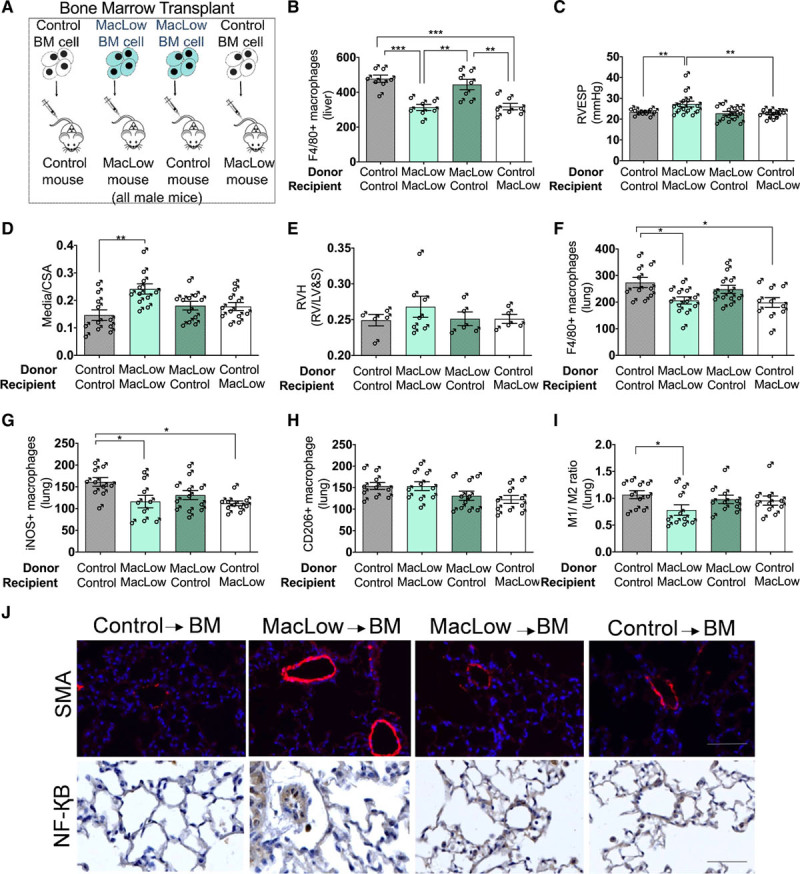
Macrophage low (MacLow)–derived bone marrow (BM) cells alone are not sufficient to induce PH in control mice: male BM chimeras were generated and fed with doxycycline-containing diet for 6 wk following a recovery period. A, Generation of male chimeric mice by BM transplant. B, Quantification of the number of F4/80+ macrophages remaining within the liver following 6-wk doxycycline treatment. Assessment of PH phenotype in male chimeric mice was performed using (C) right ventricular (RV) systolic pressure (RVSP) and (D) medial wall thickness as a ratio of total vessel size (media/cross-sectional area [CSA]). E, Right ventricular hypertrophy (RVH) index. Quantification of the number of macrophages remaining within the lung following 6-wk doxycycline treatment was performed using (F) anti-F4/80 antibody for total macrophage count, (G) anti-iNOS (inducible NO synthase) antibody for M1 macrophages, and (H) anti-CD206 (cluster of differentiation) antibody for M2 macrophages. I, M1-to-M2 ratio. J, Representative image of immunofluorescence staining (top) for anti-SMA (smooth muscle actin; red) and the cell nuclei labeled with DAPI (4′,6-diamidino-2-phenylindole; blue) and immunohistochemical staining (bottom) for anti–NF-κB (nuclear factor kappa-B). ♂, males. Bars show mean±SEM. Scale bar=50 µm. LV&S indicates left ventricular free wall plus septum; and RVESP, right ventricular end-systolic pressure. *P<0.05, **P<0.01.
Doxycycline Treatment In Vitro Leads to Macrophage Polarization Toward M2 Subtype in MacLow-Derived Macrophages
Since the PAH phenotype developed in male MacLow mice, we investigated the polarization capacity of MacLow-derived macrophages in vitro. Male MacLow BMDMs and control BMDMs were stimulated with cytokines or doxycycline before assessing cell surface marker expression to characterize M1 and M2 phenotype. No significant differences were detected in the basal expression of CD68 and F4/80 or M1/M2 markers in BMDM between untreated MacLow (not treated with doxycycline) and control mice (data not shown). Similarly, there was no significant difference between the expression profile of MacLow- and control-derived BMDM stimulated in vitro with either lipopolysaccharide (100 ng/mL) and IFNγ (50 ng/mL) for M1 activation or IL-4 (10 ng/mL) for M2 activation (Figure 5A). Interestingly, MacLow-derived macrophages stimulated with low dose of doxycycline (1 µg/mL) induced differentiation toward the alternatively activated M2 macrophage subtype as shown by increased expression of CD206+F4/80+ compared with untreated MacLow-derived cells (Figure 5A). A higher dose of doxycycline (5 ug/mL) demonstrated a significant increase in CD206+F4/80+ expression in MacLow-derived BMDM that was not observed in control BMDM (Figure 5B). This increase in polarization toward the alternatively activated M2 phenotype was comparable to that observed when stimulating either control or nondoxycycline-treated MacLow BMDM with IL-4 (canonical M2 driver). These observations were also supported by the morphological assessment of doxycycline-treated cells as they were elongated, and their morphology strongly resembles the M2 morphology (Figure 5A).
Figure 5.
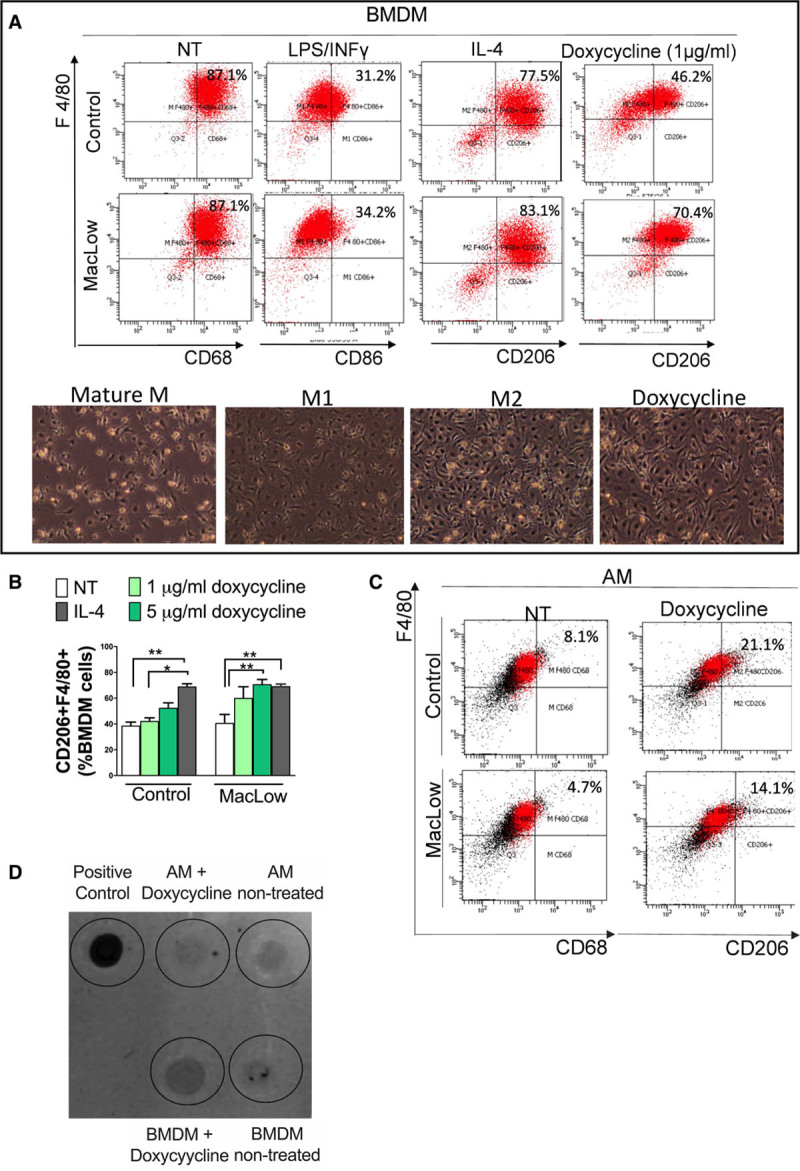
Doxycycline treatment in vitro lead to skew macrophage polarization toward M2 subtype. A, Representative flow cytometry scatter plots of bone marrow–derived macrophages (BMDMs) following culture and stimulation using anti-F4/80 vs anti-CD68 (cluster of differentiation; for nontreated), anti-CD86 (for LPS [lipopolysaccharide]/INF [interferon]-γ treated), and anti-CD206 (for IL [interleukin]-4 and doxycycline treated), with representative images of the corresponding morphology of macrophage low (MacLow)–derived macrophage population. B, Bar graph comparing the percentage of CD206+F4/80+ BMDM following stimulation with doxycycline alone (nontreated, NT) to doxycycliine and IL-4 (IL-4). Bars represent mean±SEM from 3 independent experiments. C, Representative flow cytometry scatter plot of alveolar macrophages (AMs) following harvesting and stimulation using anti-F4/80 vs anti-CD68 (for nontreated) and anti-CD206 (for doxycycline treated). D, Dot plot analysis using anti-DTA antibody comparing the level of expression of DTA protein between BMDM and AM following doxycycline stimulation in vitro. Protein extracted from the liver of a MacLow/doxycycline-treated mouse was used as a positive control. *P<0.05.
To investigate whether these phenotypic changes in BMDM were also observed in AM, AMs were isolated by bronchoalveolar lavage and stimulated with 5 µg/mL doxycycline for 48 hours as described above. In contrast to the BMDM, the expression of CD68 was found to be low in AMs from both MacLow and control mice, and no significant difference in the expression of CD68 or CD206 was observed following doxycycline stimulation (Figure 5C). These data suggest that the AM may be less susceptible to cell death via the CD68-driven DTA in the MacLow model due to lower expression levels of CD68. To test this, and the dependence of the MacLow system on CD68 for DTA expression, we assayed production of DTA by AMs following doxycycline stimulation in vitro. The expression of DTA was confirmed in cells isolated from MacLow liver (positive control; Figure 5D); however, a much lower expression of DTA was observed in MacLow BMDMs and less still in AM following doxycycline treatment (Figure 5D). These data suggest, relative to circulating sources of macrophages, AM would be less susceptible to doxy-induced killing via CD68-driven expression of DTA.
Finally, to determine whether the polarized MacLow macrophages could potentially impact on pulmonary vascular remodeling, we treated PASMC proliferation with conditioned media from MacLow and control BMDMs in vitro. We found that conditioned media from doxycycline-treated MacLow BMDMs but not control mouse–derived BMDMs stimulated PASMC proliferation (Figure 6A). Unlike BMDMs, conditioned media from AMs was unable to induce PASMC proliferation in vitro (data not shown). To determine whether tissue IL-6 levels could, in part, arise from the MacLow macrophages, we measured IL-6 levels in BMDM culture media following treatment with doxycycline by ELISA. We found that IL-6 was significantly higher in MacLow-derived BMDMs compared with control BMDMs (Figure 6B).
Figure 6.
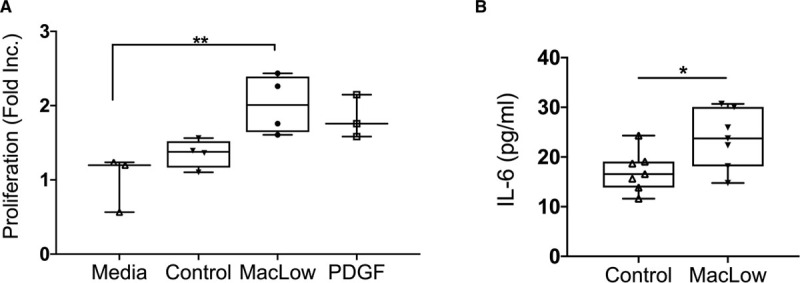
Doxycycline treatment in vitro induces PASMC proliferation and IL (interleukin)-6 release. A, Proliferation of PASMCs following stimulation with BMDM culture media for 72 h; BMDMs from control and macrophage low (MacLow) mice were pretreated with doxycycline for 48 h, presented as fold increase. B, IL-6 was measured in BMDM culture supernatant following doxycycline treatment for 48 h using ELISA. Box and whisker plots represent the interquartile range (box) with the line representing the median and whisker, the full range of the data. PDGF indicates platelet-derived growth factor. *P<0.05, **P<0.01.
MDMs From PAH Patients Show Reduced M2 Polarization Potential Following Activation With IL-4
After demonstrating that reducing numbers of CD68+ macrophages in MacLow mice led to M1/M2 imbalance, we next sought to determine whether there was evidence of an altered M1/M2 ratio in blood-derived macrophages from patients with PAH. Peripheral blood mononuclear cells isolated from HV and patients with PAH were cultured to generate MDMs and then stimulated to induce M1 or M2 polarization using 50 ng/mL INF (interferon)-γ+ 100 ng/mL lipopolysaccharide or 20 ng/mL IL-4, respectively. Morphological assessment of MDMs revealed that unstimulated macrophages (M0) exhibited a round shape, M1 macrophages predominantly retained this shape, while the majority of M2 macrophages developed a spindle-like appearance (Figure 7A). Evaluation of polarization markers in M0, M1, and M2 was performed by flow cytometric analysis using fluorophore-conjugated antibodies to detect cells expressing CD11b, CD80, and CD206 (Figure 7B). In MDMs from patients with PAH or HV, we confirmed the increase in expression of the M1 marker (CD80) in polarized M1 MDMs (≈65%) in comparison with resting M0 (Figure 7C). However, while there was a significant increase in expression of the M2 marker (CD206) following IL-4 stimulation in MDMs from HV (Figure 7D), this was not observed in MDMs from patients with PAH (Figure 7D). Similar to the observations from the MacLow mice, this also resulted in a skew in the M1/M2 ratio. Since conditioned media from male MacLow BMDMs caused proliferation of PASMCs in vitro, we next investigated whether conditioned media from human MDMs differentiated toward the M2 phenotype would similarly induce PASMC proliferation. Conditioned media was collected from both HV and PAH isolated MDM following stimulation with IL-4, applied to PASMCs, and cell proliferation assessed. Similar to the MacLow mice BMDM, M2-MDM conditioned media induced proliferation in PASMC; however, there was no significant difference between media harvested from HV or patients with PAH (Figure 7E).
Figure 7.
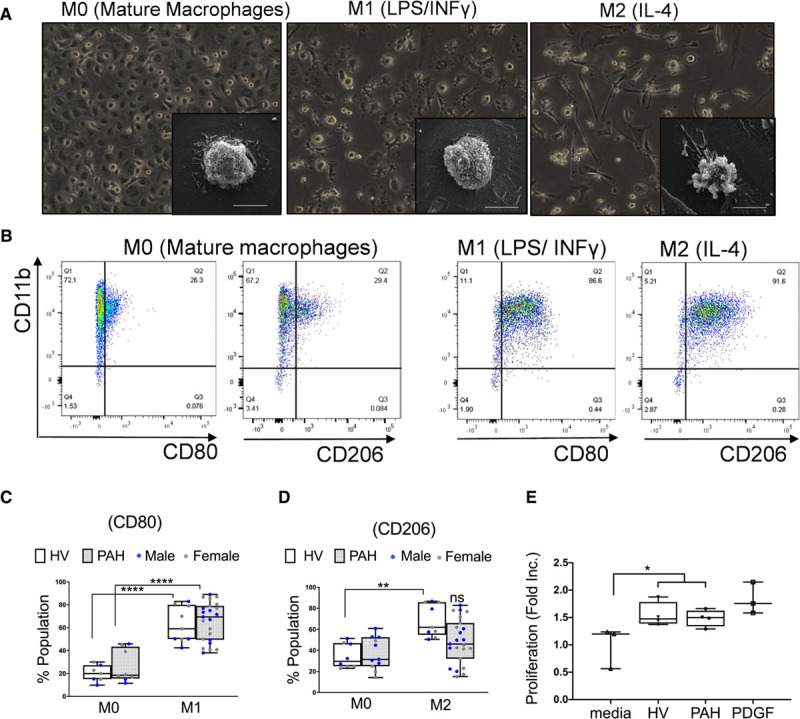
Monocyte-derived macrophages from pulmonary arterial hypertension (PAH) patients show impaired M2 polarization potential following activation with IL (interleukin)-4. A, Representative microscopy images show the morphology of mature and polarized monocyte-derived macrophages (MDMs) from patients with PAH. Qualitative assessment of morphology was performed by light microscopy with ×20 magnification (large panels) and by scanning electron microscopy for higher magnification images of individual cells (small panels/scale bar=10 µL). B, Representative flow cytometry scatter plots of MDMs following culture and stimulation using anti-CD11b (cluster of differentiation) vs anti-CD80 and anti-CD206. C, Box plot represents the percentage of CD80+ population in M0 and M1 macrophages derived from patients and healthy volunteers (HV). D, Box plot represents the percentage of CD206+ population in M0 and M2 macrophages derived from patients and HV. E, Proliferation of PASMCs following stimulation with MDM culture media for 72 h. Box and whisker plots represent the interquartile range (box) with the line representing the median and whisker, the full range of the data. INF indicates interferon; ns, nonsignificant; and PDGF, platelet-derived growth factor.
Discussion
Inflammation and the recruitment of inflammatory cells are regarded as playing a key role in the pathogenesis of PAH; however, the specific role of individual cell populations remains unclear. In this study, we demonstrate that macrophage number increases within the lung following hypoxia (as reported previously2–4). Perhaps counterintuitively, we found an association between the reduction of CD68+ macrophages and development of PAH in male but not female MacLow mice. Through the use of chimeric mice, we demonstrated that both MacLow tissue and MacLow BM is required to produce the PAH phenotype. BMDMs from male MacLow mice that develop PAH have skewed M1/M2 ratio, release IL-6, and induce proliferation of PASMC in vitro. Similarly, MDMs derived from patients with PAH also showed impaired polarization toward M2 subtype following IL-4 stimulation and altered M1/M2 balance similar to that observed in the male MacLow mice with PAH, and similar to MacLow cell, conditioned media was able to induce proliferation of PASMC in vitro. These data further support a modulatory role of macrophages in PAH.
To our knowledge, this is the first demonstration that specific CD68+ macrophage depletion using this transgenic approach leads to the development of PAH in an animal model. Previous ablation strategies have used nonspecific intravenous3 or intratracheal administration21 of liposomes containing clodronate (Cl2MBP) to deplete monocyte/macrophage number or therapeutic inhibition of SDF (stromal cell derived factor)-1 to inhibit the pulmonary vascular infiltration of CD68+, CD3, and mast cells in the monocrotaline rat model of PH.5 Interestingly, none of these studies reported a spontaneous increase in pulmonary pressure or associated remodeling in the normoxic control animals after macrophage depletion. This suggests that each macrophage depletion strategy may target different macrophage populations.
During the acute phase of inflammation, macrophages adopt a classical activated M1 phenotype driving the release of proinflammatory mediators. However, regulation of this must be controlled to prevent tissue damage, and switching toward the anti-inflammatory M2 phenotype helps mediate wound healing and tissue repair. An imbalance of M1 and M2 and switching back and forth may induce pathological consequences.13,27,28 The MacLow-induced PAH phenotype was associated with a decrease in M1/M2 ratio but with the presence of M2 macrophages within the lungs, consistent with previous literature.4,29 This may, in part, be due to the surviving macrophages switching their phenotype to the M2 alternative activation state to clear up the dead cells and cell debris.14 However, given the sex specificity of the skewing toward more M2 macrophages in the lungs of male MacLow mice and the PAH phenotype observed only under these conditions, it may be reasonable to assume a certain degree of causality. Data from the MacLow mouse data demonstrate an imbalance in M1/M2 ratio that was driven by a predominance of M2 macrophages in the lung. In contrast, MDM samples collected from patients with PAH demonstrated an impaired M2 polarization following stimulation with IL-4 in vitro. Although the imbalance in macrophage population is generated differently, both result in a skewed proportion of M1 to M2 macrophages. However, caution should be taken in directly extrapolating results from mice to humans, and in particular, the comparison of transgenic depletion of macrophages with a toxin in vivo, with the differentiation of blood-derived macrophages in vitro. Our study also proposes a link between M2 phenotype, IL-6, NF-κB, PASMC proliferation, and pathogenesis of PAH. This is in keeping with previous studies that demonstrated that lung-specific overexpression of IL-6 in transgenic mice is sufficient to induce pulmonary vascular remodeling and PH via a pro-proliferative mechanism.8 Furthermore, IL-6 release has been shown to induce M2 accumulation within the lung29 and influence the M1/M2 phenotypic change in dermatitis30 and colorectal tumors.31
Transplanting MacLow-derived BM into control animals was not sufficient to induce PAH emphasizing the contribution of lung-resident macrophages in the phenotype, although the milder phenotype observed (most likely due to the irradiation) made it difficult to interpret. It is still not completely known what happens to resident tissue macrophages in the lung after irradiation, but the removal of local stem cells residing in the alveolar spaces has previously been reported.32
Our data also highlight a potential inflammatory mechanism to contribute to the sex imbalance in PAH. Sex bias is well documented in both clinical and experimental forms of PH. In human disease, PAH is female dominant (4.1:1 female-to-male ratio in idiopathic PAH subcategory).33 However, men present with more severe disease with worse RV function and poorer survival33–35; however in contrast, in some animal models, the female sex has been shown to produce a less severe phenotype.36–38 The mechanisms behind this sex difference in PAH prevalence and survival are unclear and have resulted in the so-called sex paradox in PAH, focusing largely on the role of sex hormones.39–41 However, sex hormones alone fail to explain the sex paradox as conflicting findings also correlate female sex, estrogen, and estrogen metabolites to the development of PAH.42–44 This suggests that other factors must contribute to increasing the susceptibility to PAH. Studies of nonhormonal factors such as XY chromosome showed that hypoxic XY male developed less severe PH regardless of gonadal sex compared with XX mice.45 Moreover, regulatory T cells have a well-known role in controlling self-tolerance, and abnormal regulatory T-cell numbers have been reported in patients with PAH.46 More recently, regulatory T-cell function has been linked to the sex paradox in PAH with regulatory T-cell function shown to be a critical protective mechanism against pulmonary vascular remodeling in female rodents.46 On a cellular level, in vitro characterization of human- and mouse-derived macrophages showed no sex difference in polarization of macrophages.
Collectively, this study further highlights the important role that sex may play in the pathogenesis of PAH and further supports the concept that inflammation may be a contributing factor to this. The MacLow model provides a useful alternative to previous macrophage depletion models to further study their contribution to PAH and pulmonary vascular remodeling. Targeting this imbalance of macrophage population may be a future therapeutic target.
Acknowledgments
We would like to acknowledge participants from the Sheffield Teaching Hospitals Observational Study of Pulmonary Hypertension, Cardiovascular and Other Respiratory Diseases who donated samples. We gratefully acknowledge the support from the UK Department of Health via the Sheffield National Institute for Health Research Clinical Research Facility awarded to Sheffield Teaching Hospitals Foundation National Health Service Trust.
Sources of Funding
This research was supported by a Libyan Ministry of Higher Education and Scientific Research grant to A. Zawia and a British Heart Foundation project grant (PG/18/23/33605). A. Lawrie is a British Heart Foundation Senior Basic Science Research Fellow (FS/18/52/33808). A.A.R. Thompson is a British Heart Foundation Intermediate Clinical Fellow (FS/18/13/33281). Funding from the British Heart Foundation (PG/11/116/29288) supported collection of samples to the Sheffield Teaching Hospitals Observational Study of Pulmonary Hypertension, Cardiovascular and Other Respiratory Diseases.
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- α-SMA
- α-smooth muscle actin
- AM
- alveolar macrophage
- BM
- bone marrow
- BMDM
- bone marrow–derived macrophages
- CD
- cluster of differentiation
- DAPI
- 4′,6-diamidino-2-phenylindole
- HV
- healthy volunteer
- DTA
- diphtheria toxin A chain
- IL
- interleukin
- INF
- interferon
- iNOS
- inducible NO synthase
- LPS
- lipopolysaccharide
- MacLow
- macrophage low
- MDM
- monocyte-derived macrophage
- NF-κB
- nuclear factor kappa-B
- PAH
- pulmonary arterial hypertension
- PASMC
- pulmonary artery smooth muscle cell
- PDGF
- platelet-derived growth factor
- PH
- pulmonary hypertension
- RV
- right ventricle
- RVESP
- right ventricular end-systolic pressure
- SDF
- stromal cell derived factor
- TNFα
- tumor necrosis factor alpha
These authors are joint senior authors.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.314639.
For Sources of Funding and Disclosures, see page 444.
Contributor Information
Amira Zawia, Email: a.a.zawia@sheffield.ac.uk.
Nadine D. Arnold, Email: n.d.arnold@sheffield.ac.uk.
Laura West, Email: l.west@sheffield.ac.uk.
Josephine A. Pickworth, Email: j.a.pickworth@sheffield.ac.uk.
Helena Turton, Email: helena.turton@sheffield.ac.uk.
James Iremonger, Email: james.iremonger@gmail.com.
Adam T. Braithwaite, Email: adam.braithwaite@cardiov.ox.ac.uk.
Jaime Cañedo, Email: jcanedo1@sheffield.ac.uk.
Simon A. Johnston, Email: s.a.johnston@sheffield.ac.uk.
A.A. Roger Thompson, Email: r.thompson@sheffield.ac.uk.
Gaynor Miller, Email: g.miller@bham.ac.uk.
References
- 1.Kiely DG, Elliot CA, Sabroe I, Condliffe R. Pulmonary hypertension: diagnosis and management. BMJ. 2013;346:f2028 doi: 10.1136/bmj.f2028 [DOI] [PubMed] [Google Scholar]
- 2.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f [DOI] [PubMed] [Google Scholar]
- 3.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123:1986–1995. doi: 10.1161/CIRCULATIONAHA.110.978627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908. doi: 10.1164/rccm.201202-0335OC [DOI] [PubMed] [Google Scholar]
- 6.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–272. doi: 10.1164/rccm.201201-0164OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, Maeda Y, Urabe M, Mizukami H, Kume A, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. 2007;101:734–741. doi: 10.1161/CIRCRESAHA.107.153023 [DOI] [PubMed] [Google Scholar]
- 8.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–44, 28p following 244. doi: 10.1161/CIRCRESAHA.108.182014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol. 2015;308:L229–L252. doi: 10.1152/ajplung.00238.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978 [DOI] [PubMed] [Google Scholar]
- 12.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen TO, Schmidt H, Møller HJ, Høyer M, Bogdan Maniecki M, Sjoegren P, Jarle Christensen IB, Steiniche T. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J Clin Oncol. 2009;27:3330–3337 [DOI] [PubMed] [Google Scholar]
- 14.Weisser SB, van Rooijen N, Sly LM. Depletion and reconstitution of macrophages in mice. J Vis Exp. 2012;66:4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gheryani N, Coffelt SB, Gartland A, Rumney RM, Kiss-Toth E, Lewis CE, Tozer GM, Greaves DR, Dear TN, Miller G. Generation of a novel mouse model for the inducible depletion of macrophages in vivo. Genesis. 2013;51:41–49. doi: 10.1002/dvg.22343 [DOI] [PubMed] [Google Scholar]
- 16.Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, Di W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res. 2014;7:19 doi: 10.1186/1757-2215-7-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyachi Y, Tsuchiya K, Shiba K, Mori K, Komiya C, Ogasawara N, Ogawa Y. A reduced M1-like/M2-like ratio of macrophages in healthy adipose tissue expansion during SGLT2 inhibition. Sci Rep. 2018;8:16113 doi: 10.1038/s41598-018-34305-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu B, Zhang M, Zhao J, Zheng M, Yang H. Imbalance of M1/M2 macrophages is linked to severity level of knee osteoarthritis. Exp Ther Med. 2018;16:5009–5014. doi: 10.3892/etm.2018.6852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCubbrey AL, Barthel L, Mould KJ, Mohning MP, Redente EF, Janssen WJ. Selective and inducible targeting of CD11b+ mononuclear phagocytes in the murine lung with hCD68-rtTA transgenic systems. Am J Physiol Lung Cell Mol Physiol. 2016;311:L87–L100. doi: 10.1152/ajplung.00141.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing Lung disease?. Trends Mol Med. 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004 [DOI] [PubMed] [Google Scholar]
- 21.Žaloudíková M, Vytášek R, Vajnerová O, Hniličková O, Vízek M, Hampl V, Herget J. Depletion of alveolar macrophages attenuates hypoxic pulmonary hypertension but not hypoxia-induced increase in serum concentration of MCP-1. Physiol Res. 2016;65:763–768. doi: 10.33549/physiolres.933187 [DOI] [PubMed] [Google Scholar]
- 22.Rumney RMH, Coffelt SB, Neale TA, Dhayade S, Tozer GM, Miller G. PyMT-MacLow: a novel, inducible, murine model for determining the role of CD68 positive cells in breast tumor development. PLoS One. 2017;12:e0188591 doi: 10.1371/journal.pone.0188591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrie A, Hameed AG, Chamberlain J, Arnold N, Kennerley A, Hopkinson K, Pickworth J, Kiely DG, Crossman DC, Francis SE. Paigen diet-fed apolipoprotein E knockout mice develop severe pulmonary hypertension in an interleukin-1-dependent manner. Am J Pathol. 2011;179:1693–1705. doi: 10.1016/j.ajpath.2011.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hameed AG, Arnold ND, Chamberlain J, Pickworth JA, Paiva C, Dawson S, Cross S, Long L, Zhao L, Morrell NW, et al. Inhibition of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J Exp Med. 2012;209:1919–1935. doi: 10.1084/jem.20112716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold ND, Pickworth JA, West LE, Dawson S, Carvalho JA, Casbolt H, Braithwaite AT, Iremonger J, Renshall L, Germaschewski V, et al. A therapeutic antibody targeting osteoprotegerin attenuates severe experimental pulmonary arterial hypertension. Nat Commun. 2019;10:5183 doi: 10.1038/s41467-019-13139-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chamberlain J, Francis S, Brookes Z, Shaw G, Graham D, Alp NJ, Dower S, Crossman DC. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS One. 2009;4:e5073 doi: 10.1371/journal.pone.0005073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinet W, Verheye S, De Meyer GR. Selective depletion of macrophages in atherosclerotic plaques via macrophage-specific initiation of cell death. Trends Cardiovasc Med. 2007;17:69–75. doi: 10.1016/j.tcm.2006.12.004 [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, Minami M, Inagaki T, Miyagawa S, Sawa Y, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2015;112:E2677–E2686. doi: 10.1073/pnas.1424774112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luckett-Chastain L, Calhoun K, Schartz T, Gallucci RM. IL-6 influences the balance between M1 and M2 macrophages in a mouse model of irritant contact dermatitis. J Immunol. 2016;196:196.17–196.1726621863 [Google Scholar]
- 31.Chen L, Wang S, Wang Y, Zhang W, Ma K, Hu C, Zhu H, Liang S, Liu M, Xu N. IL-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget. 2018;9:17443–17454. doi: 10.18632/oncotarget.24734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarling JD, Lin HS, Hsu S. Self-renewal of pulmonary alveolar macrophages: evidence from radiation chimera studies. J Leukoc Biol. 1987;42:443–446 [PubMed] [Google Scholar]
- 33.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–387. doi: 10.1378/chest.09-1140 [DOI] [PubMed] [Google Scholar]
- 34.Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141:363–373. doi: 10.1378/chest.10-3114 [DOI] [PubMed] [Google Scholar]
- 35.Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A, Vonk Noordegraaf A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest. 2014;145:1230–1236. doi: 10.1378/chest.13-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240:H62–H72. doi: 10.1152/ajpheart.1981.240.1.H62 [DOI] [PubMed] [Google Scholar]
- 37.Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol. 1999;276:H699–H708. doi: 10.1152/ajpheart.1999.276.2.H699 [DOI] [PubMed] [Google Scholar]
- 38.Umar S, de Visser YP, Steendijk P, Schutte CI, Laghmani el H, Wagenaar GT, Bax WH, Mantikou E, Pijnappels DA, Atsma DE, et al. Allogenic stem cell therapy improves right ventricular function by improving lung pathology in rats with pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2009;297:H1606–H1616. doi: 10.1152/ajpheart.00590.2009 [DOI] [PubMed] [Google Scholar]
- 39.Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol. 1993;110:719–723. doi: 10.1111/j.1476-5381.1993.tb13871.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. 2010;56:696–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, van der Laarse A, Eghbali M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184:715–723. doi: 10.1164/rccm.201101-0078OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA., IIIAlterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009;34:1093–1099. doi: 10.1183/09031936.00010409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, Morecroft I, Loughlin L, McClure JD, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–1098. doi: 10.1161/CIRCULATIONAHA.111.062927 [DOI] [PubMed] [Google Scholar]
- 44.Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, Nilsen M, Loughlin L, Thomas M, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190:456–467. doi: 10.1164/rccm.201403-0483OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Umar S, Cunningham CM, Itoh Y, Moazeni S, Vaillancourt M, Sarji S, Centala A, Arnold AP, Eghbali M. The Y chromosome plays a protective role in experimental hypoxic pulmonary hypertension. Am J Respir Crit Care Med. 2018;197:952–955. doi: 10.1164/rccm.201707-1345LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamosiuniene R, Manouvakhova O, Mesange P, Saito T, Qian J, Sanyal M, Lin YC, Nguyen LP, Luria A, Tu AB, et al. A dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res. 2018;122:689–1702. doi: 10.1161/CIRCRESAHA.117.312058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.