

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis, cholesterol, fibroblast, macrophage, phenotype, smooth muscle cell
Objective:
Vascular smooth muscle cells (SMCs) dedifferentiate and initiate expression of macrophage markers with cholesterol exposure. This phenotypic switching is dependent on the transcription factor Klf4 (Krüppel-like factor 4). We investigated the molecular pathway by which cholesterol induces SMC phenotypic switching.
Approach and Results:
With exposure to free cholesterol, SMCs decrease expression of contractile markers, activate Klf4, and upregulate a subset of macrophage and fibroblast markers characteristic of modulated SMCs that appear with atherosclerotic plaque formation. These phenotypic changes are associated with activation of all 3 pathways of the endoplasmic reticulum unfolded protein response (UPR), Perk (protein kinase RNA-like endoplasmic reticulum kinase), Ire (inositol-requiring enzyme) 1α, and Atf (activating transcription factor) 6. Blocking the movement of cholesterol from the plasma membrane to the endoplasmic reticulum prevents free cholesterol–induced UPR, Klf4 activation, and upregulation of the majority of macrophage and fibroblast markers. Cholesterol-induced phenotypic switching is also prevented by global UPR inhibition or specific inhibition of Perk signaling. Exposure to chemical UPR inducers, tunicamycin and thapsigargin, is sufficient to induce these same phenotypic transitions. Finally, analysis of published single-cell RNA sequencing data during atherosclerotic plaque formation in hyperlipidemic mice provides preliminary in vivo evidence of a role of UPR activation in modulated SMCs.
Conclusions:
Our data demonstrate that UPR is necessary and sufficient to drive phenotypic switching of SMCs to cells that resemble modulated SMCs found in atherosclerotic plaques. Preventing a UPR in hyperlipidemic mice diminishes atherosclerotic burden, and our data suggest that preventing SMC transition to dedifferentiated cells expressing macrophage and fibroblast markers contributes to this decreased plaque burden.
Highlights.
Exposure to cholesterol or oxidized phospholipids triggers phenotypic switching of vascular smooth muscle cells to a macrophage/fibroblast–like cell and is associated with an unfolded protein response (UPR), involving activation of all 3 arms of endoplasmic reticulum stress: Perk (protein kinase RNA-like endoplasmic reticulum kinase), Ire (inositol-requiring enzyme) 1α, and Atf (activating transcription factor) 6.
Induction of endoplasmic reticulum stress and UPR is necessary for phenotypic switching of smooth muscle cells to macrophage/fibroblast-like cells.
Cholesterol-induced phenotypic switching is mediated primarily by the Perk-eIF2α (α-subunit of the eukaryotic elongation factor 2)-Atf4 axis of UPR.
Chemically induced UPR without cholesterol exposure is sufficient to cause phenotypic switching of smooth muscle cells to a macrophage/fibroblast-like cell.
Reanalysis of previously published single-cell RNA sequencing data shows preliminary evidence of UPR activation in modulated smooth muscle cells in vivo.
Vascular smooth muscle cells (SMCs) in healthy arteries are quiescent, differentiated cells that are characterized by expression of contractile proteins, including the smooth muscle–specific isoforms of α-SMA (α-smooth muscle actin, Acta2) and MHC (myosin heavy chain, Myh11), along with SM22α (smooth muscle 22α, Tagln) and Cnn1 (calponin, Cnn1).1 SMCs remain phenotypically pliable and can dedifferentiate in response to environmental cues, switching off contractile gene expression and increasing proliferation and migration. In preatherosclerotic arteries, SMCs form the major cellular component of a thickened intimal layer, known as diffuse intimal thickening.2 During plaque formation, increasing amounts of lipids are deposited in the inner layer of the diffuse intimal thickening around the SMCs. SMCs are also involved in preventing plaque rupture by stabilizing the fibrous cap.3,4 Atherosclerotic plaques contain cholesterol-laden macrophages (ie, foam cells), which undergo apoptosis to form necrotic lipid cores in these lesions.5 Multiple studies have also reported the presence of lipid-laden SMCs in both cell culture and whole animal studies.6–9 These SMC foam cells have been detected in advanced atherosclerotic plaques in apoE-deficient (Apoe−/−) mice, as well as in early lesions, in which the majority of the foam cells are SMC-derived.3,10,11
Lineage tracing studies have identified an expanded role of SMCs in atherosclerotic lesions. Initial studies indicated SMCs can assume a macrophage-like phenotype by dedifferentiating and initiating the expression of macrophage markers, a transition that is dependent on Klf4 (Krüppel-like factor 4) signaling.12 The transition of SMCs to a macrophage-like cell is modeled in cell culture by exposing SMCs to free cholesterol or oxidized phospholipids, which decreases expression of contractile genes and increases expression of macrophage markers (eg, Lgals3 [lectin, galactoside-binding, soluble, 3] and Cd68 [cluster of differentiation 68]).13–15 In human atherosclerotic plaques, cells expressing both SMC and macrophage markers have been detected, which underscores the clinical relevance of SMC phenotypic switching to a macrophage-like cell in atherosclerotic plaque burden.16 Recent single-cell RNA sequencing (scRNA-Seq) analyses have identified a distinct population of SMC-derived cells, termed modulated SMCs, that appear with the development of atherosclerosis in Apoe−/− mice fed a high-fat diet (HFD).17 These modulated SMCs express a few macrophage markers, including Lgals3, but also express fibroblast markers.
A number of events can overwhelm the folding capacity of the endoplasmic reticulum (ER) and trigger an unfolded protein response (UPR), including accumulation of misfolded or incorrectly folded proteins, increased expression of normal proteins, oxidative stress, impaired calcium homeostasis, and nutrient deprivation.18–21 In mammalian cells, the UPR involves 3 membrane-associated sensors: Perk (protein kinase RNA-like ER kinase) oligomerizes and phosphorylates the eIF2α (α-subunit of the eukaryotic elongation factor 2) which attenuates cellular protein synthesis and preferentially promotes translation of the Atf (activating transcription factor) 4; Ire (inositol-requiring enzyme) 1 splices X-box binding protein (Xbp1) mRNA and generates the activated transcription factor Xbp1; and the 90 kDa Atf6 translocates to the Golgi complex and is activated by cleavage to a smaller 50 kDa protein.22–24 The combined effect of activation of these UPR sensors is to reduce the overall protein load by translational inhibition, increase levels of chaperones like Bip/Grp78 (binding immunoglobulin protein), Grp94 (heat shock protein 90kDa beta member 1), and Edem (endoplasmic reticulum degradation enhancing α-mannosidase like protein) to help fold the misfolded proteins and increase apoptosis.
ER stress and the UPR contribute to the formation of atherosclerotic plaques. Free cholesterol levels are typically low in the ER and an abnormal enrichment of cholesterol in the ER can also trigger a UPR.25 Cholesterol-loaded macrophages activate a UPR, and activation of the downstream cell death effector Chop ([CCAAT-enhancer-binding protein] C/EBP homologous protein) leads to apoptosis.25 Hypercholesterolemic mice (Apoe−/−) that are also deficient in Chop (Chop−/−) have 35% less atherosclerotic lesions, indicating a pathogenic role for UPR in atherosclerosis.26 Exposure of SMCs to either free cholesterol or oxidized LDL (low-density lipoprotein) in culture induces a UPR and apoptosis, but a role of the UPR in phenotypic switching of SMCs was not pursued in this study.27 Furthermore, ER stress is present in SMCs in atherosclerotic plaques in mouse models of diabetes and hyperhomocysteinemia, based on increased expression of ER chaperones.28,29 Additionally, exposure to oxidized phospholipids can also elicit a UPR in human endothelial cells, including the induction of Atf4.30–32 Since the UPR effector protein Atf4 both increases expression and prevents proteasomal degradation of Klf4, we investigated whether UPR is responsible for phenotypic switching of cholesterol-loaded SMCs to cells with expression of macrophage and fibroblast markers, that is, the modulated SMCs that appear with atherosclerotic plaque formation.17,33
Materials and Methods
Data Availability Statement
The authors declare that all supporting data are available within the article and in the Data Supplement.
SMCs Explant and Culture Conditions, Cholesterol Exposure, and Drug Treatments
Mouse aortic SMCs were explanted from the ascending aortas of wild-type C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) mice as described earlier.34 SMCs were explanted from both male and female C57BL/6J mice and used in these studies. Aortas from male and female mice were pooled to obtain enough viable cells for maintenance and studies were performed using this pooled population. This has been a standard practice in our laboratory. The SMCs were immortalized by hTERT (human telomerase reverse transcriptase) overexpression using conditioned medium containing retrovirus from pBabe.hTERTpuro-pA317 packaging cell line. Infected cells were selected using 0.5 µg/mL puromycin for 1 week, then switched back to regular SMC maintenance medium.
SMCs were treated with indicated amounts of either free cholesterol complexed to methyl-β-cyclodextrin (methyl-β-cyclodextrin cholesterol, MBD-Chol; Millipore Sigma) or 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC; Cayman Chemical) or aggregated LDL (human LDL, Thermo Fisher Scientific) in DME containing high glucose (Cellgro), 10% FBS (Gibco), 1% antibiotic/antimycotic (Millipore Sigma), and 0.2% BSA (Fisher Scientific) for 72 hours at 37 °C and 5% CO2.13,35
SMCs were treated with U18666A, 4-phenylbutyric acid (4-PBA), Kira6 (IRE1α kinase inhibiting RNase attenuator 6; Millipore Sigma), trans-ISRIB (integrated stress response inhibitor; Thermo Fisher Scientific), tunicamycin (Calbiochem), and thapsigargin (Tocris Bioscience) for the indicated amounts of time. All drugs were dissolved in DMSO (Millipore Sigma) and DMSO was used for no-treatment controls.
Generation of shRNA Against Perk
shRNA (short hairpin RNA) oligo against Perk (CACTTTGAACTTCGGTATATT) or scramble (CCTAAGGTTA-AGTCGCCCTCG) was cloned into the pLKO.1 vector (Sigma-Aldrich) and then packed into lentiviral particles using psPAX2 and pMD2.G (Takara Bio USA) in HEK293T cells and used to infect immortalized SMCs using polybrene.
RNA Extraction, Quantitative Real-Time Polymerase Chain Reaction, and Immunoblot Analyses
RNA extraction, quantitative real-time polymerase chain reaction, and immunoblot analyses were performed using standard methods that are described in details in the Methods in the Data Supplement. Please see the Major Resources Table in the Data Supplement for detailed information on antibodies and quantitative polymerase chain reaction primer sequences.
Klf4 Transactivation Assays
Klf4 transactivation assay was performed using a Cignal Reporter Plasmid (Qiagen) and the luciferase activity was measured using a Dual-Luciferase Reporter Assay Kit (Promega) according to manufacturer’s instructions.
Phagocytosis Assay
Phagocytic activity of SMCs was estimated by their uptake of green fluorescent microspheres in response to various treatments, followed by imaging using confocal microscopy. The detailed procedure is described in the Data Supplement.
Cholesteryl Ester Formation Assay
Cholesteryl ester formation was quantified using Cholesterol/Cholesteryl Ester Quantitation Assay kit (Colorimetric/Fluorometric) from Abcam (ab65359), as per manufacturer’s instructions. Detailed description is provided in the Data Supplement.
Analysis of scRNA-Seq Data
The mouse aorta scRNA-Seq count data was downloaded from Gene Expression Omnibus under accession number GSE131776. The data was submitted to R package Seurat for cluster identification and the analysis was performed as previously described.17 Relative gene expression projected to t-stochastic neighbor embedding plot was generated by function FeaturePlot of Seurat.
Statistical Analysis
All data shown are representative of at least 3 independent experiments, unless noted otherwise, and expressed as ±SD. Data were tested for normality using Graph Pad Prism software (Graph Pad Software, Inc, San Diego, CA), and all the data passed normality. Unpaired 2-tailed Student t tests or 2-way ANOVA followed by Tukey Honest Significant Difference post hoc test were performed using Prism software. P<0.05 was considered statistically significant.
Results
Exposing SMCs to Either Free Cholesterol, Aggregated Cholesterol, or Oxidized Phospholipid Induces ER Stress and Phenotypic Changes
We initially confirmed that exposing immortalized mouse aortic SMCs to 10 µg/mL of MBD-Chol for 72 hours leads to increased mRNA expression and protein levels of the macrophage markers Cd68 and Lgals3, along with Abca1 (Abca1 was assessed because its expression is high in macrophages and controlled by intracellular cholesterol levels; Figure 1A and 1C, Figure IB in the Data Supplement) and decreased expression and levels of the SMC differentiation markers Acta2, Tagln, and Cnn1 (Figure 1B and 1C, Figure IA and IB in the Data Supplement).36,37 We repeated the MBD-Chol exposure experiments with low passage, primary SMCs, and obtained similar results to those with immortalized SMCs (Figure IIA through IIE in the Data Supplement); therefore, the remainder of the studies were performed using immortalized SMCs. Phenotypic switching of SMCs to cells with macrophage-like functions is further supported by the induction of phagocytic activity in response to cholesterol exposure, as assessed by increased uptake of fluorescently labeled beads compared with little to no uptake by SMCs not exposed to cholesterol (Figure 1D, Figure IC in the Data Supplement). MBD-Chol exposure also increases lipid droplets in SMCs, as assessed by increased Oil red O staining (Figure ID in the Data Supplement). Klf4 expression, protein levels, and transcriptional activity are also increased with exposure to MBD-Chol (Figure 1A, 1C, and 1E; Figure IB in the Data Supplement). Notably, we found that SMCs exposed to MBD-Chol activate all 3 UPR pathways as indicated by the following increases: ratio of spliced to unspliced Xbp1, Ire1α phosphorylation, eIF2α phosphorylation (eIF2α is phosphorylated by activated Perk and is, therefore, a marker of Perk pathway activation), Atf4 levels, and cleavage of Atf6 to its 50 kDa active form (Figure 1C, 1F, and 1G, Figure IB, IID, and IIE in the Data Supplement). Furthermore, downstream effectors of these pathways, including Chop and the chaperones BiP/Grp78, Grp94, and Edem, are also significantly upregulated by MBD-Chol (Figure 1F). MBD-Chol exposure also induces increased SMC apoptosis (Figure IG in the Data Supplement). Taken together, these data indicate that MBD-Chol–induced phenotypic switching of SMCs to cells with macrophage features is associated with activation of all 3 ER stress sensor pathways.
Figure 1.
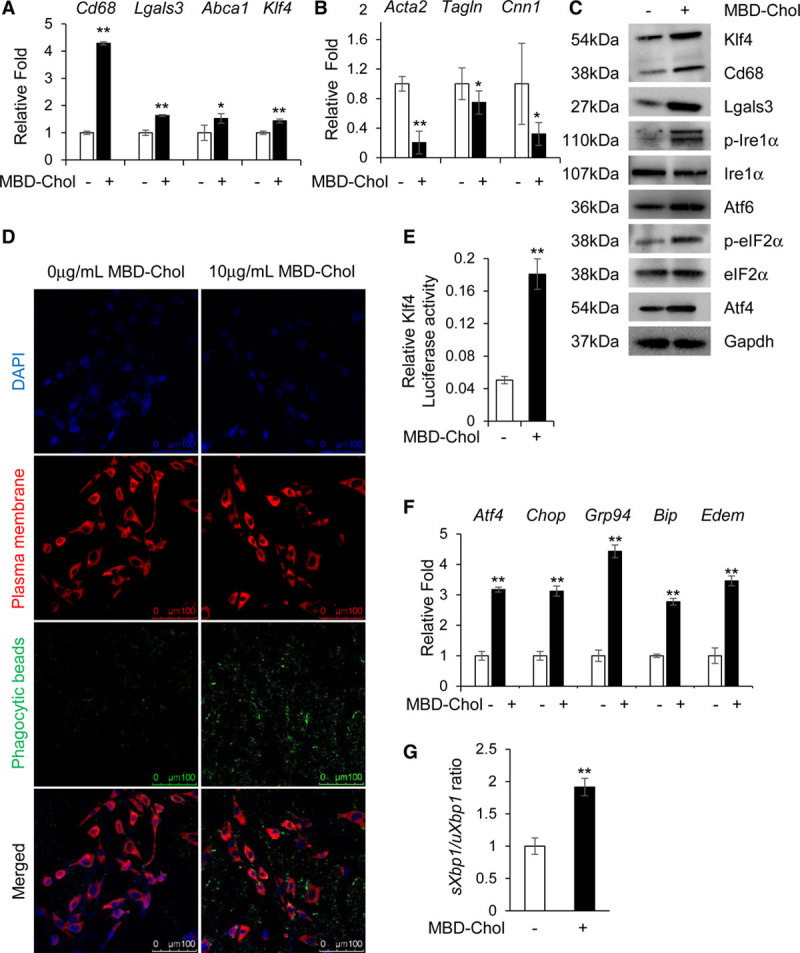
Cholesterol induces phenotypic switching and endoplasmic reticulum (ER) stress in immortalized vascular smooth muscle cells (SMCs). A and B, With exposure to 10 µg/mL methyl-β-cyclodextrin cholesterol (MBD-Chol) for 72 h, expression of macrophage markers Cd68 and Lgals3 along with Abca1 and Klf4 (Krüppel-like factor 4) are upregulated (A), while contractile marker genes Acta2, Tagln and Cnn1 are downregulated (B). C, Immunoblots confirm increased protein levels of Cd68 (cluster of differentiation 68), Lgals3 (lectin, galactoside-binding, soluble, 3), and Klf4 along with induction of ER stress including phosphorylation of Ire (inositol-requiring enzyme) 1α and eIF2α (α-subunit of the eukaryotic elongation factor 2), induction of Atf (activating transcription factor) 4 and increased cleavage of Atf6. D, Exposure to 10 µg/mL MBD-Chol for 72 h increases phagocytic activity of SMCs as evident from the increased uptake of green fluorescent phagocytic beads. E, Klf4 transcriptional activity is increased when SMCs are exposed to cholesterol. F, Cholesterol exposure upregulates ER stress effector genes like Atf4 and Chop as well as the chaperones Grp94, Bip, and Edem. G, Cholesterol treatment increases splicing of Xbp1, indicating activation of the Ire1α pathway. Each result displayed here is representative of at least 3 independent biological replicates. P values were calculated using unpaired 2-tailed Student t test. *P<0.05 or **P<0.01, vs no cholesterol treatment. DAPI indicates 4′,6-diamidino-2-phenylindole; p-eIF2α, phosphorylated eIF2α; and p-Ire1α, phosphorylated Ire1α.
SMCs were also exposed to 25 µg/mL aggregated LDL or 20 µg/mL of the oxidized phospholipid POVPC; treatment with either compound also upregulates Cd68, Lgals3, and Abca1 and downregulates Acta2, Tagln, and Cnn1 (Figure IIIA through IIID, VIA through VIC, and VIE in the Data Supplement) along with inducing phagocytic activity (Figure IVA and IVB, VIID and VIIE in the Data Supplement) and foam cell formation (Figure IVC in the Data Supplement). Phenotypic switching in response to aggregated LDL is associated with increased Klf4 transcript expression, protein levels, and transcriptional activation (Figure IIIA, IIIC, IIID, and IVD in the Data Supplement) accompanied by activation of all 3 arms of UPR (Figure IIIA, IIIC through IIIH in the Data Supplement). Additionally, treatment with aggregated LDL increases cholesteryl ester content in the cell (Figure IVE in the Data Supplement).
Inhibition of Cholesterol Transport From the Plasma Membrane to the ER Abrogates Cholesterol-Induced Phenotypic Switching
Nanomolar concentrations of the amphipathic amine U18666A selectively block transport of free cholesterol from the plasma membrane to the ER without significantly interfering with transfer to the plasma membrane.38 Studies of cholesterol-loaded macrophages showed that U18666A blocks free cholesterol movement to the ER and UPR-driven apoptosis.25 To determine if cholesterol transport from the plasma membrane to the ER is required for the UPR, we treated SMCs with MBD-Chol in the presence and absence of 70 nmol/L U18666A. Cholesterol moves from the plasma membrane through the Golgi to the ER, where it is converted to cholesteryl esters. We confirmed that U18666A treatment prevents the increase in cholesteryl ester formation with cholesterol loading in SMCs to verify that U18666A decreases the levels of cholesterol in the ER (Figure 2A). U18666A treatment prevents SMC phenotypic switching with cholesterol loading (Figure 2B and 2C; Figure VA, VB, and VE in the Data Supplement) and also blocks the increase in Klf4 transcript and protein levels, as well as transcriptional activity (Figure 2B through 2D; Figure VE in the Data Supplement). We confirmed that U18666A prevents the cholesterol-induced UPR based on the following: no increase in eIF2α phosphorylation, Atf4 levels, or Ire1α phosphorylation; lower Xbp1 splicing ratio; and no induction of Chop and the chaperones Bip/Grp78, Grp94, and Edem (Figure 2C, 2E, and 2F; Figure VC through VE in the Data Supplement). These data indicate that cholesterol trafficking to the ER is necessary for SMCs to both induce UPR and transition to a macrophage-like state with cholesterol loading. Cotreatment of SMCs with U18666A reverses POVPC-induced phenotypic switching (Figure VIA through VIC, and VIE in the Data Supplement) and increase in Klf4 levels and transcriptional activation (Figure VIA, VIC, VIE, and VIIC in the Data Supplement) and reduces phagocytic activity (Figure VIID and VIIE in the Data Supplement). U18666A successfully inhibits POVPC-induced activation of all arms of UPR (FigureVIC through VIE and VIIA and VIIB in the Data Supplement). Additionally, treatment with POVPC increases cholesteryl ester content in the cell which is reversed by U18666A (Figure VIIIF in the Data Supplement). Taken together, these data suggest that POVPC induces UPR and phenotypic switching in SMCs in a manner similar to that of free cholesterol.
Figure 2.
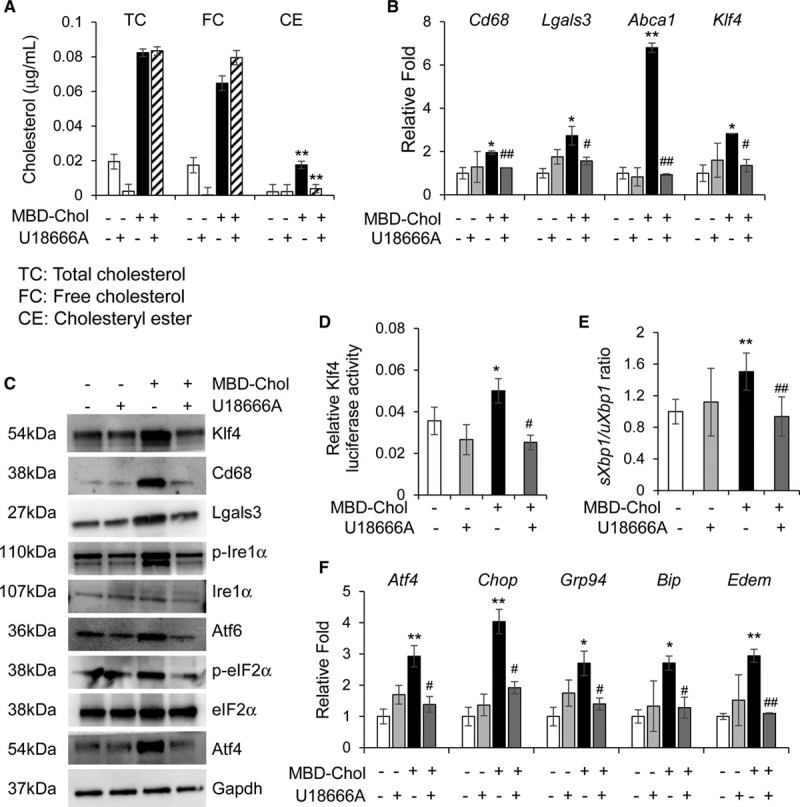
Blocking the movement of cholesterol from the plasma membrane to the endoplasmic reticulum (ER) blocks phenotypic switching and ER stress in immortalized smooth muscle cells. A, Cholesterol esterification is abrogated by 70 nmol/L U18666A, which prevents the trafficking of free cholesterol from the plasma membrane to the ER. B, Cholesterol-induced upregulation of macrophage markers and Krüppel-like factor 4 (Klf4) at the mRNA level is reduced to baseline upon concurrent exposure to 10 µg/mL methyl-β-cyclodextrin cholesterol (MBD-Chol) and 70 nmol/L U18666A for 72 h. C, Treatment with U18666A prevents cholesterol-induced increases in Klf4, macrophage marker proteins, and unfolded protein response effector proteins and signaling. D, Transcriptional activity of Klf4 is enhanced by cholesterol but reduced to baseline when U18666A blocks the movement of cholesterol from the plasma membrane to the ER. E, U18666A reduces cholesterol-induced splicing of Xbp1, a marker for Ire (inositol-requiring enzyme) 1α activation. F, Treatment with U18666A also reduces cholesterol-induced ER stress, as evidenced by decreased expression of activating transcription factor (Atf) 4, Chop, and the ER chaperones Grp94, Bip, and Edem. Each result displayed here is representative of at least 3 independent biological replicates. P values were calculated using 2-way ANOVA followed by Tukey Honest Significant Difference post hoc t test. *P<0.05 or **P<0.01 vs no cholesterol treatment. For the U18666A-treated samples, #P<0.05 or ##P<0.01 for U18666A treatment vs no treatment (DMSO). CE indicates cholesteryl esters; eIF2α, α-subunit of the eukaryotic elongation factor 2; FC, free cholesterol; Lgals3, lectin, galactoside-binding, soluble, 3; p-eIF2α, phosphorylated eIF2α; p-Ire1α, phosphorylated Ire1α; and TC, total cholesterol.
Cholesterol-Induced Phenotypic Switching of SMCs Is Prevented by Inhibition of the UPR
To confirm a role for the UPR in SMC phenotypic switching, SMCs were exposed to MBD-Chol for 72 hours and a general UPR inhibitor, 4-PBA. 4-PBA prevents the cholesterol-induced increases in macrophage marker expression and decreases in SMC differentiation markers, along with lowering the expression and transcriptional activation of Klf4 (Figure 3A through 3C, Figures VIIIA, VIIIC, and VIIIG in the Data Supplement). Interestingly, 4-PBA treatment alone increases expression and protein levels of Acta2, Tagln, and Cnn1 (Figure VIIIA and VIIIC in the Data Supplement). 4-PBA treatment successfully inhibits MBD-Chol–induced UPR, as evidenced by lower expression of all tested ER stress effectors and chaperones (Figure VIIIB through VIIIG in the Data Supplement).
Figure 3.
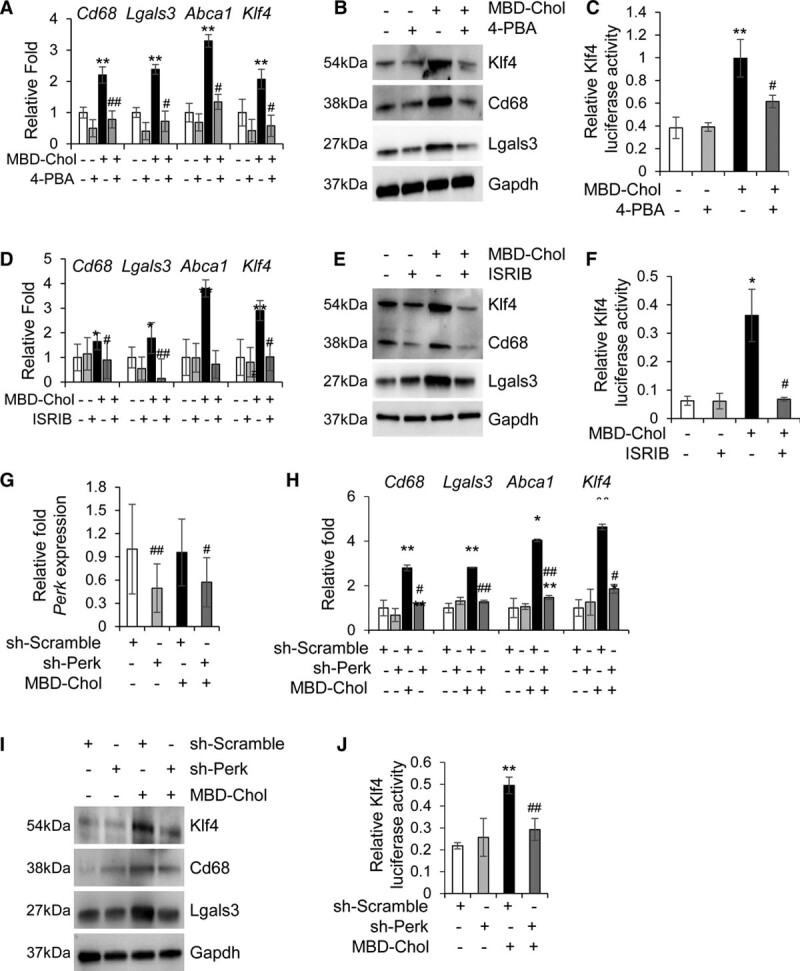
Inhibition of endoplasmic reticulum (ER) stress abrogates phenotypic switching of smooth muscle cells. A and B, Cholesterol-induced upregulation of macrophage markers is reduced to baseline upon concurrent exposure to 10 µg/mL methyl-β-cyclodextrin cholesterol (MBD-Chol) and 5 mmol/L 4-phenylbutyric acid (4-PBA) for 72 h at both the mRNA (A) and protein (B) levels. C, Transcriptional activity of Klf4 (Krüppel-like factor 4) that is enhanced by cholesterol is reduced to baseline by the ER stress inhibitor 4-PBA. D and E, Treatment with 200 nmol/L ISRIB (integrated stress response inhibitor) reverses cholesterol-induced upregulation of macrophage marker genes (D) and proteins (E). F, Exposure to ISRIB prevents the cholesterol-induced increase in Klf4 transcriptional activity. G, Quantitative polymerase chain reaction analysis demonstrates successful downregulation of protein kinase RNA-like ER kinase (Perk) mRNA by shRNA (short hairpin RNA) treatment. H, Perk knockdown reverses cholesterol-induced upregulation of macrophage marker genes. I, Immunoblots confirm downregulation of Perk abrogates cholesterol-induced increases in macrophage marker proteins. J, Downregulation of Perk also reduces cholesterol-induced increase in the transcriptional activation of Klf4. Each result displayed here is representative of at least 3 independent biological replicates. P values were calculated using 2-way ANOVA followed by Tukey Honest Significant Difference post hoc test. *P<0.05 and **P<0.01 vs no cholesterol treatment. For the 4-PBA–treated or ISRIB-treated samples, #P<0.05 or ##P<0.01 for drug treatment vs no treatment (DMSO). For the shRNA-treated samples, #P<0.05 or ##P<0.01 for comparison between scramble shRNA (sh-Scramble) and sh-Perk. Lgals3 indicates lectin, galactoside-binding, soluble, 3; sh-Perk, short hairpin RNA against Perk; and shRNA, short hairpin RNA.
To explore if a specific UPR pathway is responsible for phenotypic switching, SMCs were exposed to MBD-Chol and the following inhibitors: ISRIB to block the phosphorylated eIF2α-induced upregulation of Atf4, CA7 (ceapin A7) to block the cleavage and activation of Atf6, and Kira6 to inhibit Ire1α.39–41 ISRIB prevents the increased expression and protein levels of macrophage markers and Klf4 with MBD-Chol exposure, along with decreasing Klf4 transcriptional activity, induction of phagocytic activity, and foam cell formation (Figure 3D through 3F, Figure XA through XC in the Data Supplement). SMCs treated with ISRIB and MBD-Chol also maintain expression and levels of the SMC differentiation markers (Figure IXA, IXC, and IXG in the Data Supplement). Expectedly, ISRIB treatment blocks the Perk-eIF2α-Atf4 sensor, as indicated by the lack of cholesterol-driven increases of Atf4, but has no effect on the Ire1α or Atf6 pathways (Figure IXB through IXG in the Data Supplement). To further confirm the role of the Perk pathway in cholesterol-induced phenotypic switching, Perk levels were depleted using shRNA (sh-Perk) and the cells subjected to MBD-Chol treatment. Perk knockdown was confirmed by reduction in Perk mRNA levels compared to scramble shRNA (Figure 3G). In cells infected with shRNA against Perk (sh-Perk), MBD-Chol–induced increases in mRNA and protein levels of macrophage markers and Klf4 were prevented, as was the increase in Klf4 transcriptional activity and the decreases in SMC contractile markers (Figure 3H through 3J, Figure XIA, XIB, and XID in the Data Supplement). Efficacy and specificity of Perk knockdown were additionally confirmed by decreased eIF2α phosphorylation and Atf4 levels (Figure XIC, XIF, and XIH in the Data Supplement) with no significant effect on Ire1α or Atf6 (Figure XIC through XIG in the Data Supplement). These data suggest that the Perk arm of UPR is primarily responsible for cholesterol-induced phenotypic switching.
Exposure to an Atf6 inhibitor, CA7, does not reduce macrophage marker expression, maintain contractile marker expression, or prevent increased levels and activation of Klf4 in cholesterol-loaded SMCs (Figure XIIA, XIIB, and XIIE in the Data Supplement), indicating that the Atf6 pathway is most likely not involved in MBD-Chol–induced phenotypic switching. CA7 efficacy and specificity were confirmed by significant reduction in cleaved Atf6 levels with no effect on phosphorylated Ire1α or Atf4 levels (Figure XIIC, XIID, and XIIF in the Data Supplement). Inhibition of Ire1α with Kira6 prevents cholesterol-driven increases of Cd68 and Abca1 transcript levels but does not alter Lgals3 expression or Klf4 levels or activity (Figure XIIIA and XIIIE in the Data Supplement). Kira6 also increases the expression of contractile proteins in the absence and presence of cholesterol and, in the absence of cholesterol, this is associated with decreased Klf4 transcriptional activity (Figure XIIIB in the Data Supplement). The increased expression of contractile marker genes upon Ire1α is inhibition is in agreement with a recent study that demonstrated a reciprocal relationship between UPR and SMC differentiation through the Ire1α pathway.42 Efficacy of Kira6 is confirmed by significant reduction of Xbp1 splicing and Ire1α phosphorylation levels, whereas its specificity is confirmed by a lack of effect on Atf4 induction and Atf6 cleavage (Figure XIIIC, XIIID, and XIIIF in the Data Supplement). The data support that phenotypic switching of SMCs to macrophage-like cells is primarily driven by the Perk-eIF2α-Atf4 pathway of the UPR, which is established to increase levels of Klf4.33 The Ire1α pathway may also contribute to increased Cd68 expression, which appears to be independent of Klf4. This observation aligns with an identified role of Xbp1 activation by Ire1α in increasing CD68 expression during granulocyte differentiation.43 Importantly, when SMCs were treated with MBD-Chol and any of the inhibitors described above, there was no change in apoptosis compared with cells treated with MBD-Chol alone (Figure XIV in the Data Supplement).
SMCs Exposed to Either Tunicamycin or Thapsigargin Undergo Phenotypic Switching to a Macrophage-Like Cell
We investigated if activation of the UPR by methods other than cholesterol influx into the ER would also induce phenotypic switching of SMCs to a macrophage/fibroblast–like cell. Tunicamycin inhibits N-glycosylation of proteins in the ER and activates all 3 UPR pathways.44 Thapsigargin promotes UPR by inhibiting the transport of calcium ions from the cytosol to the ER and the sarcoplasmic reticulum, leading to depletion of ER calcium stores that are critical for protein folding.45 Treatment with either tunicamycin (5 hours) or thapsigargin (8 hours) successfully induces macrophage marker gene expression and decreases expression of SMC markers (Figure 4A, 4B, and 4D, Figures XVA, XVC, XVG, and XVIA in the Data Supplement). Tunicamycin induces Klf4 expression and increases its transcriptional activity (Figure 4A through 4C, Figure XVG in the Data Supplement), whereas thapsigargin does not affect Klf4 expression but increases its transcriptional activity (Figure 4D and 4E). Cotreatment with ISRIB reverses tunicamycin-induced upregulation of Klf4 levels and activity and macrophage marker expression, again confirming that the Perk-eIF2α-Atf4 axis of UPR is necessary to drive SMCs to a macrophage-like phenotype (Figure 4A through 4C, Figure XVC and XVG in the Data Supplement). Both tunicamycin and thapsigargin successfully induce ER stress, as shown by significant increases in the levels of markers from all 3 pathways (Figures XVB through XVG and XVIB and XVIC in the Data Supplement). Thus, chemical induction of a UPR is sufficient to induce phenotypic switching of SMCs.
Figure 4.
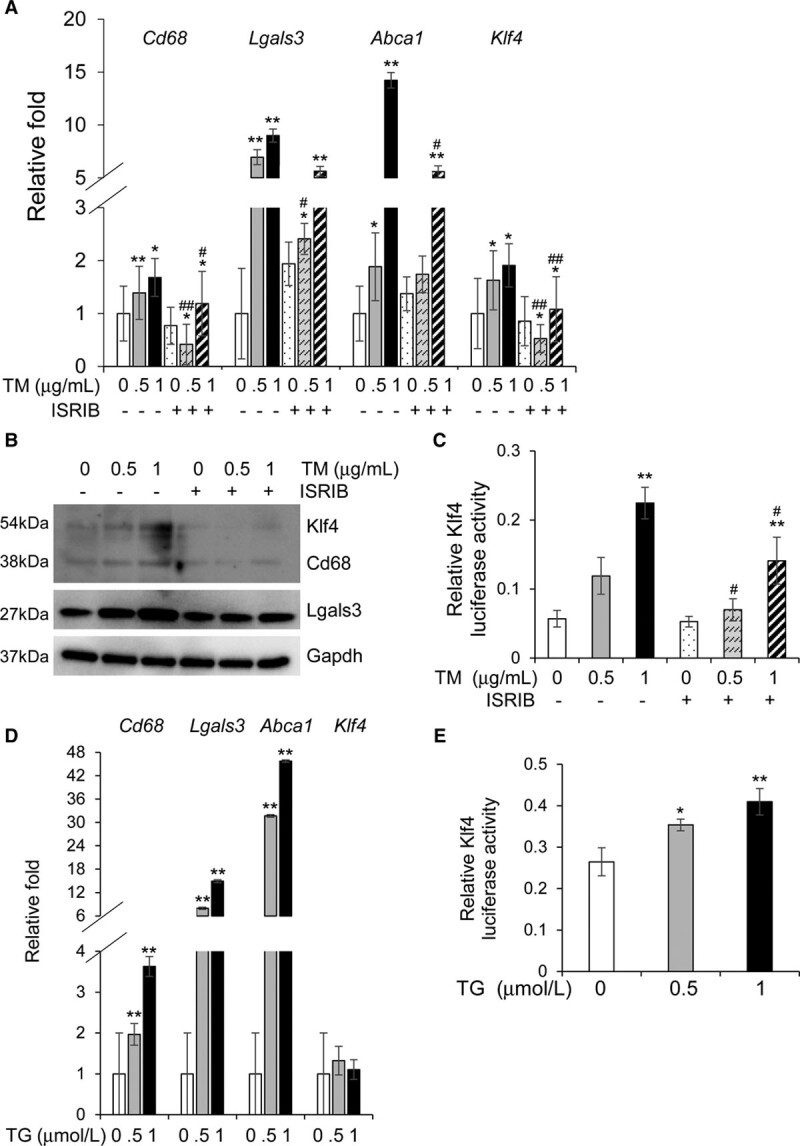
Tunicamycin or thapsigargin treatment induces phenotypic switching in vascular smooth muscle cells (SMCs) independent of cholesterol. A and B, Exposure to 0.5 or 1 µg/mL tunicamycin (TM) for 5 h upregulates macrophage markers and Krüppel-like factor 4 (Klf4), and these increases are successfully reversed by ISRIB (integrated stress response inhibitor) treatment both at the mRNA (A) and protein (B) levels. C, TM treatment increases Klf4 transcriptional activity in a dose-dependent manner and this increase is reversed by ISRIB treatment. D, Treatment with 0.5 or 1µmol/L thapsigargin (TG) for 8 h induces phenotypic switching in SMCs but does not affect Klf4 expression. E, TG treatment causes a dose-dependent increase in the transcriptional activity of Klf4. Each result displayed here is representative of at least 3 independent biological replicates. P values were calculated using 2-way ANOVA followed by Tukey post hoc test. *P<0.05 or **P<0.01 vs no drug treatment (DMSO). #P<0.05 or ##P<0.01 for TM+ISRIB treatment vs TM treatment only. Lgals3 indicates lectin, galactoside-binding, soluble, 3.
Analysis of scRNA-Seq Data Shows Increased UPR in Modulated SMCs
To validate our findings in vivo, we reanalyzed previously published scRNA-Seq data (GSE131780).17 In that article, scRNA-Seq was performed on Apoe−/− mice with an SMC lineage tracer at baseline and at 8 and 16 weeks after initiating an HFD. The resulting clusters of cells are in agreement with the published data (Figure 5A).17 The modulated SMC cluster that appears over time on HFD has decreased expression of contractile markers Acta2 (Figure 5B), Tagln, and Cnn1 (Figure XVIIA and XVIIB in the Data Supplement) when compared with other SMC clusters. Modulated SMCs also show time-dependent increased expression of the macrophage marker Lgals3 but not Cd68 (Figure 5C; Figure XVIIC and XVIID in the Data Supplement). Interestingly, Atf4 (Figure 5D) and Klf4 (Figure 5E) expression also increase in the modulated SMCs compared with other SMC clusters. Thus, these analyses provide preliminary data that UPR is involved in phenotypic switching in vivo to a modulated SMC, expressing both macrophage and fibroblast markers.
Figure 5.
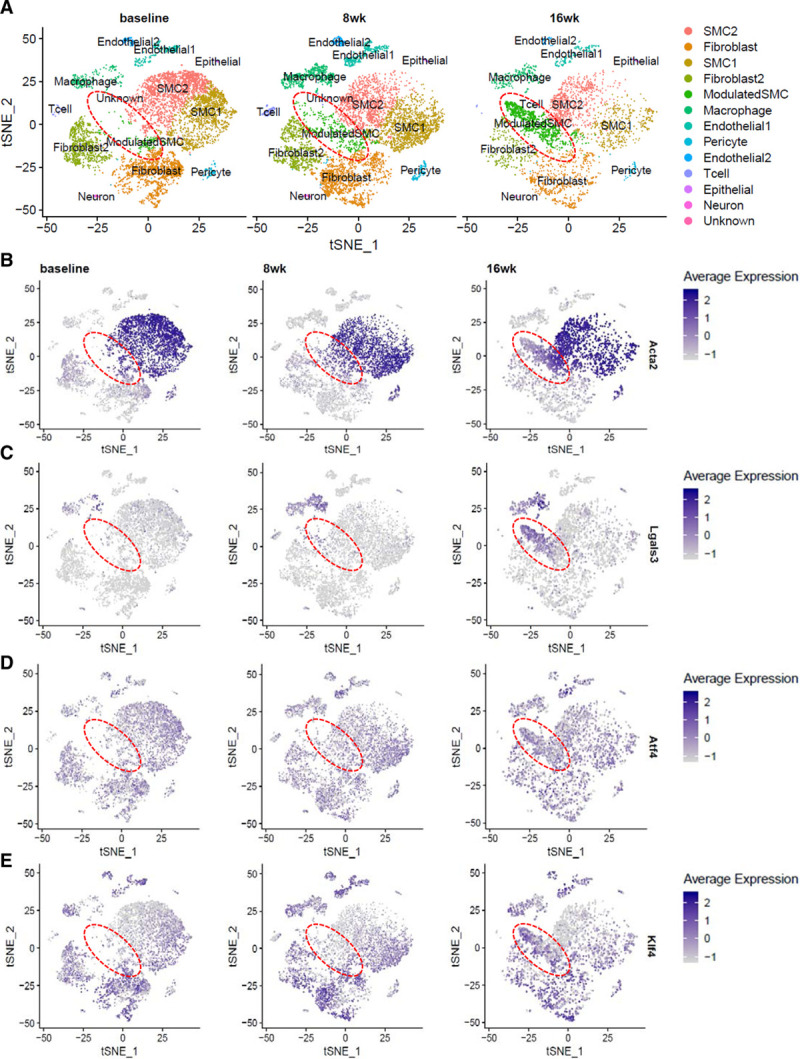
Transcriptomic profiling of mouse aortic root atherosclerotic plaque shows increased unfolded protein response (UPR) in modulated vascular smooth muscle cells (SMCs). A, t-stochastic neighbor embedding (t-SNE) representation of various cell clusters detected in mouse aortic roots at baseline, and after 8 and 16 wk of high-fat diet (HFD) feeding. The disease-specific modulated SMC cluster, as identified by Wirka et al,17 is highlighted by the dotted red circle. B–E, t-SNE visualization of cell clusters at the 3 time points overlaid with expression of various genes shows decreased expression of the contractile marker Acta2 (B) and increased expression of the macrophage marker Lgals3 (C), the UPR effector Atf4 (D), and Klf4 (E), which is known to be responsible for phenotypic switching, in the modulated SMC cluster that appears over time on HFD. The scale on the right of each gene indicates level of expression. Analysis was performed on published single-cell RNA sequencing data (GSE131780) from Wirka et al17 where n=3 mice were used at each time point. Lgals3 indicates lectin, galactoside-binding, soluble, 3.
SMCs Exposed to Cholesterol Upregulate Fibroblast Cell Markers Identified in Modulated SMCs Associated With Atherosclerosis Progression
Based on the scRNA sequencing data of atherosclerotic plaques in the root and ascending aorta of Apoe−/− mice on HFD, phenotypically altered modulated SMCs that appear with plaque formation upregulate Lgals3 but also express fibroblast cell markers.17 Therefore, we assessed whether cholesterol exposure in vitro also upregulates these fibroblast markers. Treatment with 10 µg/mL MBD-Chol for 72 hours upregulates the following fibroblast markers identified in modulated SMCs: Fn1, Ecrg4 (1500015O10Rik), Prg4, Spp1, Lcn2, Timp1, Bgn, and Dcn (Figure 6A) but not Fmod, Tnfrsf11b, and Col1a1 (data not shown). Interestingly, cotreatment with 4-PBA abrogates the MBD-Chol–induced upregulation of all of these genes except Ecrg4, indicating that a UPR is necessary for the upregulation of these markers, similar to its role in upregulating macrophage markers. To confirm if the Perk pathway is responsible for the upregulation of fibroblast markers, we cotreated SMCs with MBD-Chol and ISRIB. Inhibition of the Perk pathway by ISRIB prevents the upregulation of all the fibroblast markers except Ecrg4 (Figure 6B). Additionally, tunicamycin treatment upregulates the same fibroblast markers upregulated by cholesterol (Figure XVIII in the Data Supplement). Taken together, our data indicate that cholesterol exposure in vitro causes a phenotypic switch of SMCs to a dedifferentiated cell, expressing macrophage and fibroblast markers and resembling modulated SMCs detected in atherosclerotic plaques, and that this phenotypic transition is primarily regulated by the Perk arm of the UPR.
Figure 6.

Cholesterol upregulates a subset of fibroblast markers which are driven by unfolded protein response (UPR). A, With exposure to 10 µg/mL methyl-β-cyclodextrin cholesterol (MBD-Chol) for 72 h, expression of a subset of fibroblast marker genes identified in modulated vascular smooth muscle cells (SMCs) is upregulated, and this upregulation is abrogated for all but one of these markers when UPR is inhibited by cotreatment with 5 mmol/L 4-phenylbutyric acid (4-PBA). B, Upregulation of fibroblast markers indicative of modulated SMCs induced by 10 µg/mL MBD-Chol is also prevented by specific inhibition of the Perk (protein kinase RNA-like endoplasmic reticulum kinase) pathway with 200 nmol/L ISRIB (integrated stress response inhibitor). Each result displayed here is representative of at least 3 independent biological replicates. P values were calculated using 2-way ANOVA followed by Tukey Honest Significant Difference post hoc test. *P<0.05 or **P<0.01 vs no cholesterol treatment. For the 4-PBA- or ISRIB-treated samples, #P<0.05 or ##P<0.01 for cholesterol+drug treatment vs no treatment (DMSO).
Discussion
With exposure to free cholesterol, SMCs downregulate their differentiation markers and upregulate macrophage and fibroblast markers to assume a modulated SMC phenotype. The upregulation of macrophage makers is dependent on the transcription factor Klf4, but the exact mechanism for this activation of Klf4 has not previously been determined.12 In this study, we demonstrate that cholesterol exposure activates all 3 pathways of the UPR and induces phenotypic switching primarily via the Perk-eIF2α-Atf4 arm of ER stress. Thus, our data establish a link between cholesterol-induced UPR and phenotypic modulation of SMCs to a macrophage/fibroblast–like cell. The ER is sensitive to minimal changes in membrane cholesterol levels, and inhibiting the movement of free cholesterol from the plasma membrane to the ER prevents phenotypic switching with exposure to MBD-Chol. Furthermore, overall inhibition of ER stress response by treatment with 4-PBA also prevents SMC phenotypic switching with exposure to free cholesterol. Importantly, tunicamycin and thapsigargin exposure are known triggers for UPR, and either treatment leads to phenotypic switching of SMCs. Thus, induction of UPR via chemical agents is sufficient to cause SMC phenotypic switching independent of cholesterol loading. Tunicamycin-induced phenotypic transition is prevented by inhibiting the Perk-eIF2α-Atf4 arm, once again confirming the involvement of this pathway in SMC phenotypic switching.
Analysis of previously published scRNA-Seq data from Apoe−/− SMC lineage traced mice showed increased Atf4 and Klf4 expression in the modulated SMC clusters that appear with exposure to HFD, suggesting that UPR is active in modulated SMCs. However, further studies are needed to confirm the involvement of UPR in regulating SMC fate in atherosclerotic lesions in vivo. These modulated SMCs have significantly decreased expression of contractile markers (eg, Tagln and Cnn1) and upregulation of Lgals3 but not Cd68. The modulated SMCs also increase expression of genes that characterize fibroblasts, specifically Fn1, Tnfsrf11b, and Col1a1, along with small leucine-rich proteoglycans, Lum, Dcn, and Bgn.17 When exposed to cholesterol in culture, SMCs upregulate the majority of the fibroblast marker genes identified in these modulated SMCs, including Fn1, Ecrg4 (1500015O10Rik), Prg4, Spp1, Lcn2, Timp1, Bgn, and Dcn but not Tnfsrf11b, Fmod, Col1a1, and Lum. Interestingly, the MBD-Chol–induced upregulation of all but one of these genes is reduced by both inhibition of general UPR (4-PBA) and specifically the Perk pathway (ISRIB). Therefore, cholesterol-induced UPR in SMCs may contribute to the upregulation of a subset of the fibroblast markers. The appearance of the modulated SMCs in vivo involves the transcription factor Tcf21 (transcription factor 21), but our studies did not detect increased Tcf21 expression in MBD-Chol–treated SMCs (data not shown).17 Therefore, activation of additional pathways, including pathways increasing the expression of Tcf21, are most likely involved in phenotypic modulation of SMCs in vivo with atherosclerotic plaque formation. Similarly, we did not detect increased levels of Sca1 in our cells (data not shown), another factor that has been recently shown to be a marker for SMC phenotypic switching in healthy mouse aorta and atherosclerotic plaques.46
Although ISRIB or shRNA-mediated inhibition of the Perk-eIF2α-Atf4 pathway effectively prevents phenotypic switching with exposure to free cholesterol, blocking Ire1 signaling with Kira6 treatment significantly lowers cholesterol-induced expression of Cd68 and Abca1 but has no effect on Lgals3 and Klf4. Kira6 treatment prevents Ire1α-induced splicing of Xbp1, and spliced Xbp1 (sXbp1) has been shown to upregulate Cd68 during granulocyte differentiation.43 Consistent with our results, sXbp1 has not been shown to control Lgals3 or Klf4 expression. Intriguingly, the fact that Lgals3 and Klf4 remained high with cholesterol exposure and Kira6 treatment, but Cd68 decreased, mimics the observed expression profile of atherosclerosis-related modulated SMCs. It is important to note that the scRNA-Seq data contradicts prior publications the show SMC lineage trace positive cells expressing Cd68, as well as Lgals3, in the atherosclerotic plaques of the Apoe−/− mouse model.12 Immunohistochemical staining of human coronary arteries identified up to 40% of CD68+ cells in plaques as coexpressing smooth muscle α-actin, and CD68+ cells have been identified with an SMC lineage–specific epigenetic mark.12,16 scRNA-Seq data of human coronary artery specimens identified a similar modulated SMC cluster found in mice, but with upregulation of CD68.17 Although the expression and relevance of Cd68 remains unresolved in the field, our data suggest a possible mechanism for the suppression of Cd68 in some models of phenotypic modulation, specifically, activation of the Perk arm of UPR with suppression of Ire1α signaling.
We noted that both general (4-PBA) and each specific UPR inhibition in the absence of cholesterol loading increases expression of SMC differentiation markers. SMC differentiation markers were significantly upregulated when Ire1α was inhibited in another study, and we also found the most significant increase in these markers with Kira6 treatment alone.42 These indicate that minimizing ER stress may contribute to maintaining SMCs in a differentiated, contractile state, and factors increasing ER stress play a role in dedifferentiation of SMCs. A role for specifically Perk signaling in SMC differentiation was previously supported by studies illustrating that inhibition of Perk signaling prevents SMC proliferation and restenosis in the rat angioplasty model of restenosis.47 Conversely, overexpression of Perk exacerbates SMC dedifferentiation and proliferation.
Extensive work has explored how genetic variants altering lipid levels and inflammation contribute to atherosclerosis, but less is known about how genetic alterations affecting SMCs predispose to atherosclerosis. Our data raise the possibility that some genetic syndromes or specific variants associated with coronary artery disease potentially increase the risk for atherosclerosis by triggering UPR and switching of SMCs to macrophage/fibroblast–like cells. In Hutchinson-Gilford progeria syndrome, atherosclerosis is the major cause of mortality.48 The syndrome is due to a mutation in lamin A (LMNA) that deletes 50 amino acids and leads to the production of a truncated protein, termed progerin.49 Using 2 mouse models for Hutchinson-Gilford progeria syndrome (Apoe−/− mice expressing progerin ubiquitously or in an SMC-specific manner), a recent study demonstrated that both ER stress and UPR drive SMC death and atherosclerosis but a role of UPR augmenting phenotypic switching of SMCs to macrophage/fibroblast–like cells was not explored.50,51 Medial SMC loss and atherosclerosis were decreased using the ER stress inhibitor, tauroursodeoxycholic acid, but without addressing the possibility that this drug also decreased SMC phenotypic switching.51 Given these findings and our data, we speculate that SMC phenotypic switching dedifferentiated cell expression macrophage and fibroblast markers in Hutchinson-Gilford progeria syndrome patients and mice contributes to the early atherosclerosis associated with this condition.
ER stress triggering a UPR in macrophages and SMCs has an established role in contributing to atherosclerosis. ER stress markers, like Grp78, Grp94, and Chop, are elevated in SMCs and macrophages in human atherosclerotic plaques.52 Exposure to oxidized LDL and free cholesterol has been shown to induce ER stress and apoptosis in SMCs.27,53 Chop-deficiency protects Ldlr−/− and Apoe−/− mice from atherosclerosis.26 Furthermore, inhibition of ER stress by administering either the chemical chaperone 4-PBA in drinking water or the Perk inhibitor, GSK2606414, has been shown to reduce lesion size in Apoe−/− mice.54,55 However, previous studies focused on ER stress leading to macrophage and SMC cell death as drivers of atherosclerosis. Our data, demonstrating that cholesterol loading of SMCs also drives ER stress-associated phenotypic switching of these cells, identify a novel role for ER stress in plaque pathogenesis. Importantly, blocking SMC phenotypic switching via SMC-specific deletion of Klf4 decreases plaque burden in Apoe−/− mice.12 These data suggest that UPR-driven SMC phenotypic switching promotes atherosclerotic plaque formation and that preventing phenotypic switching via pharmacological inhibition of the UPR in SMCs may be therapeutically beneficial. To determine the role of ER stress-driven SMC phenotypic conversion in plaque burden, UPR-driven alterations in the SMC phenotype need to be distinguished from UPR-driven SMC apoptosis.
SMCs dedifferentiate and increase expression of macrophage and fibroblast markers in vivo and in vitro when exposed to free cholesterol, and this phenotypic transition is dependent on activation of a UPR. Our data add to the growing body of knowledge around SMC behavior in atherosclerosis by determining that activation of the UPR is both necessary and sufficient to induce this phenotypic switching of SMCs in vitro, and may contribute to the formation of modulated SMCs with atherosclerotic plaque formation in vivo.17 These data extend our knowledge of the role of ER stress and UPR in atherosclerotic plaque formation and further emphasize the potential of blocking ER stress in both macrophages and SMCs as a therapeutic target to prevent coronary artery disease.
Acknowledgments
Graphical abstract was created with BioRender.com.
Sources of Funding
This work was supported by the National Heart, Lung and Blood Institute (RO1 HL146583) to D.M. Milewicz and the American Heart Association (AHA18SFRN33960114) to S.A. LeMaire. A. Chattopadhyay is supported by a Victor A. McKusick Postdoctoral Fellowship from The Marfan Foundation; C.S. Kwartler was supported by an American Heart Association Postdoctoral Fellowship (17POST33670040); K. Kaw is supported by Training Interdisciplinary Pharmacology Scientists (TIPS) grant funded by the National Institutes of Health (grant T32GM120011) to The University of Texas Health Science Center at Houston, and A. Kaw by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Numbers TL1TR003169 and UL1TR003167.
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- 4-PBA
- 4-phenylbutyric acid
- Atf
- activating transcription factor
- BiP/Grp78
- binding immunoglobulin protein
- CA7
- ceapin A7
- Chop
- C/EBP homologous protein
- Cnn1
- calponin
- Edem
- endoplasmic reticulum degradation enhancing α-mannosidase like protein
- eIF2α
- α-subunit of the eukaryotic elongation factor 2
- Grp94
- heat shock protein 90 kDa beta member 1
- HFD
- high-fat diet
- hTERT
- human telomerase reverse transcriptase
- Ire
- inositol-requiring enzyme
- ISRIB
- integrated stress response inhibitor
- Klf4
- Krüppel-like factor 4
- LDL
- low-density lipoprotein
- MBD-Chol
- methyl-β-cyclodextrin cholesterol
- Myh11
- smooth muscle myosin heavy chain
- Perk
- protein kinase RNA-like endoplasmic reticulum kinase
- POVPC
- 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine
- scRNA-Seq
- single-cell RNA sequencing
- shRNA
- short hairpin RNA
- SMC
- vascular smooth muscle cell
- Tagln
- transgelin, SM-22α
- UPR
- unfolded protein response
- Xbp1
- X-box binding protein 1
For Sources of Funding and Disclosures, see page 315.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315164.
Contributor Information
Abhijnan Chattopadhyay, Email: Abhijnan.Chattopadhyay@uth.tmc.edu.
Callie S. Kwartler, Email: Callie.S.Kwartler@uth.tmc.edu.
Kaveeta Kaw, Email: Anita.Kaw@uth.tmc.edu.
Yanming Li, Email: yanming.li@bcm.edu.
Anita Kaw, Email: Anita.Kaw@uth.tmc.edu.
Jiyuan Chen, Email: Jiyuan.Chen@uth.tmc.edu.
Scott A. LeMaire, Email: slemaire@bcm.edu.
Ying H. Shen, Email: hyshen@bcm.edu.
References
- 1.Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem. 1984;259:14383–14388 [PubMed] [Google Scholar]
- 2.Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K. Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol. 2007;27:1159–1165. doi: 10.1161/ATVBAHA.106.134080 [DOI] [PubMed] [Google Scholar]
- 3.Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res. 2008;79:14–23. doi: 10.1093/cvr/cvn099 [DOI] [PubMed] [Google Scholar]
- 4.Dubland JA, Francis GA. So Much Cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol. 2016;27:155–161. doi: 10.1097/MOL.0000000000000279 [DOI] [PubMed] [Google Scholar]
- 5.Ball RY, Stowers EC, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 1995;114:45–54. doi: 10.1016/0021-9150(94)05463-s [DOI] [PubMed] [Google Scholar]
- 6.Mietus-Snyder M, Gowri MS, Pitas RE. Class A scavenger receptor up-regulation in smooth muscle cells by oxidized low density lipoprotein. Enhancement by calcium flux and concurrent cyclooxygenase-2 up-regulation. J Biol Chem. 2000;275:17661–17670. doi: 10.1074/jbc.275.23.17661 [DOI] [PubMed] [Google Scholar]
- 7.Frontini MJ, O’Neil C, Sawyez C, Chan BM, Huff MW, Pickering JG. Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ Res. 2009;104:832–841. doi: 10.1161/CIRCRESAHA.108.187302 [DOI] [PubMed] [Google Scholar]
- 8.Choi HY, Rahmani M, Wong BW, Allahverdian S, McManus BM, Pickering JG, Chan T, Francis GA. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation. 2009;119:3223–3231. doi: 10.1161/CIRCULATIONAHA.108.841130 [DOI] [PubMed] [Google Scholar]
- 9.Song L, Lee C, Schindler C. Deletion of the murine scavenger receptor CD68. J Lipid Res. 2011;52:1542–1550. doi: 10.1194/jlr.M015412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133 [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, Sin DD, Seidman MA, Leeper NJ, Francis GA. Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:876–887. doi: 10.1161/ATVBAHA.119.312434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. doi: 10.1161/CIRCRESAHA.107.152736 [DOI] [PubMed] [Google Scholar]
- 16.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015 [DOI] [PubMed] [Google Scholar]
- 17.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25:1280–1289. doi: 10.1038/s41591-019-0512-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0 [DOI] [PubMed] [Google Scholar]
- 19.Plaisance V, Brajkovic S, Tenenbaum M, Favre D, Ezanno H, Bonnefond A, Bonner C, Gmyr V, Kerr-Conte J, Gauthier BR, et al. Endoplasmic reticulum stress links oxidative stress to impaired pancreatic beta-cell function caused by human oxidized LDL. PLoS One. 2016;11:e0163046 doi: 10.1371/journal.pone.0163046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 21.Jiang P, Gan M, Lin WL, Yen SH. Nutrient deprivation induces α-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front Aging Neurosci. 2014;6:268 doi: 10.3389/fnagi.2014.00268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- 24.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035 [DOI] [PubMed] [Google Scholar]
- 26.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larroque-Cardoso P, Swiader A, Ingueneau C, Nègre-Salvayre A, Elbaz M, Reyland ME, Salvayre R, Vindis C. Role of protein kinase C δ in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. 2013;4:e520 doi: 10.1038/cddis.2013.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan MI, Pichna BA, Shi Y, Bowes AJ, Werstuck GH. Evidence supporting a role for endoplasmic reticulum stress in the development of atherosclerosis in a hyperglycaemic mouse model. Antioxid Redox Signal. 2009;11:2289–2298. doi: 10.1089/ars.2009.2569 [DOI] [PubMed] [Google Scholar]
- 29.Zhou J, Werstuck GH, Lhoták S, de Koning AB, Sood SK, Hossain GS, Møller J, Ritskes-Hoitinga M, Falk E, Dayal S, et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–213. doi: 10.1161/01.CIR.0000134487.51510.97 [DOI] [PubMed] [Google Scholar]
- 30.Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A. 2006;103:12271–12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romanoski CE, Che N, Yin F, Mai N, Pouldar D, Civelek M, Pan C, Lee S, Vakili L, Yang WP, et al. Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ Res. 2011;109:e27–e41. doi: 10.1161/CIRCRESAHA.111.241869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou AX, Wang X, Lin CS, Han J, Yong J, Nadolski MJ, Borén J, Kaufman RJ, Tabas I. C/EBP-Homologous protein (CHOP) in vascular smooth muscle cells regulates their proliferation in aortic explants and atherosclerotic lesions. Circ Res. 2015;116:1736–1743. doi: 10.1161/CIRCRESAHA.116.305602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwartler CS, Zhou P, Kuang SQ, Duan XY, Gong L, Milewicz DM. Vascular smooth muscle cell isolation and culture from mouse aorta. Bio-Protocol. 2016;6:e2045 [Google Scholar]
- 35.Grosheva I, Haka AS, Qin C, Pierini LM, Maxfield FR. Aggregated LDL in contact with macrophages induces local increases in free cholesterol levels that regulate local actin polymerization. Arterioscler Thromb Vasc Biol. 2009;29:1615–1621. doi: 10.1161/ATVBAHA.109.191882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmann T, Klucken J, Reil M, Liebisch G, Luciani MF, Chimini G, Kaminski WE, Schmitz G. Molecular cloning of the human ATP-binding cassette transporter 1 (hABC1): evidence for sterol-dependent regulation in macrophages. Biochem Biophys Res Commun. 1999;257:29–33. doi: 10.1006/bbrc.1999.0406 [DOI] [PubMed] [Google Scholar]
- 37.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419 doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 38.Underwood KW, Andemariam B, McWilliams GL, Liscum L. Quantitative analysis of hydrophobic amine inhibition of intracellular cholesterol transport. J Lipid Res. 1996;37:1556–1568 [PubMed] [Google Scholar]
- 39.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013;2:e00498 doi: 10.7554/eLife.00498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallagher CM, Walter P. Ceapins inhibit ATF6α signaling by selectively preventing transport of Atf6α to the Golgi apparatus during ER stress. eLife. 2016;5:e11880 10.7554/eLife.11880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548. doi: 10.1016/j.cell.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu B, Daoud F, Zeng S, Matic L, Hedin U, Uvelius B, Rippe C, Albinsson S, Sward K. Antagonistic relationship between the unfolded protein response and myocardin-driven transcription in smooth muscle. J Cell Physiol. 2020;235:7370–7382. doi: 10.1002/jcp.29637 [DOI] [PubMed] [Google Scholar]
- 43.Kurata M, Yamazaki Y, Kanno Y, Ishibashi S, Takahara T, Kitagawa M, Nakamura T. Anti-apoptotic function of Xbp1 as an IL-3 signaling molecule in hematopoietic cells. Cell Death Dis. 2011;2:e118 doi: 10.1038/cddis.2011.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. 1979;18:2186–2192. doi: 10.1021/bi00578a008 [DOI] [PubMed] [Google Scholar]
- 45.Rogers TB, Inesi G, Wade R, Lederer WJ. Use of thapsigargin to study Ca2+ homeostasis in cardiac cells. Biosci Rep. 1995;15:341–349. doi: 10.1007/BF01788366 [DOI] [PubMed] [Google Scholar]
- 46.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, Jørgensen HF. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun. 2018;9:4567 doi: 10.1038/s41467-018-06891-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B, Zhang M, Urabe G, Huang Y, Chen G, Wheeler D, Dornbos DJ, Huttinger A, Nimjee S, Gong S, et al. PERK inhibition mitigates restenosis and thrombosis - a potential low-thrombogenic anti-restenotic paradigm. JACC Basic Transl. Sci. 2020;19:245–263. doi: 10.1016/j.jacbts.2019.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulrich NJ, Gordon LB. Hutchinson-Gilford progeria syndrome Handb. Clin. Neurol. 2015;1332:249–264. doi: 10.1016/B978-0-444-62702-5.00018-4 [DOI] [PubMed] [Google Scholar]
- 49.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055 doi: 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- 50.Hamczyk MR, Villa-Bellosta R, Gonzalo P, Andrés-Manzano MJ, Nogales P, Bentzon JF, López-Otín C, Andrés V. Vascular Smooth Muscle-Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation. 2018;138:266–282. doi: 10.1161/CIRCULATIONAHA.117.030856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamczyk MR, Villa-Bellosta R, Quesada V, Gonzalo P, Vidak S, Nevado RM, Andres-Manzano MJ, Misteli T, Lopez-Otin C, Andres V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol Med. 2019;11:e9736. 10.15252/emmm.201809736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054 [DOI] [PubMed] [Google Scholar]
- 53.Kedi X, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis. 2009;207:123–130. doi: 10.1016/j.atherosclerosis.2009.04.019 [DOI] [PubMed] [Google Scholar]
- 54.Lynn EG, Lhoták Š, Lebeau P, Byun JH, Chen J, Platko K, Shi C, O’Brien ER, Austin RC. 4-Phenylbutyrate protects against atherosclerotic lesion growth by increasing the expression of HSP25 in macrophages and in the circulation of Apoe−/− mice. FASEB J. 2019;33:8406–8422. doi: 10.1096/fj.201802293RR [DOI] [PubMed] [Google Scholar]
- 55.Onat UI, Yildirim AD, Tufanli Ö, Çimen I, Kocatürk B, Veli Z, Hamid SM, Shimada K, Chen S, Sin J, et al. Intercepting the lipid-Induced integrated stress response reduces atherosclerosis. J Am Coll Cardiol. 2019;73:1149–1169. doi: 10.1016/j.jacc.2018.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and in the Data Supplement.