

Abstract
Objective:
Inhibition of adenosine kinase (ADK), via augmenting endogenous adenosine levels exerts cardiovascular protection. We tested the hypothesis that ADK inhibition improves microvascular dilator and left ventricle (LV) contractile function under metabolic or hemodynamic stress.
Methods and Results:
In Obese diabetic Zucker fatty/spontaneously hypertensive heart failure F1 hybrid rats, treatment with the selective ADK inhibitor, ABT-702 (1.5 mg/kg, intraperitoneal injections for 8-week) restored acetylcholine-, sodium nitroprusside-, and adenosine-induced dilations in isolated coronary arterioles, an effect that was accompanied by normalized end-diastolic pressure (in mm Hg, Lean: 3.4 ± 0.6, Obese: 17.6 ± 4.2, Obese + ABT: 6.6 ± 1.4) and LV relaxation constant, Tau (in ms, Lean: 6.9 ± 1.5, Obese: 13.9 ± 1.7, Obese + ABT: 6.0 ± 1.1). Mice with vascular endothelium selective ADK deletion (ADKVECKO) exhibited an enhanced dilation to acetylcholine in isolated gracilis muscle (lgEC50 WT: −8.2 ± 0.1, ADKVECKO: −8.8 ± 0.1, P < .05) and mesenteric arterioles (lgEC50 WT: −7.4 ± 0.2, ADKVECKO: −8.1 ± 1.2, P < .05) when compared to wild-type (WT) mice, whereas relaxation of the femoral artery and aorta (lgEC50 WT: −7.03 ± 0.6, ADKVECKO: −7.05 ± 0.8) was similar in the two groups. Wild-type mice progressively developed LV systolic and diastolic dysfunction when they underwent transverse aortic constriction surgery, whereas ADKVEC-KO mice displayed a lesser degree in decline of LV function.
Conclusions:
Our results indicate that ADK inhibition selectively enhances microvascular vasodilator function, whereby it improves LV perfusion and LV contractile function under metabolic and hemodynamic stress.
Keywords: adenosine-kinase, coronary microcirculation, heart failure with preserved ejection fraction
PERSPECTIVE
Coronary microvascular dysfunction contributes to the development of left ventricle diastolic dysfunction in patients with heart failure with preserved ejection fraction (HFpEF), although the mechanistic interrelationship remains elusive. This study demonstrates that pharmacological or endothelium selective inhibition of adenosine kinase selectively augments microvascular vasodilator function and improves left ventricle diastolic function in rodent models. Likely via a more efficient coupling of the microcirculation with metabolism adenosine kinase inhibition may provide therapeutic benefit to HFpEF patients.
1 |. INTRODUCTION
Recent clinical observations revealed that abnormalities in the coronary microcirculation are often accompanied by the development of LV diastolic dysfunction in patients with HFpEF.1–3 However, the mechanistic interrelationship between coronary microvascular dysfunction and LV diastolic dysfunction remains elusive, and hence microvascular targeted pharmacological treatment options are lacking in HFpEF.
It is known that the microcirculation plays a vital role in matching tissue perfusion with the metabolic demand to maintain the integrity of organ function.4 Alterations in coronary microvascular vasodilator function could significantly limit the blood supply to the working myocardium resulting in LV contractile dysfunction.5,6 One important mechanism to protect the heart from ischemic episodes is the augmented release of adenosine from cardiomyocytes.7 In response to ischemia adenosine is released from cardiomyocytes through nucleoside transporters to facilitate coronary artery dilation, thereby mitigating myocardial ischemia and maintaining heart pump function.8–10 Thus, an ischemic and failing heart is expected to occur with progressively increased adenosine levels. Interestingly, an early study by Meyer et al11 found that adenosine production was elevated only in the initial, compensated phase of pressure-overloaded rat hearts, but myocardial adenosine levels decreased during the phase of cardiac decompensation. Laghi-Pasini et al12 found that following administration of dipyridamole, an adenosine reuptake inhibitor, patients with idiopathic dilated cardiomyopathy actually had lower plasma adenosine levels than controls. Furthermore, an early study by Funakoshi et al13 reported that mice with LV dysfunction, which developed secondary to chronic pressure overload or after genetic overexpression of calsequestrin or tumor necrosis factor, had reduced cardiac adenosine levels by 70%. These aforementioned studies raised the possibility that the protective cardiovascular effects of adenosine become mitigated in the failing heart, but the molecular mechanism behind this phenomenon remains unknown.
It is known that adenosine is rapidly metabolized to adenosine monophosphate by ADK, which is one of the main regulator of tissue adenosine levels.14 Under hypoxia, ADK activity is rapidly reduced, which facilitates the production and interstitial release of adenosine.15 Accordingly, a previous study has shown that selective pharmacological inhibition of ADK could provide beneficial, protective effects on the myocardium.16 Interestingly, our recent study revealed that ADK expression is upregulated in the myocardium and coronary microvessels in HFpEF patients.17 We also found that pharmacological inhibition of ADK prevented the development of LV diastolic dysfunction in a rodent HFpEF model.17 However, it remained unclear if ADK inhibition acts primarily via coronary microvascular mechanisms, and if this could contribute to the improved LV perfusion and LV contractile function. Therefore, in this study we set out to test the hypothesis that pharmacologic or vascular selective genetic inhibition of ADK augments microvascular dilator function and thereby provides protection of LV contractile function under metabolic and hemodynamic stress.
2 |. MATERIALS AND METHODS
2.1 |. Obese ZSF1 rat model of LV diastolic dysfunction
The Institutional Animal Care and Use Committee at Medical College of Georgia approved all animal protocols described in this study, and the protocols were in compliance with the ARRIVE guidelines. Male obese diabetic ZSF1 hybrid rats (n = 12) rats and ZSF1 lean control rats (n = 8) were obtained from Charles River (Wilmington, MA, USA) and housed under a controlled environment (21–23°C room temperature; 12:12-h light-dark cycle) with free access to food (standard rat chow) and water.18 Twenty-four weeks old male obese ZSF1 rats (n = 4) received an IP injection of the adenosine kinase inhibitor 4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2, 3-d]pyrimidine (ABT-702, 1.5 mg/kg, i.p, TOCRIS) or vehicle (n = 4, 2.8% DMSO in saline) two times a week for eight weeks. The dose and mode of administration of ABT-702 were selected based on previous studies.19,20 Using invasive catheter-based approach LV contractility was assessed in anesthetized (1.5% isoflurane) rats. SPR-838 micro-tip pressure-volume catheter (Millar Instruments) was inserted into the LV through the right carotid artery and the heart rate, stroke volume, LV ESP, EDP, dP/dtmax, dP/dtmin, and isovolumetric relaxation time constant (Tau) were obtained (Powerlab, LabChart, ADInstruments).
2.2 |. Transverse aortic constriction in mice with endothelial-specific deletion of ADK
We employed mice with an endothelial-specific deletion of ADK, as we have established and reported before.21,22 In brief, deletion of ADK in endothelial cells (ADKVEC-KO) was achieved by crossbreeding Cdh5-Cre transgenic mice (The Jackson Laboratory) with ADKflox/flox mice (Xenogen Biosciences Corporation, Cranbury, NJ, USA) on a C57BL/6 background. In all experiments, littermates, both males and females from the same breeding pair were used as WT controls. Wild-type and ADKVEC-KO mice were subjected to TAC as previously described23 and also implemented by us.24 In brief, mice were randomly assigned to have sham or TAC surgery, which were performed under anesthesia with 1%−2% isoflurane. Partial thoracotomy was performed at the 2nd-3rd intercostal space along the midsternal line, the aorta was isolated and a 6.0 silk suture was applied against a 27-gauge needle between the innominate and left carotid artery. The needle was removed after ligation, and the chest and overlying skin were closed.
2.3 |. Echocardiographic assessment of LV function in mice
Echocardiography (VEVO 2100 digital ultrasound microimaging system, VisualSonics) was used to measure LV dimensions, including anterior and PW thickness during diastole and systole, LV volumes and ejection fraction at the parasternal short-axis view in anesthetized (1%−2% inhaled isoflurane) mice. To evaluate diastolic function, the pulsed-wave Doppler was positioned above the mitral leaflets to measure mitral flow velocity tracings (peak velocity of early (E), late (A) mitral inflow, and DT of early filling of mitral inflow).
2.4 |. Assessment of vasomotor function in isolated arteries
Using previously established methods,25–27 rat coronary arterioles, and the first-order branch of the femoral artery, as well as mouse skeletal (gracilis) muscle, mesenteric arterioles and femoral arteries were dissected, cannulated, and pressurized (70 mm Hg) in a vessel chamber (CH-1-LIN, Living Systems Instrumentation) containing two glass micropipettes filled with Krebs solution composed of (in mmol/L): 110 NaCl, 5.0 KCl, 1.25 CaCl2, 1.0 MgSO4, 1.0 KH2PO4, 5.0 glucose and 24.0 NaHCO3 equilibrated with a gas mixture of 21% O2 and 5% CO2, at pH 7.4 with a maintained temperature of 37°C. Arteries were pressurized using a pressure servo control system (Living Systems Instrumentation). Using an inverted microscope (Olympus IX70), changes in internal arteriole diameter were measured with a videocaliper and recorded (Powerlab, LabChart, ADInstruments). Diameter changes were measured in response to cumulative concentrations of endothelium-dependent agonist, ACh, the NO donor, SNP, or to adenosine. Arteries either developed a spontaneous tone (~30%) or were preconstricted with U46619, a thromboxane A2 receptor agonist to achieve ~30% vascular tone. In separate protocols mouse aorta sections (2 mm in length) were mounted on wire myograph to measure isometric force generation. The aorta was preconstricted with phenylephrine (10−6 mol/L), and relaxation was assessed in response to ACh and SNP.
2.5 |. Histology and immunofluorescence
Heart samples were fixed in 4% paraformaldehyde and embedded in paraffin. Sections were cut (8 μm) and immunolabeled with commercially available rabbit polyclonal anti-ADK (Abcam, #ab38010, 1:100). Subsequent fluorescent labeling was performed with Cy5-conjugated secondary antibody (Jackson-Immunoresearch, 1:250). 4′,6-Diamidino-2-phenylindole dihydrochloride was used for nuclear staining. Structured illumination microscopy (SIM-Apotome, AxioImagerM2, CarlZeiss) was used for immunofluorescent detection. Heart sections were also stained with Masson’s Trichrome and myocardial surface area was measured with Image J in a blinded fashion.
2.6 |. Western immunoblot
Heart samples were homogenized in radio-immunoprecipitation assay buffer (20 μL) and were mixed with 4× Laemmli sample buffer, then loaded for gel electrophoresis. After transfer to PVDF membrane and blocking, membranes were incubated with anti-ADK antibody (Abcam, #ab38010, 1:1000) followed by horseradish peroxidase-labeled secondary antibody. Adenosine kinase expression was normalized for the total loaded protein using a BIO-RAD TGX Stain-Free FastCast Acrylamide Kit (Cat. #1610183), ChemiDoc imaging system and Image Lab Software (BIO-RAD).
2.7 |. Statistical analyses
Data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism software by one-way or two-way ANOVA, as appropriate, followed by multiple comparisons with Tukey post hoc test. P < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. Adenosine kinase inhibitor, ABT-702 improves LV diastolic dysfunction in obese ZSF1 rats
As others18 and we17 have shown previously, male obese ZSF1 rats develop LV diastolic dysfunction while preserving systolic function by 18–20 weeks of age. In this study, using invasive LV pressure measurements we confirmed the presence of LV diastolic dysfunction by showing a significantly increased EDP and LV relaxation time constant, Tau in obese ZSF1 rats, when compared to lean controls (Figure 1A,B). These changes in the obese ZSF1 rats were accompanied by reduced stroke volume and cardiac output (Figure 1E,F). Eight-week treatment with the ADK inhibitor, ABT-702 significantly reduced EDP and Tau, when compared to vehicle-treated obese ZSF1 rats (Figure 1A,B). ABT-702 treatment did not significantly affect blood pressure (in mm Hg: Lean ZSF1: 148 ± 2; Obese ZSF1: 150 ± 7; Obese ZSF1 + ABT-702:143 ± 2), heart rate and indices of LV contractility (±dP/dt) (Figure 1D,G,H) of obese ZSF1 rats. We also confirmed that obese ZSF1 rats develop cardiomyocyte hypertrophy, but found that 8-week ABT-702 treatment had no significant effects on the enlarged cardiomyocyte size (Figure 1I).
FIGURE 1.
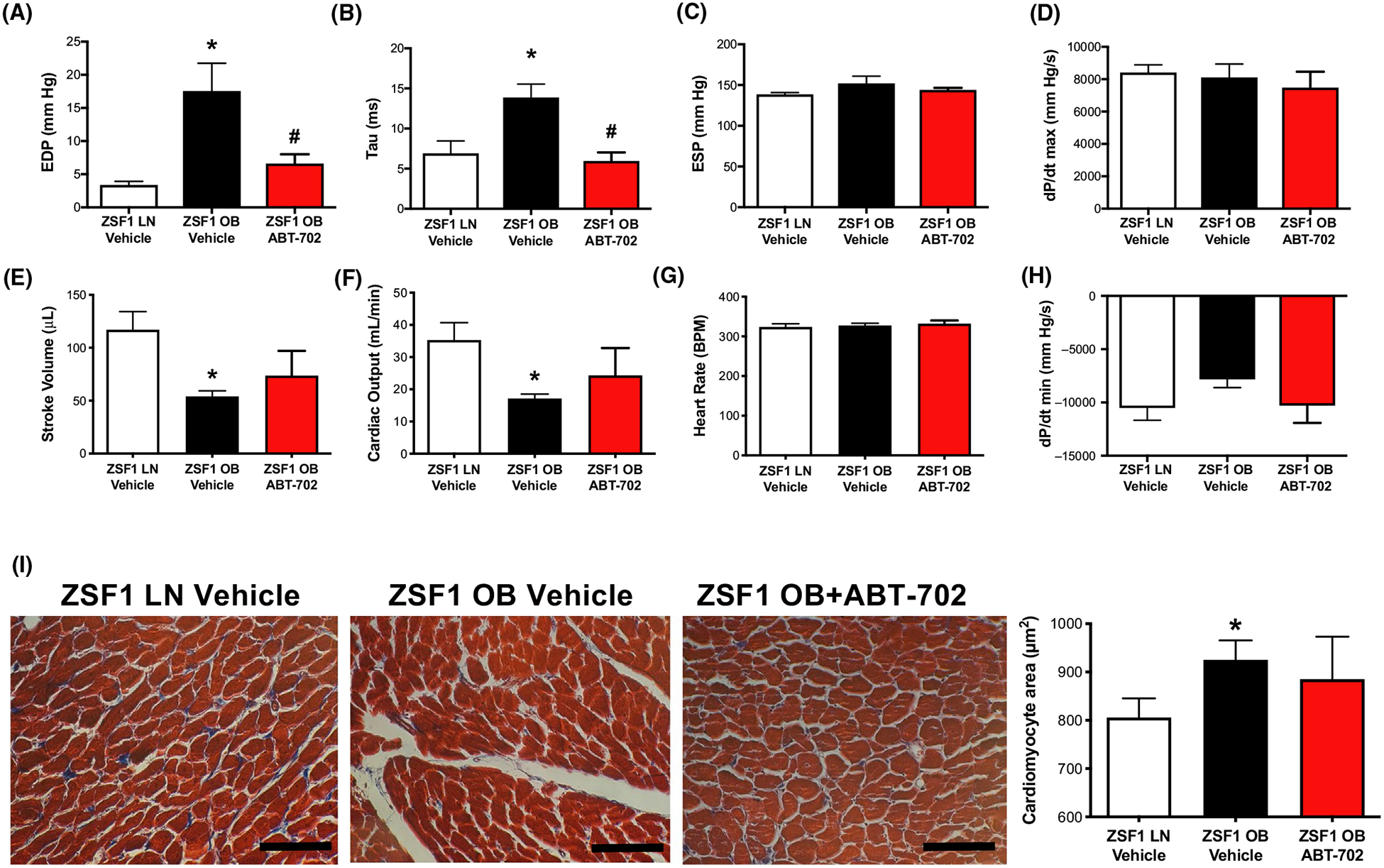
Effect of ABT-702 treatment on LV function and morphology in obese ZSF1 rats. Panels A-H, Summary data of parameters obtained during LV pressure measurement in lean (LN) and obese (OB) ZSF1 rats treated with ABT-702 or vehicle (n = 4 in each group). Panel I, Representative Masson’s Trichrome images and summary data of quantification of myocardial surface area in LV sections in lean and in obese ZSF1 rats with or without ABT-702 treatment (n = 4 in each group). Bar = 100 μm. *P < .05 lean vs obese ZSF1 rats. #P < .05 between obese vehicle and obese ABT-702 treated rats
3.2 |. ABT-702 augments coronary arteriole dilation in obese ZSF1 rats
We next tested the hypothesis that ADK inhibition improves vasodilator function of coronary arterioles in obese ZSF1 rats. We found that ADK protein expression was significantly increased in the heart of obese ZSF1 rats, when compared to lean controls (Figure 2A). Similar to what we previously found in skeletal muscle arterioles17 an abundant ADK expression was detected in coronary arterioles of obese ZSF1 rats (Figure 2B,C). In isolated and pressurized coronary arterioles (baseline diameters; lean ZSF1: 119 ± 12 μm vs. obese ZSF1: 127 ± 9 μm, spontaneously developed or after preconstriction) of obese ZSF1 rats we found that the magnitude of endothelium-dependent dilation in response to ACh was decreased (Figure 2D). In addition, coronary arterioles from the obese ZSF1 rats displayed a significantly reduced vasodilation in response to the direct NO donor, SNP and also to adenosine when compared to lean control ZSF1 rats (Figure 2E,F). Coronary arteriole from the ABT-702-treated obese ZSF1 rats exhibited a significantly augmented vasodilation to ACh, SNP, and adenosine, when compared to vehicle-treated obese ZSF1 rats (Figure 2D–F). We found no significant changes in agonist-induced vasodilation in femoral arteries (baseline diameter; lean ZSF1: 177 ± 12 μm vs. obese ZSF1: 157 ± 4 μm, after preconstriction) of obese ZSF1 rats, with or without ABT-702 treatment (Figure 2G–I).
FIGURE 2.
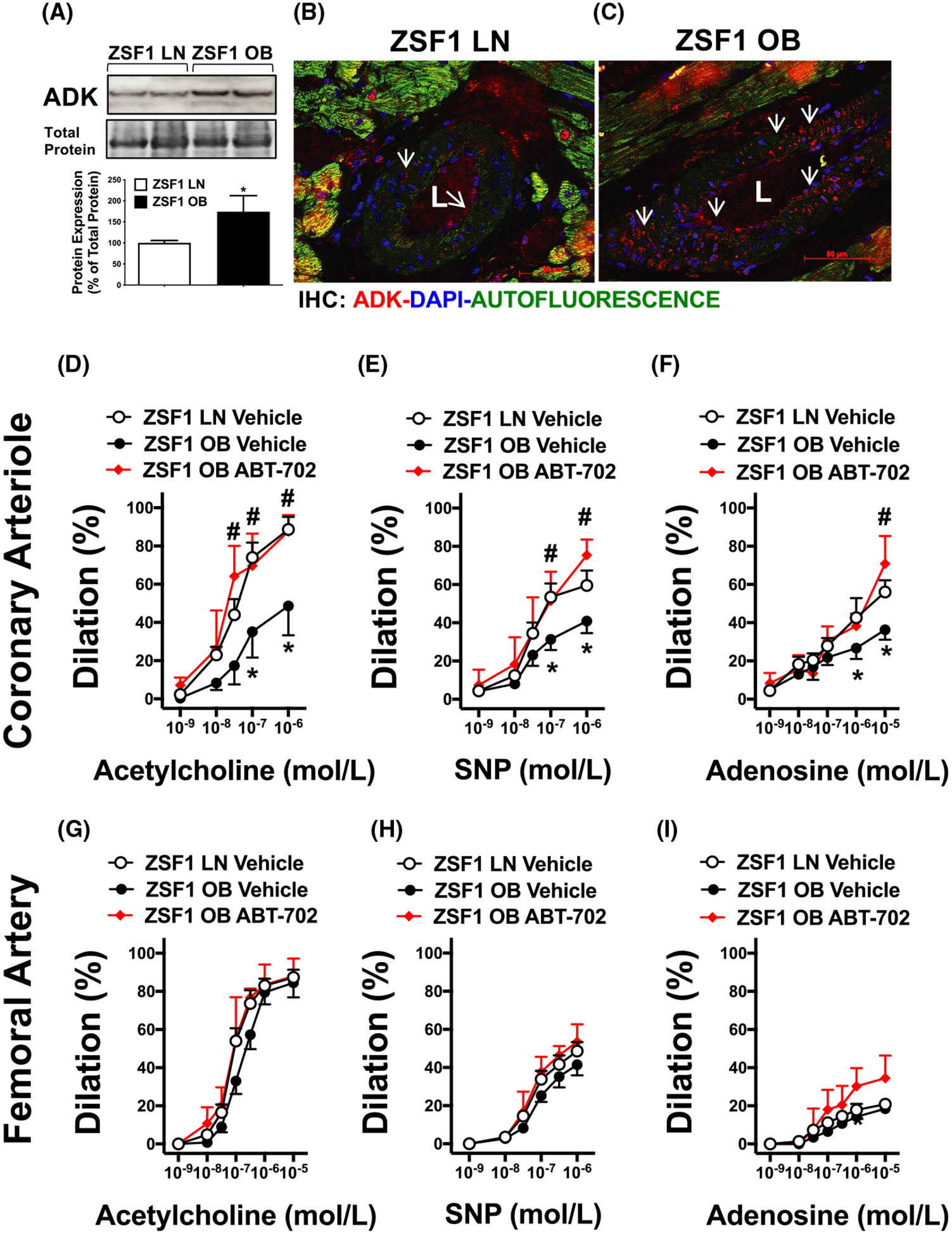
Effect of ABT-702 treatment on coronary arteriole dilation in obese ZSF1 rats. Panel A, Western immunoblot and summary data (n = 4 in each group) of ADK levels in lean (LN) and obese (OB) ZSF1 rats. Panels B&C, Representative immunofluorescence images of coronary arterioles in lean and obese ZSF1 rats (ADK in red:, 4′,6-Diamidino-2-phenylindole dihydrochloride in Blue: Green is autofluorescence). Summary data show coronary arteriole (Panels D-F) and femoral artery (Panels G-I): dilations in response to cumulative concentrations of acetylcholine (1 nmol/L-1 μmol/L, or 1 nmol/L-10 μmol/L in the femoral artery), sodium-nitroprusside (1 nmol/L-1 μmol/L) and adenosine (1 nmol/L-10 μmol/L) in lean and obese ZSF1 rats with and without in vivo ABT-702 treatments (n = 4 in each group). *P < .05 lean vs obese ZSF1 rats. #P < .05 between obese vehicle and obese ABT-702 treated rats
3.3 |. Endothelium-selective deletion of ADK augments dilation selectively in microvessels
Next, we examined to what extent the effect of ADK inhibition can be attributed to direct vascular mechanisms. To that end, we employed endothelium-specific ADKVECKO.21,22 We found that in isolated skeletal (gracilis) muscle arterioles there was no significant difference in the baseline diameter (ADKVECKO: 97 ± 14 μm vs. WT: 75 ± 4 μm, spontaneously developed) and calculated myogenic tone (ADKVECKO: 39 ± 7% vs. WT: 41 ± 6%) between ADKVECKO and WT mice. We found that dilation in response to ACh was significantly enhanced in gracilis muscle arterioles of ADKVECKO mice when compared to WT mice (lgEC50 WT: −8.2 ± 0.1, ADKVECKO: −8.8 ± 0.1, P < .05) (Figure 3A). While mesenteric arterioles (baseline diameter; ADKVECKO: 115 ± 11 μm vs. WT: 125 ± 15 μm, after preconstriction) from ADKVECKO mice also exhibited enhanced dilations to ACh (lgEC50 WT: −7.4 ± 0.2, ADKVECKO: −8.1 ± 1.2, P < .05) (Figure 3B), we found no significant changes in vasodilation to ACh in the femoral artery (baseline diameter; ADKVECKO: 187 ± 19 vs. WT: 168 ± 14 μm, after preconstriction), (lgEC50 WT: −7.5 ± 0.2, ADKVECKO: −7.6 ± 0.3) or the aorta (lgEC50 WT: −7.03 ± 0.6, ADKVECKO: −7.05 ± 0.8) in ADKVECKO and WT mice (Figure 3C,D). Vasodilation to the direct NO donor, SNP was similar between ADKVECKO and WT mice in all types of blood vessels (Figure 3E–H).
FIGURE 3.
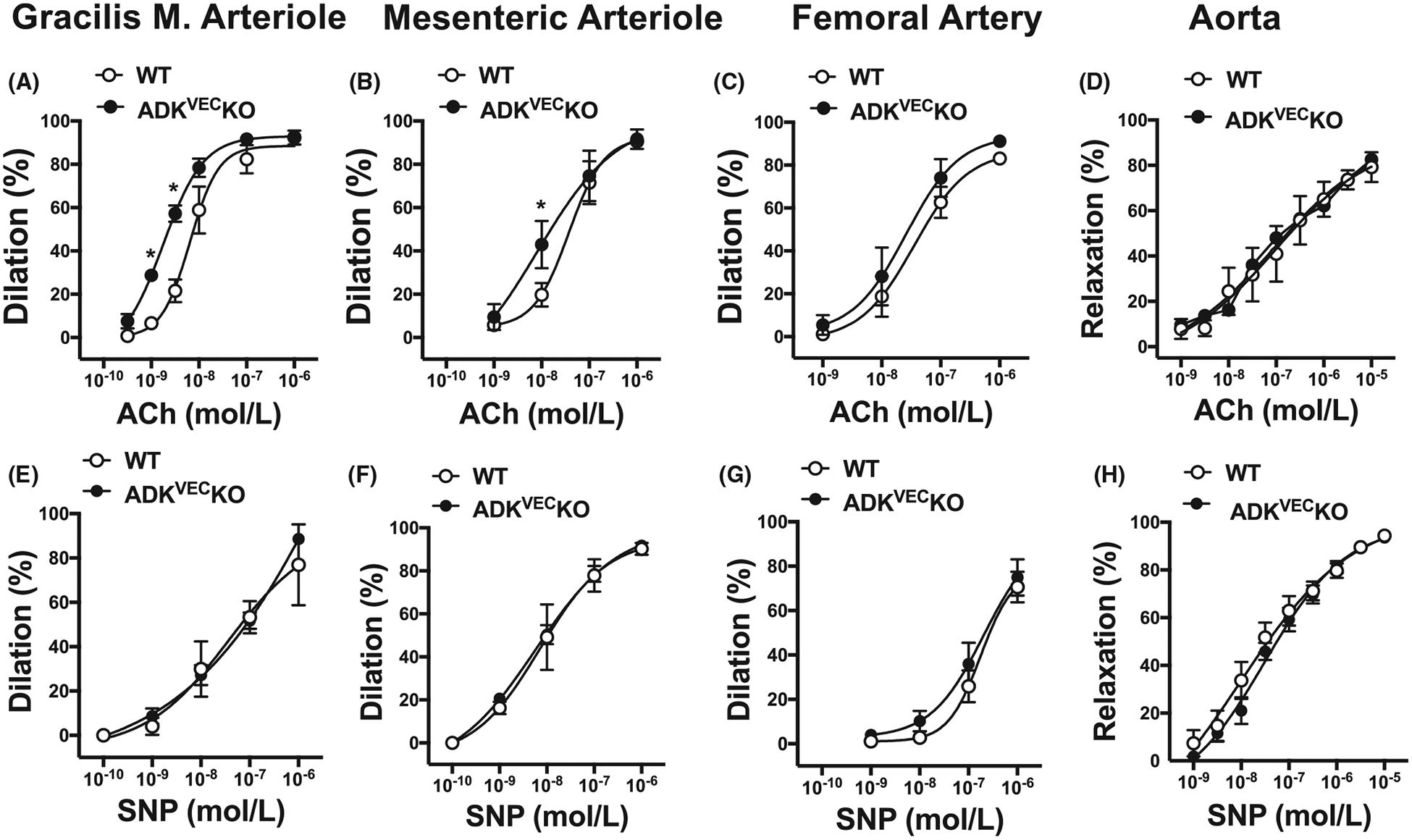
Vasodilator responses in microvessels vs conduit arteries in WT and ADKVEC-KO mice. Summary data show vasodilations in response to cumulative concentrations of ACh (0.3 nmol/L-1 μmol/L in m. gracilis arteriole, or 1 nmol/L-1 μmol/L in mesenteric arteriole and femoral arteries, and 1 nmol/L-10 μmol/L in aorta, Panels A-D) or SNP (0.1 nmol/L-1 μmol/L in m. gracilis, mesenteric and femoral arteries, and 1 nmol/L-10 μmol/L in aorta, Panels E-H) in WT and ADKVEC-KO mice (N = 5–9 in each groups). *P < .05 compared to WT mice
3.4 |. Endothelium-selective deletion of ADK protects against pressure overload-induced LV dysfunction
To assess the effect of vascular endothelium selective inhibition of ADK on LV function we employed the ADKVECKO mice, in which LV dysfunction was induced by TAC. Figure 4 shows that in WT mice TAC increases LV wall thickness and induces a progressive decline in ejection fraction, increases DT, indicating development of LV systolic and diastolic dysfunction after TAC (Figure 4). We found that ADKVEC-KO mice with TAC displayed a lesser degree in decline in EF and maintained normal DT without changes in LV wall thickness when compared to WT mice (Figure 4E–G). Moreover, we found that the myocardial surface area, measured histologically, was increased in WT TAC mice, and that ADKVEC-KO mice with TAC showed only a trend toward decreased myocardial surface area (Figure 5A,B). We have additionally assessed vasoreactivity in isolated and pressurized skeletal (gracilis) muscle arterioles of wild-type and ADKVEC-KO mice with or without TAC. We found that dilator responses to ACh, SNP, or adenosine were essentially preserved in ADKVEC-KO mice with TAC, when compared to sham operated ADKVEC-KO or wild-type mice, with or without TAC (Figure 5C–E).
FIGURE 4.
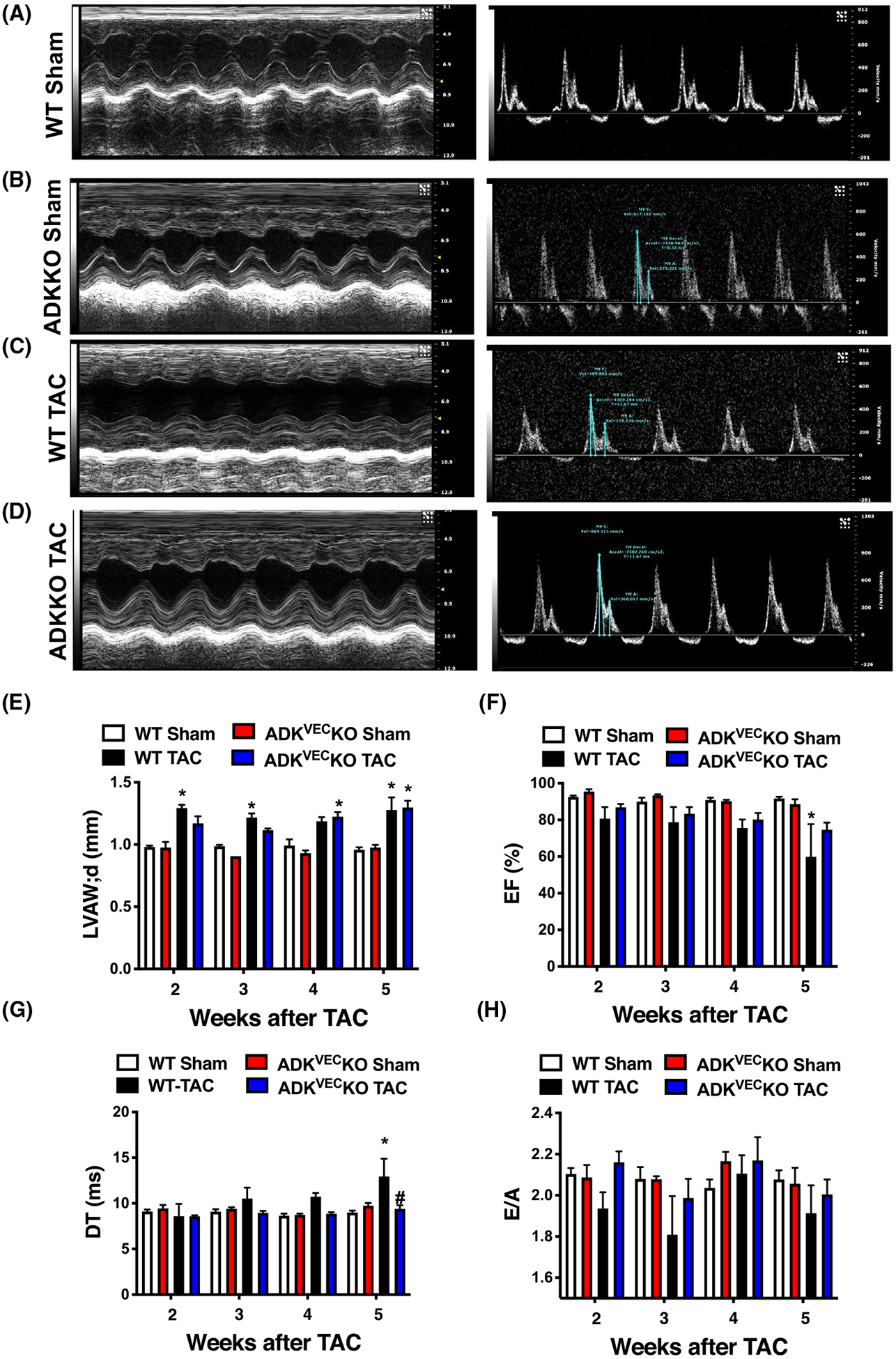
Echocardiographic assessment of WT and ADKVEC-KO mice with TAC. Representative echocardiogram images (Panels A-D) and summary data (Panels E-H) of echocardiogram parameters in WT and ADKVEC-KO mice after sham operation or after TAC surgery (N = 4–5 in each groups). *P < .05 compared to sham-operated WT mice. #P < .05 compared to WT TAC mice
FIGURE 5.
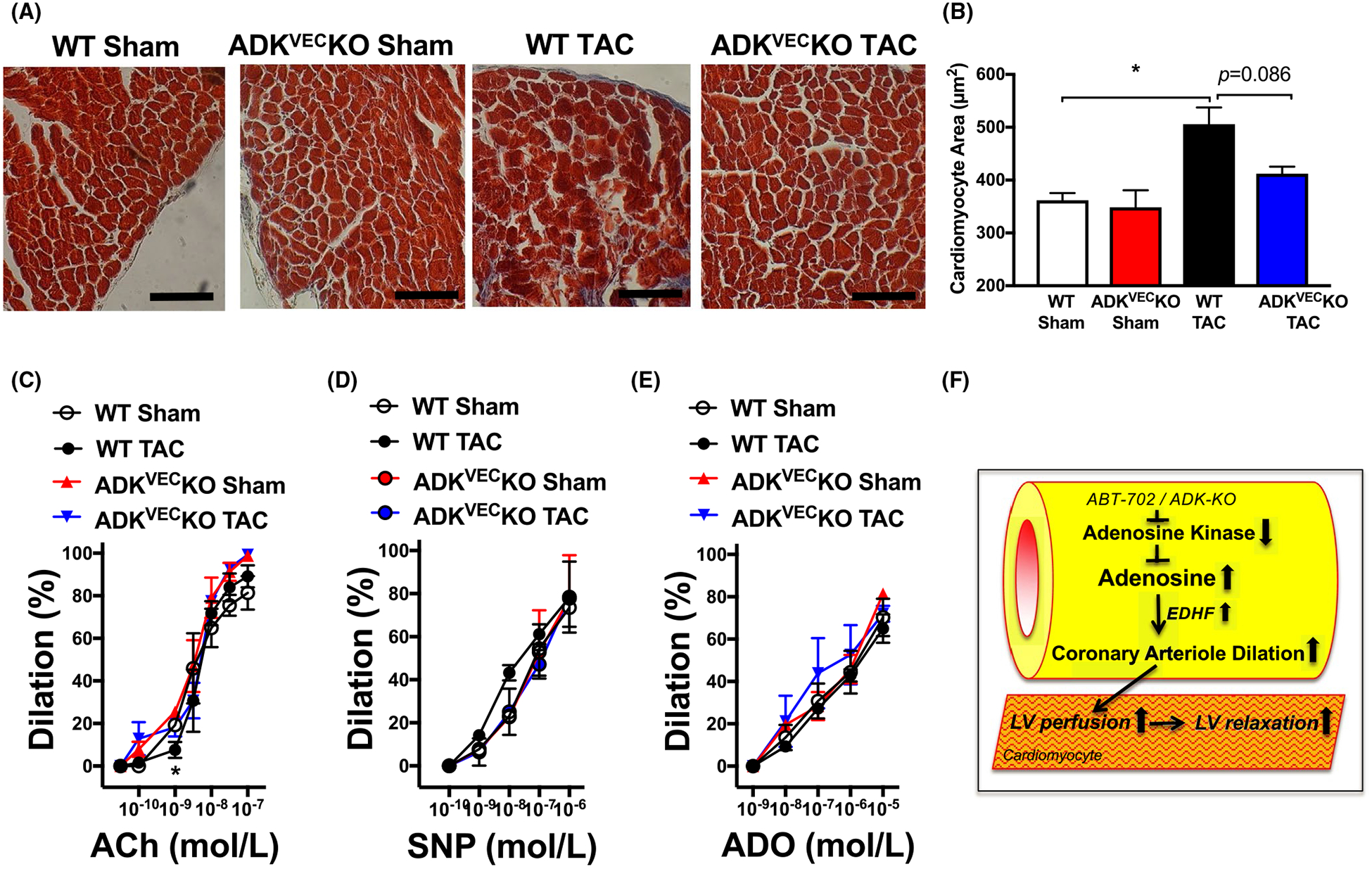
Cardiomyocyte morphology and agonist-induced vasodilations in WT and ADKVEC-KO mice with TAC. Representative Masson’s Trichrome images and summary data of quantification of myocardial surface area in LV sections (Panels A&B) and summary data of gracilis mucle arteriole (Panels C-E) dilations in response to cumulative concentrations of ACh (0.03 nmol/L-0.1 μmol/L), SNP (0.1 nmol/L-1 μmol/L) and ADO (1 nmol/L-10 μmol/L) in WT and ADKVEC-KO mice after sham operation or after TAC surgery (N = 3–5 in each groups). Bar = 100 μm. *P < .05 compared with sham-operated WT mice. Panel F, Schema depicts proposed mechanisms by which pharmacological (ABT-702) or endothelium-selective ADK inhibition augments endogenous adenosine levels and there by facilitates EDHF-mediated microvascular dilations to improve LV perfusion and restore LV diastolic function under metabolic or hemodynamic stress
4 |. DISCUSSION
The salient findings of this study are that pharmacological inhibition of ADK augments coronary microvascular dilator function, which is associated with improved LV diastolic function. Mechanistically, we found that vascular endothelium-selective genetic deletion of ADK resulted in an enhanced microvascular dilator function and protected against the development of pressure overload-induced LV dysfunction. Based on these results, we posit that microvascular-targeted ADK inhibition could be beneficial in preventing LV contractile dysfunction in the failing heart.
Adenosine mediates a prompt increase in myocardial blood flow in response to ischemia.8–10 Under hypoxia, the activity of ADK rapidly decreases and intracellular adenosine levels rise and then released to the extracellular/interstitial space to exert vasodilator effects.7 We recently found that the expression of ADK becomes upregulated in patients with diastolic heart failure (also known as heart failure with preserved ejection fraction or HFpEF).17 We postulated that an upregulated ADK could mitigate hypoxia-induced or hemodynamic stress-related protective effects by adenosine; and therefore, inhibition of ADK can provide cardioprotection, which has also been previously proposed.14,16 In this study, we aimed to specifically test the novel hypothesis that ADK inhibition acts via augmenting microvascular dilator function and thereby providing protection on LV function.
In support, our present study demonstrated that both pharmacological and genetic inhibition of ADK improves key indices of LV diastolic function under metabolic and hemodynamic stress. Because endothelium-selective genetic ADK inhibition was found to be protective on hemodynamic stress-induced LV dysfunction, we concluded that the effect is likely to be mediated by direct vascular mechanisms. Interestingly, our study also revealed that dilation in response to ACh was significantly enhanced in gracilis muscle and mesenteric arterioles of ADKVECKO mice, whereas dilations in larger conduit arteries, such as femoral artery and aorta were similar when compared to WT mice. To our best knowledge, our study is the first to report that endothelium-selective targeting of a specific signaling molecule results in an augmented microvascular but not conduit artery dilation in response to vasoactive agonist stimulation. The exact molecular mechanisms that underlie ADK inhibition-related, selectively augmented microvascular dilator function, which also could exert a direct beneficial cardiac effect have yet to be elucidated.
In this context, previously it has been found that extracellular adenosine may act as an EDHF in the human coronary arteriole.28 In line with this, results from our present and previous17 studies indicate that ADK inhibition and subsequently increased endogenous adenosine levels could have a permissive effect and amplify the EDHF response. Correspondingly, we found previously that adenosine, even at non-vasoactive, nanomolar concentration could act to amplify the hyperpolarization spread and augment spreading vasodilation in isolated skeletal muscle arterioles.17 The effect by the endogenously produced adenosine could lead to a more efficient coupling of tissue perfusion with metabolism, especially under metabolic or hemodynamic stress conditions. In this process, adenosine can activate A2B and A2A adenosine receptors expressed on both vascular smooth muscle cells and also vascular endothelial cells.8–10 It is therefore possible that A2A or A2B adenosine receptor activation, and subsequently increased intracellular cAMP levels facilitates the EDHF-type vasodilation, such as by enhancing electrotonic conduction via gap junctions.29
Moreover, we recently reported that genetic ablation of endothelial ADK, in vivo and in vitro, resulted in Akt-dependent activation of endothelial NO synthase.22 It has been proposed that LV diastolic dysfunction30 could arise from diminished vascular endothelial production of NO in HFpEF, which via a reduced soluble guanylate cyclase activation and subsequently decreased protein kinase-G-dependent phosphorylation of myocardial structural proteins, such as titin, causes stiffening in cardiomyocytes.18 Results from the present study showed that dilation of coronary arterioles, but not larger conduit blood vessels, in response to NO donor, SNP became significantly augmented after pharmacologic ADK inhibition. Thus, restoring microvascular responsiveness to exogenous NO donors by ADK inhibition also can be beneficial on cardiomyocyte function, and this effect carries important translational potential. Indeed, clinical trials, such as the NEAT-HFpEF trial of administering Isosorbide Mononitrate, a direct NO donor to HFpEF patients produced negative results.31 An intriguing possibility of restoring nitrate responsiveness after pharmacologic ADK inhibition warrants further mechanistic studies. Also, the relative contribution of EDHF- and NO-mediated microvascular mechanisms that are augmented and that could be responsible for ADK inhibition-mediated improvement in LV diastolic function has yet to be elucidated.
There are several, mainly mechanistic limitations to the present study. Results from this study provide no direct evidence for the causative link between reduced coronary microvascular vasodilator function and impaired LV diastolic dysfunction. We also note that in mice, due to technical difficulties, we could only assess the vasodilator function in skeletal muscle and mesenteric microvessels and the responses and their vasoactive mediators may differ from rat coronary arterioles. In addition, microvascular and myocardial changes are clearly different in the two rodent models of cardiometabolic and hemodynamic stress-induced LV dysfunction, which we did not address specifically in this study. In this context, our recent studies revealed that endothelium selective genetic inhibition of ADK confers protection against the development of high-fat diet-induced insulin resistance and atherosclerosis.21,22 It is therefore possible that ADK inhibition in obese ZSF1 rat model also influences glucose and lipid homeostasis and the observed beneficial cardiovascular effects can also be attributed to improved metabolic state.
In summary, this study is the first to demonstrate that inhibition of ADK selectively augments coronary arteriole dilation in a rodent HFpEF model. Likely via a more efficient coupling of tissue perfusion with metabolism ADK inhibition improves LV diastolic function in the obese ZSF1 rats (proposed mechanisms are depicted on Figure 5F). Since endothelium selective genetic knockdown of ADK was associated with enhanced microvascular vasodilator function and was found to be protective against TAC-induced LV dysfunction, we propose that selective targeting of microvascular ADK may be considered in prevention of LV diastolic dysfunction in HFpEF.
ACKNOWLEDGMENTS
The technical assistance of Marta Balogh in performing immunohistochemical and Western immunoblot experiments is gratefully acknowledged.
Funding information
The author’s studies are supported by awards from the National Institute of Aging (R01AG054651 to ZB), National Heart, Lung, and Blood Institute (F31 HL142183 to AD) and the American Heart Association (GRNT33680171 to ZB).
Abbreviations:
- ACh
acetylcholine
- ADK
adenosine kinase
- ADKVECKO
ADK knockout mice
- ADO
adenosine
- DT
deceleration time
- EDHF
endothelium-dependent hyperpolarization factor
- EDP
end-diastolic pressure
- EF
ejection fraction
- ESP
end-systolic pressure
- HFpEF
heart failure with preserved ejection fraction
- IP
intraperitoneal
- LV
left ventricle
- PW
posterior wall
- SNP
sodium nitroprusside
- TAC
transverse aortic constriction
- WT
wild-type
- ZSF1
Zucker fatty/spontaneously hypertensive heart failure F1
Footnotes
CONFLIC T OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
- 1.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 2.Franssen C, Chen S, Unger A, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016;4:312–324. [DOI] [PubMed] [Google Scholar]
- 3.Dryer K, Gajjar M, Narang N, et al. Coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2018;314:H1033–H1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy BI, Schiffrin EL, Mourad JJ, et al. Impaired tissue perfusion: a pathology common to hypertension, obesity, and diabetes mellitus. Circulation. 2008;118:968–976. [DOI] [PubMed] [Google Scholar]
- 5.Niccoli G, Scalone G, Lerman A, Crea F. Coronary microvascular obstruction in acute myocardial infarction. Eur Heart J. 2016;37:1024–1033. [DOI] [PubMed] [Google Scholar]
- 6.Henderson KK, Turk JR, Rush JW, Laughlin MH. Endothelial function in coronary arterioles from pigs with early-stage coronary disease induced by high-fat, high-cholesterol diet: effect of exercise. J Appl Physiol. 2004;97:1159–1168. [DOI] [PubMed] [Google Scholar]
- 7.Loffler M, Morote-Garcia JC, Eltzschig SA, Coe IR, Eltzschig HK. Physiological roles of vascular nucleoside transporters. Arterioscler Thromb Vasc Biol. 2007;27:1004–1013. [DOI] [PubMed] [Google Scholar]
- 8.Sato A, Terata K, Miura H, et al. Mechanism of vasodilation to adenosine in coronary arterioles from patients with heart disease. Am J Physiol Heart Circ Physiol. 2005;288:H1633–H1640. [DOI] [PubMed] [Google Scholar]
- 9.Mustafa SJ, Morrison RR, Teng B, Pelleg A. Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol. 2009;193:161–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Headrick JP, Ashton KJ, Rose’meyer RB, Peart JN. Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol Ther. 2013;140:92–111. [DOI] [PubMed] [Google Scholar]
- 11.Meyer TE, Chung ES, Perlini S, et al. Antiadrenergic effects of adenosine in pressure overload hypertrophy. Hypertension. 2001;37:862–868. [DOI] [PubMed] [Google Scholar]
- 12.Laghi-Pasini F, Guideri F, Petersen C, et al. Blunted increase in plasma adenosine levels following dipyridamole stress in dilated cardiomyopathy patients. J Intern Med. 2003;254:591–596. [DOI] [PubMed] [Google Scholar]
- 13.Funakoshi H, Zacharia LC, Tang Z, et al. A1 adenosine receptor upregulation accompanies decreasing myocardial adenosine levels in mice with left ventricular dysfunction. Circulation. 2007;115:2307–2315. [DOI] [PubMed] [Google Scholar]
- 14.Boison D Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev. 2013;65:906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–5580. [DOI] [PubMed] [Google Scholar]
- 16.Peart JN, Gross GJ. Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res Cardiol. 2005;100:328–336. [DOI] [PubMed] [Google Scholar]
- 17.Davila AC, Tian Y, Czikora I, et al. Adenosine kinase inhibition augments conducted vasodilation and prevents left ventricle diastolic dysfunction in heart failure with preserved ejection fraction. Circ Heart Fail. 2019;12(8):e005762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamdani N, Franssen C, Lourenco A, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–1249. [DOI] [PubMed] [Google Scholar]
- 19.Elsherbiny NM, Ahmad S, Naime M, et al. ABT-702, an adenosine kinase inhibitor, attenuates inflammation in diabetic retinopathy. Life Sci. 2013;93:78–88. [DOI] [PubMed] [Google Scholar]
- 20.Kowaluk EA, Mikusa J, Wismer CT, et al. ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin- 3-yl)pyrido[2,3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties. II. In vivo characterization in the rat. J Pharmacol Exp Ther. 2000;295:1165–1174. [PubMed] [Google Scholar]
- 21.Xu Y, Wang Y, Yan S, et al. Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nat Commun. 2017;8:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Yang Q, Zhang X, et al. Endothelial adenosine kinase deficiency ameliorates diet-induced insulin resistance. J Endocrinol. 2019;242(2):159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.deAlmeida AC, van Oort RJ, Wehrens HT. Transverse aortic constriction in mice. J Vis Exp. 2010;38:e1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Yue G, Ma W, et al. Ufm1-specific ligase Ufl1 regulates endoplasmic reticulum homeostasis and protects against heart failure. Circ Heart Fail. 2018;11:e004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassuto J, Dou H, Czikora I, et al. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes. 2014;63:1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fulop T, Jebelovszki E, Erdei N, et al. Adaptation of vasomotor function of human coronary arterioles to the simultaneous presence of obesity and hypertension. Arterioscler Thromb Vasc Biol. 2007;27:2348–2354. [DOI] [PubMed] [Google Scholar]
- 27.Szerafin T, Erdei N, Fulop T, et al. Increased cyclooxygenase-2 expression and prostaglandin-mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ Res. 2006;99:e12–e17. [DOI] [PubMed] [Google Scholar]
- 28.Ohta M, Toyama K, Gutterman DD, et al. Ecto-5′-nucleotidase, CD73, is an endothelium-derived hyperpolarizing factor synthase. Arterioscler Thromb Vasc Biol. 2013;33:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffith TM, Chaytor AT, Taylor HJ, Giddings BD, Edwards DH. cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc Natl Acad Sci USA. 2002;99:6392–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redfield MM, Anstrom KJ, Levine JA, et al. Isosorbide Mononitrate in heart failure with preserved ejection fraction. N Engl J Med. 2015;373:2314–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]