

Abstract
Detecting clinical grade CNV based on WES is being improved in the NGS era.
Keywords: Cabezas syndrome, CUL4B, intellectual disability, XLID
Detecting clinical grade CNV based on WES is being improved in the NGS era
1. INTRODUCTION
Cabezas syndrome, which is characterized by intellectual disability, short stature, and speech delay, together with other more variable features and caused by variants in CUL4B gene located at Xq23. We used whole‐exome sequencing (WES) data to study copy number variation (CNV). We detected a 22.5 kb deletion in CUL4B gene in a 10‐year‐old boy with severe intellectual disability and clinically undiagnosed. The increasing use of NGS for undiagnosed ID cases is helping to find a genetic explanation for rare or complex disorders.
Intellectual disability (ID) includes a set of clinically and genetically heterogeneous disorders in which brain development and/or function are compromised. ID is defined by substantial limitations in both intellectual functioning and adaptive behavior with onset before the age of 18 years. According to the phenotype, ID is usually divided into two categories: nonsyndromic, in which ID is the only clinical feature, and syndromic, associated with other neurological and/or behavioral manifestations and structural anomalies. ID affects 1%‐3% of the population and is more prevalent in males than in females. 1 This observation, along with the identification of ID families with extended X‐linked pedigrees, indicated the presence of pathogenic variants. 2 To date, pathogenic variants in more than 100 genes have been found to cause X‐linked ID (XLID). XLID is the most common cause of ID in males and accounts for 5%‐10% of all IDs. 3 Cabezas syndrome (MIM #300354) is a type of syndromic XLID characterized by ID, short stature, hypogonadism, and abnormal gait, together with other more variable features such as speech delay, prominent lower lip, and tremor. This syndrome is known to be caused by pathogenic variants in CUL4B gene (MIM #300304, NM_003588). The CUL4B gene is located on ChrX:120523858‐120560962 reverse strand. This gene contains 20 coding exons and encodes a 895‐amino acid protein that is a core component of the cullin‐RING‐based E3 ubiquitin protein ligase complex. 4 CUL4B is thought to play significant roles in cellular processes including ubiquitination of histones and control of the cell cycle through downregulation of cyclin E and β‐catenin.
Since the description of the first family by Cabezas et al, 5 several CUL4B‐associated XLID cases have been reported, sharing some of the characteristic features of the Cabezas syndrome. 3 , 4 , 6 , 7 , 8 , 9 , 10 , 11 Variants described in CUL4B included missense, frameshift, and primary truncations variants. They appear to be distributed throughout the gene and in most cases result in significantly reduced CUL4B protein expression. 12 Even noncoding deletion located in 5´UTR which eliminates CUL4B expression has been described. 2
Analysis by targeted exome sequencing of individuals with clinical ID, who previously tested negative for pathogenic variants and copy number variants (CNVs) in a subset of known ID related‐genes, reveals CUL4B as one of the most frequently mutated genes underlying XLID. More specifically, the prevalence of CUL4B variations in XLID has been estimated to be between 2% and 3%. 6 , 11 In this report, we describe the first case of Cabezas syndrome in a boy with ID and severe speech delay, caused by a 22.5 kb deletion in CUL4B gene detected by CNV analysis from whole‐exome sequencing (WES) data.
2. MATERIAL AND METHODS
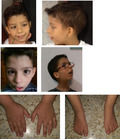
Proband was a 10‐year‐old boy, first‐born child of healthy, nonconsanguineous parents. The mother had a previous miscarriage. He was delivered at full term with a birth height of 49 cm, weight of 2.815 g, and plagiocephaly. Psychomotor development was delayed: He holds his head up at the age of 7 m.o., he sat at 1 y.o., and he never crawled. He started to walk between 1.5 and 2 y.o. He showed severe intellectual disability, pronounced language delay: He is not able to speak a single word. He suffered several behavioural problems such as anxiety, autism/autism‐like conduct, and aggressive and self‐injurious character. He had short stature, slight macrocephaly, low set ears, nasal rounded tip, strabismus, prominent lower lip, dental crowding, small feet, and broad toes (Figure 1A). He also suffered seizures, mild sensorineural hearing loss, gait abnormality, fine motor delay, and wasting lower leg muscles. First clinical suspicion was Rubinstein‐Taybi syndrome (RSTS).
Figure 1.
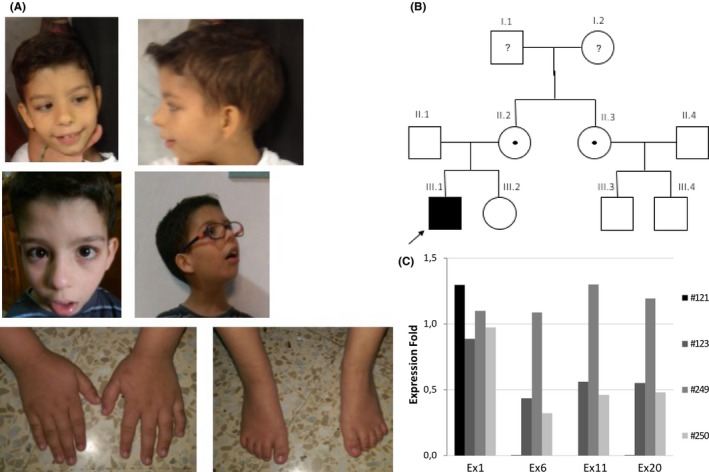
A, Patient photographs at 5 and 7 y.o., hands and feet. B, Family pedigree. C, qPCR results of exons 1, 6, 11, 20 located in CUL4B gene (#121 proband, #123 mother, #249 sister. #250 aunt)
Clinical data, samples, and photographs were obtained after written informed consent. This work has been approved by the Committee for Ethics in Clinical Research in La Rioja (CEICLAR).
Blood samples from the proband and his parents, as well as from his sister and maternal aunt, were collected in EDTA tubes. DNA was extracted using QIAamp DNA Mini Kit (QIAGEN) following the manufacture's protocol. Following the first suspicion of RSTS, MLPA of CREBBP and EP300 was performed (P313 and P333 Kit, MRC‐Holland) and panel‐based next‐generation sequencing (NGS) of CREBBP and EP300 genes was carried out. Since MLPA was normal and variants were not found in this panel, WES was performed in the patient (SureSelectXT Human All Exon V6, Agilent, Santa Clara, USA sequenced on an Illumina HiSeq sequencing system, coverage 100X). The resulting reads were mapped to the human genome hg19 using Burrows‐Wheeler Aligner (BWA). Sequence variants were called using the Genome Analysis Toolkit (GATK), and called variants were annotated with Annovar. ExAC browser of Broad Institute, 1000 Genomes database, and dbSNP138, as well as the Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD), and ClinVar databases, were checked to assess the presence/absence of detected alterations in variation repositories. A second analysis was performed to calculate CNVs with CNVkit 13 and detected CNVs were corroborated by qPCR. For this purpose, primers targeting exons 1, 6, 11, and 20 were designed, and qPCR assays were performed using LightCycler 480 SYBR Green I Master (Roche). CUL4B primer sequences and qPCR conditions are available under request.
3. RESULTS AND DISCUSSION
No pathogenic variants were detected after analysis of WES results. However, CNVs assessment revealed a 22.5 kb deletion at chromosome X position chrX: g.120526646‐120546685del (GRCh38p.13) that includes exons 4‐20 of CUL4B gene.
As a result of this molecular finding, the patient was diagnosed with Cabezas syndrome (Figure 1C).
The common phenotype, first described by Cabezas et al, 5 is shared by most patients and includes ID ranging from mild to severe, delayed psychomotor development, severe speech delay, ataxia gait, tremor, pes cavus, and seizures. Growth retardation is also a hallmark of this syndrome. After the molecular diagnosis of Cabezas syndrome in our patient, we reviewed the clinical data reveling that most of these features were present, corroborating the molecular diagnosis (Table 1): severe ID, muscle wasting, delayed growth, prominent lower lip, hypogenitalism, seizures, abnormal gait, diminished fine motor skills, and severe language deficit. No pes cavus, obesity, or tremor was found in our patient.
Table 1.
Summary of clinical data in patients with CUL4B variants
| Cabezas et al (2000) 5 | Zou et al (2007) 4 | Tarpey et al (2007) 6 | Badura‐Stronka et al (2010) 7 | Isidor et al (2010) 8 | Ravn et al (2012) 9 | Londin et al (2014) 10 | Vulto‐van Silfhout et al (2015) 11 | Okamoto et al (2017) 3 | Weissbachet al (2017) 14 | This case | Total | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. Affected | 5 | 6 | 8 families | 3 | 1 | 2 | 8 | 29 | 1 | 1 | |||
| Neurological | |||||||||||||
| Intellectual disability | 5/5 | 6/6 | 22/22 | 3/3 | 1/1 | 2/2 | 8/8 | 24/24 | 1/1 | 0/1 | + | 73/74 | 98.65% |
| Motor delay | 5/5 | 6/6 | 5/5 | na | 1/1 | 2/2 | 8/8 | 23/23 | 1/1 | 0/1 | + | 52/53 | 98.11% |
| Language/speech delay | 4/5 | 6/6 | 18/18 | 3/3 | 1/1 | 2/2 | 8/8 | 23/23 | 1/1 | 0/1 | + | 67/69 | 97.10% |
| Seizures | na | 4/5 | 8/11 | 0/3 | 0/1 | 2/2 | 1/8 | 7/22 | 1/1 | 0/1 | + | 24/55 | 43.64% |
| Tremors | 4/5 | 1/5 | 11/13 | 2/3 | 1/1 | 2/2 | na | 9/20 | 0/1 | 0/1 | − | 30/52 | 57.69% |
| Gait disturbances | 3/5 | 6/6 | 6/12 | na | 1/1 | 2/2 | 0/8 | 10/21 | 0/1 | 1/1 | + | 31/58 | 53.44% |
| Behavioral problems | 3/5 | 5/6 | 12/15 | 3/3 | 1/1 | 1/2 | na | 13/22 | 0/1 | na | + | 39/56 | 69.64% |
| Dysmorphims | |||||||||||||
| Microcephaly | 0/5 | 6/6 | – | 1/3 | 1/1 | – | 3/8 | – | – | na | − | 11/60 | 18.33% |
| Macrocephaly | 0/5 | – | 8/11 | – | – | 2/2 | – | 7/22 | 1/1 | na | − | 18/60 | 30.00% |
| Prominent lower lip | 4/5 | 5/5 | 6/17 | 2/3 | 1/1 | 2/2 | na | 18/23 | 1/1 | na | + | 40/58 | 68.96% |
| Low set ears | 0/5 | 0/6 | na | 3/3 | 1/1 | na | 8/8 | 17/19 | 1/1 | na | + | 31/44 | 70.45% |
| Muscle/Skeletal | |||||||||||||
| Short stature | 5/5 | 6/6 | 7/11 | 2/3 | 1/1 | 2/2 | 8/8 | 17/22 | 1/1 | 0/1 | + | 50/61 | 81.97% |
| Short feet | 5/5 | na | 7/14 | 3/3 | na | na | 8/8 | 14/19 | na | na | + | 38/50 | 76.00% |
| Gap between 1&2 toes | 3/5 | na | 11/13 | 3/3 | 1/1 | na | 8/8 | na | na | na | − | 26/31 | 83.87% |
| Pes cavus | na | 0/5 | 7/8 | 0/3 | 1/1 | 2/2 | na | 2/11 | na | na | − | 12/31 | 38.71% |
| Kyphosis | 4/5 | na | 3/18 | na | 1/1 | na | 6/8 | 6/17 | na | 0/1 | + | 21/51 | 41.18% |
| Muscle wasting | 5/5 | na | 7/12 | 1/3 | na | na | na | 5/11 | na | 0/1 | + | 19/33 | 57.57% |
| Other | |||||||||||||
| Genital abnormalities | 4/5 | 2/5 | 10/15 | 1/3 | 1/1 | 0/2 | 8/8 | 17/20 | 1/1 | 0/1 | + | 45/62 | 72.58% |
| Obesity | 4/5 | 0/5 | 15/19 | 2/3 | 0/1 | 2/2 | 0/8 | 11/21 | 0/1 | 0/1 | − | 34/67 | 50.75% |
Driven by the molecular finding in this patient, the presence of the deletion was also checked in some relatives by qPCR. The mother and the aunt were found to be asymptomatic carriers of the same pathogenic variant, and his sister exhibited normal doses. No Cabezas syndrome phenotype has been observed in his cousins (not studied) (Figure 1B).
As in our patient, in most reported cases, the clinical diagnosis of Cabezas syndrome was established after the identification of pathogenic variants in CUL4B, suggesting that Cabezas syndrome is usually underdiagnosed. In this line, some authors have developed useful guidelines to identify potential patients with CUL4B variants. 3 , 11
CUL4B is a component of the ubiquitin system. Ubiquitination is a chemical reaction whereby proteins are marked for degradation or transport to specific compartments within the cell. It is also involved in histone modification and, by extension, may play a role in gene expression. CUL4B is the first ubiquitin E3 ligase mutated in XLID. Abnormalities of E3 ligases, however, have been reported in other human genetic diseases. Pathogenic variants in the ubiquitin‐protein ligase E3A gene (UBE3A) underlie a subset of Angelman syndrome, in which ID is a notable clinical feature, and abnormalities in other E3 ligases have been shown to cause other diseases (recessive juvenile Parkinson disease, autoimmune polyendocrinopathy syndrome type 1, etc). 12
All the cases of Cabezas syndrome described showed ID and speech delay. This language deficiency was evident in early childhood and remained disproportionately severe given the degree of intellectual impairment; in fact, our patient, at the age of 10, was not able to speak a single word, and in most of the cases speech was very limited or nonexistent. Notably, speech impairment is also present in other syndromes caused by defects in genes involved in ubiquitination processes, such as Angelman syndrome and the X‐linked syndromic ID type Nascimento (MIM #300860) caused by UBE2A gene variants. Short stature, another typical feature of Cabezas syndrome, has also been linked to anomalies of the ubiquitination, as is the case of 3‐M syndrome caused by CUL7 pathogenic variants. 8
CUL4B gene should be considered as a first approach in males with ID, speech and motor delay, and behavior abnormalities. The increasing use of NGS for undiagnosed ID cases is helping to find a genetic explanation for rare or complex disorders. The clarification of the etiological diagnosis is necessary in order to answer the questions regarding the possibilities for therapeutic intervention and the risk of recurrence.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORS’ CONTRIBUTIONS
ML and EDG: contributed to conception or design of the work. IGC: contributed to data collection. ML, VPG, and EDG: analyzed and interpreted the data. ML, VPG, and EDG: drafted the article. ML, VPG, IGC, and EDG: revised the article. IGC and EDG: read and approved the version to be published.
ACKNOWLEDGMENTS
We are grateful to the patient and his family for their cooperation and their participation in the study. Published with written consent of the patient.
López M, Pérez‐Grijalba V, García‐Cobaleda I, Domínguez‐Garrido E. A 22.5 kb deletion in CUL4B causing Cabezas syndrome identified using CNV approach from WES data. Clin Case Rep. 2020;8:3184–3188. 10.1002/ccr3.3381
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Larson SA, Lakin KC, Anderson L, Kwak Lee N, Anderson D. Prevalence of mental retardation and developmental disabilities: estimates from the 1994/1995 National Health Interview Survey disability supplements. Am J Ment Retard. 2001;106:231‐252. [DOI] [PubMed] [Google Scholar]
- 2. Whibley AC, Plagnol V, Tarpey PS, et al. Fine‐scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87:173‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okamoto N, Watanabe M, Naruto T, et al. Genome‐first approach diagnosed Cabezas syndrome via novel CUL4B mutation detection. Hum Genome Var. 2017;4:16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zou Y, Liu Q, Chen B, et al. Mutation in CUL4B, which encodes a member of cullin‐RING ubiquitin ligase complex, causes X‐linked mental retardation. Am J Hum Genet. 2007;80:561‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cabezas DA, Slaugh R, Abidi F, et al. A new X linked mental retardation (XLMR) syndrome with short stature, small testes, muscle wasting, and tremor localizes to Xq24‐q25. J Med Genet. 2000;37:663‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tarpey PS, Raymond FL, O'Meara S, et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X‐linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet. 2007;80(2):345‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Badura‐Stronka M, Jamsheer A, Materna‐Kiryluk A, et al. A novel nonsense mutation in CUL4B gene in three brothers with X‐linked mental retardation syndrome. Clin Genet. 2010;77:141‐144. [DOI] [PubMed] [Google Scholar]
- 8. Isidor B, Pichon O, Baron S, David A, Le Caignec C. Deletion of the CUL4B gene in a boy with mental retardation, minor facial anomalies, short stature, hypogonadism, and ataxia. Am J Med Genet A. 2010;152A:175‐180. [DOI] [PubMed] [Google Scholar]
- 9. Ravn K, Lindquist SG, Nielsen K, Dahm TL, Tümer Z. Deletion of CUL4B leads to concordant phenotype in a monozygotic twin pair. Clin Genet. 2012;82:292‐294. [DOI] [PubMed] [Google Scholar]
- 10. Londin ER, Adijanto J, Philp N, et al. Donor splice‐site mutation in CUL4B is likely cause of X‐linked intellectual disability. Am J Med Genet A. 2014;164A:2294‐2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vulto‐van Silfhout AT, Nakagawa T, Bahi‐Buisson N, et al. Variants in CUL4B are associated with cerebral malformations. Hum Mutat. 2015;36(1):106‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kerzendorfer C, Hart L, Colnaghi R, et al. CUL4B‐deficiency in humans: understanding the clinical consequences of impaired Cullin 4‐RING E3 ubiquitin ligase function. Mech Ageing Dev. 2011;132(8–9):366‐373. [DOI] [PubMed] [Google Scholar]
- 13. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12(4):e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weissbach S, Reinert MC, Altmüller J, et al. A new CUL4B variant associated with a mild phenotype and an exceptional pattern of leukoencephalopathy. Am J Hum Genet. 2017;173:2803–2807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.