

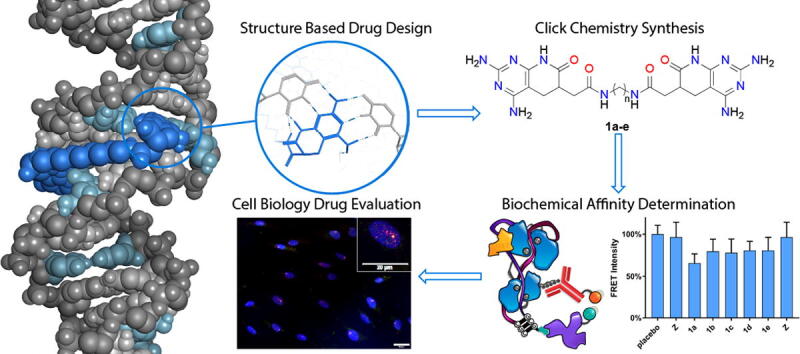
Graphical abstract
Keywords: Myotonic dystrophy, Molecular modelling, RNA targeting, Small molecule, Base recognition
Highlights
-
•
Drug design with rational methods, using structure-based methods.
-
•
Novel RNA binders based on pyrido[2,3-d]pyrimidin-7-(8H)-ones.
-
•
Myotonic Dystrophy type 1 small molecule drug discovery.
Abstract
Myotonic Dystrophy type 1 (DM1) is an incurable neuromuscular disorder caused by toxic DMPK transcripts that carry CUG repeat expansions in the 3′ untranslated region (3′UTR). The intrinsic complexity and lack of crystallographic data makes noncoding RNA regions challenging targets to study in the field of drug discovery. In DM1, toxic transcripts tend to stall in the nuclei forming complex inclusion bodies called foci and sequester many essential alternative splicing factors such as Muscleblind-like 1 (MBNL1). Most DM1 phenotypic features stem from the reduced availability of free MBNL1 and therefore many therapeutic efforts are focused on recovering its normal activity. For that purpose, herein we present pyrido[2,3-d]pyrimidin-7-(8H)-ones, a privileged scaffold showing remarkable biological activity against many targets involved in human disorders including cancer and viral diseases. Their combination with a flexible linker meets the requirements to stabilise DM1 toxic transcripts, and therefore, enabling the release of MBNL1. Therefore, a set of novel pyrido[2,3-d]pyrimidin-7-(8H)-ones derivatives (1a-e) were obtained using click chemistry. 1a exerted over 20% MBNL1 recovery on DM1 toxic RNA activity in primary cell biology studies using patient-derived myoblasts. 1a promising anti DM1 activity may lead to subsequent generations of ligands, highlighting a new affordable treatment against DM1.
1. Introduction
Trinucleotide repeat expansion diseases (TREDs) are characterized by an expansion into a disease-causing range of a short homogeneous sequence of nucleotides [1]. These microsatellite expansions may not be toxic per se. In some cases they are located in intronic or intergenic loci; however, they usually undergo transcription giving rise to aberrant transcripts and, in some cases, if these mRNAs are successfully exported outside the nuclei, they may trigger repeat-associated non-AUG (RAN) translation [2]. One representative case of TRED is myotonic dystrophy type 1 (DM1). DM1 is the most common adult-onset muscular dystrophy, an incurable neuromuscular disorder characterized by symptoms such as progressive muscle weakening, myotonia, cardiac arrhythmias, cognitive dysfunction, and cataracts [3]. To date, there is no effective treatment against this disease. This fact, along with the increasing number of cases due to the improvement of diagnostic tools, is raising awareness of DM1-like diseases [4]. The expansion of dCTG repeats within the 3′ untranslated (3′-UTR) region of the dystrophia myotonica protein kinase (DMPK) gene constitutes the molecular basis of the disease [5]. Somatic instability related to this expansion causes progressive phenotypic worsening as the pathogenic CTG expansion tends to grow with age [6]. Upon that, mutant DMPK transcription produces aberrant non-coding CUG trinucleotide pre-mRNAs [r(CUG)EXP]. These transcripts fail to exit to the cytosol and tend to accumulate in the nuclei and sequester many essential RNA-binding proteins (RBPs), such as Muscleblind-like 1 (MBNL1) [7], forming complex inclusion bodies called ribonuclear foci [8]. These gel-like structures are able to arrest different RBP when forming foci or being in their diffuse state. Concomitant to MBNL1 sequestration, CUGBP Elav-like family member 1 (CELF1), an MBNL1 antagonist, is upregulated in DM1 patients [9]. Both MBNL1 and CELF1 proteins are essential alternative splicing factors and their alterations in DM1 maintain a fetal splicing pattern of several muscle transcripts in adults. Therefore, the fetal isoforms are produced instead of the adult ones. Hence, the unbalance in splicing pattern originates disease symptoms. Most phenotypic features are explained through MBNL1 protein sequestration in the foci, albeit DM1 is a multifactorial disease [10]. Therefore, relieving MBNL1 off the foci, and thus recovering the normal splicing pattern, is a remarkable goal in drug development against DM1.
To recover MBNL1 and balance the splicing pattern, many strategies have been described: [11], [12] including gene editing, transcription repression or chemotherapy (i.e.: antisense oligonucleotides, peptides, and small molecules) [13], [14], [15], [16], [17], [18]. Currently, the small molecule approach is undoubtedly the preferred pharmacological strategy to treat any kind of disease due to its robustness and cost-effectiveness ratio. In the last few years, some molecular entities have been described to exert a significant activity against DM1. They can be classified into few major groups based on their characteristic mechanisms of action: compounds able to bind DNA and repress transcription, inhibitors able to recover missplicing events by CELF1 repression, and compounds able to recover MBNL1 from the foci by targeting RNA [18], [19], [20]. Focusing on the foci, only three strategies have led to compounds able to achieve selectivity and potency: groove binding, base intercalation, and base recognition [18], [19], [21]. Among them, and given the characteristic regular structure of DM1 transcripts, base recognizers represent the most revolutionary strategy in terms of having a comprehensive action mechanism. They act mimicking nucleotides and stablishing hydrogen bond interactions with the target nucleic acids. This leads to a secondary structure stabilization in double-stranded conformation of CUG. As previously described, stabilization of r(CUG)EXP triggers MBNL1 recovery as MBNL1 colocalizes with CUG in foci [22]. Such an accomplishment has also been reported via uridine replacement, antisense oligonucleotides treatment [15].
In order to adapt base recognition strategy to exogenous drug-like structures a common paradigm is used. Candidates must be able to bridge non-canonical Watson-Crick base pairs, the so-called Janus-Wedge interaction (JW) [23]. The interaction of these molecules with the foci can turn the unstructured gel-like inclusion bodies to regular double stranded conformation (dsCUG). An example of base recognizers is the Zimmerman’s bisamidinium linker (Z), which is included in this study as reference [24]. The target nucleobases to fit in the recognizers are the U•U mismatches that are generated when dsCUG conformation is achieved [25]. By stabilizing the irregular structure of the foci, the toxic r(CUG)EXP may return to its soluble and non-pathogenic form, relieving MBNL1.
We applied a de novo computer-aided drug design procedure to predict the ability of novel compounds to bind to r(CUG)EXP. The consideration of Janus-Wedge interactions as the base mechanism for CUG recognition led us to the identification of a novel active core based on pyrido[2,3-d]pyrimidine derivatives. Pyrido[2,3-d]pyrimidine is a privileged scaffold with notable biological activity in fields such as cancer or viral diseases [26], [27]. Herein, we report a set of new multivalent compounds, composed by two pyrido[2,3-d]pyrimidin-7-(8H)-ones units linked by an aliphatic spacer (Fig. 1a), able to target two U•U mismatches in r(CUG)EXP structures via Janus-Wedge interactions (Fig. 1b). The effect of the spacer length on the ability to rescue MBNL1 was assessed by both computational and biological techniques.
Fig. 1.

General structure of ligands proposed on current work. a) All structures are characterized for bearing a pyrido[2,3-d]pyrimidine scaffold at both terminal ends separated by a series of aliphatic spacers of different lengths. Both terminal scaffolds are bound to the central spacer with amide bond. b) Janus Wedge interaction mechanism described between an U•U mismatch and the introduced pyrido[2,3-d]pyrimidine scaffold, allowing base recognition of singular nucleic acids such as dsCUG.
2. Materials and methods
2.1. Computer-aided molecular design
2.1.1. Receptor preparation
The receptor used in the current study is a CUG16 RNA model previously developed [28]. The flexible behavior of the system was assessed by molecular dynamics simulations. The system was initially heated to 300 K within 500 ps restraining the position of the RNA with a 2.0 kcal/mol·Å2 force constant. These restraints were gradually reduced along the system’s equilibration stage, performed at constant temperature and pressure (1.0 bar, 300 K correspondingly). A final NPT production stage was conducted during 100 ns without restraints. The Particle Mesh Ewald method was used for electrostatics interactions under periodic boundary conditions and SHAKE [29] algorithm was applied for hydrogen atoms. The time step was fixed to 2 fs and simulations were conducted using AMBER18 software (University of California, San Francisco, CA). Upon that, a set of frames exhibiting opening values similar to the ones expected from alternate U•U mismatches were selected using the cpptraj [30] module.
2.1.2. Pharmacophore model and docking
A pharmacophore model was developed using Molecular Operating Environment (MOE) software (Chemical Computing Group, Montreal, QC), to evaluate the ability of the ligands to establish a Janus-Wedge interaction pattern with two U•U mismatches. The generated model involved three consecutive U•U mismatches, each of them led to 5 pharmacophore features (three H-bond donors and two H-bond acceptors) (Fig. S1). Thereafter, pharmacophore guided docking was performed using MOE under induced fit conditions. We considered two different score functions (London dG and GBVI/WSA dG) for the docking study. The best conformation for each ligand was selected for the subsequent calculations.
2.1.3. Molecular dynamics and data analysis
Ligands were prepared in UCSF-Chimera [31], calculating AM1-BCC atomic charges and using the GAFF [32] forcefield. Complex structures were generated using tleap, defining the Amber14 and Rochester torsions forcefields [33] for the RNA. Complexes were neutralized and solvated using the OPC water model in a truncated octahedral box [34]. All molecular dynamics simulations were conducted using pmemd.cuda module in Amber18.[35], [36], [37] and run through the following steps. The system was initially heated to 300 K within 500 ps restraining the position of the RNA with a 2.0 kcal/mol·Å2 force constant. These restraints were gradually reduced along the system’s equilibration stage, performed at constant temperature and pressure (1.0 bar, 300 K correspondingly). A final NPT production stage was conducted during 100 ns without restraints. The Particle Mesh Ewald method was used for electrostatics interactions under periodic boundary conditions and SHAKE [29] algorithm was applied for hydrogen atoms. The time step was set to 2 fs. RMSD analysis for the full-length trajectories was conducted using ccptraj module and MMPBSA binding free energies were calculated for the last 20 ns of simulation. RNA structural parameters analysis was performed using the X3DNA software [38]. Data analysis and plotting were performed using R (R Foundation for Statistical Computing, Vienna, AT).
2.2. Chemistry
2.2.1. General considerations
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all solvents and chemicals were purchased from commercial vendors (Sigma Aldrich, ABCR, Fluorochem, Apollo scientific and ACROS Organics) and used without further purification. 1H and 13C and spectra were recorded on a Varian 400-MR spectrometer (1H NMR at 400 MHz and 13C NMR at 100.5 MHz). Chemical shifts were reported in parts per million (δ) and are referenced to the residual signal of the solvent DMSO‑d6 2.50 ppm or in 1H NMR spectra and to the residual signal of the solvent DMSO‑d6 39.5 ppm in 13C NMR). Coupling constants are reported in Hertz (Hz). Standard and peak multiplicities are designed as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublets of doublets; t, triplet; tt, triplet of triplets; br, broad signal. IR spectra were recorded in a Thermo Scientific Nicolet iS10 FTIR spectrophotometer with Smart iTr. Wavenumbers (ν) are reported in cm−1. MS data (m/z (%), EI, 70 eV) were obtained by using an Agilent Technologies 5975. HRMS data were obtained by using a Bruker micrOTOF (ESI-FIA-TOF). Elemental microanalyses were obtained on a EuroVector Instruments Euro EA 3000 elemental analyzer. The melting points were determined with a Büchi-Tottoli 530 capillary apparatus and are uncorrected. Automatic flash chromatography was performed in an Isco Teledyne Combiflash Rf medium pressure liquid chromatograph with RediSep® silica gel columns (35–70 µm) using a suitable mixture of solvents as eluent. Microwave irradiation experiments were carried out in an Biotage Initiator™ microwave apparatus, operating at a frequency of 2.45 GHz with continuous irradiation power from 0 to 400 W. Reactions were carried out in 5, or 20 mL glass tubes, sealed with aluminum/Teflon crimp tops. Compound 3 and Z were prepared according to literature [24], [39].
dimethyl 2-methylenesuccinate (3)0.4 purified by flash chromatography (Cy: AcOEt, 80:20) 2.94 g (19 mmol, 48% yield) was afforded as a colorless oil. 1H NMR (CDCl3, 400 MHz): δ 6.18 (s, 1H), 5.60 (s, 1H), 3.66–3.61 (m, 3H), 3.59–3.54 (m, 3H), 3.21 (s, 2H). Spectroscopic data are consistent with those reported in reference.
dimethyl 2-(2,2-dicyanoethyl)succinate (6). NaH (0.13 g, 5.41 mmol) was dissolved in anhydrous THF (5 mL) and the resulting mixture was stirred for 10 min at room temperature. Then, malononitrile (0.21 g, 2.14 mmol) was dissolved in anhydrous THF (5 mL) and added to the mixture that was further stirred for 20 min at room temperature. After that, 4 (0.50 g, 3.16 mmol) was dissolved in anhydrous THF (5 mL) and added to the mixture that was stirred for 2 h at 40 °C. After this period, water was added. The resulting mixture was acidified with concentrated acetic acid and extracted with dichloromethane (3 × 15 mL). The organic layer was dried over MgSO4, filtered, and evaporated under reduced pressure to afford 6 (0.61 g, 2.7 mmol, 87% yield) as a brown oil. 1H NMR (400 MHz, CDCl3): δ 4.23 (dd, J = 9.8, 5.7 Hz, 1H), 3.74 (s, 3H), 3.70 (s, 3H), 3.09 – 2.99 (m, 1H), 2.74 (ddt, J = 22.8, 17.2, 6.0 Hz, 2H), 2.50 (ddd, J = 13.9, 10.2, 5.7 Hz, 1H), 2.20 (ddd, J = 13.9, 9.8, 4.0 Hz, 1H); 13C NMR (100.6 MHz, CDCl3): δ 172.5, 171.0, 112.5, 112.3, 52.8, 52.3, 38.0, 35.5, 32.1, 21.3; IR (KBr) νmax(cm−1): 2959, 2928, 1735, 1440, 1376, 1214, 1170.
methyl 2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidin-6-yl)acetate (4). Compound 3 (1.6 g, 10.3 mmol), malononitrile (0.35 g, 5.2 mmol) and guanidine carbonate (0.93 g, 5.2 mmol) were added to a microwave vial with 20 mL of anhydrous MeOH. The mixture was heated at 140 °C under microwave irradiation for 15 min. The resultant solid was collected by filtration, washed with H2O and Et2O and oven dried to afford 0.93 g (3.7 mmol, 71% yield) of 4 as yellow solid.
Or alternatively, guanidine carbonate (1.08 g, 6.02 mmol) and sodium methoxide (650 mg, 12.0 mmol) were refluxed in 4.5 mL of anhydrous MeOH under Ar atmosphere for 15 min. The mixture was filtered to eliminate the formed sodium carbonate. To the resulting solution, 6 (900 mg, 4.0 mmol) was added. Then, the mixture was refluxed under Ar atmosphere overnight. The formed solid was filtered and washed with water, EtOH and Et2O. Methyl 2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidin-6-yl)acetate (4) (520 mg, 2.1 mmol, 52% yield) was obtained as a yellow solid. mp = 276.8 °C; 1H NMR (400 MHz, DMSO‑d6): δ 10.29 (s, 1H), 6.17 (s, 2H), 5.85 (s, 2H), 3.62 (s, 3H), 2.89–2.70 (m, 3H), 2.47–2.43 (m, 1H), 2.26 (dd, J = 15.3, 13.0 Hz); 13C NMR (100.6 MHz, DMSO‑d6): δ 172.4, 172.0, 161.7, 156.4, 83.7, 51.4, 36.8, 34.0, 23.0; IR (KBr) νmax(cm−1): 3381, 3346, 3189, 1717, 1683, 1634, 1570, 1493, 1445, 1391, 1364, 1296, 1237, 1223, 1159; MS (EI) m/z: 251.1 (7%) [M + 1]+, 178.1 (100%).
2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidin-6-yl)acetic acid (5). Compound 4 (0.500 g, 2.0 mmol) was dissolved in NaOH 0.5 M (7 mL) and the solution was stirred for 21 h. Reaction mixture was acidified with conc. HCl, then, the mixture was filtered and the resulting solid was washed with H2O and oven dried to afford 2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidin-6-yl)acetic acid (0.298 g, 1.26 mmol, 63% yield) as a white solid. mp > 300 °C; 1H NMR (400 MHz, DMSO‑d6): δ 12.25 (s, 1H), 10.74 (s, 1H), 7.74 (s, 2H), 7.53 (s, 2H), 2.94–2.80 (m, 2H), 2.71 (dd, J = 16.9, 4.9 Hz, 1H), 2.43 (dd, J = 17.0, 6.6 Hz, 1H), 2.35 (dd, J = 14.8, 12.9 Hz, 1H); 13C NMR (100.6 MHz, DMSO‑d6): δ 178.2, 175.6, 159.3, 154.7, 83.1, 41.1, 37.5, 24.9; IR (KBr) νmax(cm−1): 3444, 3368, 3160, 1698, 1646, 1397; HRMS (m/z): [M + 1]+ calcd. for C9H12N5O3: 238.0935; found: 238.0935.
2.2.2. General procedure for the synthesis of ligands 1a-e
1 eq 5, 1 eq of the corresponding aliphatic diamine, 2.2 eq EDC·HCl and 2.2 eq DMAP are dissolved in anhydrous DMF (2.5 mL) and the mixture is stirred for 24 h at room temperature. After that, the reaction mixture is concentrated under reduced pressure. The resulting solid is resuspended in HCl 0.5 M to afford the desired diamides by filtration.
N,N'-(pentane-1,5-diyl)bis(2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d] pyrimidin-6-yl)acetamide) (1a). 60 mg (0.11 mmol, 44% yield). mp > 300 °C; 1H NMR (400 MHz, DMSO‑d6): δ 10.48 (s, 2H), 7.98 (t, J = 5.5 Hz, 2H), 7.36 (s, 4H), 7.07 (s, 4H), 3.10–2.99 (m, 4H), 2.92–2.84 (m, 2H), 2.73 (dd, J = 15.6, 7.0 Hz, 2H), 2.60 (dd, J = 15.4, 4.5 Hz, 2H), 2.30–2.19 (m, 4H), 1.44–1.37 (m, 4H), 1.44–1.23 (m, 2H); 13C NMR (100.6 MHz, DMSO‑d6): δ 172.8, 169.7, 154.9, 83.5, 38.6, 36.2, 35.2, 28.8, 23.9, 22.2; IR (KBr) νmax(cm−1): 3361, 2932, 1648, 1569, 1495, 1462, 1395, 1329, 1274, 1226, 1164, 1088, 762, 600; HRMS (m/z): [M + 1]+ calcd. for C23H33N12O4, 541.2742; found, 541.2743.
N,N'-(heptane-1,7-diyl)bis(2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d] pyrimidin-6-yl)acetamide) (1b). 80 mg (0.14 mmol, 57% yield). mp > 300 °C;1H NMR (400 MHz, DMSO‑d6): δ 10.63 (s, 2H), 7.88 (t, J = 5.6 Hz, 4H), 7.56 (s, 4H), 7.36 (s, 4H), 3.10–2.96 (m, 4H), 2.91–2.85 (m, 2H), 2.74 (dd, J = 15.4, 6.9 Hz, 2H), 2.60 (dd, J = 15.5, 4.5 Hz, 2H), 2.32–2.19 (m, 4H), 1.40–1.37 (m, 4H), 1.25 (s, 6H); 13C NMR (100.6 MHz, DMSO‑d6): δ 172.8, 169.6, 153.5, 83.4, 39.1, 36.0, 35.0, 29.1, 28.5, 26.4, 22.0; IR (KBr) νmax(cm−1): 3383, 2927, 2853, 1642, 1497, 1387, 1318, 1270, 1226, 1162, 1086, 818, 761, 534; HRMS (m/z): [M + 1]+ calcd. for C25H37N12O4, 569.3055; found, 569.3052.
N,N'-(octane-1,8-diyl)bis(2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d] pyrimidin-6-yl)acetamide) (1c). 76 mg (0.13 mmol, 52% yield). mp > 300 °C; 1H NMR (400 MHz, DMSO‑d6): δ 10.14 (s, 2H), 7.82 (t, J = 3.8 Hz, 2H), 6.14 (s, 4H), 5.82 (s, 4H), 3.09–2.99 (m, 4H), 2.85–2.78 (m, 2H), 2.70–2.59 (m, 4H), 2.26–2.13 (m, 4H), 1.40–1.38 (m, 4H), 1.25 (s, 8H); 13C NMR (100 MHz, DMSO‑d6) δ 173.3, 170.0, 161.8, 161.6, 156.3, 83.7, 38.6, 36.9, 35.7, 29.1, 28.8, 26.5, 23. IR (KBr) νmax (cm−1): 3372, 3192, 2925, 1643, 1598, 1569, 1506, 1458, 1387, 1277, 1225, 1093, 1015, 815, 760, 678, 536, 436; HRMS (m/z): [M + 1]+ calcd. for C26H39N12O4, 583.3212; found, 583.3211.
N,N'-(nonane-1,9-diyl)bis(2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d] pyrimidin-6-yl)acetamide) (1d). 80 mg (0.14 mmol, 57% yield). mp > 300 °C; 1H NMR (400 MHz, DMSO‑d6): δ 10.20 (s, 2H), 7.80 (t, J = 5.5 Hz, 2H), 6.15 (s, 4H), 5.85 (s, 4H), 3.10–2.98 (m, 4H), 2.87–2.79 (m, 2H), 2.67–2.55 (m, 2H), 2.26–2.12 (m, 4H), 1.40– 1.38(m, 4H), 1.25 (s, 10H); 13C NMR (100 MHz, DMSO‑d6) δ 172.8, 169.6, 164.1, 83.5, 38.6, 36.1, 35.1, 29.1, 28.8, 26.5; IR (KBr) νmax (cm−1): 3338, 2925, 2852, 1646, 1565, 1494, 1465, 1389, 1320, 1269, 1226, 1165, 1087, 818, 762, 668, 535; HRMS (m/z): [M + 1]+ calcd. for C27H41N12O4, 597.3368; found, 597.3370.
N,N'-(decane-1,10-diyl)bis(2-(2,4-diamino-7-oxo-5,6,7,8-tetrahydropyrido[2,3-d] pyrimidin-6-yl)acetamide) (1e). 80 mg (0.14 mmol, 57% yield). mp > 300 °C; 1H NMR (400 MHz, DMSO‑d6): δ 10.54 (s, 2H), 7.88 (t, J = 5.6 Hz, 2H), 7.34 (s, 4H), 7.10 (s, 4H), 3.12–2.95 (m, 4H), 2.90–2.84 (m, 2H), 2.76–2.70 (m, 2H), 2.63–2.58 (m, 2H), 2.28–21.8 (m, 4H), 1.39 (t, J = 6.8 Hz, 4H), 1.25 (s, 12H); 13C NMR (100 MHz, DMSO‑d6) δ 173.0, 169.7, 155.8, 83.5, 38.7, 36.3, 35.2, 29.1, 29.0, 28.8, 26.5, 22.3; IR (KBr) νmax (cm−1): 3382, 2923, 2851, 1641, 1566, 1461, 1387, 1320, 1271, 1224, 1161, 1087, 816, 762, 534; HRMS (m/z): [M + 1]+ calcd. for C28H43N12O4, 611.3525; found, 611.3525.
2.3. Biochemistry
2.3.1. Protein purification
Competent BL21(DE3)pLysS E. coli (Invitrogen, Thermo Scientific, Carlsbad, CA) were transformed with the pGEX-6P-MBNL1-Δ105-His plasmid (GE Healthcare & Invitrogen) [40]. A single colony of the transfected BL21 was picked into 10 mL of LB with 100 μg/mL ampicillin and grown for 16 h at 37 °C and 200 rpm. 1 mL of the resulting culture mixture was inoculated to 1 L of LB with 100 μg/mL ampicillin and incubated at 37 °C and 150 rpm in a shaking incubator until OD600nm reached 0.5–0.6. The cell culture was then induced with 500 μM IPTG for 3 h at 30 °C. Thereafter, the culture media was centrifuged at 4000 xg and 4 °C for 30 min*. The supernatant was discarded and the pellet was resuspended in 50 mL of cell lysis buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.00), 5 mM imidazole, 0.1% IGEPAL CA-630, 5% glycerol and freshly added: 0.1 M PMSF, 5 mM benzamidine, 1 EDTA-free complete protease inhibitors tablet, 1 mM DNAse and 1 mg/mL lysozyme). The cell culture was lysed by alternating 15 cycles of sonication (1 min sonication – 1 min icing). To remove cell debris, the sample was centrifuged at 10,000 xg and 4 °C for 50 min and the supernatant was collected for purification. The supernatant was added to 2 mL of a previously equilibrated TALON® Metal Affinity Resin (Takara Bio, Kusatsu, JP) and incubated at 4 °C for 1 h. The resin was washed three times with 10 column volumes (30 mL × 3 times) of washing buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.00), 25 mM imidazole, 0.1% IGEPAL CA-630, 5% glycerol). The protein was eluted with 5 mL of elution buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8.00), 250 mM imidazole, 0.1% IGEPAL CA-630, 5% glycerol). The eluates were added to PD-10 Desalting Columns (GE Healthcare, Chicago, IL) that was previously preequilibrated using fluorescence test buffer (25 mM HEPES pH 7.4, 110 mM KCL, 10 mM NaCl, 1 mM MgCl2, 15 μM ZnCl2, 0.02%Tween-20). Protein purity was assessed via Comassie staining of SDS-PAGE electrophoresis 10% polyacrylamide gel (Figure S4). Finally, GST-MBNL1-His6 was quantified via direct absorbance spectroscopy at 280 nm using the ExPASy Protparam (protein parameter) tool to assess extinction coefficient (57300 M−1·cm−1) [41]. GST-MBNL1-His6 samples were stored at −80 °C with 50% glycerol. Prior to each use, samples were ultracentrifuged at 150,000 xg, 4 °C for 1 h and quantified to avoid aggregation issues.
2.3.2. AID 2675
The previously described assay was adapted to RNase pretreated 96-well plates (Greiner, Kremsmünster, AT). Reagents were premixed and added to the wells containing compounds solution (20 nM Biot-(CUG)6, 20 nM MBNL1-His6, 0,11 ng/mL anti-His-Tb, 10 µM SA-XL665 and 0,1 µM tested compound). All reagents were diluted in binding buffer composed of 25 mM HEPES pH 7.4, 110 mM KCl, 10 mM NaCl, 1 mM MgCl2, 15 μM ZnCl2, 0.02% Tween-20, 0.1% BSA and 5 mM freshly added DTT. After 60 min incubation at room temperature, the fluorescence was measured using a BMG Labtech Fluostar Optima microplate reader. Samples were irradiated at 340 nm, and fluorescence was read at 545 and FRET at 665 nm with a time gap of 30 µs and an integration time of 1500 µs.
2.4. Cell biology
2.4.1. Cell culture
Immortalized human myoblasts, control and DM1 derived from primary dermal fibroblasts of control and DM1 affected individuals respectively, were used as described in previous work [13]. Fibroblasts were sequentially infected with retroviral vectors carrying TERT and Hygromycin selection (Addgene #1773, Cambridge, MA) [42], and with retroviruses carrying estrogen-inducible mouse Myod1 and puromycin selection (Addgene #13494) [43]. Cells were propagated in dishes coated with collagen from rat tail at 5 µg/cm2 (Gibco, Thermo Fisher Scientific, Waltham, MA), using DMEM without phenol red (Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 15% FBS (Sigma-Aldrich, St. Louis, MO) and incubated for 48 h. Differentiation to myotubes was induced by growing cells to confluency and replacing the proliferation medium with differentiation medium consisting of DMEM without phenol red supplemented with 10-7 M β-estradiol (Sigma-Aldrich) and 2% horse serum and incubating for 24 h. All cells were incubated under a 5% CO2 atmosphere at 37 °C. For PCR and WB experiments 35 mm dishes were seeded at 1.3·103 cells/cm2 whereas for FISH and IF cells where seeded on glass coverslips at 3.2·103 cells/cm2. DM1 cells were treated with the compounds at 10 µM for 48 h in all experiments.
2.4.2. FISH IF
Treated cells were washed twice in PBS for 5 min. After that, cells were fixed with 2% formaldehyde for 30 min at 4 °C. After fixation, coverslips were washed 3 times in PBS, and permeabilized in 0.4% Triton™ X-100 (Sigma-Aldrich) in PBS for 10 min. After washing with PBS 3 times, coverslips were incubated in 40% formamide and 2x SSC for 10 min at room temperature and then hybridized with (CAG)6 probe labeled with Texas Red at the 5′ end (IDT, Coralville, IA) (1 ng/μL for 2 h at 37 °C in 30% formamide, 2x SSC, 0.02% BSA, 67 ng/μL yeast tRNA, 2 mM vanadyl ribonucleoside complex). Cells were then washed first in 40% formamide and 2x SSC at 45 °C for 30 min. After that, coverslips were washed sequentially in 2x SSC and 1x SSC at 45 °C for 15 min each. Then, samples were incubated with anti-MBNL1 3A4 antibody (Santa Cruz Biotechnology) solution in PBS at room temperature for 1 h. After that samples were washed 3 times with PBS and incubated using goat anti-mouse IgG-Alexa Fluor 488 (Invitrogen, Thermo Scientific, Carlsbad, CA). After that, samples were incubated with Hoechst 33,258 (Invitrogen) in PBS at room temperature for 10 min. Finally, coverslips were washed 3 times in PBS, water and mounted on glass slides with ProLong™ Gold Antifade Mountant (Invitrogen). Samples were examined with an Olympus AX70 immunofluorescence microscope. Images were recorded on an Olympus XM10 camera and processed using the Olympus CellSens Standard 1.8.1 software. Cell quantification was conducted using ImageJ software.
2.4.3. Western blot
Treated cells were washed twice with PBS for 5 min. After that, cells were ice cooled and lysed by adding RIPA buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 0.5% Sodium deoxycholate, 0.1% SDS, 1% NP40, 1 mM EDTA, 5 mM NaF, 5 mM Na3VO4 and Protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). After 5 min of incubation at 0 °C, cells were scraped, and the lysate was transferred to an Eppendorf tube. The extract was centrifuged at 14,000 xg, 4 °C for 10 min to remove cell debris. The supernatant was transferred to a new Eppendorf tube and total protein concentration was assessed using the Micro BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Homogeneous protein samples mixed with Laemmli sample buffer and Novex Nupage sample reducing agent (Invitrogen, Thermo Scientific, Carlsbad, CA) were loaded and separated using Bolt 4–20% Tris-glycine SDS-PAGE (Invitrogen, Thermo Scientific, Carlsbad, CA). Gels were run in Novex Nupage MOPS buffer (Invitrogen, Thermo Scientific, Carlsbad, CA) setting voltage to 220 V for 40 min. After gel running, proteins were transferred to PVDF or nitrocellulose membranes using a stack soaked in transfer buffer (25 mM Tris, 190 mM glycine, 10% methanol) and setting current at 350 mA for 90 min at 4 °C. After checking correct transferring using ponceau staining, membranes were blocked for 1 h with 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween 20 (TBST). After 3 washes with TBST, membranes were then exposed to primary antibodies overnight at 4 °C (MBNL1, CUGBP1, myogenin, myosin). Subsequently, membranes were washed 3 times in TBST and incubated in horseradish-peroxidase-conjugated anti-mouse antibody solution at room temperature for 1 h. Finally, Chemiluminescence detection was achieved using ETA C 2.0 and NOVA 2.0 reagents (Cyanagen, Bologna, Italy) in a ChemiDoc XRS+ (Bio-Rad, Hercules, CA). Imaging and quantitation of the bands were carried out by the ChemiDoc XRS Western Blot Imaging System using the ImageLab 4.0 software (Bio-Rad, Hercules, CA). Equal loading of proteins onto the gel was confirmed by immunodetection of vinculin using Monoclonal Ab αVinculin (Sigma-Aldrich, St. Louis, MO) at room temperature for 1 h.
2.4.4. PCR
RNAs from both control and patient cell lines were extracted with TRIzol reagent (Invitrogen, Thermo Scientific, Carlsbad, CA). Cells were washed twice with PBS and frozen at −80 °C for 30 min. After that, TRIzol reagent was added and cells were scraped off the plates, transferred into Eppendorf tubes and incubated at room temperature for 5 min. Chloroform was added and the resulting mixture was vigorously shaken and incubated 3 min at room temperature. The aqueous phase was separated by centrifugation, and RNA precipitated by adding IPA. The sample was centrifuged at 12,000 xg for 10 min at 4 °C. The supernatant was discarded, and the RNA was washed twice with ethanol 80% prechilled at −20 °C. After drying, samples were dissolved in RNAse free water, incubated 5 min at 60 °C, cooled in ice and quantified using Nanodrop ND-1000 (Nanodrop Technologies, Wilmington, DE). After that, RNA samples were retro transcribed with the GoScript Reverse Transcription System (Promega, Fitchburg, WI). Oligo (dT) and random primers were preincubated with RNA for 5 min at 70 °C. The resulting mixture was chilled in ice for 5 min. Next, reactions were prepared by adding 3 mM MgCl2, PCR Nucl. Mix, Rnasin, Reverse Transcriptase and placed in the thermal cycler at 42 °C for 1 h, 70 °C for 15 min and finally chilled. The resulting cDNA samples were amplified with GoTaq Flexi DNA Polymerase (Promega, Fitchburg, WI) for 35 cycles using the specific primers (Table S2). Upon reaction ending, samples were loaded on agarose gel containing Midori staining (Nippon Genetics, Düren, GE). Quantitation of amplified bands on gel images was performed using UVITec1D software. Exon inclusion was quantified as the percentage of the total intensity of both isoforms.
2.4.5. Statistics
All biological experiments were performed at least in triplicate, and pictures represent typical examples. Data are presented as mean ± standard deviation (SD). A two-tailed unpaired t-test was applied for assessment of the statistical significance of differences between two groups. A p-value lower than 0.05 was deemed statistically significant. Statistical analyses were conducted using GraphPad Prism version 7.01 (GraphPad Software, Inc., La Jolla, CA, USA).
3. Results
3.1. Rational design
The binding mechanism of 1a-1e compounds was assessed by combining molecular docking and molecular dynamics simulations, using a short hairpin CUG16 (shCUG) model as receptor [28]. Although allowing moderate motion of the side chains, flexible docking does not guarantee the generation of Janus-Wedge arrangement. Given the difficulty in dealing with static structures, molecular dynamics simulations were applied to generate an ensemble of shCUG conformations. The length of the hairpin model ensures a sufficiently large double-stranded conformation for bivalent ligands to bind consecutive or non-consecutive uridine units. It is also short enough to allow long production times in MDs, which is a critical point in this study. Dynamics of non-canonical U•U pairs were therefore analyzed to capture the preferred conformation able to stabilize JW interactions. Docking was pharmacophore-guided considering that base recognizers tend to fall in base pairs via major groove (Fig S1) [24]. The pyrido[2,3-d]pyrimidine unit fulfils the requirements to act as uracil recognizer, establishing the desired hydrogen bond (Fig. 2a).
Fig. 2.

Molecular mechanics results summary. a) Representative snapshot taken from MD showing the interaction mechanism between 1a compound and shCUG b) MM-PBSA values of free Gibbs energy binding calculated over 20 ns of each ligand in their respective MD. c) Structural parameter analysis of opening values. Full-length MD is displayed in boxplot graph format.
The stability of docked complexes was assessed by molecular dynamics (MD, Fig. S2). The spacer length determines the binding mechanism of pyrido[2,3-d]pyrimidine derivatives and their ability to act as bivalent ligands (targeting two U•U pairs); shorter linkers would not be long enough to reach both mismatches while longer ones could trigger swapping the interaction to a more distant base pair. The stability of all complexes was assessed by studying the structural parameters and quantified through MMPBSA analysis.
Variations in the binding mechanism would explain the difference in Poisson-Boltzmann Gibbs free energy of binding (Δ°GPB), predicted by MMPBSA (Fig. 2b). Results show a clear dependence of the spacer length, highlighting 1a and 1e (-37 ± 5 kcal/mol and −50 ± 4 kcal/mol respectively). The pharmacophore allows both consecutive and non-consecutive binding for the ligands. Short ligands (1a) can achieve higher binding energies via consecutive mismatch interaction whereas longer ones reach alternate mismatches (1d, 1e). The nature of the spacer would account for que significant energy gap between pyrido[2,3-d]pyrimidine derivatives (1a-1e) and the positive control (Z). The bisamidinium linker gave not only the desired distance between active sites of the molecule but also a boost in unspecific binding energy while linker moieties used in the current work lack of any functional groups. As this was the first time that this set of structures have been evaluated against DM1, it is desirable to assess whether the primary scaffolds are active or not, without any further interactions from the moiety core. For this reason, linkers were maintained as simple as possible although it is a drawback in terms of total binding energy.
The analysis of structural parameters in the hairpin base-pairs evidenced that most of the compounds under study increase the stability of the system by reducing the dispersion of structural parameters in the U•U mismatch and its vicinities (Fig. S3), particularly of the opening value (Fig. 2c). From all the structural parameters considered, opening singularly represents the instability of a generated U•U mismatch and it gives valuable data when a Janus-Wedge interaction mechanism is established (inducing an opening value shift about 60°).
3.2. Synthesis
A standard procedure to synthesize the pyrido[2–3-d]pyrimidine scaffold [44] has been adapted to fulfil the structural features required for Janus-Wedge interaction: (a) leave the pyrimidinic amino groups unsubstituted (since these amines are meant to play a critical role in hydrogen bonding between ligand and receptor) and (b) to be able to derivate the core with a reactive group, to bind the linker, without compromising the amines. We chose to introduce a carboxylic acid group at the pyridone ring due to the widely reported robustness of the click-chemistry reactions between carboxylic acids and aliphatic amines even in complex systems as our extremely polar pyrido[2–3-d]pyrimidines. Upon that, a full novel synthetic pathway was designed and performed to achieve the desired compounds (Fig. 3).
Fig. 3.

General synthetic pathway to attain structures 1a-e. a) H2SO4/MeOH reflux 21 h. b) Guanidine carbonate, malononitrile in MeOH MW 140 °C 15 min. c) NaOH 0.5 M RT 21 h + conc HCl. d) aliphatic diamine, EDC·HCl, DMAP, and K2CO3 in DMF RT 24 h.
The above-mentioned synthesis leads to compounds 1a-e in a 4-step synthesis. Starting from the commercially available itaconic acid (2) a Fischer esterification using MeOH and H2SO4 renders the methyl diester 3 in 48% yield. The next step is to build the heterocyclic core using our two-step synthetic protocol. First a Michael addition of malononitrile to the α, β-unsaturated ester 3 and the resulting adduct is reacted with guanidine to obtain the corresponding pyridopyrimidine 4 (45% aggregate yield). However, the α, β-unsaturated ester 3 is also suitable to use our microwave-assisted multicomponent reaction to obtain pyrido[2,3-d]pyrimidines. Thus, 3 is placed in a sealed vial together with 0.5 eq. of malononitrile and 0.5 eq of guanidine carbonate in anhydrous methanol and brought to 140 °C for 15 min to give pyrido[2,3-d]pyrimidine 4 in 71% yield. Once the main base-pair recognizing moiety is formed, the next steps are to prepare and bind the aliphatic linker. First, the carboxymethyl group is saponified to achieve the corresponding carboxylic acid. Thus, 4 is dissolved in 0.5 M aqueous NaOH and stirred at room temperature for 21 h, and the reaction mixture is acidified to afford 5 in 63% yield. Ligands and linkers are bound via Steglich amidation using EDC·HCl as coupling agent and DMAP as catalyst. Given the fact that amino groups at C2 and C4 of the pyrimidine ring of 5 are much less nucleophilic than the aliphatic primary amines, the only expected by-products of the coupling were mono-aminated derivatives.
Pyridopyrimidines 1a-e hamper classical purification techniques (i.e. flash chromatography) due to its high MW and polarity. The sidechain unsubstituted diamino pyridopyrimidines tend to form stable hydrogen bonds with other pyridopyrimidines giving stable microcrystalline structures that are barely soluble in water. For this reason, most reagents and reaction by-products must be eliminated via washes instead of classical flash column chromatography. This plays a critical role in a double amidation where potential multiple species can be obtained (product, by-products, excess of reactants). Despite that, compounds 1a-e were successfully synthetized, and their structure confirmed by NMR and high-resolution mass spectrometry in 44–57% yields (see Supporting Information).
3.3. Biochemistry
One of the main challenges of dealing with a rare disease in drug development stages is the lack of any commercial kits or suppliers for assessing the potency of novel drug candidates. For this reason, we chose to reproduce the already described Förster Resonance Energy Transfer (FRET) based AID 2675 assay [40]. This fluorescent test can measure the ability of candidates to free MBNL1 of an DM1-like oligo-RNA. The ability of MBNL1 (and the recombinant form used) to recognize and bind to small GC rich structures is the key factor of the AID 2675 molecular basis. Two fluorophores bind to both protein and RNA (via antibody recognition and avidin–biotin interaction respectively) making FRET possible when the complex is formed (Fig. 4a). If the complex is broken by a candidate, fluorescence intensity falls as the fluorophores are no longer near enough. Thus, FRET does not occur.
Fig. 4.

a) Graphical representation of AID2675 FRET test molecular basis and its principal components: 5′-biotinylated RNA oligo Biot-(CUG)12 and the recombinant GST-MBNL1-His6 form a stable complex. FRET donor anti-His6-Terbium cryptate and FRET acceptor streptavidin-conjugated XL665 bind to His tag and Biotin, respectively. While no active compound is present in the reaction media, 340 nm beam light hits Tb conjugate and this is close enough to transfer the energy to XL665 conjugate emitting at 665 nm. If a compound can free the protein no FRET occurs, and Tb fluorophore emits at 545 nm. b) Normalized FRET intensity measured as a relationship between the fluorescence intensity of the signals of 545 and 665 nm (average of three measures). Final concentrations of studied candidates were set at 0.1 µM and 20 nM of protein-RNA complex. Z is included for comparison.
To obtain the recombinant MBNL1, transfected E. coli were grown, and the purified recombinant protein was harvested prior its use in the test. Minor changes to the original protocol allowed achieving the final recombinant protein with fewer impurities and to avoid aggregation issues (Fig. S4). The whole preparation process was conducted in RNase free conditions and all reagents were RNase free grade to avoid synthetic oligo CUG to degrade before measuring the protein displacement.
Compound concentration was set at a very low range to avoid fluorescent interactions and possible precipitation issues. However, it is interesting to see that even in these conditions structure 1a can bind tightly to RNA displacing recombinant MBNL1 and therefore reducing the FRET intensity a 33% compared to the negative control (Fig. 4b). The rest of the candidates also exhibit some MBNL1 displacing potency. These results encouraged us to further study these candidates in patient-derived cells.
3.4. Cell biology
In order to determine the drug efficacy and gain understanding of the behavior of compounds 1a-e in a complex system, we performed the biological characterization of the candidates. Dermal fibroblasts of both healthy and DM1 affected individuals were used to run the experiments. Both cell lines were previously immortalized by infection a with retroviral vector carrying the human telomerase (hTERT) gene. Moreover, cells were also retrovirally infected with vector producing an inducible myogenic differentiation 1 (MYOD1) transcription factor fused to the estrogen receptor (ER) hormone-binding domain (MYOD1-ER) [43]. On the one hand, TERT ensures a homogeneous proliferation over cycles of duplication with no evidence of genetic affection related upon infection. On the other hand, MYOD1 allows, via estradiol addition to the culture media, the expression of myogenic factors leading to differentiation into myoblasts and finally forming mature myotubes. All the characteristic events of DM1 such as CUG nuclear foci, splicing factor sequestration and missplicing events were maintained in the DM1 patient-derived cells in culture and absent in control cells derived from healthy individuals [13]. Original fibroblasts from DM1 patient had 290 repeats. Nonetheless, further expansion occurs in culture. Thus, and despite the cell population's heterogeneity, the most abundant length is in PCR amplification is larger than 1000 CTG repeats. The biological activity of the compounds on these DM1 features was tested on these cell models. Being skeletal muscle the most affected tissue in DM1 patients, the use of myogenic cell models offers the advantage to test the effect of the designed drugs on both DM1 hallmarks and differentiation capacity of these cells, in the perspective of a human therapy. To evaluate 1a-1e and Z in cell culture some considerations were made. Some of these candidates underwent crystallization in the culture media at concentrations over 50 µM in previous studies. For this reason, and to make all candidates comparable, their concentration was set to 10 µM. These test concentrations are lower than the ones used in previously reported studies [24]. Nevertheless, it is compatible with a possible in vivo use of the candidates.
In order to directly see whether MBNL1 was sequestered in CUG foci, RNA Fluorescent In Situ Hybridization (RNA-FISH) analysis combined to immunofluorescent staining (IF) of MBNL1 protein with a specific antibody were performed as described previously (Fig. 5, extended in Fig. S5) [45]. Ligands 1a-e were designed to specifically bind to CUG motifs and obtained FISH results are consistent with this prediction. Nevertheless, a reduction in the total number of nuclear foci was not expected as that means transcription inhibition. The staining with the (CAG)6 RNA-FISH probe (red) showed no significant difference between untreated and treated cells in the foci count per cell ratio for 1a-1e compounds. This is consistent with the prediction since the specific binding of these ligands to CUG motifs is not expected to affect the expression level of the mutant transcripts. However, some compounds were able to reduce the amount of colocalized MBNL1 in those nuclei. That was observed via IF using anti-MBNL1 mAb (green). Interestingly, 1a induces an important depletion of MBNL1 sequestered in nuclear foci of above 23%, which means that this compound can selectively attach to CUG and competitively displace MBNL1, eventually reducing the number of nuclear foci containing the protein in a significant manner (Fig. 5b).
Fig. 5.

a) FISH images using a (CAG)6 probe labelled with Texas Red at the 5′ end in combination with immunofluorescence staining. To verify the co-localization of MBNL1 in ribonuclear inclusions, following the last post-hybridization wash, cells were stained sequentially with antibodies to MBNL1 and with goat anti-mouse antibody conjugated with Alexa Fluor 488. Nuclei were visualized with Hoechst 33,258 dye. Insets show zoomed-in images (scale bars 20 µm). b) Quantitative FISH analysis of total foci per cell and colocalized MBNL1 in foci. Results are presented as mean (±SD) of n = 3 experiments. Statistical significance was assessed from a two-tailed Student's t-test (**p < 0.01, *p < 0.05).
Another important event in DM1 is missplicing deficiencies of a large number of gene transcripts. To study possible recoveries in the splicing pattern we chose two gene transcripts known to be affected in DM1: the sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (SERCA1) and the insulin receptor (INSR). The inclusion or not of exon 22 of SERCA1 and exon 11 of INSR was monitored via semiquantitative RT-PCR to check whether missplicing events were modulated by test compounds present in the media. The results showed that at the studied conditions (see Supporting information) the missplicing of these genes was not significantly affected by the addition to the culture media of either ligands 1a-e or Z (Fig. S6).
To check if structures 1a-e affected the expression levels of DM1-related splicing factors or myogenic differentiation markers, Western blot analysis was performed. The effect of compounds was studied on 4 protein levels: MBNL1, CELF1, myosin and myogenin (Fig. S7). Ligands 1a-e were observed not to affect the total amount of MBNL1 or CELF1, which is in fact a positive result, as these proteins are essential and any anti-DM1 drug candidate aims to recover the natural balance between these antagonists. On the other hand, myosin and myogenin are crucial muscle differentiation proteins [46], [47]. A decrease in their total cell concentration may alter cell differentiation. Therefore, looking at the state of differentiation is an important indication of a possible toxic effect of a tested compound. Our findings with Western blot analysis show that only in the case of ligand 1e a visible decrease of Myogenin levels is observed while no effect at all was observed for the remaining compounds.
4. Discussion and conclusion
Base recognition has been one of the main trends in small molecule drug development in the DM1 field. This is due to the implementation of a comprehensive structure–activity relationship [18], [48]. The triangular shape of pyrido[2,3-d]pyrimidin-7(6H)-ones, combined with the spatial disposition of hydrogen bond donors and acceptors in such core, resulted in a potential U•U recognizer interaction.
Computer-aided rational design studies have been described in the literature to study ligand-RNA interaction in recent years [48], [49], [50]. It is worth noting that studies describing a rational approach, leaned on ligand-based drug design strategies, are commonly applied on commercially available chemical libraries [51]. This study provides tools to design novel compounds with structure-based methods, using the Janus-Wedge recognition as strategy to stabilize CUG in its non-toxic conformation. Selectivity is one of the key points when designing small molecules to target the DM1 pathogenic RNA transcripts. Around 5% of DM1 patients do not have a pure CTG repeat, including interrupting CCG or CGG triplets, which may alter RNA’s secondary structure [52]. Thus, the molecular modeling approach herein described could be adapted to embrace other kind of transcripts.
A set of 5 novel chemical entities based on two pyrido[2,3-d]pyrimidin-7(6H)-one scaffold separated by an aliphatic linker of variable length was studied (1a-e). These candidates would exhibit different interaction mechanisms with CUG repeats depending on their length, according to their predicted Gibbs free energy of binding. Simulations point to the 5-carbon length linker for an optimal recognition of consecutive U·U mismatches the 10-carbon length linker and for alternate mismatches the 10-carbon linker. Moreover, the analysis of base pair opening showed a reduction in data variance for 1a stating that the candidate can stabilize the CUG environment and maintain the JW interaction during the whole simulation. Interestingly, this observation was validated in biological studies that proved the ability of 1a for CUG binding and triggering MBNL1 recovery in vitro (AID2675 test). It is worth of attention that 1a also exhibited these features in DM1 patient-derived myoblasts, reducing the amount of observed MBNL1 in FISH-IF microscopy. These findings not only provide valuable data about the length needed to reach two consecutive U•U mismatches for the pyrido[2,3-d]pyrimidines scaffold suggested in this study. Moreover, results confirm the usefulness of molecular modelling for the rational design of JW base recognizers with potential anti-DM1 activity.
These results allow a better understanding of the biological interaction of pyrido[2,3-d]pyrimidine structures and set a starting point in future drug discovery studies.
Funding
This work was supported by the Plan Estatal de I+D+i 2013–2016, and the Instituto de Salud Carlos III - Subdirección General de Evaluación y Fomento de la Investigación (FIS13-0386, including funds from FEDER). Roger Estrada-Tejedor thankfully acknowledge the financial support of l’Obra Social “La Caixa”.
CRediT authorship contribution statement
Raul Ondono: Conceptualization, Methodology, Formal analysis, Investigation, Writing - original draft, Visualization. Ángel Lirio: Methodology, Software, Investigation, Data curation. Carlos Elvira: Methodology, Investigation, Supervision. Elena Álvarez-Marimon: Methodology, Investigation, Supervision. Claudia Provenzano: Methodology, Investigation, Supervision. Beatrice Cardinali: Methodology, Investigation, Supervision. Manuel Pérez-Alonso: Supervision, Funding acquisition. Alex Perálvarez-Marín: Supervision. José I. Borrell: Supervision. Germana Falcone: Methodology, Investigation, Supervision, Writing - review & editing. Roger Estrada-Tejedor: Conceptualization, Supervision, Funding acquisition, Project administration, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Authors thank professor R. Artero from University of Valencia for his valuable contribution in revising the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.11.053.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hummerich H., Lehrach H. Trinucleotide repeat expansion and human disease. Electrophoresis. 1995;16(1):1698–1704. doi: 10.1002/elps.11501601282. [DOI] [PubMed] [Google Scholar]
- 2.Cleary J.D., Ranum L.P.W. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet. 2013;22(R1):R45–R51. doi: 10.1093/hmg/ddt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas J.D., Oliveira R., Sznajder Ł.J., Swanson M.S. Myotonic dystrophy and developmental regulation of RNA processing. Compr Physiol. 2018;8:509–553. doi: 10.1002/cphy.c170002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chong-Nguyen C., Wahbi K., Algalarrondo V., Bécane H.M., Radvanyi-Hoffman H., Arnaud P., Furling D., Lazarus A., Bassez G., Béhin A., Fayssoil A., Laforêt P., Stojkovic T., Eymard B., Duboc D. Association Between Mutation Size and Cardiac Involvement in Myotonic Dystrophy Type 1: An Analysis of the DM1-Heart Registry. Circ Cardiovasc Genet. 2017;10(3) doi: 10.1161/CIRCGENETICS.116.001526. [DOI] [PubMed] [Google Scholar]
- 5.André LM, Van Cruchten RTP, Willemse M, Wansink DG. (CTG)n repeat-mediated dysregulation of MBNL1 and MBNL2 expression during myogenesis in DM1 occurs already at the myoblast stage. PLoS One 2019;14:1–17. https://doi.org/10.1371/journal.pone.0217317. [DOI] [PMC free article] [PubMed]
- 6.Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum Mol Genet 2012;21:3558–67. https://doi.org/10.1093/hmg/dds185. [DOI] [PubMed]
- 7.Jain A., Vale R.D. RNA phase transitions in repeat expansion disorders. Nature. 2017;546(7657):243–247. doi: 10.1038/nature22386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mankodi A., Urbinati C.R., Yuan Q.-P., Moxley R.T., Sansone V., Krym M. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 9.Philips AV, Timchenko LT, Cooper TA. Disruption of Splicing Regulated by a CUG-Binding Protein in Myotonic Dystrophy. Science (80-) 1998;280:737–41. https://doi.org/10.1126/science.280.5364.737. [DOI] [PubMed]
- 10.Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A., Swanson M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci. 2006;103(31):11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overby S.J., Cerro-Herreros E., Llamusi B., Artero R. RNA-mediated therapies in myotonic dystrophy. Drug Discovery Today. 2018;23(12):2013–2022. doi: 10.1016/j.drudis.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Konieczny P., Selma-Soriano E., Rapisarda A.S., Fernandez-Costa J.M., Perez-Alonso M., Artero R. Myotonic dystrophy: candidate small molecule therapeutics. Drug Discovery Today. 2017;22(11):1740–1748. doi: 10.1016/j.drudis.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Provenzano C., Cappella M., Valaperta R., Cardani R., Meola G., Martelli F., Cardinali B., Falcone G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol Ther Nucleic Acids. 2017;9:337–348. doi: 10.1016/j.omtn.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo Scrudato M., Poulard K., Sourd C., Tomé S., Klein A.F., Corre G., Huguet A., Furling D., Gourdon G., Buj-Bello A. Genome Editing of Expanded CTG Repeats within the Human DMPK Gene Reduces Nuclear RNA Foci in the Muscle of DM1 Mice. Mol Ther. 2019;27(8):1372–1388. doi: 10.1016/j.ymthe.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jauvin D., Chrétien J., Pandey S.K., Martineau L., Revillod L., Bassez G., Lachon A., MacLeod A.R., Gourdon G., Wheeler T.M., Thornton C.A., Bennett C.F., Puymirat J. Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol Ther Nucleic Acids. 2017;7:465–474. doi: 10.1016/j.omtn.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Lopez A., Llamusi B., Orzaez M., Perez-Paya E., Artero R.D. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc Natl Acad Sci. 2011;108(29):11866–11871. doi: 10.1073/pnas.1018213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rzuczek S.G., Colgan L.A., Nakai Y., Cameron M.D., Furling D., Yasuda R., Disney M.D. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat Chem Biol. 2017;13(2):188–193. doi: 10.1038/nchembio.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J.Y., Bai Y., Chembazhi U.V., Peng S., Yum K., Luu L.M., Hagler L.D., Serrano J.F., Chan H.Y.E., Kalsotra A., Zimmerman S.C. Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc Natl Acad Sci USA. 2019;116(18):8709–8714. doi: 10.1073/pnas.1820827116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coonrod L.A., Nakamori M., Wang W., Carrell S., Hilton C.L., Bodner M.J., Siboni R.B., Docter A.G., Haley M.M., Thornton C.A., Berglund J.A. Reducing Levels of Toxic RNA with Small Molecules. ACS Chem Biol. 2013;8(11):2528–2537. doi: 10.1021/cb400431f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palomo V., Perez D.I., Roca C., Anderson C., Rodríguez-Muela N., Perez C., Morales-Garcia J.A., Reyes J.A., Campillo N.E., Perez-Castillo A.M., Rubin L.L., Timchenko L., Gil C., Martinez A. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J Med Chem. 2017;60(12):4983–5001. doi: 10.1021/acs.jmedchem.7b00395.s002. [DOI] [PubMed] [Google Scholar]
- 21.Jahromi A.H., Nguyen L., Fu Y., Miller K.A., Baranger A.M., Zimmerman S.C. A Novel CUG exp ·MBNL1 Inhibitor with Therapeutic Potential for Myotonic Dystrophy Type 1. ACS Chem Biol. 2013;8(5):1037–1043. doi: 10.1021/cb400046u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.deLorimier E., Hinman M.N., Copperman J., Datta K., Guenza M., Berglund J.A. Pseudouridine Modification Inhibits Muscleblind-like 1 (MBNL1) Binding to CCUG Repeats and Minimally Structured RNA through Reduced RNA Flexibility. J Biol Chem. 2017;292(10):4350–4357. doi: 10.1074/jbc.M116.770768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Branda N., Kurz G., Lehn J.-M. JANUS WEDGES: a new approach towards nucleobase-pair recognition. Chem Commun. 1996;(21):2443. doi: 10.1039/cc9960002443. [DOI] [Google Scholar]
- 24.Wong C.-H., Nguyen L., Peh J., Luu L.M., Sanchez J.S., Richardson S.L., Tuccinardi T., Tsoi H.o., Chan W.Y., Chan H.Y.E., Baranger A.M., Hergenrother P.J., Zimmerman S.C. Targeting Toxic RNAs that Cause Myotonic Dystrophy Type 1 (DM1) with a Bisamidinium Inhibitor. J Am Chem Soc. 2014;136(17):6355–6361. doi: 10.1021/ja5012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, et al. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res 2007;35:5474–86. https://doi.org/10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed]
- 26.Camarasa M., Puig de la Bellacasa R., González À.L., Ondoño R., Estrada R., Franco S., Badia R., Esté J., Martínez M.Á., Teixidó J., Clotet B., Borrell J.I. Design, synthesis and biological evaluation of pyrido[2,3-d]pyrimidin-7-(8H)-ones as HCV inhibitors. Eur J Med Chem. 2016;115:463–483. doi: 10.1016/j.ejmech.2016.03.055. [DOI] [PubMed] [Google Scholar]
- 27.Recasens-Zorzo C., Cardesa-Salzmann T., Petazzi P., Ros-Blanco L., Esteve-Arenys A., Clot G., Guerrero-Hernández M., Rodríguez V., Soldini D., Valera A., Moros A., Climent F., González-Barca E., Mercadal S., Arenillas L., Calvo X., Mate J.L., Gutiérrez-García G., Casanova I., Mangues R., Sanjuan-Pla A., Bueno C., Menéndez P., Martínez A., Colomer D., Tejedor R.E., Teixidó J., Campo E., López-Guillermo A., Borrell J.I., Colomo L., Pérez-Galán P., Roué G. Pharmacological modulation of CXCR4 cooperates with BET bromodomain inhibition in diffuse large B-cell lymphoma. Haematologica. 2019;104(4):778–788. doi: 10.3324/haematol.2017.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González ÀL, Teixidó J, Borrell JI, Estrada-Tejedor R. On the applicability of elastic network models for the study of RNA CUG trinucleotide repeat overexpansion. PLoS One 2016;11:1–20. https://doi.org/10.1371/journal.pone.0152049. [DOI] [PMC free article] [PubMed]
- 29.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 30.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9(7):3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 33.Aytenfisu A.H., Spasic A., Grossfield A., Stern H.A., Mathews D.H. Revised RNA Dihedral Parameters for the Amber Force Field Improve RNA Molecular Dynamics. J Chem Theory Comput. 2017;13(2):900–915. doi: 10.1021/acs.jctc.6b00870.s002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izadi S., Anandakrishnan R., Onufriev A.V. Building Water Models: A Different Approach. J Phys Chem Lett. 2014;5(21):3863–3871. doi: 10.1021/jz501780a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salomon-Ferrer R., Götz A.W., Poole D., Le Grand S., Walker R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput. 2013;9(9):3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 36.Le Grand S., Götz A.W., Walker R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun. 2013;184(2):374–380. doi: 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- 37.Götz A.W., Williamson M.J., Xu D., Poole D., Le Grand S., Walker R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8(5):1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu X.-J., Olson W.K. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3(7):1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gualandi A., Mazzarella D., Ortega-Martínez A., Mengozzi L., Calcinelli F., Matteucci E., Monti F., Armaroli N., Sambri L., Cozzi P.G. Photocatalytic Radical Alkylation of Electrophilic Olefins by Benzylic and Alkylic Zinc-Sulfinates. ACS Catal. 2017;7(8):5357–5362. doi: 10.1021/acscatal.7b01669.s001. [DOI] [Google Scholar]
- 40.Chen C.Z., Sobczak K., Hoskins J., Southall N., Marugan J.J., Zheng W., Thornton C.A., Austin C.P. Two high-throughput screening assays for aberrant RNA–protein interactions in myotonic dystrophy type 1. Anal Bioanal Chem. 2012;402(5):1889–1898. doi: 10.1007/s00216-011-5604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Artimo P., Jonnalagedda M., Arnold K., Baratin D., Csardi G., de Castro E., Duvaud S., Flegel V., Fortier A., Gasteiger E., Grosdidier A., Hernandez C., Ioannidis V., Kuznetsov D., Liechti R., Moretti S., Mostaguir K., Redaschi N., Rossier G., Xenarios I., Stockinger H. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40(W1):W597–W603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Counter C.M., Meyerson M., Eaton E.N., Ellisen L.W., Caddle S.D., Haber D.A., Weinberg R.A. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene. 1998;16(9):1217–1222. doi: 10.1038/sj.onc.1201882. [DOI] [PubMed] [Google Scholar]
- 43.Hollenberg S.M., Cheng P.E.I.F., Weintraub H. Trans-Activation and Muscle Determination. Proc Natl Acad Sci U S A. 1993;90:8028–8032. doi: 10.1073/pnas.90.17.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mont N., Teixidó J., Kappe C.O., Borrell J.I. A one-pot microwave-assisted synthesis of pyrido[2,3-d]pyrimidines. Mol Divers. 2003;7(2-4):153–159. doi: 10.1023/B:MODI.0000006808.10647.f8. [DOI] [PubMed] [Google Scholar]
- 45.Cardani R, Mancinelli E, Sansone V, Rotondo G, Meola G. Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsy. Eur J Histochem 2004;48:437–42. https://doi.org/10.4081/918. [DOI] [PubMed]
- 46.Sharma A, Agarwal M, Kumar A, Kumar P, Saini M, Kardon G, et al. Myosin Heavy Chain-embryonic is a crucial regulator of skeletal muscle development and differentiation. BioRxiv 2018:261685. https://doi.org/10.1101/261685.
- 47.Flynn JM, Meadows E, Fiorotto M, Klein WH. Myogenin regulates exercise capacity and skeletal muscle metabolism in the adult mouse. PLoS One 2010;5. https://doi.org/10.1371/journal.pone.0013535. [DOI] [PMC free article] [PubMed]
- 48.Li J., Nakamori M., Matsumoto J., Murata A., Dohno C., Kiliszek A. A Dimeric 2,9-Diamino-1,10-phenanthroline Derivative Improves Alternative Splicing in Myotonic Dystrophy Type 1 Cell and Mouse Models. Chem - A Eur J. 2018;24:18115–18122. doi: 10.1002/chem.201804368. [DOI] [PubMed] [Google Scholar]
- 49.Velagapudi S.P., Gallo S.M., Disney M.D. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat Chem Biol. 2014;10(4):291–297. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong C.H., Richardson S.L., Ho Y.J., Lucas A.M.H., Tuccinardi T., Baranger A.M. Investigating the Binding Mode of an Inhibitor of the MBNL1{dot operator}RNA Complex in Myotonic Dystrophy Type 1 (DM1) Leads to the Unexpected Discovery of a DNA-Selective Binder. ChemBioChem. 2012;13:2505–2509. doi: 10.1002/cbic.201200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez ÀL, Konieczny P, Llamusi B, Delgado-Pinar E, Borrell JI, Teixidó J, et al. In silico discovery of substituted pyrido[2,3-d] pyrimidines and pentamidine-like compounds with biological activity in myotonic dystrophy models. PLoS One 2017;12:1–23. https://doi.org/10.1371/journal.pone.0178931. [DOI] [PMC free article] [PubMed]
- 52.Braida C, Stefanatos RK, Adam B, Mahajan N, Smeets HJ, Niel F, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients l. Hum Mol Genet. 2010;19:1399–412. doi: 10.1093/hmg/ddq015 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.