

Graphical abstract

Keywords: Tyrosinase; 2-thioxooxazoline-4-one; Anti-melanogenesis; Kojic acid; Docking simulation; β-phenyl-α,β-unsaturated carbonyl scaffold
Abstract
The β-phenyl-α,β-unsaturated carbonyl (PUSC) scaffold confers tyrosinase inhibitory activity, and in the present study, 16 (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one analogues containing this scaffold were synthesized. Mushroom tyrosinase inhibitory activities were examined. Compound 1c (IC50 = 4.70 ± 0.40 μM) and compound 1j (IC50 = 11.18 ± 0.54 μM) inhibited tyrosinase by 4.9 and 2.1-fold, respectively, and did so more potently than kojic acid (IC50 = 23.18 ± 0.11 μM). Kinetic analysis of tyrosinase inhibition revealed that 1c and 1j inhibited tyrosinase competitively. Results of docking simulation with mushroom tyrosinase using four docking programs suggested that 1c and 1j bind more strongly than kojic acid to the active site of tyrosinase and supported kinetic findings that both compounds are competitive inhibitors. The docking results of human tyrosinase homology model indicated that 1c and 1j can also strongly inhibit human tyrosinase. EZ-cytox assays revealed 1c and 1j were not cytotoxic to B16F10 melanoma cells. The effects of 1c and 1j on cellular tyrosinase activity and melanin production were also investigated in α-MSH- and IBMX-co-stimulated these cells. Both compounds significantly and dose-dependently reduced tyrosinase activity, and at 10 µM were more potent than kojic acid at 20 µM. Compounds 1c and 1j also inhibited melanogenesis, which suggested that the inhibitory effects of these compounds on melanin production were mainly attributable to their inhibitions of tyrosinase. These results indicate that compounds 1c and 1j with the PUSC scaffold have potential use as whitening agents for the treatment of hyperpigmentation-associated diseases.
1. Introduction
Melanin largely determines the color of bird feathers and the skins of mammals and affects the browning of apples, potatoes, and mushrooms [1], [2], [3], [4], [5]. In humans, melanin colors hair, eyes, and skin. Melanin protects skin by blocking UV radiation, which produces reactive oxygen species (ROS) [6], [7], [8], [9], causes DNA damage, and can cause skin cancer [10], [11], [12], [13], [14], [15].
Skin color is considered important in the contexts of fashion and beauty, but the overproduction of melanin due to, for example, excessive UV exposure results in hyperpigmentation, spots, freckles, and melasma [16], [17], [18], [19], [20]. Melanin is biosynthesized in the melanosomes of melanocytes by complicated enzymatically driven processes. Three enzymes are primarily involved, namely, tyrosinase and two tyrosinase-related proteins, TYRP1 and TYRP2 [7]. Tyrosinase is the key rate-limiting enzyme of the conversion of L-tyrosine to dopaquinone via L-dopa, and thus, influences melanin biosynthesis in melanocytes [6], [21]. Depending on the presence of thiols, such as glutathione and L-cysteine, dopaquinone can act in two different ways [22]. First, it can react with thiols by a Michael addition and be converted to pheomelanin, which is yellow–red colored [22]. Alternatively, in the absence of thiols, dopaquinone is transformed to eumelanin, which is brown-black colored [23], [24]. Thus, human skin color is largely determined by the proportions and amounts of eumelanin and pheomelanin in skin [25], [26].
Numerous attempts have been made to reduce the production of melanin. These approaches include tyrosinase inhibition, the suppression of melanin transfer from melanocytes to keratinocytes, and interventions that target intracellular signals for melanogenesis [27]. Although many different natural and synthetic substances have exhibited meaningful anti-melanogenic effects in cell-based assays [28], [29], [30], [31], [32], [33], [34], the majority have side effects, such as an insufficient potency, carcinogenic effects, permanent depigmentation, and dermatitis, in vivo, in animal models and in humans [35]. Although hydroquinone and kojic acid are used as whitening agents at limited concentrations in a few countries, these skin-lightening agents are prohibited in most countries due to the risks of undesirable side effects, such as possible carcinogenic effects in thyroid [7], nephrotoxicity [6], genotoxicity [36], and cytotoxic effects on melanocytes [6]. Arbutin is α-D-glucopyranoside of hydroquinone that occurs naturally in the bearberry plant of the genus Arctostaphylos. Due to its lesser side effects, arbutin is more widely used as a whitening agent than hydroquinone, and its anti-melanogenic effect is known to be due to tyrosinase inhibition. Arbutin is hydrolyzed to hydroquinone and D-glucose by skin microflora, such as Staphylococcus epidermidis and Staphylococcus aureus [22], by enzymes like α-glycosidase, and by temperature (10% decomposition after 5 days at 20 °C) [37]. Therefore, there is a need for novel tyrosinase inhibitors that are non-carcinogenic and more clinically effective.
Many researchers are trying to discover new tyrosinase inhibitors by exploring new scaffolds and repositioning of old scaffolds including thiourea [38]. Compounds with the 2-thioxooxazolidin-4-one template exhibit many biological activities such as anticancer [39], HIV-1 fusion inhibiting [40], 17β-hydroxysteroid dehydrogenase type 3 inhibiting [41], and glucose and triglyceride-lowering activities [42], but have not been reported to exhibit tyrosinase inhibitory activity. Over the past ten years, we reported a variety of compounds with β-phenyl-α,β-unsaturated carbonyl (PUSC) scaffold that have demonstrated excellent inhibition of tyrosinase in vitro and in vivo [34], [43], [44], [45], [46], [32], [47]. Therefore, we were interested in an incorporating a 2-thioxooxazolidin-4-one template with different benzaldehydes to produce a PUSC scaffold (Fig. 1). According to our accumulated structure–activity relationship data [45], [46], [48], [49], [50], [51], [52], [53], [54], derivatives with a hydroxyl group on the β-phenyl ring of the PUSC scaffold generally have high tyrosinase inhibitory activities. Furthermore, the numbers and positions of hydroxyl groups appear to be closely responsible for the tyrosinase inhibitory efficacy. Accordingly, in the present study, we designed derivatives with various substituents at different positions on the β-phenyl ring of the PUSC scaffold, and subsequently synthesized a series of (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives. We then evaluated the abilities of derivatives to inhibit mushroom tyrosinase and to suppress tyrosinase activity and melanin production in B16F10 melanoma cells, and also performed kinetic studies and docking simulations to determine how these derivatives interact with tyrosinase.
Fig. 1.

Synthetic strategy of (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives possessing the (E)-β-phenyl-α,β-unsaturated carbonyl scaffold.
2. Results and discussion
2.1. Chemistry
The strategy used to synthesize the desired derivatives, (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-ones, involved the use of 3-phenyl-2-thioxooxazolidin-4-one as a key template for the construction of the PUSC scaffold (Fig. 1). As depicted in Scheme 1, reaction of ethyl glycolate with phenyl isothiocyanate gave the key intermediate, 3-phenyl-2-thioxooxazolidin-4-one (2), in a 29% yield. Sixteen benzaldehydes were refluxed with this intermediate in the presence of acetic acid and sodium acetate to give the final compounds 1a–1p in yields of 18–85%. None of these compounds, with the exception of 1i, which is a 17β-hydroxysteroid dehydrogenase type 3 inhibitor, has been previously described [41]. Thermodynamically more stable Z-isomers were predominately generated. As described by Nair and co-workers [55], the E-/Z-configuration was determined using vicinal 1H, 13C-coupling constants in proton-coupled 13C spectra. As shown in Fig. 2, vicinal coupling constants between the amide carbonyl C-atom C(1) and the olefinic H-atom at C(3) depend on the geometry of the double bond ((E)-isomer: 3Jcis = 6.8 Hz, (Z)-isomer: 3Jtrans = 11.5 Hz). Different vicinal 1H, 13C-coupling constants of geometric isomers are observed in many compounds, including 5-membered and 6-membered exocyclic compounds. The values of 3Jcis range from 3.6 to 6.4 Hz, whereas the range of values of 3Jtrans values is roughly twice as large (generally > 10 Hz). 13C NMR of compound 1b was measured in proton-coupled 13C mode, and the 3J value of C4 in 1b was 3.5 Hz (Refer to Fig. S52 in Supplementary data), suggesting a (Z)-configuration.
Scheme 1.

Synthesis of (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives 1a–1p. Reagents and conditions: (a) Et3N, CH2Cl2, rt, 42 h, 29.3%; (b) NaOAc, AcOH, reflux, 9–25 h, 18–85%.
Fig. 2.

Relationship between the C,H-spin-coupling constants over three bonds indicated by the arrow and the geometry of the double bond.
Chemical shifts of vinylic protons were analyzed based on consideration of the NMR solvent used and the positions and types of substituents on the β-phenyl ring. The chemical shifts of vinylic protons appeared at 6.76 ~ 7.10 ppm. The chemical shifts (6.76 ~ 6.81 ppm) of vinylic protons without a substituent at the 2-position of the β-phenyl ring generally appeared up-field in CDCl3 compared to those (6.82 ~ 6.95 ppm) in DMSO‑d6. The presence of highly electronegative elements, such as hydroxyl, methoxyl, and fluoro group at the 2-position of the β-phenyl ring moved the chemical shift of the vinylic proton down-field (1c, 1 h, 1j and 1p vs. the remaining compounds). In the case of compound 1p, which had a highly electronegative fluoro substituent at the 2-position of the β-phenyl ring, the chemical shift of the vinylic proton in CDCl3 solvent appeared at 7.08 ppm, which was 0.27 ~ 0.32 ppm higher than the chemical shifts of vinylic protons in other compounds measured in the same solvent. On the other hand, the two hydroxyl groups of 1c moved 6′-H down-field (7.82 ppm), and the two fluoro substituents of 1p moved 6′-H more strongly down-field (8.28 ppm). The 1H NMR spectrum of 1p showed characteristic coupling patterns of the two fluoro substituents. 6′-H appeared as a triplet of doublets due to coupling by the two fluorine atoms and 5′-H. 3′-H and 5′-H also appeared as a triplet of doublets by coupling to the fluorine atoms and vicinal hydrogen atoms. The results of characteristic coupling by fluorine atoms were observed in 13C NMR spectrum of 1p. All carbon peaks of β-phenyl rings attached to fluorine atoms appeared as doublet of doublets and the ββ-carbon of the PUSC scaffold was also split into a doublet by the 2-fluorine atom.
2.2. Inhibitory activities of thioxazolidinedione derivatives 1a–1p against mushroom tyrosinase
To select derivatives for cell-based assays of anti-melanogenic and tyrosinase-inhibitory effects, the inhibitory activities of the sixteen synthesized (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one analogues, 1a–1p, were examined using mushroom tyrosinase and kojic acid (the positive control with known potent tyrosinase inhibitory activity). All compounds and kojic acid were tested at a concentration of 25 μM. Tyrosinase inhibitory results are shown in Table 1.
Table 1.
Mushroom tyrosinase inhibition of the synthesized (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives 1a–1p and kojic acid.
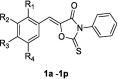 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Tyrosinase inhibition (%)a |
| 1a | H | H | OH | H | 10.25 ± 2.36 |
| 1b | H | OH | OH | H | 30.57 ± 1.77 |
| 1c | OH | H | OH | H | 78.05 ± 4.03 |
| 1d | H | OMe | OH | H | NIb |
| 1e | H | OEt | OH | H | NI |
| 1f | H | OH | OMe | H | 13.39 ± 9.67 |
| 1g | H | H | OMe | H | NI |
| 1h | OMe | H | OMe | H | 4.09 ± 3.66 |
| 1i | H | OMe | OMe | H | 7.47 ± 5.52 |
| 1j | OH | H | H | H | 71.12 ± 0.71 |
| 1k | H | OMe | OMe | OMe | NI |
| 1l | H | OMe | OH | OMe | 10.22 ± 4.82 |
| 1m | H | t-Bu | OH | t-Bu | 8.53 ± 2.25 |
| 1n | H | Br | OH | H | 24.96 ± 4.72 |
| 1o | H | Br | OH | Br | NI |
| 1p | F | H | F | H | NI |
| Kojic acid | 58.09 ± 5.82 | ||||
aTyrosinase inhibitions of the synthesized compounds and kojic acid were evaluated at 25 μM using L-tyrosine as a substrate. bNI: no inhibition. Results are expressed as means ± SEMs.
Of the sixteen synthesized compounds, two derivatives 1c (78.05 ± 4.03% inhibition) with a 2,4-dihydroxyl substituent and 1j (71.12 ± 0.71% inhibition) with a 2-hydroxyl substituent exhibited more potent tyrosinase-inhibition than kojic acid (58.09 ± 5.82% inhibition), and compound 1b exhibited a moderate inhibitory effect (30.57 ± 1.77% inhibition). Although compound 1a (10.25 ± 2.36% inhibition) with a 4-hydroxyphenyl only weakly inhibited tyrosinase, the introduction of an additional 3-hydroxyl group into the β-phenyl ring enhanced tyrosinase inhibitory activity to 30.57% (see compound 1b). Furthermore, the insertion of an additional 2-hydroxyl group into the β-phenyl ring of 1a increased the tyrosinase inhibitory activity to 78% (1a vs. 1c). On the contrary, the introduction of methoxyl or ethoxyl at the 3-position of the β-phenyl ring eliminated tyrosinase inhibitory activity (1a vs. 1d and 1e). Derivatives (1 h, 1i, and 1 k) without a hydroxyl group on the β-phenyl ring exhibited very weak or no inhibition. The insertion of a 3-bromo group into the β-phenyl ring of 1a increased tyrosinase inhibitory activity (1a vs. 1n) to 25%, while the introduction of substituents (3,5-dimethoxy, 3,5-di-tert-butyl, and 3,5-dibromo substituents) at both 3- and 5-positions in the β-phenyl ring did not enhance inhibitory activities (1a vs. 1 l, 1 m, and 1o). These results indicate hydroxyl groups on the β-phenyl ring are necessary for tyrosinase inhibition and that the number and position of substituents, especially hydroxyl groups, markedly influence tyrosinase inhibitory activity.
Derivative 1c with a 2,4-dihydroxyphenyl group was the most potent inhibitor of tyrosinase. To investigate whether the hydrogen bond donor or acceptor characteristics of the hydroxyl groups are associated with tyrosinase inhibition, compound 1p with a 2,4-difluorophenyl group was synthesized as a congener of 1c, which possessed a 2,4-dihydroxyphenyl. Unlike a hydroxyl group, fluorine can only serve as a hydrogen bond acceptor. Tests showed 1p had no mushroom tyrosinase inhibitory activity. Compound 1 h, which had 2,4-dimethoxyl groups that only act as hydrogen bond acceptors, exhibited very weak tyrosinase inhibition. These results suggest substituents that can act as hydrogen bond acceptors and donors at the 2- and 4-positions of the β-phenyl group, or that can serve as hydrogen bond donors, may play a more important role in tyrosinase enzyme inhibition than substituents capable of acting as hydrogen bond acceptors only at same positions. This suggestion is supported by the pharmacophore results obtained using LigandScout based on AutoDock 4, which showed hydroxyl substituents at the 2- and 4-positions of the β-phenyl group form a hydrogen bond with amino acid residues of Asn260 or Met280 at the active site of tyrosinase, respectively, and that the hydroxyls serve as hydrogen bond donors (Fig. 5). Taken together, the hydroxyl substituents at the 2- and 4-positions of the β-phenyl group play a key role in tyrosinase enzyme inhibition, and the effect on the tyrosinase inhibitory activity is due to the ability of the hydrogen bond donor of hydroxyls.
Fig. 5.

Docking simulation of the (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives 1c, and 1j and of kojic acid with Agaricus bisporus tyrosinase using AutoDock 4, Dock 6 and Schrödinger suite and pharmacophore analysis. (a) Pharmacophore results for 1c, 1j, and kojic acid obtained using LigandScout 4.3 based on AutoDock 4 showed possible hydrophobic (yellow), π-π stacking (violet arrow), and hydrogen bonding (green arrow) interactions between tyrosinase amino acid residues and the three ligands. (b) Docking scores for tyrosinase interactions with 1c, 1j, and kojic acid (PDB code: 2Y9X). (c) Binding interactions between mushroom tyrosinase and 1c, 1j, and kojic acid obtained by Schrödinger suite. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We also investigated the IC50 values of compounds 1c and 1j, which more potently inhibited mushroom tyrosinase than kojic acid, and compound 1b, which moderately inhibited tyrosinase. All four compounds dose-dependently inhibited mushroom tyrosinase (data not shown). The low IC50 values of 1c (4.70 ± 0.40 μM) and 1j (11.18 ± 0.54 μM) indicated that these compounds inhibited tyrosinase activity more strongly than kojic acid (IC50 = 23.18 ± 0.11 μM), more specifically, the inhibitory potencies of 1c and 1j were 5- and 2-fold greater, respectively, than that of kojic acid. On the other hand, the IC50 value of compound 1b, which possessed a catechol group, was 53.38 ± 0.39 μM, indicating poorer mushroom tyrosinase inhibitory activity than kojic acid.
2.3. Studies on the action of mode of compounds 1c, and 1j
To examine the inhibitory modes of action of compounds 1c and 1j, we performed a kinetic study on mushroom tyrosinase in the presence of 1c or 1j using L-tyrosine as a substrate. Mechanisms of tyrosinase inhibition were investigated by Lineweaver-Burk plot analysis as depicted in Fig. 3. Lineweaver-Burk plots of 1c or 1j were similar (Fig. 3a vs. b). For each compound, lines produced at different concentrations converged at one point on the y-axis. As the concentrations of 1c and 1j were increased, KM values for tyrosinase dose-dependently increased without changing Vmax values. These results imply that 1c and 1j competitively inhibited tyrosinase and that they bind to the same binding pocket as the tyrosinase substrate, L-tyrosine. The kinetics of these inhibitions (Table 2) showed the following: for 1c; Ki = 3.02 × 10−6, 2.51 × 10−6, and 2.19 × 10−6 M at 2.5, 5.0, and 10.0 μM, respectively; and for 1j; Ki = 5.85 × 10−6, 4.51 × 10−6, and 3.22 × 10−6 M at 5.0, 10.0, and 20.0 μM, respectively. KM values of 1c at concentrations of 2.5, 5.0, and 10.0 μM were 6.56, 10.72, and 19.93 mM, respectively, and those of 1j at concentrations of 5.0, 10.0 and 20.0 μM were 5.47, 9.48, and 21.26 mM, respectively. Compounds 1c, and 1j had similar Vmax values of 4.0 × 10−2 and 2.7 × 10−2 mM/min, respectively, regardless of concentration.
Fig. 3.

Lineweaver-Burk plots for the inhibition of mushroom tyrosinase activity by (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives (a) 1c and (b) 1j. Inhibition types were investigated using Lineweaver-Burk plots. Data are shown as mean values of 1/V, defined as the inverse of increased absorbance at 475 nm per min as determined by three independent experiments at five different L-tyrosine concentrations. Concentrations of 1c were 0 μM (filled circles), 2.5 μM (unfilled circles), 5.0 μM (filled triangles), and 10.0 μM (unfilled triangles) and the concentrations of 1j were 0 μM (filled circles), 5.0 μM (unfilled circles), 10.0 μM (filled triangles), and 20.0 μM (unfilled triangles). The modified Michaelis-Menten equation was used: 1/Vmax = (1/KM)(1 + [S]/Ki), where Vmax is maximum reaction rate, KM is a Michaelis-Menten constant, [S] is L-tyrosine concentration, and Ki is the inhibition constant. All experiments were independently performed in triplicate.
Table 2.
Kinetic analysis of compounds 1c, and 1j.
| Inhibitor | |||||||
|---|---|---|---|---|---|---|---|
|
1c |
1j |
||||||
| Conc. | Vmax (mM/min) | KM (mM) | Ki (M) | Conc. | Vmax (mM/min) | KM (mM) | Ki (M) |
| 2.5 μM | 4.0 × 10−2 | 6.56 | 3.02 × 10−6 | 5.0 μM | 2.7 × 10−2 | 5.47 | 5.85 × 10−6 |
| 5.0 μM | 4.0 × 10−2 | 10.72 | 2.51 × 10−6 | 10.0 μM | 2.7 × 10−2 | 9.48 | 4.51 × 10−6 |
| 10.0 μM | 4.0 × 10−2 | 19.93 | 2.19 × 10−6 | 20.0 μM | 2.7 × 10−2 | 21.26 | 3.22 × 10−6 |
Data are mean values of 1/V (inverse of the increase in absorbance at a wavelength of 475 nm per min (ΔA475/min)), of three independent experiments conducted using different L-tyrosine concentrations. The Lineweaver-Burk plot equation is: 1/V = 1/Vmax + KM/Vmax × 1/[S] and the modified Michaelis-Menten equation is 1/Vmax = (1 + [I]/Ki) × 1/KM, where V is the reaction rate, Vmax is the maximum reaction rate, KM is the Michaelis-Menten constant, [S] is substrate concentration, [I] is inhibitor concentration, and Ki is the inhibition constant.
2.4. Docking simulation studies of compounds 1c and 1j and kojic acid with mushroom tyrosinase
To investigate whether the synthesized (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-ones can bind directly to the active site of tyrosinase, docking simulations were performed using AutoDock Vina 1.1.2 software (developed by The Scripps Research Institute). Two compounds, 1c, and 1j, with highest inhibitory activity against mushroom tyrosinase were selected as ligands for the docking simulation. After energy minimization of 2D-structures using Chem3D Pro 12.0 software (CambridgeSoft Corporation), 3D-structures of the two compounds were created. Tyrosinase of Agaricus bisporus (a species of mushroom) was utilized as the 3D-structure [Protein Data Bank (PDB) ID: 2Y9X] for docking simulation. Although the correlation between the binding affinities of the two ligands and their abilities to inhibit mushroom tyrosinase was not perfect, both derivatives had much stronger binding affinities (−7.3 ~ −7.5 kcal/mol) than the kojic acid (−5.7 kcal/mol) (Fig. 4d). LigandScout 4.3 software was utilized to determined which amino acid residues of tyrosinase interacted with 1c and 1j. Three amino acids (His259, His263, and Met280) of tyrosinase were found to interact with kojic acid (Fig. 4c). The branched hydroxyl group of kojic acid formed two hydrogen bonds with amino acid residues His259 and His263 and the ring hydroxyl formed a hydrogen bond with Met280. Both hydroxyl groups of kojic acid acted as hydrogen bonding donors. Compounds 1c and 1j both interacted hydrophobically with five amino acid residues (Val248, Met257, Phe264, Val283, and Ala286) (Fig. 4a and b) without hydrogen bonding. Docking simulation results suggested although the amino acids that interacted with kojic acid and those that interacted with 1c and 1j differed all three ligands bind to the active site of tyrosinase. However, LigandScout results based on AutoDock Vina docking simulations showed kojic acid appeared to bind more strongly to the active site of tyrosinase than 1c or 1j, which was contrary to the results of the binding affinity obtained from AutoDock Vina.
Fig. 4.

Docking simulation of the (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives 1c and 1j and of kojic acid with Agaricus bisporus tyrosinase using AutoDock Vina and pharmacophore analysis. (a-c) Pharmacophore results for 1c, 1j, and kojic acid obtained using LigandScout 4.3 showed possible hydrophobic (yellow), π-π stacking (violet arrow), and hydrogen bonding (green arrow) interactions between tyrosinase amino acid residues and the three ligands. Docking simulation 3D-results indicated hydrophobic (yellow sphere), π-π stacking (violet ring), and hydrogen bonding (green sphere) regions on the ligands. (d) Docking scores for interactions between tyrosinase and 1c, 1j, and kojic acid (PDB code: 2Y9X). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Two more docking simulation software packages, that is, AutoDock 4 and Dock 6, were utilized to increase the reliability of docking simulation results. The same tyrosinase species used for the AutoDock Vina simulation were used. According to AutoDock 4 and Dock 6, the binding affinities of 1c were −7.41 and −30.70 kcal/mol and for 1j were −7.19 and −32.42 kcal/mol, respectively (Fig. 5b), which were greater than those of kojic acid (−4.2 and −27.59 kcal/mol, respectively). Furthermore, these results were consistent with experimental data for mushroom tyrosinase inhibition. However, according to Dock 6, 1j had greater binding affinity than 1c, while in AutoDock 4, the reverse was the case. Compound 1c which showed greater inhibitory activity against mushroom tyrosinase than 1j showed higher binding affinity to tyrosinase than 1j in AutoDock 4. Thus, LigandScout results based on AutoDock 4 were examined (Fig. 5a). These results showed kojic acid formed one hydrogen bond with Met280 and that its ring interacted with His263 by π-π stacking. Compound 1c which showed stronger binding affinity than compound 1j formed two hydrogen bonds with Asn260 and Met280 using its two hydroxyls and interacted hydrophobically with Val248, Val283, and Ala286 through its two phenyl rings. On the other hand, compound 1j interacted hydrophobically with Val 248, Met257, Phe264, Val283, and Ala 286 and by π-π stacking interacted with His263. These LigandScout results agreed well with AutoDock 4 binding affinity results. Summarized, the observations above indicate that the resorcinol (2,4-dihydroxyphenyl) moiety plays an important role in ligand binding to the active site of tyrosinase by forming two hydrogen bonds and participating in two hydrophobic interactions.
Another docking software Schrödinger suite was used to further improve our understanding of the tyrosinase binding interactions of 1c, 1j and kojic acid. The binding interactions of kojic acid, 1c and 1j are shown in 2D and 3D structures in Fig. 5c. Kojic acid was found to form one hydrogen bond with Gly281 and a π-π stacking with His263. Compound 1c formed metal coordination and salt bridges with Cu400 and Cu401 and its 2,4-dihydroxyphenyl ring interacted with His259 and His263 by π-π stacking. Compound 1j formed one hydrogen bond with Asn260 and interacted with His263 by π-π stacking. As can be seen in Fig. 6b, the docking scores obtained by the Schrödinger suite indicated that 1c more strongly inhibited mushroom tyrosinase than kojic acid and 1j, which may be due to the metal coordination and salt bridges of 1c with copper ions.
Fig. 6.

Predicted binding mode of 1c, 1j and kojic acid at the active site of human tyrosinase homology model and docking score using Schrödinger suite. (a) Pharmacophore results are represented in 2D and 3D structures. (b) Docking scores of 1c, 1j and kojic acid using mushroom tyrosinase (PDB: 2Y9X) and human tyrosinase homology model are represented, respectively.
2.5. Human tyrosinase homology model for the study of docking simulations of compounds 1c and 1j and kojic acid
To further validate the nature and mode of binding interactions of compound 1c and 1j in human tyrosinase, a homology model based on human tyrosinase related protein 1 (TRP1) was used. The entire sequence of human tyrosinase (P14679) was imported from the UniProt database [56] to build the 3D homology model of human tyrosinase. The X-ray structure of TRP1 (PDB ID:5M8Q) [57] was used as a protein template due to its 45.81% sequence identity with the human tyrosinase. The co-crystal ligand and zinc ions were reflected in the newly constructed homology model of human tyrosinase. The 3D structure of the newly constructed human tyrosinase homology model was further prepared using Schrödinger suite and docked to compounds 1c and 1j and kojic acid. The homology model of the newly constructed human tyrosinase and the aligned protein sequence are shown in Figure S53.
2.6. Docking score and binding mode of compounds 1c and 1j and kojic acid at the active site of human tyrosinase homology model
The binding mode of 1c, 1j and kojic acid was predicted based on the nature of the binding interactions with amino acids in the active site of the human tyrosinase homology model using Schrödinger suite (Fig. 6a). Kojic acid formed a hydrogen bond with Ser375 and interacted with His367 through π-π stacking. In addition, kojic acid produced a metal coordination with one of the two zinc ions (Zn7). The binding affinity of kojic acid to human tyrosinase was measured with a docking score of −4.15 kcal/mol (Fig. 6b), which was weaker than that of mushroom tyrosinase (−4.65 kcal/mol). Compound 1c was observed to generate metal coordination and salt bridges with zinc ions (Zn6 and Zn7) in the same way that 1c interacted with mushroom tyrosinase. Compound 1c also interacted with His367 through π-π staking. The combined effect of these interactions on 1c resulted in a docking score of −5.36 kcal/mol, indicating a stronger binding affinity than kojic acid. However, the binding affinity of 1c was slightly weaker in human tyrosinase than in mushroom tyrosinase (−5.65 kcal/mol). On the other hand, compound 1j did not interact with zinc ions, but created a salt bridge with Lys306 and interacted with His202 through π-π staking. In addition, 1j formed a hydrogen bond with Val377. The calculated docking score for 1j is −4.57 kcal/mol in human tyrosinase. These docking results imply that both compounds 1c and 1j may effectively inhibit mushroom tyrosinase as well as human tyrosinase.
2.7. Binding analysis of 1c, 1j and kojic acid in human tyrosinase homology model and mushroom tyrosinase enzyme
To compare the interactions of compounds 1c, 1j and kojic acid, we performed docking studies using Schrödinger suite on both the human tyrosinase homology model and mushroom tyrosinase and found interesting results. As shown in Figure S54, the distance between the two copper ions and the hydroxyl group of kojic acid in mushroom tyrosinase is 3.14 Å and 3.07 Å, and the distance between the zinc ions and the hydroxyl group of kojic acid in human tyrosinase is 2.55 Å and 2.13 Å. However, the hydroxyl groups of kojic acid that interacted with the metal (Cu or Zn) were different in the two tyrosinases. In mushroom tyrosinase, the phenolic hydroxyl group participated in metal coordination and salt bridges, whereas in human tyrosinase, the alcoholic hydroxyl group was involved in these interactions. The hydrogen bond length of kojic acid was 2.13 Å in mushroom tyrosinase and 1.75 Å in human tyrosinase model. Similarly, the distances between the metal ions (Cu or Zn) and the 4-hydroxyl group of 1c were recorded as 2.31 Å and 2.35 Å in the mushroom tyrosinase and 2.12 Å and 2.48 Å in the human tyrosinase model, respectively. The distance between the hydroxyl group of 1j and the copper ions in mushroom tyrosinase was recorded as 5.23 Å and 4.49 Å, indicating the absence of metal coordination and salt bridges between the metal ions and the hydroxyl group. Interestingly, in human tyrosinase, 1j was located at the active site in a different arrangement from that in mushroom tyrosinase. Unlike in mushroom tyrosinase, the 2-hydroxyphenyl ring of 1j was far from the zinc ions in human tyrosinase. Instead, the N-phenyl ring of 1j was placed close to the zinc ions with 3.72 Å and 3.30 Å away. The order of decreasing binding affinity for human tyrosinase was 1c, 1j and kojic acid, and 1c, kojic acid and 1j for mushroom tyrosinase. Regardless of the type of tyrosinase, 1c showed the strongest binding affinity to tyrosinase. Although compound 1j showed lower binding affinity than kojic acid in mushroom tyrosinase docking simulations using Schrödinger suite, results obtained with other docking programs such as AutoDock Vina, AutoDock 4 and Dock 6 indicated that 1j had a higher binding affinity than kojic acid. These results imply that compounds 1c and 1j may effectively inhibit both mushroom tyrosinase and human tyrosinase.
2.8. Cytotoxic effects of 1c and 1j in B16F10 melanoma cells
An EZ-cytox assay was used to investigate the cytotoxic effects of compounds 1c and 1j. After murine B16F10 melanoma cells had been cultured for 24 h, they were treated with different concentrations (0, 5, 10, or 20 µM) of 1c and 1j for 24 h in a humidified atmosphere. Optical densities were measured on a microplate reader.
Cell viability results are shown in Figure S55. Compounds 1c and 1j did not have a significant cytotoxic effect on B16F10 melanoma cells at concentrations up to 20 µM, which indicated both compounds were non-toxic to these cells at concentrations below 20 µM. Thus, cell-based assays on cellular tyrosinase inhibition and melanin production were performed using 1c and 1j at concentrations of ≤ 20 µM.
2.9. Cellular tyrosinase inhibitory activities of compounds 1c and 1j in α-MSH- and IBMX-co-stimulated B16F10 melanoma cells
Cellular tyrosinase inhibitory effect of compounds 1c and 1j was investigated by using murine B16F10 melanoma cells co-stimulated with 3-isobutyl-1-methylxanthine (IBMX) and α-melanocyte-stimulating hormone (α-MSH). B16F10 melanoma cells were cultured for 24 h and then pretreated with 20 μM of kojic acid or compounds 1c or 1j at concentrations of 0, 5, 10, or 20 μM for 3 h. Cells were then co-treated with α-MSH 1 μM and IBMX 200 µM for 48 h to increase tyrosinase activity. Tyrosinase-inhibitory effects were accessed by measuring optical densities using a microplate reader.
α-MSH and IBMX co-treatment increased cellular tyrosinase activity level 2.65-fold (Fig. 7). Treatments with 1c or 1j significantly and concentration-dependently decreased cellular tyrosinase activity enhanced by α-MSH and IBMX co-treatment. Compound 1c at 10 μM and compound 1j at 5 μM both inhibited cellular tyrosinase more than kojic acid at 20 μM, and 1j at 20 μM resulted in cellular tyrosinase activity similar to that of the control group. Considering that 1c and 1j had no observable cytotoxic effect at concentrations below 20 μM (Figure S55), these tyrosinase-inhibitory activities were attributed to direct tyrosinase inhibition by 1c and 1j. This notion was supported by our docking simulation finding that 1c and 1j bind strongly to the active site of tyrosinase.
Fig. 7.

Cellular tyrosinase inhibitory activities of 1c and 1j on murine B16F10 cells. In the presence of 1 μM α-MSH and 200 μM IBMX, B16F10 cells were treated with different concentrations (0, 5, 10, and 20 μM) of 1c, 1j, or kojic acid (20 μM) for 24 h. Results are percentages of untreated controls, and columns represent the means ± SEMs of three determinations. ###p < 0.001 versus untreated controls; **p < 0.01, ***p < 0.001 versus α-MSH and IBMX co-treated cells.
2.10. Inhibitory effects of compounds 1c and 1j on melanin production in α-MSH and IBMX co-stimulated B16F10 melanoma cells
Melanin assays were conducted on α-MSH and IBMX co-stimulated B16F10 melanoma cells to investigate the inhibitory effects of compounds 1c and 1j on melanin production. Murine B16F10 melanoma cells were seeded in 6-well culture plates and pretreated with 20 μM of kojic acid or compounds 1c or 1j at 0, 5, 10, or 20 μM for 3 h. Cells were then co-treated with α-MSH and IBMX for 48 h to increase melanin contents. The inhibitory effects of 1c, 1j, and kojic acid on melanin production were determined by measuring optical densities using a microplate reader.
The effects of 1c, 1j, and kojic acid on melanin production are shown in Fig. 8. Compounds 1c and 1j significantly and potently decreased melanin contents in α-MSH and IBMX co-stimulated B16F10 melanoma cells in a concentration-dependent manner. Interestingly, 1c at 10 μM and 1j at 5 μM inhibited melanin production more than kojic acid at 20 µM. These results indicate that these suppressions of melanin production were mainly caused by the inhibition of tyrosinase activity (Fig. 7, Fig. 8).
Fig. 8.

Effect of compounds 1c and 1j on melanogenesis in murine B16F10 cells in the presence of 1 μM α-MSH and 200 μM IBMX. B16F10 cells were treated with varying concentrations (0, 5, 10, and 20 μM) of 1c and 1j or kojic acid (20 μM) for 48 h in the presence of α-MSH and IBMX. Melanin contents were measured at 405 nm and columns represent the means ± SEMs of three experiments. Results are expressed as percentages of untreated controls. ###p < 0.001, versus untreated controls; **p < 0.01, ***p < 0.001 versus cell co-treated with α-MSH and IBMX.
3. Conclusion
In summary, sixteen (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one analogues 1a–1p containing the β-phenyl-α,β-unsaturated carbonyl scaffold, which has been reported to play an important role in conferring tyrosinase inhibitory activity, were synthesized by reacting 3-phenyl-2-thioxooxazolidin-4-one (2) with different benzaldehydes. Configurations of the exocyclic double bond of synthesized products were determined using C,H-spin-coupling constants. The inhibitory activities of the synthesized compounds against mushroom tyrosinase were evaluated at 25 μM. Of the sixteen compounds, compounds 1c (78% inhibition) and 1j (71% inhibition) were found to be more potent than kojic acid (58% inhibition, the positive control). Compounds 1c and 1j also had lower IC50 values than kojic acid (4.70 ± 0.40 and 11.18 ± 0.54 μM, respectively, vs. 23.18 ± 0.11 μM). Lineweaver-Burk plots showed 1c and 1j were competitive inhibitors of tyrosinase. Docking simulations of 1c or 1j with mushroom tyrosinase were performed using AutoDock Vina, AutoDock 4, Dock 6, and Schrödinger suite. All docking simulations confirmed that compounds 1c and 1j bind to the active site of tyrosinase more strongly than kojic acid. Pharmacophore analysis using LigandScout and Schrödinger suite confirmed that the 2,4-dihydroxyphenyl moiety of 1c formed two hydrogen bonds with the amino acids of tyrosinase at the active site, or metal coordination and salt bridges with the copper ions. Docking simulation results with the human tyrosinase homology model obtained using Schrödinger suite supported the possibility that 1c and 1j might strongly inhibit human tyrosinase. Our investigation of the tyrosinase activity and melanin production in α-MSH and IBMX co-stimulated murine B16F10 melanoma cells revealed that 1c and 1j effectively inhibited tyrosinase activity and melanogenesis concentration-dependently and more potently than kojic acid without evidence of cytotoxicity. The similarity between the inhibitions of cellular tyrosinase activity and melanin production by 1c and 1j suggests their anti-melanogenic effects are the result of tyrosinase inhibition. Furthermore, the observed anti-melanogenic effects of 1c and 1j demonstrate these agents have promising potential as skin-lightening therapeutics for the treatment of hyperpigmentation diseases.
4. Experimental section
4.1. General methods
All chemicals and reagents were obtained commercially (Sigma-Aldrich and Alfa Aesar) and used without further purification, and all anhydrous solvents were distilled over CaH2 or Na/benzophenone before use. Reactions were monitored by thin-layer chromatography (TLC) on glass plates coated with silica gel using a fluorescent indicator (TLC Silica Gel 60 F254, Merck, Germany) and column chromatography was conducted on MP Silica 40–63, 60 Å. High resolution mass spectroscopy data was obtained on an Agilent Accurate Mass Q-TOF (quadruple-time of flight) liquid chromatography mass spectrometer (Agilent, Santa Clara, CA, USA) in negative ESI mode. Low-resolution mass data were obtained in ESI negative or positive mode on an Expression CMS spectrometer (Advion Ithaca, NY, USA). Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Unity INOVA 400 spectrometer or a Varian Unity AS500 spectrometer (Agilent Technologies, Santa Clara, CA, USA) at 400 MHz and 500 MHz 1H NMR, respectively, and on a Varian Unity INOVA 400 spectrometer for 100 MHz 13C NMR. DMSO‑d6 (δH 2.50 ppm and δC 39.7 ppm) or CDCl3 (δH 7.24 ppm and δC 77.0 ppm) was used as solvents for NMR samples. Coupling constants (J) and chemical shifts were measured in hertz (Hz) and parts per million (ppm), respectively. The abbreviations used for 1H NMR data are; s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), td (triplet of doublets), m (multiplet), and brs (broad singlet).
4.1.1. General procedure used for the synthesis of (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one analogues (1a–1p)
Trimethylamine (1.03 mL, 7.39 mmol) and phenyl isothiocyanate (1.76 mL, 14.74 mmol) were added slowly to a stirred solution of ethyl glycolate (1.4 mL, 14.79 mmol) in dichloromethane (20 mL) at 0 ℃. The reaction mixture was stirred at ambient temperature for 42 h and partitioned between dichloromethane and water. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated in vacuo. The resultant residue was purified by silica gel column chromatography using hexane and ethyl acetate (5:1) as eluant to give 3-phenyl-2-thioxooxazolidin-4-one (2, 834.7 mg, 29.3%). A solution of 2 (100 mg, 0.52 mmol), a benzaldehyde (1.0 equiv.), and sodium acetate (139 mg, 1.69 mmol) in acetic acid (1.0 mL) was refluxed for 9–25 h. After cooling, water was added to the reaction mixture and the mixture was stirred for 1 h. The precipitate generated was filtered and washed with water to give (Z)-5-(substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives (1a–1p) as solids in yields of 18–85%.
4.1.1.1. 3-Phenyl-2-thioxooxazolidin-4-one (2)
1H NMR (400 MHz, CDCl3) δ 7.53–7.46 (m, 3H, 3-H, 4-H, 5-H), 7.29 (d, 2H, J = 8.4 Hz, 2-H, 6-H), 4.97 (s, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 190.2, 170.5, 132.2, 130.1, 129.8, 127.7, 70.5.
4.1.1.2. (Z)-5-(4-Hydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1a)
Yellow solid; reaction time, 22 h, yield, 52%; 1H NMR (500 MHz, DMSO‑d6) δ 10.34 (brs, 1H, OH), 7.79 (d, 2H, J = 9.0 Hz, 2′-H, 6′-H), 7.54 (t, 2H, J = 7.0 Hz, 3-H, 5-H), 7.49 (t, 1H, J = 7.0 Hz, 4-H), 7.46 (d, 2H, J = 7.5 Hz, 2-H, 6-H), 6.92 (s, 1H, vinylic H), 6.92 (d, 2H, J = 9.0 Hz, 3′-H, 5′-H); 13C NMR (100 MHz, DMSO‑d6) δ 183.8, 162.1, 160.9, 137.9, 134.2, 133.5, 130.1, 129.8, 128.7, 122.7, 117.0, 114.5; LRMS (ESI-) m/z 296 (M−H)−.
4.1.1.3. (Z)-5-(3,4-Dihydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1b)
Green solid; reaction time, 10 h, yield, 70%; 1H NMR (500 MHz, DMSO‑d6) δ 9.84 (s, 1H, OH), 9.49 (s, 1H, OH), 7.53 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.48 (t, 1H, J = 7.0 Hz, 4-H), 7.46–7.44 (m, 3H, 2-H, 6-H, 2′-H), 7.22 (d, 1H, J = 7.5 Hz, 6′-H), 6.85 (d, 1H, J = 8.0 Hz, 5′-H), 6.82 (s, 1H, vinylic H); 13C NMR (100 MHz, DMSO‑d6) δ 183.8, 162.1, 149.9, 146.5, 137.7, 133.5, 130.1, 129.8, 128.7, 125.9, 123.1, 118.3, 116.8, 115.1; LRMS (ESI-) m/z 312 (M−H)−.
4.1.1.4. (Z)-5-(2,4-Dihydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1c)
Brown solid; reaction time, 22 h, 18%; 1H NMR (500 MHz, DMSO‑d6) δ 10.52 (s, 1H, OH), 10.25 (s, 1H, OH), 7.82 (d, 1H, J = 8.5 Hz, 6′-H), 7.53 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.48 (t, 1H, J = 7.0 Hz, 4-H), 7.45 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 7.05 (s, 1H, vinylic H), 6.45 (dd, 1H, J = 8.0, 2.0 Hz, 5′-H), 6.41 (d, 1H, J = 2.0 Hz, 3′-H); 13C NMR (100 MHz, DMSO‑d6) δ 183.6, 162.8, 162.1, 160.2, 136.9, 133.5, 132.8, 130.1, 129.8, 128.7, 110.3, 109.6, 109.0, 103.0; LRMS (ESI-) m/z 312 (M−H)−; HRMS (ESI + ) m/z C16H12NO4S (M + H)+ calcd 314.0482, obsd 314.0483, m/z C16H11NNaO4S (M + Na)+ calcd 336.0301, obsd 336.0304.
4.1.1.5. (Z)-5-(4-Hydroxy-3-methoxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1d)
Yellow solid; reaction time, 21 h, 18%; 1H NMR (500 MHz, CDCl3) δ 7.55 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.50 (t, 1H, J = 7.0 Hz, 4-H), 7.47 (s, 1H, 2′-H), 7.41–7.39 (m, 3H, 2-H, 6-H, 6′-H), 7.00 (d, 1H, J = 8.5 Hz, 5′-H), 6.78 (s, 1H, vinylic H), 6.06 (s, 1H, OH), 3.98 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ 182.4, 161.8, 149.1, 147.1, 137.5, 132.5, 129.9, 129.6, 127.6, 127.3, 123.6, 115.5, 115.4, 113.2, 56.3; LRMS (ESI-) m/z 326 (M−H)−, 311 (M−H−CH3)−.
4.1.1.6. (Z)-5-(3-Ethoxy-4-hydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1e)
Green solid; reaction time, 22 h, 55%; 1H NMR (500 MHz, DMSO‑d6) δ 9.91 (s, 1H, OH), 7.53 (t, 2H, J = 7.0 Hz, 3-H, 5-H), 7.50–7.43 (m, 5H, 2-H, 4-H, 6-H, 2′-H, 6′-H), 6.94 (d, 1H, J = 8.0 Hz, 5′-H), 6.90 (s, 1H, vinylic H), 4.05 (q, 2H, J = 6.5 Hz, OCH2CH3), 1.34 (t, 3H, J = 6.5 Hz, OCH2CH3); 13C NMR (100 MHz, DMSO‑d6) δ 183.7, 162.1, 150.9, 147.7, 137.9, 133.4, 130.1, 129.8, 128.7, 126.5, 123.1, 117.2, 117.1, 114.8, 64.7, 15.3; LRMS (ESI-) m/z 340 (M−H)−, 311 (M−H−C2H5)−.
4.1.1.7. (Z)-5-(3-Hydroxy-4-methoxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1f)
Yellow solid; reaction time, 21 h, 85%; 1H NMR (500 MHz, CDCl3) δ 7.55 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.52 (s, 1H, 2′-H), 7.49 (t, 1H, J = 7.5 Hz, 4-H), 7.42 (d, 1H, J = 8.5 Hz, 6′-H), 7.39 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 6.93 (d, 1H, J = 8.5 Hz, 5′-H), 6.76 (s, 1H, vinylic H), 5.68 (s, 1H, OH), 3.97 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ 182.5, 161.9, 149.4, 146.2, 137.9, 132.6, 129.9, 129.6, 127.6, 125.6, 124.5, 117.3, 115.1, 111.1, 56.3; LRMS (ESI-) m/z 326 (M−H)−, 311 (M−H−CH3)−.
4.1.1.8. (Z)-5-(4-Methoxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1g)
Yellow solid; reaction time, 21 h, 68%; 1H NMR (500 MHz, CDCl3) δ 7.85 (d, 2H, J = 8.5 Hz, 2′-H, 6′-H), 7.55 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.50 (t, 1H, J = 7.5 Hz, 4-H), 7.40 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 6.99 (d, 2H, J = 8.5 Hz, 3′-H, 5′-H), 6.81 (s, 1H, vinylic H), 3.88 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ 182.5, 162.2, 161.9, 137.6, 133.8, 132.6, 129.9, 129.6, 127.6, 123.7, 115.1, 115.0, 55.7; HRMS (ESI + ) m/z C17H14NO3S (M + H)+ calcd 312.0689, obsd 312.0686.
4.1.1.9. (Z)-5-(2,4-Dimethoxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1h)
Yellow solid; reaction time, 16 h, 65%; 1H NMR (500 MHz, DMSO‑d6) δ 7.97 (d, 1H, J = 9.0 Hz, 6′-H), 7.54 (t, 2H, J = 7.0 Hz, 3-H, 5-H), 7.49 (t, 1H, J = 7.0 Hz, 4-H), 7.45 (d, 2H, J = 7.5 Hz, 2-H, 6-H), 7.01 (s, 1H, vinylic H), 6.78 (dd, 1H, J = 9.0, 1.5 Hz, 5′-H), 6.69 (d, 1H, J = 1.5 Hz, 3′-H), 3.90 (s, 3H, OCH3), 3.85 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO‑d6) δ 183.7, 164.0, 162.2, 160.7, 138.3, 133.4, 132.7, 130.1, 129.8, 128.7, 112.8, 108.0, 107.2, 99.1, 56.8, 56.4; HRMS (ESI + ) m/z C18H16NO4S (M + H)+ calcd 342.0795, obsd 342.0796.
4.1.1.10. (Z)-5-(3,4-Dimethoxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1i)
Yellow solid; reaction time, 12 h, 58%; 1H NMR (500 MHz, DMSO‑d6) δ 7.58–7.53 (m, 4H, 3-H, 5-H, 2′-H, 6′-H), 7.49 (t, 1H, J = 7.0 Hz, 4-H), 7.46 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 7.15 (d, 1H, J = 8.5 Hz, 5′-H), 6.95 (s, 1H, vinylic H), 3.83 (s, 3H, OCH3), 3.79 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO‑d6) δ 183.7, 162.1, 152.0, 149.5, 138.6, 133.4, 130.1, 129.8, 128.7, 126.1, 124.4, 115.0, 114.1, 113.0, 56.4, 56.3; HRMS (ESI + ) m/z C18H16NO4S (M + H)+ calcd 342.0795, obsd 342.0796, m/z C18H15NNaO4S (M + Na)+ calcd 364.0614, obsd 364.0624.
4.1.1.11. (Z)-5-(2-Hydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1j)
Green solid; reaction time, 25 h, 28%; 1H NMR (500 MHz, DMSO‑d6) δ 10.55 (s, 1H, OH), 7.94 (d, 1H, J = 8.0 Hz, 6′-H), 7.54 (t, 2H, J = 7.0 Hz, 3-H, 5-H), 7.49 (t, 1H, J = 7.0 Hz, 4-H), 7.47 (d, 2H, J = 7.5 Hz, 2-H, 6-H), 7.32 (t, 1H, J = 7.5 Hz, 4′-H), 7.10 (s, 1H, vinylic H), 6.98 (t, 1H, J = 7.5 Hz, 5′-H), 6.97 (d, 1H, J = 7.5 Hz, 3′-H); 13C NMR (100 MHz, DMSO‑d6) δ 183.9, 162.2, 158.0, 139.3, 133.4, 133.2, 131.2, 130.2, 129.8, 128.7, 120.6, 118.5, 116.7, 107.5; HRMS (ESI + ) m/z C16H12NO3S (M + H)+ calcd 298.0532, obsd 298.0537, m/z C16H11NNaO3S (M + Na)+ calcd 320.0352, obsd 320.0362.
4.1.1.12. (Z)-3-Phenyl-2-thioxo-5-(3,4,5-trimethoxybenzylidene)oxazolidin-4-one (1k)
Yellow solid; reaction time, 25 h, 64%; 1H NMR (500 MHz, CDCl3) δ 7.55 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.50 (t, 1H, J = 7.5 Hz, 4-H), 7.40 (d, 2H, J = 7.5 Hz, 2-H, 6-H), 7.13 (s, 2H, 2′-H, 6′-H), 6.76 (s, 1H, vinylic H), 3.93 (s, 9H, 3XOCH3); 13C NMR (100 MHz, CDCl3) δ 182.2, 161.8, 153.7, 141.4, 138.4, 132.5, 129.9, 129.7, 127.6, 126.2, 114.9, 109.3, 61.2, 56.5; HRMS (ESI + ) m/z C19H18NO5S (M + H)+ calcd 372.0900, obsd 372.0906, m/z C19H17NNaO5S (M + Na)+ calcd 394.0720, obsd 394.0722.
4.1.1.13. (Z)-5-(4-Hydroxy-3,5-dimethoxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1l)
Yellow solid; reaction time, 25 h, 54%; 1H NMR (500 MHz, CDCl3) δ 7.55 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.50 (t, 1H, J = 7.5 Hz, 4-H), 7.40 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 7.16 (s, 2H, 2′-H, 6′-H), 6.76 (s, 1H, vinylic H), 5.96 (s, 1H, OH), 3.97 (s, 6H, 2XOCH3); 13C NMR (100 MHz, CDCl3) δ 182.3, 161.8, 147.6, 138.5, 137.7, 132.5, 129.9, 129.7, 127.6, 122.5, 115.5, 109.1, 56.7; LRMS (ESI-) m/z 356 (M−H)−, 341 (M−H−CH3)−.
4.1.1.14. (Z)-5-(3,5-Di-tert-butyl-4-hydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1m)
Yellow solid; reaction time, 20 h, 34%; 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 2H, 2′-H, 6′-H), 7.52 (t, 2H, J = 7.6 Hz, 3-H, 5-H), 7.46 (t, 1H, J = 7.2 Hz, 4-H), 7.37 (d, 2H, J = 7.2 Hz, 2-H, 6-H), 6.80 (s, 1H, vinylic H), 5.71 (s, 1H, OH), 1.46 (s, 18H, 2Xt-Bu); 13C NMR (100 MHz, CDCl3) δ 182.5, 162.0, 157.2, 137.2, 137.1, 132.7, 129.8, 129.7, 129.6, 127.7, 122.8, 116.6, 34.6, 30.3; LRMS (ESI-) m/z 408 (M−H)−.
4.1.1.15. (Z)-5-(3-Bromo-4-hydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1n)
Yellow solid; reaction time, 25 h, 56%; 1H NMR (500 MHz, DMSO‑d6) δ 11.17 (s, 1H, OH), 8.08 (d, 1H, J = 1.5 Hz, 2′-H), 7.79 (dd, 1H, J = 8.5, 1.5 Hz, 6′-H), 7.53 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.49 (t, 1H, J = 7.0 Hz, 4-H), 7.45 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 7.10 (d, 1H, J = 8.5 Hz, 5′-H), 6.93 (s, 1H, vinylic H); 13C NMR (100 MHz, DMSO‑d6) δ 183.7, 162.0, 157.2, 138.7, 136.6, 133.4, 132.8, 130.2, 129.8, 128.7, 124.4, 117.7, 112.6, 110.8; LRMS (ESI-) m/z 374 (M−H)−, 376 (M + 2-H)−.
4.1.1.16. (Z)-5-(3,5-Dibromo-4-hydroxybenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1o)
Yellow solid; reaction time, 20 h, 77%; 1H NMR (500 MHz, DMSO‑d6) δ 10.84 (brs, 1H, OH), 8.11 (s, 2H, 2′-H, 6′-H), 7.54 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.50 (t, 1H, J = 7.0 Hz, 4-H), 7.45 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 6.94 (s, 1H, vinylic H); 13C NMR (100 MHz, DMSO‑d6) δ 183.6, 161.9, 153.5, 139.7, 135.4, 133.3, 130.2, 129.9, 128.6, 126.2, 112.9, 110.5; LRMS (ESI-) m/z 452 (M−H)−, 454 (M + 4-H)−, 456 (M + 6-H)−.
4.1.1.17. (Z)-5-(2,4-Difluorobenzylidene)-3-phenyl-2-thioxooxazolidin-4-one (1p)
Ivory solid; reaction time, 20 h, 40%; 1H NMR (500 MHz, CDCl3) δ 8.28 (td, 1H, J = 8.0, 6.5 Hz, 6′-H), 7.56 (t, 2H, J = 7.5 Hz, 3-H, 5-H), 7.51 (t, 1H, J = 7.0 Hz, 4-H), 7.39 (d, 2H, J = 7.0 Hz, 2-H, 6-H), 7.08 (s, 1H, vinylic H), 7.07 (td, 1H, J = 8.0, 2.5 Hz, 5′-H), 6.92 (td, 1H, J = 9.5, 2.5 Hz, 3′-H); 13C NMR (100 MHz, CDCl3) δ 182.0, 164.7 (dd, J = 246.2, 32.3 Hz), 162.1 (dd, J = 244.8, 12.0 Hz), 161.4, 139.6, 133.2 (dd, J = 10.0, 2.7 Hz), 132.3, 130.1, 129.7, 127.6, 115.9 (dd, J = 12.3, 4.1 Hz), 113.0 (dd, J = 21.6, 3.7 Hz), 104.8 (dd, J = 15.6, 10.3 Hz), 104.5 (d, J = 25.5 Hz); HRMS (ESI + ) m/z C16H10F2NO2S (M + H)+ calcd 318.0395, obsd 318.0395.
4.2. Biological evaluation
4.2.1. Mushroom tyrosinase inhibition assay
(Z)-5-(Substituted benzylidene)-3-phenyl-2-thioxooxazolidin-4-one derivatives 1a–1p were evaluated for mushroom tyrosinase inhibitory activity as previously described [58]. Briefly, 10 µL of 1a–1p (final concentration: 25 µM), 20 µL of tyrosinase solution, and 170 µL of substrate solution [potassium phosphate buffer 14.7 mM + 293 µM L-tyrosine solution (1:1, v/v)] were added to a 96-well microplate. The microplate was then wrapped with aluminum foil and incubated for 0.5 h at 37 0C. Optical densities were measured using a microplate reader (VersaMaxTM, Molecular Devices, Sunnyvale, CA, USA) at 475 nm. Kojic acid (25 µM) was used as the positive control. The experiments were performed independently three times. Mushroom tyrosinase inhibitions were calculated using the following equation.
where A is the absorbance of the test compound and B is the absorbance of the blank.
To determine the IC50 values of 1c, 1j, and kojic acid, dose-dependent inhibition experiments were carried out in triplicate. Logarithmic percentage inhibitions observed at 3–5 concentrations per experiment were plotted and curve-fitting equations were derived. Individual IC50 values were then defined as the concentrations that achieved 50% inhibition.
4.2.2. Kinetic studies of tyrosinase inhibition
Mushroom tyrosinase solution (20 µL, 150 units) and 10 μL of test compounds 1c or 1j (final concentrations: 2.5, 5, or 10 µM for 1c and 5, 10, or 20 µM for 1j) were added to a 96-well plate containing a mixture (170 μL) consisting of an aqueous solution of L-tyrosine at concentrations of 0.5, 1.0, 2.0, 4.0, or 8.0 mM, 50 mM potassium phosphate buffer (pH 6.5), and distilled water in the ratio 10:10:9. The initial rate of formation of dopachrome in the reaction mixture was determined by measuring increases in absorbance at 475 nm (ΔOD475/min) using a microplate reader. Michaelis constant (KM) and maximal velocity (Vmax) of tyrosinase activity were determined using Lineweaver-Burk plots (inverse of reaction velocity (1/V) versus the inverse of substrate concentration (1/[S])) obtained using different concentrations of L-tyrosine. Inhibitory mechanisms were determined using points of convergence of plot lines.
4.2.3. Docking studies of compounds 1c, and 1j, and tyrosinase
Binding energies between tyrosinase and compounds 1c or 1j or kojic acid were determined as previously reported with slight modification [59], [5]. Briefly, Chem3D Pro 12.0 software was used to create 3D structures of the two compounds and the 3D structure of Agaricus bisporus tyrosinase was imported from PDB (ID: 2Y9X). Docking scores were calculated using AutoDock Vina 1.1.2, AutoDock 4 [59], Dock 6 [5], and Chimera software. LigandScout 4.3 was used to generate a pharmacophore model to show possible interactions between ligands and amino acid residues of tyrosinase.
4.2.4. Homology model of human tyrosinase and docking studies of compounds 1c, and 1j, and tyrosinase
The homology model of human tyrosinase was built using the Swiss-Model online server and the Schrödinger Suite 2020–2 release. The full sequence of human tyrosinase (P14679) was obtained from the UnitPro database, and due to the highest sequence identity with human tyrosinase, the crystal structure of human tyrosinase-related protein 1 (PDB ID: 5M8Q) was used as the protein template. The homology model structure of human tyrosinase was downloaded from the Swiss-Model server and further processed by the Schrödinger suite using the Protein Preparation Wizard. The homologous structure of human tyrosinase has also been validated in Prime, a homology modeling tool from the Schrödinger Suite. The structures of compounds 1c, and 1j and kojic were imported to the entry list in CDXML format and prepared for docking using LigPrep [60]. The prepared structures were docked with the homology model using Schrödinger Suite's Ligand Docking Wizard, and 2D and 3D confirmations of the ligand protein complex were prepared [61].
4.2.5. B16F10 cell culture
Murine B16F10 melanoma cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and used for cell viability, cellular tyrosinase activity, and melanin content assays. B16F10 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS (fetal bovine serum), and 1% streptomycin at 37 °C in a 5% CO2 environment, and these cells were then used in cell viability, cellular tyrosinase activity, and melanin content assays in 96-well plates or 6-well dishes. All experiments were independently performed in triplicate.
4.2.6. Cell viability assay: EZ-cytox assay
Cell viability was determined using the EZ-cytox assay. Briefly, B16F10 cells were seeded in a 96-well plate at a density of 5 × 104 cells/well and incubated at 37 °C in a humidified 5% CO2 environment overnight. Cells were treated with compounds 1c or 1j at 0, 5, 10 or 20 μM/well for 24 h, and then EZ-cytox solution (10 μL/well, Daeil Lab Service, Seoul) was added to treated and control cells and incubated at 37 °C for 3 h. Cell viabilities were determined by measuring absorbances at 450 nm using a microplate reader. All experiments were independently performed in triplicate.
4.2.7. Cellular tyrosinase inhibition assays of 1c and 1j in B16F10 melanoma cells
Cellular tyrosinase inhibition assays were conducted as previously described with minor changes [62]. Pre-cultured cells were pretreated with compounds 1c or 1j at 0, 5, 10, or 20 μM for 3 h. α-MSH and IBMX were then added to final concentrations of 1 μM and 200 µM, respectively, and cells were incubated for 48 h. Cells were then washed with PBS two or three times, lysed with 100 μL of buffer [90 μL of PBS (50 mM), 5 μL of PMSF (0.1 mM), and 5 μL of 1% Triton X-100], and lysates were frozen (−80 °C for 0.5 h) then centrifuged (12,000 rpm for 30 min) at 4 °C. Lysate supernatants (80 µL) in 96-well microplates, were mixed with 20 µL of L-dopa (2 mg/mL) and kept in an incubator at 37 °C for 15 min. Optical densities were measured at 475 nm using a microplate reader (Tecan, Männedorf, Switzerland). All experiments were independently performed in triplicate.
4.2.8. Effects of 1c and 1j on melanin contents in B16F10 melanoma cells
The effects of 1c and 1j on melanin production in B16F10 cells were evaluated as previously described with minor changes [63]. Briefly, pre-cultured cells were pretreated with 1c or 1j at 0, 5, 10 or 20 μM and incubated for 3 h and then 1 μM of α-MSH and 200 µM of IBMX were added. After incubation of 48 h, cells were washed 2 to 3 times with PBS buffer and then incubated in 100 μL of 1 N NaOH for 1 h at 60 °C to dissolve the melanin. Optical densities were measured at 405 nm using a microplate reader to determine melanin contents. All experiments were independently performed in triplicate.
4.2.9. Statistical analysis
The significances of differences between groups were determined by one-way ANOVA and Tukey's test. Graph Pad Prism 5 was used for the analysis and results are expressed as means ± SEMs. Two-sided P-values of <0.05 were considered statistically significant.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2020R1A2C1004198).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.12.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Ando H., Kondoh H., Ichihashi M., Hearing V.J. Approaches to identify inhibitors of melanin biosynthesis via the quality control of tyrosinase. J Invest Dermatol. 2007;127(4):751–761. doi: 10.1038/sj.jid.5700683. [DOI] [PubMed] [Google Scholar]
- 2.Chedekel M.R., Post P.W., Deibel R.M., Kalus M. Photodestruction of phaeomelanin. Photochem Photobiol. 1977;26(6):651–653. doi: 10.1111/j.1751-1097.1977.tb07546.x. [DOI] [PubMed] [Google Scholar]
- 3.Germain P., Staels B., Dacquet C., Spedding M., Laudet V. Overview of Nomenclature of Nuclear Receptors. Pharmacol Rev. 2006;58(4):685–704. doi: 10.1124/pr.58.4.2. [DOI] [PubMed] [Google Scholar]
- 4.Kumar K.J.S., Vani M.G., Wang S.-Y., Liao J.-W., Hsu L.-S., Yang H.-L., Hseu Y.-C. In vitro and in vivo studies disclosed the depigmenting effects of gallic acid: A novel skin lightening agent for hyperpigmentary skin diseases. BioFactors. 2013;39(3):259–270. doi: 10.1002/biof.1064. [DOI] [PubMed] [Google Scholar]
- 5.Moustakas D.T., Lang P.T., Pegg S., Pettersen E., Kuntz I.D., Brooijmans N., Rizzo R.C. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20(10-11):601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 6.Afshin A. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med. 2017;377(1):13–27. doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.tyrosinase retardant. 2013, Google Patents.
- 8.Eun Lee K., Bharadwaj S., Yadava U., Gu Kang S. Evaluation of caffeine as inhibitor against collagenase, elastase and tyrosinase using in silico and in vitro approach. J Enzyme Inhib Med Chem. 2019;34(1):927–936. doi: 10.1080/14756366.2019.1596904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costin G.-E., Hearing V.J. Human skin pigmentation: melanocytes modulate skin color in response to stress. FASEB J. 2007;21(4):976–994. doi: 10.1096/fj.06-6649rev. [DOI] [PubMed] [Google Scholar]
- 10.Lu H., Yang K.e., Zhan L., Lu T., Chen X., Cai X., Zhou C., Li H.e., Qian L., Lv G., Chen S. Optimization of Flavonoid Extraction in Dendrobium officinale Leaves and Their Inhibitory Effects on Tyrosinase Activity. Int J Anal Chem. 2019;2019:1–10. doi: 10.1155/2019/7849198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W.-C. Discovery of highly potent tyrosinase inhibitor, T1, with significant anti-melanogenesis ability by zebrafish in vivo assay and computational molecular modeling. Sci Rep. 2015;5:7995. doi: 10.1038/srep07995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh E, et al. A Novel Role of Serotonin Receptor 2B Agonist as an Anti-Melanogenesis Agent. Int J Molecular Sci, 2016;17(4):546. [DOI] [PMC free article] [PubMed]
- 13.Lee J.-Y., Choi H.-J., Chung T.-W., Kim C.-H., Jeong H.-S., Ha K.-T. Caffeic Acid Phenethyl Ester Inhibits Alpha-Melanocyte Stimulating Hormone-Induced Melanin Synthesis through Suppressing Transactivation Activity of Microphthalmia-Associated Transcription Factor. J Nat Prod. 2013;76(8):1399–1405. doi: 10.1021/np400129z. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y. Artopithecins A-D, Prenylated 2-Arylbenzofurans from the Twigs of Artocarpus pithecogallus and Their Tyrosinase Inhibitory Activities. Chem Pharm Bull (Tokyo) 2018;66(12):1199–1202. doi: 10.1248/cpb.c18-00523. [DOI] [PubMed] [Google Scholar]
- 15.Chu H.-L., Wang B.-S., Duh P.-D. Effects of Selected Organo-sulfur Compounds on Melanin Formation. J Agric Food Chem. 2009;57(15):7072–7077. doi: 10.1021/jf9005824. [DOI] [PubMed] [Google Scholar]
- 16.Brotzman N., Xu Y., Graybill A., Cocolas A., Ressler A., Seeram N.P., Ma H., Henry G.E. Synthesis and tyrosinase inhibitory activities of 4-oxobutanoate derivatives of carvacrol and thymol. Bioorg Med Chem Lett. 2019;29(1):56–58. doi: 10.1016/j.bmcl.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee B., Moon K.M., Lee B.-S., Yang J.-H., Park K.I., Cho W.-K., Ma J.Y. Swertiajaponin inhibits skin pigmentation by dual mechanisms to suppress tyrosinase. Oncotarget. 2017;8(56):95530–95541. doi: 10.18632/oncotarget.20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juang L.-J., Gao X.-Y., Mai S.-T., Lee C.-H., Lee M.-C., Yao C.-L. Safety assessment, biological effects, and mechanisms of Myrica rubra fruit extract for anti‐melanogenesis, anti‐oxidation, and free radical scavenging abilities on melanoma cells. J Cosmet Dermatol. 2019;18(1):322–332. doi: 10.1111/jocd.12505. [DOI] [PubMed] [Google Scholar]
- 19.Kim S, et al. Optimization of extraction condition of bee pollen using response surface methodology: correlation between anti-melanogenesis, antioxidant activity, and phenolic content. Molecules, 2015;20(11):19764–74. [DOI] [PMC free article] [PubMed]
- 20.Li H, et al. Identification of anti-melanogenesis constituents from Morus alba L. leaves. Molecules, 2018;23(10):2559. [DOI] [PMC free article] [PubMed]
- 21.Alberti K.G.M.M., Zimmet P., Shaw J. Metabolic syndrome-a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23(5):469–480. doi: 10.1111/j.1464-5491.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- 22.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). Jama, 2001;285(19):2486–97. [DOI] [PubMed]
- 23.Faccio G., Kruus K., Saloheimo M., Thöny-Meyer L. Bacterial tyrosinases and their applications. Process Biochem. 2012;47(12):1749–1760. doi: 10.1016/j.procbio.2012.08.018. [DOI] [Google Scholar]
- 24.Bourquelot E., Bertrand G. Le bleuissement et le noircissement des champignons. CR Soc Biol. 1895;47:582–584. [Google Scholar]
- 25.Feng L., Shi N., Cai S., Qiao X., Chu P., Wang H., Long F., Yang H., Yang Y., Wang Y., Yu H. De Novo Molecular Design of a Novel Octapeptide That Inhibits In Vivo Melanogenesis and Has Great Transdermal Ability. J Med Chem. 2018;61(15):6846–6857. doi: 10.1021/acs.jmedchem.8b00737.s002. [DOI] [PubMed] [Google Scholar]
- 26.<Urolithin patent.pdf>.
- 27.Arndt K.A., Fitzpatrick T.B. Topical use of hydroquinone as a depigmenting agent. JAMA. 1965;194(9):965–967. [PubMed] [Google Scholar]
- 28.Chang TS. An updated review of tyrosinase inhibitors. Int J Molecular Sci, 2009;10(6):2440–75. [DOI] [PMC free article] [PubMed]
- 29.Cabanes J, Chazarra S, Garcia‐Carmona F. Kojic acid, a cosmetic skin whitening agent, is a slow‐binding inhibitor of catecholase activity of tyrosinase. J Pharmacy Pharmacol, 1994;46(12):982–5. [DOI] [PubMed]
- 30.Khatib S., Nerya O., Musa R., Shmuel M., Tamir S., Vaya J. Chalcones as potent tyrosinase inhibitors: the importance of a 2,4-substituted resorcinol moiety. Bioorg Med Chem. 2005;13(2):433–441. doi: 10.1016/j.bmc.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Jung H.J., Lee M.J., Park Y.J., Noh S.G., Lee A.K., Moon K.M., Lee E.K., Bang E.J., Park Y.J., Kim S.J., Yang J., Ullah S., Chun P., Jung Y.S., Moon H.R., Chung H.Y. A novel synthetic compound, (Z)-5-(3-hydroxy-4-methoxybenzylidene)-2-iminothiazolidin-4-one (MHY773) inhibits mushroom tyrosinase. Biosci Biotechnol Biochem. 2018;82(5):759–767. doi: 10.1080/09168451.2018.1445518. [DOI] [PubMed] [Google Scholar]
- 32.Kim S.J., Yang J., Lee S., Park C., Kang D., Akter J., Ullah S., Kim Y.-J., Chun P., Moon H.R. The tyrosinase inhibitory effects of isoxazolone derivatives with a (Z)-β-phenyl-α, β-unsaturated carbonyl scaffold. Bioorg Med Chem. 2018;26(14):3882–3889. doi: 10.1016/j.bmc.2018.05.047. [DOI] [PubMed] [Google Scholar]
- 33.Do Hyun Kim SJK, et al. Design, synthesis, and antimelanogenic effects of (2-substituted phenyl-1, 3-dithiolan-4-yl) methanol derivatives. Drug design, development and therapy, 2017;11:827. [DOI] [PMC free article] [PubMed]
- 34.Yun H.Y. Design, synthesis, and anti-melanogenic effects of (E)-2-benzoyl-3-(substituted phenyl) acrylonitriles. Drug design, development and therapy. 2015;9:4259. doi: 10.2147/DDDT.S89976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halaouli S., Asther M., Sigoillot J.-C., Hamdi M., Lomascolo A. Fungal tyrosinases: new prospects in molecular characteristics, bioengineering and biotechnological applications. J Appl Microbiol. 2006;100(2):219–232. doi: 10.1111/j.1365-2672.2006.02866.x. [DOI] [PubMed] [Google Scholar]
- 36.Barneda-Zahonero B., Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6(6):579–589. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.N.P. Seeram, D.H., Therapeutic uses of urolithins. 2007. p. WO 2007127263 A2.
- 38.Choi J, Jee JG. Repositioning of Thiourea-Containing Drugs as Tyrosinase Inhibitors. Int J Molecular Sci, 2015;16(12):28534–48. [DOI] [PMC free article] [PubMed]
- 39.Campos Júlia Furtado, Pereira Michelly Cristiny, de Sena Wanessa Layssa Batista, de Barros Martins Caio Gomes, de Oliveira Jamerson Ferreira, da Cruz Amorim Cezar Augusto, de Melo Rêgo Moacyr Jesus Barreto, da Rocha Pitta Marina Galdino, de Lima Maria do Carmo Alves, da Rocha Pitta Maira Galdino, da Rocha Pitta Ivan. Synthesis and in vitro anticancer activity of new 2-thioxo-oxazolidin-4-one derivatives. Pharmacol Rep. 2017;69(4):633–641. doi: 10.1016/j.pharep.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Shibo, Tala Srinivasa R., Lu Hong, Zou Peng, Avan Ilker, Ibrahim Tarek S., Abo-Dya Nader E., Abdelmajeid Abdelmotaal, Debnath Asim K., Katritzky Alan R. Design, synthesis, and biological activity of a novel series of 2,5-disubstituted furans/pyrroles as HIV-1 fusion inhibitors targeting gp41. Bioorg Med Chem Lett. 2011;21(22):6895–6898. doi: 10.1016/j.bmcl.2011.08.081. [DOI] [PubMed] [Google Scholar]
- 41.Harada Koichiro, Kubo Hideki, Tanaka Akio, Nishioka Kazuhiko. Identification of oxazolidinediones and thiazolidinediones as potent 17β-hydroxysteroid dehydrogenase type 3 inhibitors. Bioorg Med Chem Lett. 2012;22(1):504–507. doi: 10.1016/j.bmcl.2011.10.095. [DOI] [PubMed] [Google Scholar]
- 42.Mourão R.H., Silva T.G., Soares A.L.M., Vieira E.S., Santos J.N., Lima M.C.A., Lima V.L.M., Galdino S.L., Barbe J., Pitta I.R. Synthesis and Biological Activity of Novel Acridinylidene and Benzylidene thiazolidinediones. Eur J Med Chem. 2005;40(11):1129–1133. doi: 10.1016/j.ejmech.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Chung Ki Wung, Jeong Hyoung Oh, Jang Eun Ji, Choi Yeon Ja, Kim Dae Hyun, Kim So Ra, Lee Kyung Jin, Lee Hye Jin, Chun Pusoon, Byun Youngjoo, Moon Hyung Ryong, Chung Hae Young. Characterization of a small molecule inhibitor of melanogenesis that inhibits tyrosinase activity and scavenges nitric oxide (NO) Biochimica et Biophysica Acta (BBA) - General Subjects. 2013;1830(10):4752–4761. doi: 10.1016/j.bbagen.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Choi Yeon Ja, Uehara Yohei, Park Ji Young, Kim Seong Jin, Kim So Ra, Lee Hee Won, Moon Hyung Ryong, Chung Hae Young. MHY884, a newly synthesized tyrosinase inhibitor, suppresses UVB-induced activation of NF-κB signaling pathway through the downregulation of oxidative stress. Bioorg Med Chem Lett. 2014;24(5):1344–1348. doi: 10.1016/j.bmcl.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 45.Kim Hye Rim, Lee Hye Jin, Choi Yeon Ja, Park Yun Jung, Woo Youngwoo, Kim Seong Jin, Park Min Hi, Lee Hee Won, Chun Pusoon, Chung Hae Young, Moon Hyung Ryong. Benzylidene-linked thiohydantoin derivatives as inhibitors of tyrosinase and melanogenesis: importance of the β-phenyl-α,β-unsaturated carbonyl functionality. Med Chem Commun. 2014;5(9):1410–1417. doi: 10.1039/C4MD00171K. [DOI] [Google Scholar]
- 46.Kim So Hee, Ha Young Mi, Moon Kyoung Mi, Choi Yeon Ja, Park Yun Jung, Jeong Hyoung Oh, Chung Ki Wung, Lee Hye Jin, Chun Pusoon, Moon Hyung Ryong, Chung Hae Young. Anti-melanogenic effect of (Z)-5-(2,4-dihydroxybenzylidene) thiazolidine-2,4-dione, a novel tyrosinase inhibitor. Arch Pharm Res. 2013;36(10):1189–1197. doi: 10.1007/s12272-013-0184-5. [DOI] [PubMed] [Google Scholar]
- 47.Son Sujin, Kim Haewon, Yun Hwi Young, Kim Do Hyun, Ullah Sultan, Kim Seong Jin, Kim Yeon-Jeong, Kim Min-Soo, Yoo Jin-Wook, Chun Pusoon, Moon Hyung Ryong. (E)-2-Cyano-3-(substituted phenyl)acrylamide analogs as potent inhibitors of tyrosinase: A linear β-phenyl-α,β-unsaturated carbonyl scaffold. Bioorg Med Chem. 2015;23(24):7728–7734. doi: 10.1016/j.bmc.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 48.Kang K.H. (Z)-2-(Benzo [d] thiazol-2-ylamino)-5-(substituted benzylidene) thiazol-4 (5H)-one Derivatives as Novel Tyrosinase Inhibitors. Biol Pharm Bull. 2015;38(8):1227–1233. doi: 10.1248/bpb.b15-00300. [DOI] [PubMed] [Google Scholar]
- 49.Chung Ki Wung, Park Yun Jung, Choi Yeon Ja, Park Min Hi, Ha Young Mi, Uehara Yohei, Yoon Jung Hyun, Chun Pusoon, Moon Hyung Ryong, Chung Hae Young. Evaluation of in vitro and in vivo anti-melanogenic activity of a newly synthesized strong tyrosinase inhibitor (E)-3-(2,4 dihydroxybenzylidene)pyrrolidine-2,5-dione (3-DBP) Biochimica et Biophysica Acta (BBA) - General Subjects. 2012;1820(7):962–969. doi: 10.1016/j.bbagen.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 50.Han Yu Kyeong, Park Yun Jung, Ha Young Mi, Park Daeui, Lee Ji Yeon, Lee Naree, Yoon Jeong Hyun, Moon Hyung Ryong, Chung Hae Young. Characterization of a novel tyrosinase inhibitor, (2RS,4R)-2-(2,4-dihydroxyphenyl)thiazolidine-4-carboxylic acid (MHY384) Biochimica et Biophysica Acta (BBA) - General Subjects. 2012;1820(4):542–549. doi: 10.1016/j.bbagen.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Ullah Sultan, Park Yujin, Park Chaeun, Lee Sanggwon, Kang Dongwan, Yang Jungho, Akter Jinia, Chun Pusoon, Moon Hyung Ryong. Antioxidant, anti-tyrosinase and anti-melanogenic effects of (E)-2,3-diphenylacrylic acid derivatives. Bioorg Med Chem. 2019;27(11):2192–2200. doi: 10.1016/j.bmc.2019.04.020. [DOI] [PubMed] [Google Scholar]
- 52.Ullah Sultan, Akter Jinia, Kim Su J., Yang Jungho, Park Yujin, Chun Pusoon, Moon Hyung R. The tyrosinase-inhibitory effects of 2-phenyl-1,4-naphthoquinone analogs: importance of the (E)-β-phenyl-α,β-unsaturated carbonyl scaffold of an endomethylene type. Med Chem Res. 2019;28(1):95–103. doi: 10.1007/s00044-018-2267-9. [DOI] [Google Scholar]
- 53.Ullah Sultan, Kang Dongwan, Lee Sanggwon, Ikram Muhammad, Park Chaeun, Park Yujin, Yoon Sik, Chun Pusoon, Moon Hyung Ryong. Synthesis of cinnamic amide derivatives and their anti-melanogenic effect in α-MSH-stimulated B16F10 melanoma cells. Eur J Med Chem. 2019;161:78–92. doi: 10.1016/j.ejmech.2018.10.025. [DOI] [PubMed] [Google Scholar]
- 54.Ullah Sultan, Park Yujin, Ikram Muhammad, Lee Sanggwon, Park Chaeun, Kang Dongwan, Yang Jungho, Akter Jinia, Yoon Sik, Chun Pusoon, Moon Hyung Ryong. Design, synthesis and anti-melanogenic effect of cinnamamide derivatives. Bioorg Med Chem. 2018;26(21):5672–5681. doi: 10.1016/j.bmc.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 55.Vogeli U. C-13-Nmr Spectroscopy. 19. Structures of Addition-Products of Acetylene-Dicarboxylic Acid-Esters with Various Dinucleophiles - Application of C, H-Spin-Coupling Constants. Helv Chim Acta. 1978;61(2):607–617. [Google Scholar]
- 56.The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2016;45(D1):D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai Xuelei, Wichers Harry J., Soler‐Lopez Montserrat, Dijkstra Bauke W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew Chem Int Ed. 2017;56(33):9812–9815. doi: 10.1002/anie.201704616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hyun Sook Kyung, Lee Won-Hee, Jeong Da Mi, Kim Youngsoo, Choi Jae Sue. Inhibitory Effects of Kurarinol, Kuraridinol, and Trifolirhizin from Sophora flavescens on Tyrosinase and Melanin Synthesis. Biol Pharm Bull. 2008;31(1):154–158. doi: 10.1248/bpb.31.154. [DOI] [PubMed] [Google Scholar]
- 59.Morris Garrett M., Goodsell David S., Halliday Robert S., Huey Ruth, Hart William E., Belew Richard K., Olson Arthur J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 60.Friesner R.A. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 61.Farid Ramy, Day Tyler, Friesner Richard A., Pearlstein Robert A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem. 2006;14(9):3160–3173. doi: 10.1016/j.bmc.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 62.Bae Sung Jin, Ha Young Mi, Kim Jin-Ah, Park Ji Young, Ha Tae Kwun, Park Daeui, Chun Pusoon, Park Nam Hee, Moon Hyung Ryong, Chung Hae Young. A Novel Synthesized Tyrosinase Inhibitor: (E)-2-((2,4-Dihydroxyphenyl)diazenyl)phenyl 4-Methylbenzenesulfonate as an Azo-Resveratrol Analog. Biosci Biotechnol Biochem. 2013;77(1):65–72. doi: 10.1271/bbb.120547. [DOI] [PubMed] [Google Scholar]
- 63.Chen Lih-Geeng, Chang Wei-Ling, Lee Chia-Jung, Lee Lain-Tze, Shih Chwen-Ming, Wang Ching-Chiung. Melanogenesis Inhibition by Gallotannins from Chinese Galls in B16 Mouse Melanoma Cells. Biol Pharm Bull. 2009;32(8):1447–1452. doi: 10.1248/bpb.32.1447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.