

Summary
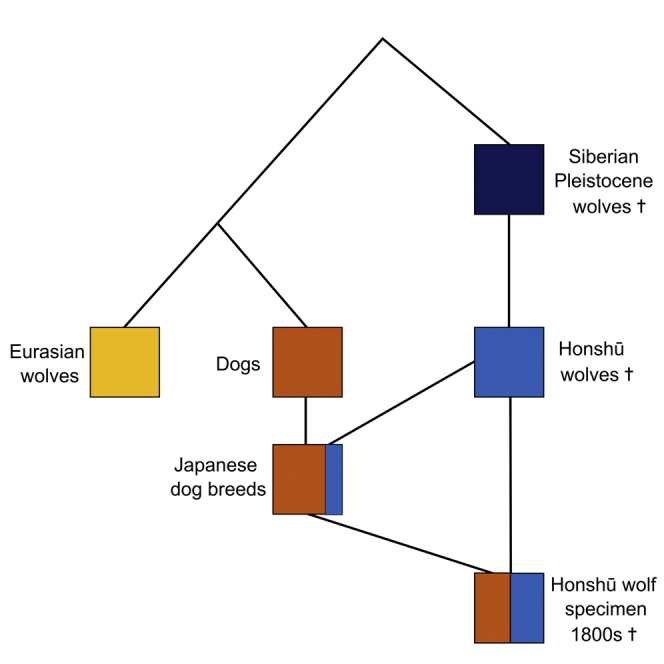
The Japanese or Honshū wolf was one the most distinct gray wolf subspecies due to its small stature and endemicity to the islands of Honshū, Shikoku, and Kyūshū. Long revered as a guardian of farmers and travellers, it was persecuted from the 17th century following a rabies epidemic, which led to its extinction in the early 20th century. To better understand its evolutionary history, we sequenced the nuclear genome of a 19th century Honshū wolf specimen to an average depth of coverage of 3.7✕. We find Honshū wolves were closely related to a lineage of Siberian wolves that were previously believed to have gone extinct in the Late Pleistocene, thereby extending the survival of this ancient lineage until the early 20th century. We also detected significant gene flow between Japanese dogs and the Honshū wolf, corroborating previous reports on Honshū wolf dog interbreeding.
Subject areas: Biological Sciences, Genetics, Phylogenetics, Evolutionary Biology
Graphical Abstract
Highlights
-
•
Generated 3.7✕ nuclear genome of the extinct Honshu wolf
-
•
The Honshū wolf belonged to the lineage of Siberian Pleistocene wolves
-
•
There was gene flow between Honshū wolves and Japanese dogs
Biological Sciences; Genetics; Phylogenetics; Evolutionary Biology
Introduction
Researchers have long debated the evolutionary origin of present-day dogs and Eurasian wolves, as well as their relationships to each other. While it is now clear from analyses based on genome-scale data sets that they are reciprocally monophyletic sister clades and thus dogs are not derived from any as yet identified extant Eurasian wolf lineage (Freedman et al., 2014), from what wolf population dogs were domesticated, and what the ancestral homeland was of their common ancestors, remains debated (Larson and Bradley, 2014). A further open question is the relationship of both lineages to other wolf groups. One key example is the enigmatic Pleistocene Beringian wolves that may have specialized on hunting megafauna (Leonard et al., 2007), several of whose genomes have recently been sequenced from subfossil materials, and found to represent lineages that fall outside of modern dogs and wolves (Ramos-Madrigal et al., 2020, Skoglund et al., 2015). Indeed, studies of both mitochondrial (Koblmüller et al., 2016; Loog et al., 2020) and nuclear genomes (Loog et al., 2020; Skoglund et al., 2015) recovered from Pleistocene and modern Eurasian wolves, suggest the Pleistocene lineages were lost and replaced by the modern lineages around the Pleistocene-Holocene transition in Siberia (Loog et al., 2020) - possibly associated with the extinction of much of the megafauna upon which the Pleistocene lineages may have depended (Leonard et al., 2007). One curious observation from such studies is the lack of evidence for admixture between these Pleistocene wolves and the ancestors of modern Eurasian wolves and most dog breeds (with the exception of some Asian dogs and Arctic sled dogs (Sinding et al., 2020, Skoglund et al., 2015). This is particularly striking given reports of widespread admixture between other coexisting species within the crown Canids (Gopalakrishnan et al., 2018), and suggests that the Pleistocene ancestors of present-day Eurasian wolves and dogs must have been physically isolated from the Siberian Pleistocene wolf lineages during the Last Glacial Maximum (LGM), prior to replacing them following their extinction after 14,000 years ago (Loog et al., 2020), likely at the transition at the Holocene (Leonard et al., 2007). This in turn raises the obvious, and currently unanswered, question, as to where this isolation could have occured?
The Japanese archipelago is one potential candidate for the LGM refugium of the ancestors of modern wolves and dogs, as land bridges between the Korean peninsula and Japan's largest island, Honshū, formed during the Pleistocene and the beginning of the Holocene (Ohshima, 1990). Hokkaidō, the second largest and northernmost island of Japan, was also connected to the Eurasian continent during periods of low sea level, which occurred for instance in the Late Pleistocene (Ohshima, 1990). Until their extinction at the beginning of the 20th century, Japan was inhabited by two highly phenotypically distinct endemic wolf subspecies: the Japanese or Honshū wolf (Canis lupus hodophilax), and the Ezo wolf (Canis lupus hattai). While the Honshū wolf could be found on Honshū, Kyūshū, and Shikoku, the habitat of the Ezo wolf was restricted to Hokkaidō and Sakhalin (Figure 1) (Ishiguro et al., 2009). The Honshū wolf was among the smallest gray wolf subspecies in the world and appreciated in medieval Japan for killing crop-destroying wildlife (Fritts et al., 2003). A rabies epidemic in the 17th century caused an increase in wolf attacks, setting the human persecution of the Honshū wolf in motion, which culminated in their extinction by 1905 (Walker, 2009).
Figure 1.

Geographical distribution of the extinct Honshū and Ezo wolf
The Tsugaru strait separates the former habitat of the Ezo wolf—Sakhalin and Hokkaidō—and the former habitat of the Honshū wolf—the islands of Honshū, Shikoku, and Kyūshū. Map created in ArcGIS, wolf shapes are in relative size (the wither height of the Honshu and Ezo wolf is 56-58cm and 70-80cm, respectively; Ishiguro, 2012)
The deep Tsugaru strait between Honshū and Hokkaidō is a major zoogeographical barrier between the two islands, also known as Blakiston's Line (Dobson, 1994). As a result, the fauna on Honshū, with its snow macaques (Macaca fuscata) and Asian black bears (Ursus thibetanus), has similarities to Southeastern Asia, while the fauna on Hokkaidō, which includes the Ussuri brown bear (Ursus arctos lasiotus), resembles the biological diversity in Northeastern Asia. As a consequence of this barrier, there is no evidence for an overlap between the habitats of the Japanese and the Ezo wolf that most likely colonized the Japanese archipelago from the Korean peninsula and Siberia, respectively.
The exact phylogenetic placement of both subspecies is speculative, as apart from osteological comparisons noting the striking morphological differences between Honshū wolves, Ezo wolves, and present-day wolves (Imaizumi, 1970; Ishiguro et al., 2010), only the mitochondrial genomes have been sequenced in previous studies (Matsumura et al., 2014; Koblmüller et al., 2016). These suggest a basal phylogenetic placement of the Honshū wolf to all modern wolves, and a placement of the Ezo wolf in the North American wolf clade. The mitochondrial genome is however only one marker, and it does not allow the quantification of admixture, which is especially of interest given that both subspecies are potential candidate populations that link Pleistocene wolves and present-day Eurasian wolves.
We sequenced the nuclear genome of one of the two subspecies, the Honshū wolf (Canis lupus hodophilax), to reassess the relationship between Honshū wolves and other wolves and test the hypothesis that Japan was the LGM refugium for the ancestors of present-day wolves.
Results
Gene flow between Honshū wolf, Pleistocene wolves, and present-day wolves and dogs
We resequenced the nuclear genome of a Honshū wolf sample provided by the Natural History Museum, London, to 3.7✕ coverage. The specimen had been shot in the wild in the 1800s in Chichibu District, Kotsuki, Northwest of Tokyo, Japan. We confirmed the authenticity of the ancient DNA based on the misincorporation and read length patterns (Figure S1) and determined the chromosomal sex of the specimen to be male (Figure S2). First, we investigated the evolutionary relationship between the historic Honshū wolf and other wolves and dogs with a whole-genome admixture analysis using NGSadmix (Figure 2A, see also Figure S3). The Honshu wolf specimen was found to have indistinguishable admixture profiles as Pleistocene wolves in the data panel (see Table S1), across all tested numbers of assumed ancestral populations. In contrast to all other modern wolf populations, we find that Pleistocene wolves contributed substantially to the Honshū wolf genome. Irrespective of the number of ancestry clusters inferred in the NGSadmix analysis, the Honshū wolf always derives the majority of its ancestry from the same cluster as the Pleistocene wolves (Figure S4).
Figure 2.

Gene flow between Honshū wolf, Pleistocene wolves, and present-day wolves and dogs
(A) Admixture plot for K = 6 ancestry components. Vertical bars represent single individuals. Different colors indicate the estimated ancestry components. The Honshū wolf forms a cluster with all other Pleistocene wolves (see also Figure S4).
(B) D-statistics scatterplot for the Portugese wolf in H1, samples from the reference data set in H2 (X), and Honshū wolf (y axis) or the Pleistocene wolf Bunge-Toll (x axis) in H3. Vertical and horizontal error bars correspond to three standard errors for the tests in the y- and x axis, respectively. The test involving samples with error bars that intersect the gray dotted line differ insignificantly between the Honshū wolf and Bunge-Toll in H3.
To further explore the admixture landscape between the Honshū wolf and ancient and present-day wolf and dog populations, we used D-statistics to formally test for gene flow between these groups. The D-statistics provide support for excess allele sharing between the Honshū wolf and Greenland dogs, Asian dogs, Pleistocene wolves, and Chinese wolves (Figure 2B). We already observed shared genetic ancestry between Pleistocene wolves and the Honshū wolf in the NGSadmix analysis, so to further investigate wolf and dog populations that might be more genetically similar to the Honshū wolf than other Pleistocene wolves, we created a scatterplot with the D-statistics test with the Honshū wolf and Pleistocene wolf in H3, the Portuguese wolf in H1, and dogs and wolves in the panel in H2 (Figure 2B). The results suggest that the Honshū wolf and Pleistocene wolves are symmetrically related to modern Eurasian and North American wolves, with the exception of some Chinese wolves that share more alleles with the Honshū wolf than they do with any Pleistocene wolf. A potential explanation for this is the substantial admixture between East Asian wolves and dogs (Fan et al., 2016).
All dog individuals included in the D-statistics analysis share significantly more alleles with the Honshū wolf than with the Siberian Pleistocene wolf, with Japanese dogs and Greenland dogs having the closest genetic affinity with the Honshū wolf. Additionally, analysis using qpWave showed that while the Siberian Pleistocene wolves can be explained as a single migration stream equally related to dogs and wolves, the Honshu wolf needs ancestry from the dog lineage. We therefore hypothesize that our Honshū wolf individual was most likely admixed with Japanese dogs, as the excess of shared alleles with the Greenland dogs can be explained by the introgression from Pleistocene wolves to Arctic dogs (Skoglund et al., 2015), and the Chinese dogs could likewise be shown to have a significant wolf contribution (Fan et al., 2016).
Haplotype-aware clustering of Honshu wolf, Pleistocene wolves, and present-day dogs and wolves
In order to more robustly identify population structure among the wolf and dog samples, we used the haplotype-aware clustering tool fineSTRUCTURE (Lawson et al., 2012; see also Tables S2 and S3), which has been previously used on low-coverage ancient individuals (Martiniano et al., 2016, 2017). In the dendrogram based on a similarity matrix, the Honshū wolf was positioned in the same clade as three other Pleistocene wolves – Tumat, Yana, and Bunge-Toll, further corroborating our earlier findings (Figure 3A, see also Figure S5). To further verify our findings of genetic affinity of the Honshū wolf to the Pleistocene Siberian wolves, we performed unsupervised dimension reduction on the haplotype data using principal component analysis (PCA). The Honshū wolf formed an incline together with all Pleistocene wolves along the first principal component (PC1). Among all the wolves included in the analysis, it placed closest to the dog cluster in the first two principal components (Figure 3B, see also Figure S6).
Figure 3.

Haplotype-aware clustering methods modeling the ancestry of Honshū wolf and Japanese/Korean dogs
(A) Heatmap and dendrogram based on shared chromosome segments. Bottom left of the coancestry matrix is based on the unlinked model, i.e. not using genetic linkage information, while the top right shows values for the linked model (see also Figure S5). Higher values in the scale express a higher relatedness. Colored boxes at the axes indicate an individual's population (legend upper left).
(B) Principal component analysis based on the fineSTRUCTURE coancestry matrix. The Honshū wolf formed an incline with all other Pleistocene wolves in Principal component 2 (see also Figure S6).
(C) Estimated proportional contribution (y axis) from the surrogate populations (right) to the respective target population (x axis).
Japanese dog genome modeled as mixture of Honshū wolf and Chinese dog
To further examine the population history of the Honshū wolves, we tested eight putatively related populations: Japanese dogs, Chinese dogs, Greenland dogs, sled dogs, Honshū wolves, Pleistocene wolves, Eurasian wolves, and North American wolves. The chromosomes of a subset of each of these populations were then painted with the best fitting haplotypes of all remaining individuals. The resulting chromosome paintings could then be used as input for GLOBETROTTER (Hellenthal et al., 2014), which uses the haplotype sharing information to describe and date admixture events involving pre-defined populations (surrogate populations) leading to the population of interest (target population).
As GLOBETROTTER requires the data of multiple individuals in the target population to infer admixture dates, we were unable to use the Honshū wolf as a target population. Instead, we chose to run GLOBETROTTER with Japanese dogs as the target population in order to potentially detect gene flow between the Honshū wolves and local dog populations. Using the Chinese dogs, Greenland dogs, sled dogs, Honshū wolves, Pleistocene wolves, Eurasian wolves, and North American wolves as surrogate populations, we estimated that the modern Japanese dog genome can be best described as a mixture of 93% Chinese dog and 7% Honshū wolf. The most likely scenario leading to this admixed Japanese dog population is a single admixture event, occurring approximately 25 generations ago, between a population that is 9% Chinese dog and 91% Honshū wolf, and a population that is 100% Chinese dog. While these preliminary results indicate that Honshū wolves significantly contributed to modern Japanese dog genomes, it is most likely that the large contribution of Japanese dogs to the Honshū wolf genome confounds the results. Further studies with larger sample sizes of Honshū wolves is therefore needed to positively determine the introgression from Honshū wolves to Japanese dog breeds.
Japanese wolf genome modeled as mixture of Pleistocene wolf and Japanese/Korean dog
Finally, using the Markov chain Monte Carlo algorithm implemented in SOURCEFIND, we modeled each of the eight populations used in the GLOBETROTTER analysis—Japanese dogs, Chinese dogs, Greenland dogs, sled dogs, Honshū wolves, Pleistocene wolves, Eurasian wolves, and North American wolves—as a mixture of the remaining seven populations, i.e. all the populations except the one being modeled. The chromosome painting of the population of interest was split into 100 subsections, and each subsection was assigned to the best fitting counterpart from one of the other populations.
Using this method, we estimated that the Honshū wolf genome can be partitioned into a 52% contribution from Pleistocene wolves, 47% contribution from dogs, and a 1% contribution from present-day Eurasian wolves. Furthermore, we detected a 15% contribution from the Honshū wolf to the Japanese/Korean dog breed cluster but found no evidence for haplotype sharing between the Honshū wolf and Chinese dogs (Figure 3C). As explained above, the inference of shared ancestry in highly admixed and ill-defined populations such as wolves and dogs is computationally challenging, and the inclusion of more Honshū wolf genomes is necessary to obtain more statistically sound estimates of gene flow between dogs and the Honshū wolf. That being said, a previous mitochondrial study also documented the introgression from the Honshū wolf to some Japanese dogs (Ishiguro et al., 2009).
Discussion
The results of our analyses show that the recently extinct Honshū wolf is not in the same phylogenetic clade as present-day Eurasian wolves and that only insubstantial gene flow occurred between present-day wolves and the Honshū wolf. We therefore deem it unlikely that the habitat of Honshū wolves was an LGM refugium for the common ancestor of modern wolves and dogs, as the colonization of Japan by the Honshū wolf is estimated to predate the LGM.
However, we made the unexpected discovery that the Honshū wolf specimen we sampled can be best described as a hybrid between Pleistocene wolves and Japanese dogs. Until now, Pleistocene wolves were thought to have gone extinct around the beginning of the Holocene, but the strong genetic affinity between Honshū wolves (Canis lupus hodophilax) and Pleistocene wolves suggests rather that the Japanese archipelago had been a refugium for Pleistocene wolves for thousands of years, where their descendants only went extinct about 100 years ago.
As the Honshū wolf specimen was one of the last of its kind after centuries of human persecution, which resulted in a drastic population decline in the 19th century, it is more than likely that the extent of dog introgression we detected was significantly lower in the Honshū wolf population before they were actively hunted. It is therefore necessary to sequence and analyze the genomes of additional Honshū wolf specimens, especially those that predate the population decline, to obtain a more accurate representation of the genetic makeup of the Honshū wolf. As of now, the high proportion of dog variants in the Honshū wolf specimen hinders our ability to quantify the extent of Honshū wolf introgression into Japanese dog breeds.
Finally, Hokkaidō and Sakhalin island remain potential candidates for LGM refugia, as our analyses only covered the more southern islands Honshū, Shikoku, and Kyūshū. Analyzing the yet understudied Ezo wolf genome might therefore be the key to resolve the mystery of the absent ancestors of present-day dogs and wolves.
Limitations of the study
This study is based on the genome of one Honshū wolf specimen, which we found to be admixed with Japanese dogs. The admixed nature of this individual limits our ability to quantify the genetic contribution from Honshū wolves to Japanese dog breeds.
Resource availability
Lead contact
Further information, requests, and inquiries should be directed to and will be fulfilled by the Lead Contact, Jonas Niemann (jonas@palaeome.org).
Materials availability
This study did not generate new materials.
Data and code availability
The project accession number for the sequencing data project reported in this paper is ENA: PRJEB41490. The sample accession number reported in this paper is ENA: ERS5374233.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
This research was funded by the European Union's Horizon 2020 research and innovation programme under grant agreement no. 676154 (ArchSci2020). The authors thank the Danish National High-throughput Sequencing Center for assistance in generating the sequencing data. We thank the London National History Museum and its curators Louise Tomsett and Richard Sabin for assisting with the sample collection.
Author contributions
M.T.P.G, M.-H.S.S. conceived the study. M.-H.S.S. did the ancient DNA lab work. J.N, S.G. performed the bioinformatic analysis. M.-H.S.S. contributed with sample collection. J.R.-M. provided computation expertise. J.N., M.-H.S.S., S.G., N.W., M.T.P.G. supervised the work. J.N, M.-H.S.S., S.G., J.R.-M., N.Y., M.T.P.G. interpreted the results. J.N, M.-H.S.S., S.G., M.T.P.G. wrote the manuscript with input from all authors. All authors read and approved the manuscript.
Declaration of interests
The authors declare no conflict of interest.
Published: January 22, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101904.
Contributor Information
Jonas Niemann, Email: jonas@palaeome.org.
Mikkel-Holger S. Sinding, Email: mhssinding@gmail.com.
Supplemental information
References
- Dobson M. Patterns of distribution in Japanese land mammals. Mammal Rev. 1994;24:91–111. [Google Scholar]
- Fan Z., Silva P., Gronau I., Wang S., Armero A.S., Schweizer R.M., Ramirez O., Pollinger J., Galaverni M., Ortega Del-Vecchyo D. Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 2016;26:163–173. doi: 10.1101/gr.197517.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman A.H., Gronau I., Schweizer R.M., Ortega-Del Vecchyo D., Han E., Silva P.M., Galaverni M., Fan Z., Marx P., Lorente-Galdos B. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10:e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritts S.H., Stephenson R.O., Hayes R.D., Boitani L. Wolves and humans. In: David Mech L., Boitani Luigi, editors. Wolves: Behavior, Ecology, and Conservation. University of Chicago Press; 2003. pp. 289–316. [Google Scholar]
- Gopalakrishnan S., Sinding M.-H.S., Ramos-Madrigal J., Niemann J., Samaniego Castruita J.A., Vieira F.G., Carøe C., Montero M.de M., Kuderna L., Serres A. Interspecific gene flow shaped the evolution of the genus Canis. Curr. Biol. 2018;28:3441–3449. doi: 10.1016/j.cub.2018.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellenthal G., Busby G.B.J., Band G., Wilson J.F., Capelli C., Falush D., Myers S. A genetic atlas of human admixture history. Science. 2014;343:747–751. doi: 10.1126/science.1243518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi Y. ニホンオオカミの系統的地位について. J. Mammalogical Soc. Jpn. 1970;5:27–32. [Google Scholar]
- Ishiguro N. 絶滅した日本のオオカミの遺伝的系統. 日本獣医師会雑誌. 2012;65:225–231. [Google Scholar]
- Ishiguro N., Inoshima Y., Shigehara N. Mitochondrial DNA analysis of the Japanese wolf (Canis lupus hodophilax Temminck, 1839) and comparison with representative wolf and domestic dog haplotypes. Zoolog. Sci. 2009;26:765–770. doi: 10.2108/zsj.26.765. [DOI] [PubMed] [Google Scholar]
- Ishiguro N., Inoshima Y., Shigehara N., Ichikawa H., Kato M. Osteological and genetic analysis of the extinct Ezo wolf (Canis lupus hattai) from hokkaido island, Japan. Zoolog. Sci. 2010;27:320–324. doi: 10.2108/zsj.27.320. [DOI] [PubMed] [Google Scholar]
- Koblmüller S., Vilà C., Lorente-Galdos B., Dabad M., Ramirez O., Marques-Bonet T., Wayne R.K., Leonard J.A. Whole mitochondrial genomes illuminate ancient intercontinental dispersals of grey wolves (Canis lupus) J. Biogeogr. 2016;43:1728–1738. [Google Scholar]
- Larson G., Bradley D.G. How much is that in dog years? The advent of canine population genomics. PLoS Genet. 2014;10:e1004093. doi: 10.1371/journal.pgen.1004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D.J., Hellenthal G., Myers S., Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8:e1002453. doi: 10.1371/journal.pgen.1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J.A., Vilà C., Fox-Dobbs K., Koch P.L., Wayne R.K., Van Valkenburgh B. Megafaunal extinctions and the disappearance of a specialized wolf ecomorph. Curr. Biol. 2007;17:1146–1150. doi: 10.1016/j.cub.2007.05.072. [DOI] [PubMed] [Google Scholar]
- Loog L., Thalmann O., Sinding M.H.S., Schuenemann V.J., Perri A., Germonpré M., Bocherens H., Witt K.E., Samaniego Castruita J.A., Velasco M.S., Lundstrøm I.K. Ancient DNA suggests modern wolves trace their origin to a Late Pleistocene expansion from Beringia. Mol. Ecol. 2020;29:1596–1610. doi: 10.1111/mec.15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiniano R., Caffell A., Holst M., Hunter-Mann K., Montgomery J., Müldner G., McLaughlin R.L., Teasdale M.D., van Rheenen W., Veldink J.H. Genomic signals of migration and continuity in Britain before the Anglo-Saxons. Nat. Commun. 2016;7:10326. doi: 10.1038/ncomms10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiniano R., Cassidy L.M., Ó’Maoldúin R., McLaughlin R., Silva N.M., Manco L., Fidalgo D., Pereira T., Coelho M.J., Serra M. The population genomics of archaeological transition in west Iberia: investigation of ancient substructure using imputation and haplotype-based methods. PLoS Genet. 2017;13:e1006852. doi: 10.1371/journal.pgen.1006852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S., Inoshima Y., Ishiguro N. Reconstructing the colonization history of lost wolf lineages by the analysis of the mitochondrial genome. Mol. Phylogenet. Evol. 2014;80:105–112. doi: 10.1016/j.ympev.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Ohshima K. The history of straits around the Japanese Islands in the late-Quaternary. Quat. Res. (Daiyonki-kenkyu) 1990;29:193–208. [Google Scholar]
- Ramos-Madrigal J., Sinding M.H.S., Carøe C., Mak S.S., Niemann J., Castruita J.A.S., Fedorov S., Kandyba A., Germonpré M., Bocherens H. Genomes of Pleistocene Siberian Wolves Uncover Multiple Extinct Wolf Lineages. Curr. Biol. 2020 doi: 10.1016/j.cub.2020.10.002. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinding M.H.S., Gopalakrishnan S., Ramos-Madrigal J., de Manuel M., Pitulko V.V., Kuderna L., Feuerborn T.R., Frantz L.A., Vieira F.G., Niemann J., Castruita J.A.S. Arctic-adapted dogs emerged at the Pleistocene–Holocene transition. Science. 2020;368:1495–1499. doi: 10.1126/science.aaz8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P., Ersmark E., Palkopoulou E., Dalén L. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr. Biol. 2015;25:1515–1519. doi: 10.1016/j.cub.2015.04.019. [DOI] [PubMed] [Google Scholar]
- Walker B.L. University of Washington Press; 2009. The Lost Wolves of Japan. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The project accession number for the sequencing data project reported in this paper is ENA: PRJEB41490. The sample accession number reported in this paper is ENA: ERS5374233.