

Severe acute respiratory syndrome coronavirus 2 pandemic capacity is derived from the unique structural features on its spike protein: fast viral surfing over the epithelium with flat N‐terminal domain, tight binding to ACE2 entry receptor, and furin protease utilization. In addition, the possible involvement of other components such as lipid rafts, CLRs, and neuropilin is, in combination, mediating the accelerated cell entry and other critical steps in its overwhelming contagious capacity and pandemy.
Keywords: COVID‐19, furin protease, receptor‐binding domain, SARS‐CoV‐2, sialic acid‐binding domain
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the causative agent of the pandemic coronavirus disease 2019 (COVID‐19) that exhibits an overwhelming contagious capacity over other human coronaviruses (HCoVs). This structural snapshot describes the structural bases underlying the pandemic capacity of SARS‐CoV‐2 and explains its fast motion over respiratory epithelia that allow its rapid cellular entry. Based on notable viral spike (S) protein features, we propose that the flat sialic acid‐binding domain at the N‐terminal domain (NTD) of the S1 subunit leads to more effective first contact and interaction with the sialic acid layer over the epithelium, and this, in turn, allows faster viral ‘surfing’ of the epithelium and receptor scanning by SARS‐CoV‐2. Angiotensin‐converting enzyme 2 (ACE‐2) protein on the epithelial surface is the primary entry receptor for SARS‐CoV‐2, and protein–protein interaction assays demonstrate high‐affinity binding of the spike protein (S protein) to ACE‐2. To date, no high‐frequency mutations were detected at the C‐terminal domain of the S1 subunit in the S protein, where the receptor‐binding domain (RBD) is located. Tight binding to ACE‐2 by a conserved viral RBD suggests the ACE2‐RBD interaction is likely optimal. Moreover, the viral S subunit contains a cleavage site for furin and other proteases, which accelerates cell entry by SARS‐CoV‐2. The model proposed here describes a structural basis for the accelerated host cell entry by SARS‐CoV‐2 relative to other HCoVs and also discusses emerging hypotheses that are likely to contribute to the development of antiviral strategies to combat the pandemic capacity of SARS‐CoV‐2.
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- CD209L
cluster of differentiation 209 Liver
- CLRs
C‐type lectin receptors
- COVID‐19
coronavirus disease 2019
- DSIGN
dendritic cell‐specific ICAM‐3 grabbing nonintegrin
- GAGs
glycosaminoglycans
- HCoV
human coronavirus
- HIV‐1
human immunodeficiency virus 1
- HSPGs
heparan sulfate proteoglycans
- L‐SIGN
liver/lymph node‐specific ICAM‐3grabbing nonintegrin
- matriptase
trypsin‐like integral‐membrane serine peptidase
- NRP1
neuropilin 1
- NTD
N‐terminal domain
- PC1
serine endoprotease proprotein convertase 1
- RBD
receptor‐binding domain
- S protein
spike protein
- SARS‐CoV
severe acute respiratory syndrome Coronavirus
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- TMPRSS2
transmembrane protease, serine 2
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the causative agent of the current pandemic COVID‐19. SARS‐CoV‐2 has a contagious capacity greater than any other previous HCoV. It is the first pandemic‐causing HCoV with unique spike protein (S protein) features and host tropism patterns [1]. However, specific questions remain about why and how this CoV could cause a pandemic, unlike the six previous HCoVs [1, 2].
We propose here a pandemic capacity model based on the unique structural features of the SARS‐CoV‐2 virion, which is a spherical single‐stranded RNA virus of positive polarity with average diameters of 65 (short), 86 (medium), and 97 (long axis) nm [3]. Its envelope structure contains an average 26 S protein trimers protruding from the envelope surface with an average distance of 15 nm to each other [3]. The approximately 10 nm‐long trimeric S protein contains 1273 amino acids and consists of S1 and S2 subunits [3, 4]. The S1 subunit can be further divided into the N‐terminal domain (NTD) and the C‐terminal domain [4]. According to the model presented herein, the highly contagious nature of SARS‐CoV‐2 can be explained based on the structural features of the S protein described below. As a starting point, the reader is referred to Fig. 1, a summary of the structural features of SARS‐CoV‐2 S protein that are unique to this virus, as compared to the six previous HCoVs that did not reach pandemic potential. Briefly, these include (a) the high binding affinity of the receptor‐binding domain (RBD) to angiotensin‐converting enzyme‐2 (ACE‐2); (b) the flat and nonsunken sialic acid‐binding domain, and (c) a four‐amino acid long insert that serves as a cleavage site for furin and other proteases. Each of these is detailed more in the discussions to follow.
Fig. 1.
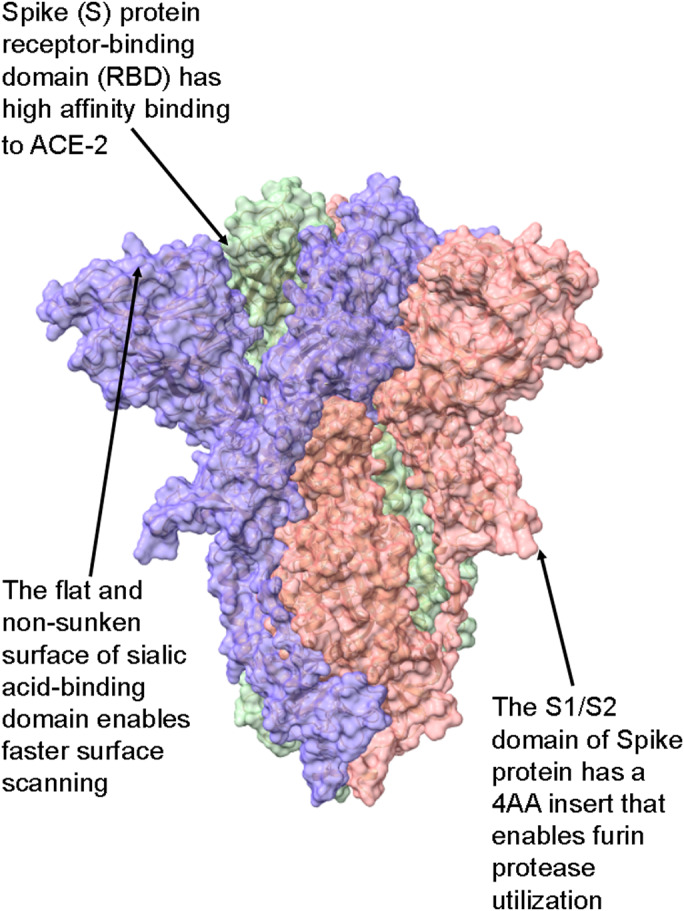
Summary of the S protein structural features unique to SARS‐CoV‐2 over other HCoVs. (A) The binding affinity of the spike RBD to its primary cellular receptor ACE‐2 is more than 10‐fold higher than that of the SARS‐CoV spike RBD. (B) The flat, nonsunken sialic acid‐binding domain is in conflict with that of all other HCoVs, which are sunken in accord with the canyon hypothesis. (C) The S1/S2 domain of SARS‐CoV‐2 S protein contains a four‐amino acid long insert that constitutes a cleavage site for furin proteases, abundant in respiratory epithelia. See text for details. The protein model was S protein, and downstate (PDB ID 6X2C) was modeled with ChimeraX [41].
Structural features of SARS‐CoV‐2 unique among the HCoVs
Tight binding to entry receptor ACE2
After exploring the cell surface, SARS‐CoV‐2 preferentially binds to the entry receptor ACE2, utilizing the critical S protein S1 subdomain residues K417, G446, Y449 L455 F486 N487 Y489 Q493 Q498 T500 N501 G502, and Y505 (Figs 2B, 3, and 4) [5]. In addition, both structural models and mass spectroscopic analysis suggested that the N‐glycans at residues N165 and N234 promote a configuration change in the S protein from the ‘down’ state to the receptor‐accessible ‘up’ state [4, 6]. The SARS‐CoV‐2's S protein has a tighter binding affinity for ACE2 compared with the genetically closest BatCoV RaTG13 and SARS‐CoV [7, 8]. The tight binding is also evident from the mutational analysis of more than 100 000 sequences of SARS‐CoV‐2, which did not demonstrate any high‐frequency mutation on the RBD of the S protein (bigd.big.ac.cn/ncov/). The S protein of other CoVs has high‐frequency hot spots for amino acid replacements that are referred to as positive selection sites subject to changes during host tropism, resistance to antibodies, or immune evasion [9]. Interestingly, in clinical isolates of SARS‐CoV‐2, the only reported S protein mutation was the D614G mutation, which increased host cell entry via ACE2 and transmembrane protease serine 2 (TMPRSS2), but decreased susceptibility to antisera neutralization [10].
Fig. 2.
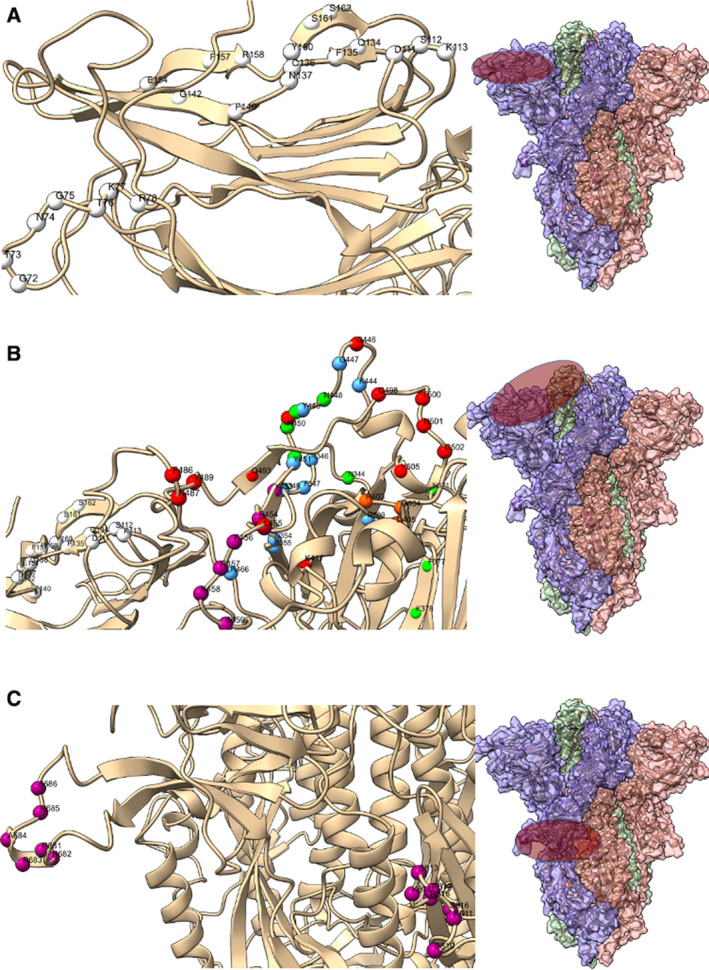
The flat sialic acid‐binding domain. The epithelium is covered with a sialic acid layer. The flat sialic acid‐binding domain of SARS‐CoV‐2 leads to a more effective first contact and interaction with the epithelium, which allows faster viral ‘surfing’ of the epithelial surface and entry receptor scanning. (A) The flat sialic acid, ganglioside, and other sugar‐binding domains (white spheres, left panel) are localized on the left side protomer of the S protein (purple oval, right panel). (B) Entry receptor ACE‐2‐binding domains (red spheres, left panel), C‐type lectin receptor (CLR, green), HSPG (heparan sulfate proteoglycan)‐binding domains based on charge interaction (blue), and HSPG‐binding domains based on GAG (glycosaminoglycan) motif (purple) are localized on the top of the peptomer adjacent to the sialic acid‐binding domain of the S protein (purple oval, right panel); (C) S1/S2 section of the S protein has a unique insert (purple spheres, left panel) that enables it to be cleaved by furin and other proteases, for example, TMPRSS2, PC1, trypsin, matriptase, cathepsin B, and cathepsin L. This domain is localized over the central section of the S protein (purple oval, right panel). Further, another region of the S2 subunit (purple spheres, left panel) has HSPG‐interaction domains (right side of left panel). In addition, the insert and downstream amino acids form a motif capable of binding NRP1 for cellular entry. See text for details. The protein model was S protein, and downstate (PDB ID 6X2C) was modeled with ChimeraX [41].
Fig. 3.
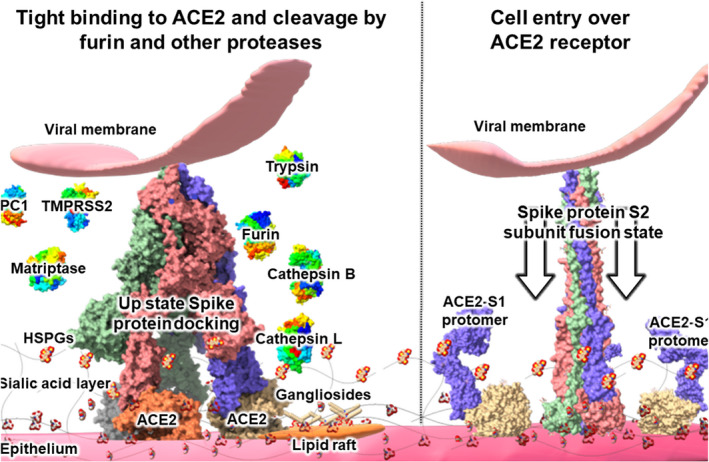
Cleavage of viral S protein by proteases. After viral surfing over the epithelium sialic acid layer, the SARS‐CoV‐2 S protein binds tightly to its entry receptor ACE‐2. A role of sialic acid‐rich gangliosides was also suggested. Unlike other HCoVs, the SARS‐CoV‐2 S1/S2 protein subdomain contains a four‐amino acid insert that constitutes an enzymatic cleavage site for furin and other proteases, which are abundant on respiratory epithelia. Compared to SARS‐CoV, a wider variety of proteases (displayed in left side of Fig. 3) is capable of cleaving the S1/S2 subunit domains of SARS‐CoV‐2, which is believed to form screw‐like S2 fusion conformations composed of spiral trimeric protomers (see right side of Fig. 4) that facilitate host cell entry by SARS‐CoV‐2. See text for details. The protein models were S protein, S protein‐ACE2 complex (PDB ID 7A98), S protein S1 subunit with ACE2 (PDB ID 7A92), S2 subunit postfusion state (PDB ID 6M3W), furin (PDB ID 5JXG), trypsin (PDB ID 3MI4), matriptase (PDB ID 4R0I), cysteine proteases cathepsin B (PDB ID 3MOR), and cathepsin L (PDB ID 3OF9). The models of TMPRSS2 (UniProtKB—O15393), PC1 (GenBank NP_000430.3), were constructed using Swiss‐model (https://swissmodel.expasy.org/). The structures were modeled with ChimeraX and visualized over Microsoft Paint 3D [41].
Fig. 4.
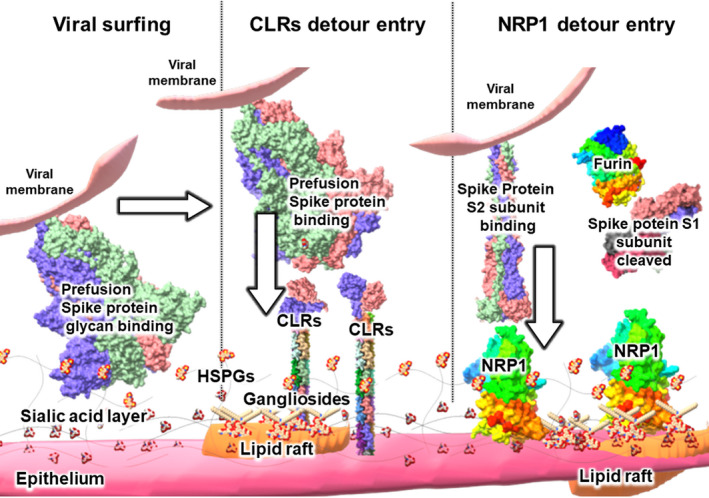
Hypothetical roles for HSPGs, gangliosides, CLRs, NRP1, and lipid rafts in viral S protein –epithelial cell interactions. In a proposed cell entry mechanism based on CLRs, the S protein moves over epithelium with its flat sialic acid‐binding domain (at left of Fig. 4), interacting with free state sialic acids, gangliosides, and various motifs on HSPGs, primarily though relatively weak interactions such as hydrogen bonding. CLRs localized either over the plasma membrane or over lipid rafts (at center of Fig. 4) interact with the S protein and may bypass ACE‐2 and promote virion entry into the cell. Some viruses are known to use CLRs for cell entry to avoid degradation by phagocytes, a process sometimes referred to as the ‘viral escape’ mechanism. Lipid rafts are known to contain many of these components, and thus may also be involved in viral entry. After protease action on the S2 subunit, NRP1 localized either over the plasma membrane or over lipid rafts (at right of Fig. 4) may interact with the S protein and help bypass ACE‐2 to promote virion entry into the cell. See text for details. The protein models were S protein, and downstate (PDB ID 6X2C), S protein S1 subunit (PDB ID 7CHF), S2 subunit postfusion state (PDB ID 6M3W), CLRs (PDB IDs 1XAR with 3JQH), furin (PDB ID 5JXG), and Neuropilin‐1 (PDB ID 2QQM) were modeled with ChimeraX and visualized over Microsoft Paint 3D [41].
In contrast, within SARS‐CoV‐2 isolates, the S protein's NTD and RBD genomic compositions are continuous, which means that these regions of the S protein are not subject to any high‐frequency mutations. Therefore, a significant part of the viral population has the same amino acid composition of the RBD as the first Wuhan isolate, despite millions of infections. This is likely due to the preservation of an optimal ACE2 receptor‐RBD binding interaction and is postulated to be the reason for low‐frequency mutations in this region of the S protein [1].
Flat sialic acid‐binding domain of SARS‐CoV‐2
Sialic acids are the common name of a group of acidic sugars, usually located at the end of epithelial glycoconjugates and carbohydrate chains [11]. A plethora of viruses use sialic acid as part of their infection cycle [12]. Typically, the S protein NTD of HCoVs mediates weak and reversible interactions via low‐affinity hydrogen bonds with the surface glycans such as sialic acid, thereby promoting viral surfing (Fig. 3) [13]. In many other viruses, the sialic acid‐binding site is masked under the S protein surface or even sacrificed to evade host cell immunity [12]. In SARS‐CoV, S protein NTD has amino acid substitutions, compared to its putative parent virus Bat CoV, which has no known sialic acid‐binding activity [14]. In contrast, the SARS‐CoV‐2 NTD has a structural ribbon formation similar to that of human galectins (galactose‐binding lectins), which are sugar‐binding proteins [12].
Therefore, the flat surface of the 290 amino acid residue‐long NTD of SARS‐CoV‐2 S protein may enhance or promotes the prefusion state of S protein sialic acid‐binding capacity, and thereby promotes faster viral surfing compare to other HCoVs (Figs 2, 3, 4) [15, 16]. Indeed, Fantini et al. [16] proposed that SARS‐CoV‐2 is binding not only to the free state sialic acid reisdues over the epithelium, but it also interacts with sialoproteins, glycoproteins, and gangliosides that contain sialic acid domains covered predominantly with Neu5Ac. Additionally, the dual and even triple binding of SARS‐CoV‐2 to ACE‐2 receptor with gangliosides present over lipid rafts, which might form a trimolecular complex with the ACE‐2 receptor for more effective entry [16].
The S protein NTD of SARS‐CoV‐2 is localized at residues 14–305 in the S protein, and the most critical residues for sialic acid‐binding were projected as D111, S112, K113, Q134, F135, C136, N137, F140, G142, E156, F157, R158, Y160, S161, and S162 (Fig. 2A) [16]. It was further noted that SARS‐CoV‐2 S protein variants Q134, F135, and N137, also named as ‘QFN triad residues’ bind to the sialic acid domain of gangliosides [17]. Additionally, SARS‐CoV‐2 S protein NTD has been suggested to have another sugar receptor‐interacting motif G72, T73, N74, G75, T76, K77, and R78 is present in other viruses (Fig. 2A) [18]. In addition, SARS‐CoV‐2 NTD residues E154, F157, and Y160 have similarity to the bovine coronavirus (BCoV) S protein NTD sugar‐binding site, which interacts explicitly with sialic acid derivate Neu5,9Ac2 (Fig. 2A) [18]. The flat surface of the sialic acid‐binding domain is likely a critical pandemic potential factor that accelerates the initial interaction and viral surfing motion of SARS‐CoV‐2 over the epithelial surface and promotes more effective interaction with proteins having sialic acid domains such as gangliosides. Since the S protein sways over the virion membrane with a major tilt angle of 40°, it is believed that the initial interaction point of the S protein with the epithelium is the NTD [3]. Table 1 summarizes the active S protein residues involved.
Table 1.
Interactive residues of SARS‐CoV‐2 S protein.
| Function | Note | Interaction Residues on S protein | Citations |
|---|---|---|---|
| ACE2 | Entry receptor | K417, G446, Y449, L455, F486, N487, Y489, Q493, Q498, T500, N501, G502 and Y505 | [5] |
| Sialic acid binding | On glycoprotein gangliosides and possibly cell surfaces | D111, S112, K113, Q134, F135, C136, N137, F140, G142, E156, F157, R158, Y160, S161 and S162 | [16] |
| Sugar binding | Sugar receptor‐interacting motif | G72, T73, N74, G75, T76, K77, and R78 | [19] |
| Sugar binding | BcoV sugar‐binding domain | E154, F157, and Y160 | [19] |
| HSPGs binding | Positively charged amino acids motif | R346, R355, K444, R466, and R509 | [22] |
| HSPGs binding | Supporting residues | F347, S349, N354, G447, Y449, and Y451 | [22] |
| HSPGs binding | GAG‐binding motif | Y453, R454, L455, F456, R457, K458 and S459 | [23] |
| HSPGs binding | XBBXBX GAG‐binding motif | S1/S2 site P681, R682, R683, A684, R685, and S686 | [23] |
| HSPGs binding | GAG‐binding motif on S2 fusion | S810, K811, P812, S813, K814, R815, and S816 | [23] |
| CLRs binding | CD209L/L‐SIGN CD209/DSIGN binding and detour entry receptor | Over the receptor binding domain and hypothetically binding sites are N344, N371, T376, F377, K378, C379, Y380, G381, N448, Y449, N450, Y451 | [27, 28] |
| NRP1 binding | C‐terminal RXXROH motif, binding and detour entry receptor | S1/S2 site, T676, Q677, T678, N679, S680, P681, R682, R683, A684, and R685 | [24] |
| Protease cleavage | Furin, TMPRSS2, trypsin, matriptase cathepsin B‐L, PC1 | P681, R682, R683, A684, R685, S686, and V687 | [36] |
Cleavage by proteases
After the S protein of SARS‐CoV‐2 binds to the receptor ACE‐2, the S protein is cleaved by proteases that act on its S1/S2 domains, leading to the separation of S1 and S2 domains and formation of screw‐like S2 fusion conformations composed of a spiral of trimeric protomers (Fig. 4) [19]. SARS‐CoV‐2 has a 4‐amino acid long insert (PRRA) on its S1/S1 site that enables furin protease cleavage over the residues P681 to S686 (Fig. 2C) [20]. The action of TMPRSS2, the primary serine protease in many epithelial cells, enhances the cell fusion and entry capacity of SARS‐CoV‐2 compared with the closest genetically related bat virus, RaTG13, which lacks a furin cleavage sequence in its S protein [1, 20].
It has been suggested that the leading proline (P) residue of the SARS‐CoV‐2 S1/S2 domain improves the protease active site accessibility and thereby promotes cleavage, not only by furin but by other proteases [21]. These authors also showed that the SARS‐CoV‐2 S protein could be cleaved by other proteases that cannot cleave the SARS‐CoV S proteins [21]. In addition to furin proteases, SARS‐CoV‐2 can be cleaved by TMPRSS2, serine endoprotease proprotein convertase 1 (PC1), trypsin, trypsin‐like integral‐membrane serine peptidase (matriptase), cysteine proteases cathepsin B, and cathepsin L [21]. SARS‐CoV‐2 S protein's capacity to be cleaved by a wider variety of proteases compared with SARS‐CoV is a key factor in its host cell entry through the ACE‐2 pathway and is likely an essential factor in its pandemic capacity (Figs 3, 4 and Table 1).
Emerging hypotheses on the structural basis of SARS‐CoV‐2 pandemic capacity
Binding to heparan sulfate proteoglycans
In addition to sialic acids, the epithelial surface contains negatively charged linear polysaccharide heparan sulfate proteoglycans (HSPGs) used by viruses for cell surface attachment [22]. Both the prefusion and ACE2‐bound states of SARS‐CoV‐2 were suggested to be interacting with HSPG for cell surface attachment and cellular entry. Clausen et al. reported the HSPGs as a cofactor for SARS‐CoV‐2 S protein binding to the ACE2 entry receptor. It was proposed that positively charged amino acid residues on S protein RBD, close to the ACE2‐binding site, are potential heparin‐binding sites [22]. Software predictions based on heparin–protein contacts and energy contributions implicated the positively charged residues on S protein of R346, R355, K444, R466, and R509. Other residues contributing to heparin binding include F347, S349, N354, G447, Y449, and Y451 (Fig. 2B). Therefore, HSPGs increase the system proton load and trigger the S protein transition from the ‘closed’ inactive RBD conformation to the ‘open’ state that facilitates ACE2 binding [22, 23]. Indeed, preparations of heparin, unfractionated heparin, non‐anticoagulant heparin, heparin lyases, and lung heparan sulfate all can block S protein binding to ACE‐2 and SARS‐CoV‐2 infection in vitro. These results underscore the vital role of HSPGs for COVID‐19 disease progression [22]. The same authors also noted that the S protein RBD of SARS‐CoV does not have an electropositive surface like that observed in SARS‐CoV‐2.
However, another molecular interaction model was predicted for SARS‐CoV‐2 and the oversulfated polysaccharides termed glycosaminoglycans (GAGs,) with such GAG‐binding being promoted by the presence of the GAG‐binding‐like motifs [23]. Kim et al predicted sites of SARS‐CoV‐2 S protein that can be involved in HSPG binding. The first location overlaps with that predicted by Clausen et al. [23] on the residues Y453, R454, L455, F456, R457, K458, and S459 (Fig. 2B), when the S protein is in the ‘up’, open or receptor‐accessible conformation. However, Kim et al. also proposed an additional function of the furin binding insert RRAR forming an XBBXBX (PRRARS) GAG‐binding motif on the S1/S2 domain on S protein. This motif includes the residues P681, R682, R683, A684, R685, and S686 (Fig. 2B) [23]. The insert P681 to A684 is a polybasic site that provides a structural interaction region on S protein to negatively charged HSPGs and possibly to the sialic acids. These polybasic sites on the SARS‐CoV‐2 S protein are, in a sense, analogous to the phosphorylation sites of proteins being activated with ATP.
This constitutes another unique functionality of the SARS‐CoV‐2 subgenus, in addition to its furin cleavage site. Some of the GAG‐binding proteins contain specific amino acid sequences ‘XBBXBX' and 'XBBBXXBX', also known as Cardin–Weintraub motifs correspond to the furin cleavage motif BBXBB of SARS‐CoV‐2 and is not present in SARS‐CoV or Middle East respiratory syndrome [23]. Further, the third HSPG‐binding site was predicted on the S protein S2 domain, immediately before the fusion peptide residues S810, K811, P812, S813, K814, R815, and S816 on the S2 proteolytic cleavage site that is functional in cell entry (Fig. 2C) [23]. When combined with the model of Clausen et al., HSPGs might also increase the negative charge of the GAG‐binding domain of the S2 and promote cell entry [22, 23]. The SARS‐CoV‐2 S protein has two different perfusion states; one is the receptor‐accessible ‘up’ configuration, and the other is the receptor‐inaccessible ‘down’ configuration state [19]. It is speculated that a SARS‐CoV‐2 virion with structurally available surface areas and polybasic domains could use negatively charged sialic acid and heparan sulfate molecules with free electrons to transition into the receptor‐accessible ‘up’ state more effectively, and this might be another factor underlying its pandemic capacity (Figs 2, 3, 4).
Neuropilin‐1‐dependent host cell entry without ACE2 receptors
Another functionality of the S1/S2 insert unique to the SARS‐CoV‐2 S protein is the presence of a C‐terminal end (residues 676‐TQTNSPRRAR‐685) RXXROH motif downstream of the furin cleavage. This motif was shown to be the binding site for neuropilin 1 (NRP1), a ‘detour’ entry receptor over olfactory neuronal cells of the nasal epithelium that functions without using ACE2 [24]. Epstein–Barr virus enters nasopharyngeal epithelial cells through NRP1 with macropinocytosis and lipid raft‐dependent endocytosis; therefore, SARS‐CoV‐2 might also use NRP1 associated with the lipid rafts for its host cell entry [25].
Viral escape pathway based on C‐type lectin receptors
As aforementioned, many viruses have structural features designed to limit their interaction with host surface sialic acids to prevent detection by the immune receptors such as C‐type lectin receptors (CLRs), which are specialized for pathogenic sialic acid interaction [12, 26]. As viruses are essentially protein/nucleic acid complexes with no active mobility, they utilize their hosts' surface components such as sialic acids, protease enzymes, and other proteins for entry into their host cells. However, perhaps the most clever and self‐serving strategy employed by viruses to capitalize on host elements is the use of CLRs for cell entry [26]. After endocytosis, some viruses intended for viral degradation find a way to survive by using immune receptors as a pathway for infection, a process is known as ‘viral escape’ [26]. The uniquely flat sialic acid interaction of SARS‐CoV‐2, which we hypothesize allows faster viral surfing, might also provide an unexpected advantage to the virus through the CLRs promoting a higher rate of not only cell entry, but also viral escape.
Watanabe et al. [6] proposed that the relatively fewer N‐linked glycosylation sites of the S protein of SARS‐CoV‐2, compared to HIV‐1 Env and Lassa virus (LASV) S proteins, could be associated with higher viral detection by CLRs and might explain the lower fatality of SARS‐CoV‐2 relative to those viruses. SARS‐CoV‐2 was found to interact with CLRs, cluster of differentiation 209 (CD209L) /liver/lymph node‐specific ICAM‐3 grabbing nonintegrin (L‐SIGN), and CD209 /dendritic cell‐specific ICAM‐3‐grabbing nonintegrin (DSIGN) as ‘detour’ entry receptors, all acting through the S protein RBD [27]. Similar interactions were detected in SARS‐CoV bound to the DC‐SIGN receptors over the S protein residues 363–368, N‐linked glycosylation sites N330 and N357, and residues 435–439 [28]. Amraie et al. [27] did not investigate the putative DSIGN interacting residues; however, based on the ClustalW alignment, it suggested that the SARS‐CoV residues 363–368 (TFKCYG) are identical to SARS‐CoV‐2 residues 376–381. The N‐linked glycosylation site N330 of SARS‐CoV is identical to N344 of SARS‐CoV‐2, N357 of SARS‐CoV is identical to N371 of SARS‐CoV‐2, and the SARS‐CoV residues on 435–439 NYNY are identical to SARS‐CoV‐2 residues on 448–451. These similarities suggest that the conserved interacting residues with DSIGN CLRs act within the Sarbecoronavirus subgenus (Fig. 2B).
Strikingly, S protein glycosylation at the residues N227 and N699 (residues N234 and N707 in SARS‐CoV‐2) increased DC/L‐SIGN‐mediated pseudovirus entry into cultured cells and was suggested to have a role in SARS‐CoV host tropism from civets to humans [29]. The analogous residues of SARS‐CoV‐2 that are important for increasing the DC/L‐SIGN‐mediated cell entry are all oligomannosylated [6, 27, 28]. This was expected because DC/L‐SIGN preferentially recognizes oligomannose [29]. Therefore, it seems plausible that the combination of effective sialic acid‐binding and the CLR‐dependent viral entry mechanism of SARS‐CoV‐2 may constitute a distinct pathway that does not require ACE‐2 receptors or protease cleavage and might be an essential factor underlying the pandemic capacity of SARS‐CoV‐2 (Fig. 3). Indeed, Fantini et al. [16] proposed the dual binding of the virus to gangliosides and adjacent ACE‐2; however, DSIGN CLRs are also found on lipid rafts, and therefore, the lipid raft might also be an essential contributor to the viral escape entry pathway [30]. In light of the finding that type II alveolar cells of the lung, respiratory dendritic cells, and associated endothelial cells are rich in CLRs, it seems plausible to theorize that CLRs are related to the pandemic capacity of SARS‐CoV‐2 [31].
With potential relevance to central nervous system pathology in COVID‐19, previous studies have shown the direct involvement of the mannose receptor, a type of CLRs, in HIV‐1 astrocyte infection, and suggested that the interaction of HIV‐1 with this receptor plays an important role in the neuropathogenesis of HIV‐1 [32]. In another study, it was established that the phagocytic pathway dependent on the binding of mannose‐rich carbohydrates of gp120 to the MR expressed in the microglia leads to the nonreplicative entry of HIV‐1 [33]. It is currently widely known that HIV‐1 infection in the brain usually occurs in monocytes/macrophages (migrating) and microglia in an MR‐mediated manner. In contrast, a less‐vigorous infection occurs in astrocytes, which are also highly reactive in producing compounds toxic to neurons. It is known that HIV‐1 does not infect neurons [34, 35].
On the other hand, it cannot be ruled out that during infection of SARS‐CoV‐2 in the brain, the MR may be involved through its expression in microglia and astrocytes. In a hypothetical scenario, some of the neurological damage related to COVID‐19 could be due to interaction between SARS‐CoV‐2 and glial cells through the expression of MR or other pattern recognition receptors (PRRs). Numerous reports reinforce brainstem infection as the leading cause of respiratory failure in patients affected by COVID‐19 [36]. Since CLRs and other PRRs are recognized to play a critical role in the innate immune signaling pathways, their involvement in COVID‐19‐related systemic hyperinflammation could be influencing neurovascular endothelial function, breakdown of the blood–brain barrier, and activation of the CNS innate immune response. All these mechanisms might contribute to CNS complications associated with SARS‐CoV‐2 infection [36]. However, the cellular and molecular components of these disorders still need to be clarified.
Hypothetical lipid raft‐dependent endocytosis
The lipid rafts are sphingolipids and cholesterol‐rich domains on the fluid mosaic model of the plasma membrane [37]. Cellular and/or exogenous proteins use lipid rafts for mobility over cell surface. Lipid rafts are usually associated with signal receptor domains (signalosomes) of the plasma membrane [37]. Many viruses use lipid rafts for binding, cell entry, assembly, and exit [38], and SARS‐CoV cell entry through the lipid rafts was detected on Vero cells [39]. Lipid raft‐dependent structural model of virus‐host interactions has been developed by Fantini et al. for human immunodeficiency virus 1 (HIV‐1) on CD4 T cells [37]. In the model, HIV‐1 surface envelope glycoprotein gp120 complex with CD4 membrane protein interacted with adjacent coreceptors over the membrane such as chemokine receptors CXCR4 and CCR5 [37]. The dual interaction between the HIV‐1 gp120 with CD4 and coreceptor first triggers lipid raft‐mediated repulsion over the surface of the T cell and a conformational changes in the HIV‐1, thus unmasking fusion protein gp41 [37]. Therefore, SARS‐CoV‐2 may experience similar membrane tension with lipid raft‐mediated repulsion through S protein ACE‐2 especially when lipid raft contains gangliosides or even other lipid raft localized signal receptors such as CLRs, NRP1, and perhaps integrins [16, 17, 37]. Considering the mobile nature of lipid rafts through the mosaic membrane, SARS‐CoV‐2 S protein ganglioside‐rich lipid rafts over the epithelium surface could act as a surf board during the viral surfing [16, 17, 25, 30, 37]. Therefore, it is quite plausible that SARS‐CoV‐2 might use lipid rafts for viral surfing over the epithelium and lipid raft‐dependent endocytosis (Fig. 3). Table 1 summarizes the S proteins and host cell features involved.
Conclusion
The model described herein of the pandemic capacity of SARS‐CoV‐2 is based on the following structural features unique to SARS‐CoV‐2: (a) Flat sialic acid‐binding domain enables faster viral surfing over the epithelial surface before receptor interaction; (b) tight and almost perfect binding to the ACE2 entry receptor; (c) the capacity to use furin and other proteases for cell entry. Other factors which are likely to contribute to the pandemic capacity of SARS‐CoV‐2 included (d) binding to HSPGs through several different sites; (e) binding to CLRs for viral escape‐based ‘detour’ entry without cleavage by protease; (f) NRP1‐based detour entry without ACE2 involvement; (g) possible lipid raft‐dependent endocytosis through gangliosides and CLRs (Figs 3, 4). SARS‐CoV‐2 can move rapidly over the cell surface by a significant interaction with surface sugars and receptors and may hypothetically be using three or more different pathways for cell entry. Therefore, these unique interactions of the SARS‐CoV‐2 are believed to be the critical pandemic capacity factors. On this basis, targets for treatment options may include antibodies against S protein NTD to limit sialic acid‐binding such as neutralizing antibody (4A8), ganglioside‐binding agents such as hydroxychloroquine, exogenous heparin against HSPGs, protease inhibitors, ganglioside mimics such as azithromycin, and cholesterol‐depleting agents targeting lipid rafts such as cyclodextrin [16, 17, 19, 23, 38, 40]. Clearly, more research is needed to explore each of these possibilities.
Conflict of interest
MS has a utility model application with the file number of GM 63/2020 to the Österreichisches Patentamt.
Author contributions
MS and BDU compiled the text. SSH, VNU, PPC, KT, KL, AAAA, DP, DA, PA, SG, GG, TMAE, AGS, RK, GKA, SPS, WB, GP, AMB, and ASA edited the manuscript; and NR and MT proofread the manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.15651
[Correction added on 12 February 2021, after first online publication: URL for peer review history has been corrected.]
Acknowledgements
This work supported by internal funds from all the authors' institutions. The author consortium thanks coauthors KT for optimization of the figures, BU for compiling author contributions and final editing, and especially MS for lead authorship, conceptualization, and modeling.
All authors are member of the Self‐Assembled COVID Research and Education Directive (SACRED) Consortium
References
- 1. Seyran M, Pizzol D, Adadi P, El‐Aziz TMA, Hassan SS, Soares A, Kandimalla R, Lundstrom K, Tambuwala M, Aljabali AA et al. (2020) Questions concerning the proximal origin of SARS‐CoV‐2. J Med Virol. 10.1002/jmv.26478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elrashdy F, Redwan EM & Uversky VN (2020) Why COVID‐19 transmission is more efficient and aggressive than viral transmission in previous coronavirus epidemics? Biomolecules 10, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, Zhang J, Weng T, Zhang Z & Wu Z (2020) Molecular architecture of the SARS‐CoV‐2 virus. Cell 183, 730–738 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casalino L, Gaieb Z, Goldsmith JA, Hjorth CK, Dommer AC, Harbison AM, Fogarty CA, Barros EP, Taylor BC & McLellan JS (2020) Beyond shielding: the roles of glycans in the SARS‐CoV‐2 spike protein. ACS Cent Sci 6, 1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A & Li F (2020) Structural basis of receptor recognition by SARS‐CoV‐2. Nature 581, 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watanabe Y, Allen JD, Wrapp D, McLellan JS & Crispin M (2020) Site‐specific glycan analysis of the SARS‐CoV‐2 spike. Science 369, 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walls AC, Park Y‐J, Tortorici MA, Wall A, McGuire AT & Veesler D (2020) Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181, 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ & Gamblin SJ (2020) SARS‐CoV‐2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin‐cleavage effects. Nat Struct Mol Biol 27, 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Forni D, Cagliani R, Clerici M & Sironi M (2017) Molecular evolution of human coronavirus genomes. Trends Microbiol 25, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ozono S, Zhang Y, Ode H, Seng TT, Imai K, Miyoshi K, Kishigami S, Ueno T, Iwatani Y & Suzuki T (2020) Naturally mutated spike proteins of SARS‐CoV‐2 variants show differential levels of cell entry. BioRxiv. [PREPRINT] 10.1101/2020.06.15.151779. [DOI] [Google Scholar]
- 11. Schauer R & Kamerling JP (2018) Exploration of the sialic acid world. In Advances in Carbohydrate Chemistry and Biochemistry (Baker DC, ed), pp. 1–213, Elsevier, Edinburgh. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li F (2015) Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol 89, 1954–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burckhardt CJ & Greber UF (2009) Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog 5, e1000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T & Hu J (2020) Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caldas LA, Carneiro FA, Higa LM, Monteiro FL, da Silva GP, da Costa LJ, Durigon EL, Tanuri A & de Souza W (2020) Ultrastructural analysis of SARS‐CoV‐2 interactions with the host cell via high resolution scanning electron microscopy. Sci Rep 10, 16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fantini J, Di Scala C, Chahinian H & Yahi N (2020) Structural and molecular modeling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS‐CoV‐2 infection. Int J Antimicrob Agents 55, 105960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fantini J, Chahinian H & Yahi N (2020) Synergistic antiviral effect of hydroxychloroquine and azithromycin in combination against SARS‐CoV‐2: what molecular dynamics studies of virus‐host interactions reveal. Int J Antimicrob Agents 56, 106020. [DOI] [PubMed] [Google Scholar]
- 18. Behloul N, Baha S, Shi R & Meng J (2020) Role of the GTNGTKR motif in the N‐terminal receptor‐binding domain of the SARS‐CoV‐2 spike protein. Virus Res 286, 198058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benton DJ, Wrobel AG, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ & Gamblin SJ (2020) Receptor binding and priming of the spike protein of SARS‐CoV‐2 for membrane fusion. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffmann M, Kleine‐Weber H & Pöhlmann S (2020) A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell 78, 779–784.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaimes JA, Millet JK & Whittaker GR (2020) Proteolytic cleavage of the SARS‐CoV‐2 spike protein and the role of the novel S1/S2 site. Iscience 23, 101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, Narayanan A, Majowicz SA, Kwong EM & McVicar RN (2020) SARS‐CoV‐2 infection depends on cellular heparan sulfate and ACE2. Cell 183, 1043–1057.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim SY, Jin W, Sood A, Montgomery DW, Grant OC, Fuster MM, Fu L, Dordick JS, Woods RJ & Zhang F (2020) Characterization of heparin and severe acute respiratory syndrome‐related coronavirus 2 (SARS‐CoV‐2) spike glycoprotein binding interactions. Antiviral Res 181, 104873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cantuti‐Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T & Anastasina M (2020) Neuropilin‐1 facilitates SARS‐CoV‐2 cell entry and infectivity. Science 370, 856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H‐B, Zhang H, Zhang J‐P, Li Y, Zhao B, Feng G‐K, Du Y, Xiong D, Zhong Q & Liu W‐L (2015) Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nature Commun 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bermejo‐Jambrina M, Eder J, Helgers LC, Hertoghs N, Nijmeijer BM, Stunnenberg M & Geijtenbeek TB (2018) C‐type lectin receptors in antiviral immunity and viral escape. Frontiers Immunol 9, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amraei R, Napoleon M, Yin W, Berrigan J, Suder E, Zhao G, Olejnik J, Gummuluru S, Muhlberger E & Chitalia V (2020) CD209L/L‐SIGN and CD209/DC‐SIGN act as receptors for SARS‐CoV‐2 and are differentially expressed in lung and kidney epithelial and endothelial cells. BioRxiv. [PREPRINT]. 10.1101/2020.06.22.165803. [DOI] [Google Scholar]
- 28. Shih Y‐P, Chen C‐Y, Liu S‐J, Chen K‐H, Lee Y‐M, Chao Y‐C & Chen Y‐MA (2006) Identifying epitopes responsible for neutralizing antibody and DC‐SIGN binding on the spike glycoprotein of the severe acute respiratory syndrome coronavirus. J Virol 80, 10315–10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han DP, Lohani M & Cho MW (2007) Specific asparagine‐linked glycosylation sites are critical for DC‐SIGN‐and L‐SIGN‐mediated severe acute respiratory syndrome coronavirus entry. J Virol 81, 12029–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cambi A, de Lange F, van Maarseveen NM, Nijhuis M, Joosten B, van Dijk EM, de Bakker BI, Fransen JA, Bovee‐Geurts PH & van Leeuwen FN (2004) Microdomains of the C‐type lectin DC‐SIGN are portals for virus entry into dendritic cells. J Cell Biol 164, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brufsky A & Lotze MT (2020) DC/L‐SIGNs of hope in the COVID‐19 pandemic. J Med Virol 92, 1396–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, Blum J & He JJ (2004) CD4‐independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol 78, 4120–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trujillo JR, Rogers R, Molina RM, Dangond F, McLane MF, Essex M & Brain JD (2007) Noninfectious entry of HIV‐1 into peripheral and brain macrophages mediated by the mannose receptor. Proc Nat Acad Sci USA 104, 5097–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lutgen V, Narasipura SD, Barbian HJ, Richards M, Wallace J, Razmpour R, Buzhdygan T, Ramirez SH, Prevedel L & Eugenin EA (2020) HIV infects astrocytes in vivo and egresses from the brain to the periphery. Plos Pathog 16, e1008381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holder GE, McGary CM, Johnson EM, Zheng R, John VT, Sugimoto C, Kuroda MJ & Kim W‐K (2014) Expression of the mannose receptor CD206 in HIV and SIV encephalitis: a phenotypic switch of brain perivascular macrophages with virus infection. J Neuroimmune Pharmaco 9, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Najjar S, Najjar A, Chong DJ, Pramanik BK, Kirsch C, Kuzniecky RI, Pacia SV & Azhar S (2020) Central nervous system complications associated with SARS‐CoV‐2 infection: integrative concepts of pathophysiology and case reports. J Neuroinflammation 17, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fantini J, Garmy N, Mahfoud R & Yahi N (2002) Lipid rafts: structure, function and role in HIV, Alzheimer's and prion diseases. Expert Rev Mol Med 4, 1–22. [DOI] [PubMed] [Google Scholar]
- 38. Bukrinsky MI, Mukhamedova N & Sviridov D (2020) Lipid rafts and pathogens: the art of deception and exploitation. J Lipid Res 61, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu Y, Liu DX & Tam JP (2008) Lipid rafts are involved in SARS‐CoV entry into Vero E6 cells. Biochem Biophys Res Commun. 369, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fantini J, Chahinian H & Yahi N (2020) Leveraging coronavirus binding to gangliosides for innovative vaccine and therapeutic strategies against COVID‐19. Biochem BiophysRes commun. 10.1016/j.bbrc.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH & Ferrin TE (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]