

Summary
IFI16 is an important player of the host intrinsic immune response. Among others, it has been reported to sense intermediate products of HIV-1 reverse transcription in the cytosol and to sequester the transcription factor Sp1 in the nucleus to attenuate viral gene expression. Here, we present three different methods to reduce IFI16 protein expression levels in HIV-1 primary target cells. These techniques can be adapted for the investigation of other cellular factors in primary macrophages and CD4+ T lymphocytes.
For complete details on the use and execution of this protocol, please refer to Hotter et al. (2019) and Bosso et al. (2020).
Subject Areas: Immunology, Molecular Biology
Graphical Abstract
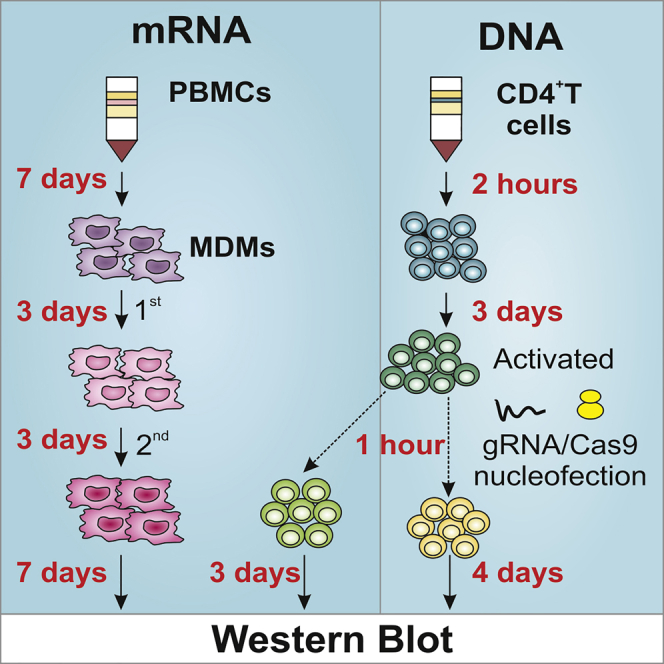
Highlights
-
•
IFI16 knockdown by siRNA achieves about 50% efficiency in primary macrophages
-
•
shRNA-mediated IFI16 knockdown in lymphocytes reduces protein levels by about 40%
-
•
Nucleofection of gRNA/Cas9 reduces IFI16 expression in lymphocytes with high efficacy
-
•
Knockdown of IFI16 by gRNA/Cas9 nucleofection did not cause off-target effects
IFI16 is an important player of the host intrinsic immune response. Among others, it has been reported to sense intermediate products of HIV-1 reverse transcription in the cytosol and to sequester the transcription factor Sp1 in the nucleus to attenuate viral gene expression. Here, we present three different methods to reduce IFI16 protein expression levels in HIV-1 primary target cells. These techniques can be adapted for the investigation of other cellular factors in primary macrophages and CD4+ T lymphocytes.
Before you begin
Isolation of peripheral blood mononucleate cells (PBMCs) from buffy coats
Timing: 2 h
-
1.Decide how many macrophages you will need for the experiment and adapt the volume of buffy coat you prepare, accordingly. Usually, 12.5∗106 cells are sufficient to perform one experiment in technical duplicates. Three donors are sufficient for one complete experiment.
-
a.Dilute 1:1 the buffy coat in 1× PBS (pH 7.2).
-
b.Prepare n 50 mL Falcons (according to the required amount of diluted buffy coat) with 15 mL Bicoll in each.
-
c.Carefully layer 15 mL of buffy coat/PBS dilution on top of the Bicoll.
-
d.Centrifuge 20 min at 1,200 rcf at 22°C–23°C (no brake, acceleration 5).
-
e.Collect the white blood cell ring from the tubes and transfer the cells from the tubes into new 50 mL Falcons (approximately 15 mL each).
-
f.Fill up with PBS to 50 mL.
-
g.Centrifuge 10 min at 453 rcf at 22°C–23°C (acceleration 9, brake 9).
-
h.Remove supernatant by pouring.
-
i.Resuspend the cell pellet by scratching the Falcons on the grid of the flow bench and add 10 mL EDTA (1 mM) in PBS.
-
j.Fill up with PBS to 50 mL.
-
k.Centrifuge 5 min at 453 rcf at 22°C–23°C (acceleration 9, brake 9).
-
l.Remove supernatant, resuspend, and add 50 mL PBS.
-
m.Centrifuge 5 min at 453 rcf at 22°C–23°C (acceleration 9, brake 9).
-
n.Discard supernatant, resuspend cell pellet, pool cells from the same donor within one tube and add 10 mL RPMI (supplemented with 10% (v/v) heat-inactivated FCS, L-glutamine (2 mM), streptomycin (100 μg/mL) and penicillin (100 U/mL)) (RPMIxxx).
-
o.Dilute cells 1:100 and count them.
-
a.
Differentiation of PBMCs into monocyte-derived macrophages (MDMs)
-
2.
Dilute cells with medium RPMIxxx supplemented with 10% (v/v) heat-inactivated human serum from AB donors and 15 ng/mL M-CSF to a final concentration of 2.5∗106 cells/mL.
-
3.Seed cells in a 12 well plate (NUNC) (1 mL/well).
-
a.On day 3 (72 h post plating) replace the medium with MDM medium (DMEM(0xx)) + 10% (v/v) heat-inactivated human AB serum + 15 ng/mL M-CSF.
-
a.
-
4.
Cells will be ready for siRNA transfection on day 7 after isolation.
Isolation of CD4+ T lymphocytes from buffy coats
-
5.Isolation of CD4+ T lymphocytes from buffy coat
-
a.Bring to room temperature (22°C–25°C ) PBS + 2% (v/v) FCS and Biocoll separating solution (Biochrom).
-
b.Dilute buffy coats with equal amount of PBS + 2% (v/v) FCS and gently mix.
-
c.For each mL of blood add 50 μL of RosetteSep Human CD4+ T cell Enrichment Cocktail and gently mix.
-
d.Incubate 20 min at 22°C–23°C.
-
e.Prepare density medium, 15 mL in 50 mL tube.
-
f.Dilute sample with equal amount of PBS + 2% (v/v) FCS and mix gently.
-
g.Layer 15 mL blood sample on top of the density medium.
-
h.Centrifuge 20 min, 1,200 rcf, acceleration 5, no breaking, 22°C–23°C.
-
i.Remove the enriched CD4+ T cell layer (between the Plasma on top and density medium under).
-
j.Wash with PBS + 2% (v/v) FCS.
-
k.Remove supernatant and add 5 mL of ACK Lysing Buffer for 5 min to remove contaminating erythrocytes, wash with PBS + 2% (v/v) FCS.
-
l.Resuspend cells in 10 mL RPMIxxx 20% (v/v) FCS.
-
m.Count the cells.
-
a.
Stimulation of CD4+ T lymphocytes
-
6.
Stimulate cells with IL-2 (10 ng/mL) and 25 μL of anti-CD3/CD28 magnetic beads per 1 million cells (1 bead/cell).
-
7.
Incubate stimulated CD4+ T lymphocytes in a concentration of 1 million cells/mL in RPMIxxx 20% (v/v) FCS.
-
8.Cultivate for 3 days at 37°C, in a 5% CO2 incubator.
-
a.Regularly check the cell culture under the light microscope to monitor cell growth and avoid overgrowth.
-
a.
Note: use magnetic beads for the lymphocyte’s stimulation. They will be easy to remove after 3 days once the activation of the cells is completed.
Amplification and isolation of shRNA
-
9.Transformation of competent Escherichia coli XL-2 blue
-
a.Mix 5 μL of competent Escherichia coli XL-2 blue with 0.5 μg/μL of shRNA.
-
b.Incubate 20 min on ice.
-
c.Heat shock 42°C for 45 s.
-
d.Incubate on ice 2 min.
-
e.Add 200 μL SOC medium.
-
f.Incubate at 37°C with gently shaking for 30 min.
-
g.Plate 30 μL on agar plate with selective antibiotic.
-
h.Incubate for 14 h at 37°C.
-
i.Select one colony and inoculate liquid broth (LB) medium, incubate for 14 h at 37°C.
-
j.For the isolation of shRNA use the QIAGEN midi kit (Cat# 12145); it is critical to use a DNA isolation kit that gives high purity DNA!
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| IRDye 680RD goat anti-mouse IgG (H + L), 1:20,000 | LI-COR | Cat# 926-68070 RRID:AB_10956588 |
| IRDye 800CW goat anti-mouse IgG (H + L), 1:20,000 | LI-COR | Cat# 926-32210 RRID:AB_621842 |
| IRDye 680RD goat anti-rat IgG (H + L), 1:20,000 | LI-COR | Cat# 926-68076 RRID: AB_10956590 |
| IRDye 800CW goat anti-rat IgG (H + L), 1:20,000 | LI-COR | Cat# 926-32219, RRID: AB_1850025 |
| Mouse monoclonal anti-IFI16, 1:150 | Santa Cruz | Cat# sc-8023 RRID: AB_627775 |
| Rat monoclonal anti-GAPDH, 1:1,000 | BioLegend | Cat# 607902 RRID: AB_2734503 |
| Rabbit monoclonal anti-actin, 1:5,000 | Abcam | Cat# ab8226; RRID: AB_306371 |
| Bacterial and virus strains | ||
| XL2-Blue MRF′ ultracompetent cells | Agilent Technologies | Cat# 200151 |
| Biological samples | ||
| Human: peripheral blood mononuclear cells | DRK-Blutspendedienst Baden-Württemberg-Hessen, Ulm, Germany | N/A |
| Oligonucleotides | ||
| Non-targeting (NT) gRNA: ACGGAGGCTAAGCGTCGCAA |
IDT | N/A |
| IFI16 gRNA: GACCAGCCCTATCAAGAAAG |
IDT | N/A |
| CTRL siRNA: UUCUCCGAACGUGUCACGUdTdT |
Eurofins | N/A |
| IFI16 siRNA (1); GGUGCUGAACGCAACAGAAUCAUUU AAAUGAUUCUGUUGCGUUCAGCACC |
Cat# IFI16HSS105205 | |
| IFI16 siRNA (2): GGAUUGAUUAGAAGUGCCAGCGUAA UUACGCUGGCACUUCUAAUCAAUCC |
Invitrogen | Cat# IFI16HSS105206 |
| IFI16 siRNA (3): UUAGUUUGUGAGCAAAGCUGAUUGA UCAAUCAGCUUUGCUCACAAACUAA |
Invitrogen | Cat# IFI16HSS105207 |
| IFI16 shRNA | Origine | Cat# TR312254 |
| Chemicals and reagents | ||
| Amaxa 4D-Nucleofector Human Activated T cell P3 Lonza Kit | Lonza | Cat# V4XP-3032 |
| Phosphate-buffer saline (PBS) | Thermo Fisher | Cat#14190144 |
| ACK lysis buffer (1×) | Lonza | Cat# 10-548E |
| HiFi Cas9 nuclease V3 | IDT | Cat# 1081061 |
| L-Glutamine | Pan Biotech | Cat# P04-80100 |
| Dulbecco’s modified Eagle’s medium (DMEM) | Gibco | Cat# 41965039 |
| Opti-MEM | Gibco | Cat# M3942 |
| Roswell Park Memorial Institute (RPMI) | Gibco | Cat# 22400089 |
| Penicillin-streptomycin | Thermo Fisher | Cat# 15140122 |
| FCS | Gibco | Cat# 10270106 |
| β-Mercaptoethanol | Sigma-Aldrich | Cat# 444203 |
| Recombinant human IL-2 | MACS Miltenyi Biotec | Cat# 170076147 |
| Lipofectamine RNAiMAX | Thermo Fisher | Cat# 13778075 |
| Anti-CD3/CD28 magnetic beads | Life Technologies | Cat# 11131D |
| Biocoll separating solution | Biochrom | Cat# L6113 |
| RosetteSep human CD4+ T cell enrichment cocktail | STEMCELL Technologies | Cat# 15022 |
| 4× protein sample loading buffer | LI-COR | Cat# 928-40004 |
| Complete ULTRA inhibitor cocktail tablet | Roche | Cat# 05892791001 |
| Human M-CSF | R&D Systems | Cat# 216-MC-01M |
| Plasmid midi kit | QIAGEN | Cat# 12145 |
| SOC medium | Invitrogen/Life Technologies | Cat# 15544034 |
| Software and algorithms | ||
| Corel DRAW 2017 | Corel Corporation | https://www.coreldraw.com/ |
| GraphPad Prism version 8 | GraphPad Software |
https://www.graphpad.com RRID: SCR_002798 |
| LI-COR Image Studio Lite version 5.0 | LI-COR |
www.licor.com/ RRID: SCR_013715 |
Materials and equipment
Alternatives: isolation of MDMs can be performed using the RosetteSep Human Monocyte Enrichment Cocktail, following the manufacturer instructions.
Step-by-step method details
siRNA transfection of macrophages
This step explains the procedure to transfect siRNAs in MDMs.
-
1.
Remove medium from plated macrophages and add 500 μL fresh DMEMxxx (supplemented with 10% (v/v) heat-inactivated FCS, L-glutamine (2 mM), streptomycin (100 μg/mL) and penicillin (100 U/mL))
-
2.Transfect with siRNA (45 nM final molarity) using Lipofectamine RNAimax (preferentially done during late afternoon to reduce Lipofectamine-derived cellular toxicity)
-
a.Prepare (scale according to the number of samples) the following master mixes
-
i.Tube 1: 3 μL Lipofectamine + 75 μL OptiMEM
-
ii.Tube 2: 1.46 μL siRNA (stock concentration: 20 μM) + 75 μL OptiMEM
-
i.
-
b.Add contents of tube 1 to tube 2 and mix by pipetting
-
c.Incubate for 10 min at 22°C–23°C
-
d.Drop 150 μL of the Lipofectamine-siRNA complex solution onto the cells in a dropwise manner. When all the required wells have been transfected, gently rock the plate to evenly distribute the liposome complexes.
-
a.
-
3.
Morning of the following day: remove medium and add 750 μL fresh MDM medium (DMEM0xx + 10% (v/v) human serum + 15 ng/ mL M-CSF) (this step is roughly done at 14 h post siRNA transfection)
-
4.Early morning of three days post transfection, remove medium from cultured macrophages and add 500 μL fresh DMEMxxx
-
a.Repeat point 2.
-
a.
-
5.
Incubate cells for 1 week at 37°C in a 5% CO2 incubator.
CRITICAL: always consider doing siRNA transfection in technical duplicates. siRNA master mixes have to be prepared under the flow hood. Disinfect the working area with 70% isopropanol and inactivate any residual nucleases with RNase away.
Validation of knockdown efficiency
This step illustrates processing of cells for western blot analysis
-
6.At 1 week post transfection, harvest and lyse cells for western blot
-
a.Remove supernatants.
-
b.Add 500 μL PBS.
-
c.Remove PBS and add 100 μL western blot lysis buffer (150 mM NaCl, 50 mM HEPES, 5 mM EDTA, 0.1% NP40, 500 μM Na3VO4, 500 μM NaF, pH 7.5) supplemented with Complete ULTRA protease inhibitor cocktail tablet (1 pill/10 mL) (this step should be done one plate at the time).
-
d.Leave the plate for 3 min on ice.
-
e.Carefully rinse the well and pool cell lysates from the same technical replicates into a 1.5 mL Eppendorf tube.
-
f.Vortex 1 s and incubate on ice for 20 min.
-
g.Vortex 1 s and centrifuge lysates at 20,817 rcf for 20 min at 4°C.
-
h.Add 90 μL of supernatants to 30 μL of loading dye (90% (v/v) 4× loading dye + 10% (v/v) β-mercaptoethanol).
-
i.Heat at 95°C for 5 min.
-
j.Freeze at −20°C.
-
a.
-
7.SDS-PAGE Immunoblot
-
k.Load 15–20 μL of cell lysates on a NuPAGE 4%–12% Bis-Tris gel (Invitrogen) and run them for 90 min at 120 V.
-
l.Transfer proteins on an Immobilion-FL Transfer PVDF Membranes (Merck Millipore) with the Trans-Blot SD Semi-Dry Transfer Cell for 2 h.
-
m.Block the membrane with 5% milk (w/v) in PBS-T (0.02% Tween (v/v)).
-
k.
Stain membranes with anti-IFI16 (1G7) mouse (stock concentration: 200 μg/mL; working dilution: 1:150) and anti-GAPDH (W17079A) rat (stock concentration: 0.5 μg/μL; working dilution: 1:1,000) antibodies.
-
8.
Quantify band intensities using Image Studio Lite Ver 5.0.
Transfection of CD4+ T lymphocytes with shRNA
Nucleofection of CD4+ T lymphocytes with shRNA to downregulate expression of IFI16.
-
9.Remove the anti-CD3/CD28 magnetic beads from CD4+ T lymphocytes
-
a.Resuspend cells thoroughly to separate cells from the beads.
-
k.Transfer cells in an appropriate tube size and place it on a magnet.
-
l.Wait 10 min.
-
m.Transfer cells into a fresh tube.
-
k.
-
a.
-
10.
Count the cells.
-
11.
Prepare 12 well plate with 1.5 mL of RPMI 20% (v/v) FCS, IL-2 (10 ng/mL) and L-glutamine (2 mM) without antibiotics (RPMIxx)/well. Place it into the cell culture incubator (37°C, 5% CO2).
-
12.
Pre-warm Human T cell nucleofector solution with added supplement (delivered together with the kit).
-
13.
Prepare cuvettes.
-
14.
Take 6 million CD4+ T lymphocytes per sample.
-
15.
Centrifuge cells at 393 rcf for 3 min at 22°C–23°C and remove the supernatant as much as possible.
-
16.
Add 100 μL of T cell nucleofector solution, add 1.5 μg of IFI16 shRNA, gently mix, and transfer into the cuvette.
-
17.
Place the cuvette into the Amaxa device and pulse the sample with U15 program.
-
18.
Immediately transfer the sample into the medium in the 12 well plate (one sample/well).
Validation of knockdown efficiency
This step describes how to determine the knockdown efficiency via western blot analysis.
-
19.Harvest and lyse cells
-
a.72 h after transfection, harvest one million cells and centrifuge them at 393 rcf for 5 min at 22°C–23°C.
-
b.Remove the medium and add 500 μL of PBS.
-
c.Centrifuge at 393 rcf for 5 min at 22°C–23°C.
-
d.Remove the supernatant and add 100 μL of western blot lysis buffer (150 mM NaCl, 50 mM HEPES, 5 mM EDTA, 0.1% (v/v) NP40, 500 μM Na3VO4, 500 μM NaF, pH 7.5) supplemented with protease inhibitor.
-
e.Vortex and incubate on ice for 10 min.
-
f.Centrifuge for 20 min at 20,817 rcf at 4°C.
-
g.Transfer the supernatant in a new 1.5 mL Eppendorf tube.
-
h.Measure protein concentration using the Nanodrop. Normalize protein concentration for each donor with CoIP lysis buffer.
-
i.Transfer 30 μL of supernatants in a new 1.5 mL Eppendorf tube and add 10 μL of loading dye (90% (v/v) 4× loading dye + 10% (v/v) β-mercaptoethanol).
-
j.Heat at 95°C for 10 min.
-
k.Freeze at −20°C.
-
a.
-
20.
Perform western blot analysis as described in the siRNA section (points 8 and 9).
gRNA/Cas9 nucleofection of CD4+ T lymphocytes
This step accomplishes the gRNA/Cas9 nucleofection
-
21.
Mix 80 pmol Cas9 and 240 pmol sgRNA in a final volume of 5 μL.
-
22.
Incubate the complex for 20 min at 22°C–23°C.
-
23.Prior to nucleofection, prepare the nucleofector master mix.
-
a.For the mix use 82% (v/v) of the Lonza P3 Primary Cell Solution and 18% (v/v) of the supplement 1 for a total volume of 20 μL for one nucleofection.
-
a.
-
24.In the meantime, remove the anti-CD3/CD28 magnetic beads from the CD4+ T lymphocyte culture and transfer 1 million cells in a new 1.5 mL Eppendorf tube (use one tube per reaction).
-
a.Spin down the cells and carefully remove the medium.
-
a.
-
25.
Add 20 μL of nucleofector master mix and straight afterward the gRNA/Cas9 complex to the cells, mix gently, and transfer it in the nucleofection strip. Avoid making the bubbles.
-
26.
Insert the strip in the 4D Nucleofector Core Unit and chose the pulse code EO115.
-
27.
Add 80 μL warm RPMIxx supplemented with 20% (v/v) FCS and IL-2 (10 ng/mL) to the strip and incubate in cell culture incubator for 10 min.
-
28.
Add 200 μL warm RPMIxx with 20% (v/v) FCS and IL-2 (10 ng/mL) in a 48 well plate, transfer the electroporated cells in the plate and culture for 14 h at 37°C, 5% CO2.
-
29.
On the following day, add 300 μL of warm RPMIxx with 20% (v/v) FCS and IL-2 (10 ng/mL). Transfer cells in a 24 well plate. (See Troubleshooting 1)
-
30.
48 h post nucleofection, supplement the culture with 500 μL fresh RPMIxx with 20% (v/v) FCS and IL-2 (10 ng/mL). If necessary, transfer the cells in a bigger culture format.
Proving knockout efficiency via western blot
This step describes how to determine the knockout efficiency via western blot analysis.
-
31.Harvest and lyse cells
-
a.96 h after nucleofection, harvest one million cells and centrifuge them at 393 rcf for 5 min at 22°C–23°C.
-
b.Remove the medium and add 500 μL of PBS.
-
c.Centrifuge at 393 rcf for 5 min at 22°C–23°C.
-
d.Remove the supernatant and add 100 μL of western blot lysis buffer (150 mM NaCl, 50 mM HEPES, 5 mM EDTA, 0.1% (v/v) NP40, 500 μM Na3VO4, 500 μM NaF, pH 7.5) supplemented with protease inhibitor.
-
e.Vortex and incubate on ice for 10 min.
-
f.Centrifuge for 20 min at 20,817 rcf at 4°C.
-
g.Transfer the supernatant in a new 1.5 mL Eppendorf tube.
-
h.Measure protein concentration using the Nanodrop. Normalize protein concentration for each donor with CoIP lysis buffer.
-
i.Transfer 30 μL of supernatants in a new 1.5 mL Eppendorf tube and add 10 μL of loading dye (90% (v/v) 4× loading dye + 10% (v/v) β-mercaptoethanol).
-
j.Heat at 95°C for 10 min.
-
k.Freeze at −20°C.
-
a.
-
32.
Perform western blot analysis as described in the siRNA section (points 8 and 9). (See Troubleshooting 2)
Expected outcomes
According to the siRNA transfection process, it is expected to see a reduction of IFI16 expression levels by an average of 50% (Figure 1).
Figure 1.

IFI16 knockdown in primary macrophages
Human monocyte-derived macrophages were treated with IFI16-specific or control siRNA and knockdown efficiencies were determined by western blot analysis (indicated below) (Hotter et al., 2019)
An efficient shRNA transfection results in a 40% reduction of IFI16 protein levels. Figure 2 is an example of an IFI16 knockdown in primary CD4+ T lymphocytes.
Figure 2.

IFI16 knockdown in primary CD4+ T lymphocytes
Human CD4+ T lymphocytes isolated from three different donors were transfected with either a control or an IFI16-specific shRNA. Knockout efficiency was assessed by western blot. The right panel shows the quantification of IFI16 and PYHIN1 band intensities in the presence of the non-targeting (nt) control or an IFI16-specific shRNA. Data are represented as means ± SD.
A successful gRNA/Cas9 complex nucleofection in CD4+ T lymphocytes should result in a 70%–90% knockout. The IFI16 gRNA should specifically target IFI16 and not cross react with any other PYHIN protein.
Figure 3 depicts an example of western blot showing an efficient IFI16 knockout using gRNA/Cas9.
Figure 3.

IFI16 knockout in primary CD4+ T lymphocytes
Human CD4+ T lymphocytes isolated from three different donors were nucleofected with either a control or an IFI16-specific gRNA and Cas9. Knockout efficiency was assessed by western blot (left panel). Quantification of IFI16 and PYHIN1 band intensity (right panel) verified the specificity of the IFI16 gRNA. Data are represented as means ± SD.
Quantification and statistical analysis
Western blot band intensities were quantified using the LI-COR Image Studio Software.
Statistical analyses were performed using GraphPad PRISM 8 (GraphPad Software). P-values were determined using a two-tailed Student’s t test. Unless otherwise stated, data are shown as the mean of at least three independent experiments ± SD. Significant differences are indicated as: ∗p <0.05; ∗∗p <0.01; ∗∗∗p <0.001.
Limitations
Cell culture viability represents the main obstacle concerning experiments with primary macrophages as this is donor-dependent. On day 7 post macrophages isolation, cells should be fully attached to the bottom of the wells and have either a fibroblast-like or rounded shape. If cells are still floating, most likely the differentiation did not work properly and it is pointless to carry on the experiment. From our experience, this is very often linked to over-counting mistakes, leading to a reduced cell density in the well. Using an automatic cell counter would help in preventing this.
Concerning the knockdown approaches, using either IFI16 siRNA or shRNA may result in potential off-target effect with PYHIN1-mRNA. To undergo this limitation, we suggest to use the gRNA/Cas9 approach. Off-targets of siRNA and shRNAs are mainly due to their instability. On the other hand, gRNAs are known to be much more stable due to their structure which includes R-loop dynamics (Mullally et al., 2020).
However, the gRNA/Cas9 approach may have economical restrictions given the high costs of both reagents but still remains the preferable approach, given its very high efficiency compared to the mRNA-targeting techniques.
Troubleshooting
Problem 1
Low CD4+ T lymphocytes viability upon nucleofection.
Potential solution
Compared to chemical transfection, nucleofection induces membrane-pores formation via an external voltage. The main drawback is represented by a partial successful membrane repair, which could be overcome by some easy steps:
Make sure that the anti-CD3/CD28 magnetic beads were completely removed;
Do not leave the cells for more than 15 min in the Lonza (P3 Primary Cell Solution or Human T cell nucleofector solution);
After nucleofection immediately add medium or transfer the cells in to the medium without antibiotics;
Avoid cells overgrowth by splitting the cells in a bigger plate format; make sure to use 20% (v/v) FCS in the RPMI culture medium.
Problem 2
Low IFI16 knockdown/knockout efficiency.
Potential solution
As these procedures involve working with RNA, it is important to work in a nuclease free environment. This can be achieved by carefully inactivating DNases and RNases with RNase away and by using sterile filter tips. Concerning the IFI16 gRNA, multiple freeze and thaw cycles will reduce its stability. Therefore, it is recommended to aliquot the gRNA upon resuspension in nuclease free water.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frank Kirchhoff (frank.kirchhoff@uni-ulm.de).
Materials availability
No materials were generated in this study.
Data and code availability
No data or code were generated in this study.
Acknowledgments
This work was supported by grants from the German Research Foundation (DFG CRC 1279 and SPP 1923) to F.K.
Author contributions
M.B., C.P.B., and M.V. developed the protocols and prepared this manuscript. F.K. supervised the studies and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Matteo Bosso, Email: matteo.bosso@uni-ulm.de.
Frank Kirchhoff, Email: frank.kirchhoff@uni-ulm.de.
References
- Bosso M., Prelli Bozzo C., Hotter D., Volcic M., Stürzel C.M., Rammelt A., Ni Y., Urban S., Becker M., Schelhaas M. Nuclear PYHIN proteins target the host transcription factor Sp1 thereby restricting HIV-1 in human macrophages and CD4+ T cells. PLoS Pathog. 2020;16:e1008752. doi: 10.1371/journal.ppat.1008752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotter D., Bosso M., Jønsson K.L.K.L., Krapp C., Stürzel C.M.C.M., Das A., Littwitz-Salomon E., Berkhout B., Russ A., Wittmann S. IFI16 targets the transcription factor Sp1 to suppress HIV-1 transcription and latency reactivation. Cell Host Microbe. 2019;25 doi: 10.1016/j.chom.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullally G., van Aelst K., Naqvi M.M., Diffin F.M., Karvelis T., Gasiunas G., Siksnys V., Szczelkun M.D. 5′ modifications to CRISPR–Cas9 gRNA can change the dynamics and size of R-loops and inhibit DNA cleavage. Nucleic Acids Res. 2020;48:6811–6823. doi: 10.1093/nar/gkaa477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data or code were generated in this study.