

Summary
Immune cells, such as macrophages, reprogram their lipid metabolism in response to the activation of pattern recognition receptors (e.g., TLRs, NLRs) and cytokine receptors (e.g., interferons, interleukins). Profiling these changes can be achieved with shotgun mass spectrometry. This protocol provides step-by-step instructions on the generation and stimulation of bone marrow-derived macrophages (BMDMs), sample collection, and lipid extraction for profiling the macrophage lipidome.
For complete details on the use and execution of this protocol, please refer to Hsieh et al. (2020).
Subject Areas: Cell culture, High Throughput Screening, Immunology, Metabolism, Model Organisms, Mass Spectrometry
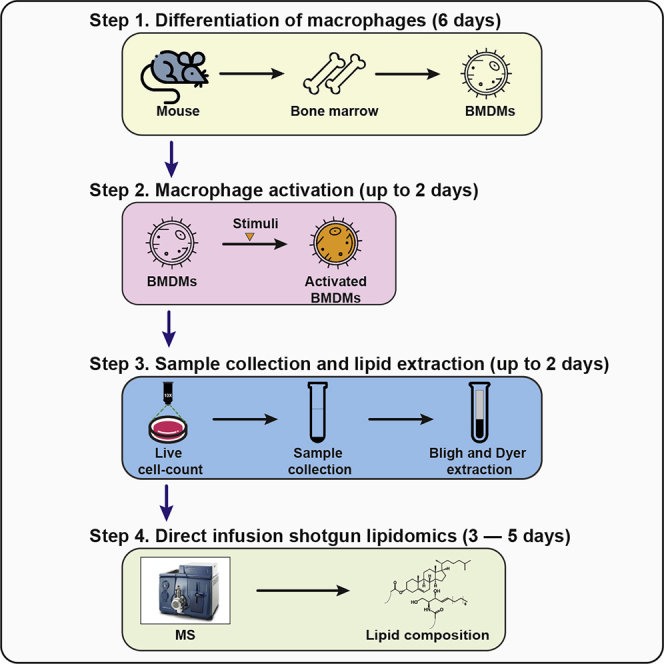
Graphical Abstract
Highlights
-
•
Protocol for profiling mouse macrophage lipidome with direct infusion mass spectrometry
-
•
Provides quantitative measurements of immune cell lipid composition
-
•
Includes cell culture, cell imaging, sample preparation, and data output analysis
-
•
Can be adapted for different lipidomics-mass spectrometry platforms
Immune cells, such as macrophages, reprogram their lipid metabolism in response to the activation of pattern recognition receptors (e.g., TLRs, NLRs) and cytokine receptors (e.g., interferons, interleukins). Profiling these changes can be achieved with shotgun mass spectrometry. This protocol provides step-by-step instructions on the generation and stimulation of bone marrow-derived macrophages (BMDMs), sample collection, and lipid extraction for profiling the macrophage lipidome.
Before you begin
A lipidomics experiment with BMDMs takes approximately two weeks to complete. As such, preparation of all the necessary reagents and equipment is important for success. A full list of reagents is provided in the Key resources table, with every reagent (including the plasticwares and the serum) tested experimentally to maximize quality and reproducibility (see Figure S1 in Hsieh et al., 2020).
Macrophages are highly sensitive to the presence of lipopolysaccharide (LPS), a major source of contamination. Unwanted activation of naïve macrophage during differentiation will negatively impact cell differentiation, morphology, and lipid composition. It is highly recommended to clean and disinfect Class II biological safety cabinets every time prior to a macrophage lipidomics experiment. Likewise, CO2 cell culture incubator should be cleaned and disinfected every four months to reduce the likelihood of cross-contamination.
Contamination of reagents (e.g., fetal bovine serum (FBS) and M-CSF containing conditioned media) by mycoplasma or endotoxin will negatively impact macrophage experiments. As such, routine testing of reagents is recommended.
Reagent quality (e.g., solvents, water) is important to reduce the chance of contamination from external fatty acid sources. The use of pre-made solutions is recommended.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Phosphate buffered saline (PBS) | Cytiva | Cat. SH30256.01 |
| 40 μm cell strainer | Fisher Scientific | Cat. 08-771-1 |
| Red blood cell lysing buffer | Sigma-Aldrich | Cat. R7757 |
| DMEM, high glucose | Fisher Scientific | Cat. 11-965-118 |
| Sodium pyruvate | Thermo Fisher | Cat. 11360-070 |
| L-Glutamine | Thermo Fisher | Cat. 25030081 |
| Penicillin-streptomycin | Thermo Fisher | Cat. 15140122 |
| Hyclone characterized FBS | Cytiva | Cat. SH30071.03 |
| Recombinant M-CSF generated from CMG14-12 culture supernatant | N/A | N/A |
| Recombinant mouse M-CSF | PeproTech | Cat. 315-02 |
| 0.22 μm sterile vacuum filtration cup | Millipore-Sigma | Cat. S2GPU05RE |
| Cell lifter | Fisher Scientific | Cat. 07-200-364 |
| 6-well polystyrene microplates | Fisher Scientific | Cat. 08-772-1 |
| Calcein-AM | Santa Cruz Biotechnology | Cat. SC-203865 |
| 10 mL Kimble conical-bottom glass centrifuge tubes | Fisher Scientific | Cat. 05-569-2 |
| Kimble PTFE black phenolic screw-thread closures | Fisher Scientific | Cat. 05-569-5 |
| Water (Optima grade) | Fisher Scientific | Cat. W6-4 |
| Methanol (Optima grade) | Fisher Scientific | Cat. A454-4 |
| Dichloromethane (HPLC grade) | Fisher Scientific | Cat. AC610050040 |
| Ammonium Acetate (Optima grade) | Fisher Scientific | Cat. A11450 |
| Chromacol 9 mm screw-thread vial 300 μL, fused insert | Thermo Scientific | Cat. 03-FISV |
| 9 mm autosampler vial screw-thread caps | Thermo Scientific | Cat. 9-SCKB-ST1 |
| Internal Standards Kit for Lipidyzer Platform | Sciex | Cat. 5040156 |
| SPLASH LIPIDOMIX mass spec standard | Avanti Polar Lipids | Cat. 330707 |
| Ultimate SPLASH ONE LIPIDOMIX standards | Avanti Polar Lipids | N/A |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
Materials and equipment
BMDM differentiation medium (4°C storage, 37°C at use)
| Reagent | Final concentration (mM or μM) | Stock concentration | Example volume (mL) |
|---|---|---|---|
| DMEM | N/A | N/A | 412.5 |
| Fetal bovine serum (FBS) | 10% v/v | N/A | 50 |
| L-Glutamine | 2 mM | 200 mM | 5 |
| Sodium pyruvate | 0.5 mM | 100 mM | 2.5 |
| Penicillin-streptomycin | 50,000 U penicillin, 50,000 μg streptomycin | 10,000 units/mL of penicillin and 10,000 μg/mL | 5 |
| CMG conditioned media or recombinant M-CSF | 5% v/v or (50 ng/mL recombinant M-CSF) | N/A | 25 |
| Total | n/a | n/a | 500 |
BMDM experimental medium (4°C storage, 37°C at use)
| Reagent | Final concentration (mM or μM) | Stock concentration | Example volume (mL) |
|---|---|---|---|
| DMEM | N/A | N/A | 437.5 |
| Fetal bovine serum (FBS) | 10% v/v | N/A | 25 |
| L-Glutamine | 2 mM | 200 mM | 5 |
| Sodium pyruvate | 0.5 mM | 100 mM | 2.5 |
| Penicillin-streptomycin | 50,000 U penicillin, 50,000 μg streptomycin | 10,000 units/mL of penicillin and 10,000 μg/mL | 5 |
| CMG conditioned media or recombinant M-CSF | 5% v/v or (50 ng/mL recombinant M-CSF) | N/A | 25 |
| Total | n/a | n/a | 500 |
1× running solution specific for Sciex Lipidyzer platform (20°C storage)
| Reagent | Final concentration (mM or μM) | Volume (mL) |
|---|---|---|
| 1 M ammonium acetate dissolved in methanol | 10 mM | 5 |
| Methanol | 50% v/v | 245 |
| Dichloromethane | 50% v/v | 250 |
| Total | n/a | 500 |
All other reagents in this protocol are listed in the Key resources table.
Equipment
Imaging device for cell count
In this protocol, live cell count was assessed using Calcein-AM staining and imaged with a ImageXpress XL instrument (Molecular Devices). Calcein-AM staining for live cell count is compatible with other imaging systems, including Operetta imaging system (PerkinElmer) (tested by the author) and IncuCyte Live Cell Imaging (Sartorius).
Lipidyzer
The Sciex Lipidyzer platform consists of a QTRAP 5500 with SelexION Ion Mobility Technology and a Shimadzu Nexera X2 system consisting of a controller (CMB-20A), two HPLC pumps (LC-30AD), autosampler (SIL-30AC), and column oven (CTO-20A). The pumps are configured to run an isocratic running solution (1× running solution). The autosampler is configured for a 50 μL infusion. This system is control by Analyst software (version 1.6.3) and the Lipidyzer Workflow Manager (version 1.0). This software supervises the running of methods to tune the SelexIon device, test system functioning, acquire data from samples in two successive direction infusion methods, and analyze the resulting data. The Lipidyzer measures 1,070 lipid species from 13 lipid classes.
CRITICAL: Dichloromethane is harmful if swallowed and/or inhaled, and can cause eye, skin, and respiratory tract irritation. This substance has caused adverse reproductive and fetal effects in animals. This chemical must be handled a chemical fume hood. As stated in the MSDS, methylene chloride is metabolically converted to carbon monoxide after systemic absorption, which yields increased concentrations of carboxyhemoglobin in the blood. As stated in the MSDS, methanol is poisonous and flammable. Ingestion may be fatal or cause blindness. It causes eye, skin, and respiratory tract irritation. May cause central nervous system depression. Cannot be made non-poisonous.
Alternatives: Murine M-CSF containing conditioned media can be generated by collecting the spent medium after culture of CMG14-12 or L929. Recombinant M-CSF can also be used to induce macrophage differentiation. The Sciex Lipidyzer Platform Mixture (containing undiluted internal standards) is designed to work exclusively with the Lipidyzer Internal Standard. The Lipidyzer Internal Standard Mixture is a master mix comprised of 13 lipid classes and includes labeled molecular species for each class. When using other direct infusion mass spec platform, user should select appropriate internal standards to that platform and to the quality of quantification requirement. Two potential alternative standard mixture are SPLASH LIPIDOMIX Mass Spec Standard and UltimateSPLASH ONE LIPIDOMIX Standards from Avanti Polar Lipids. Both alternatives are listed in the Key resources table.
Step-by-step method details
Differentiating bone marrow-derived macrophages (BMDMs)
Timing: 1–1.5 h for bone marrow preparation, 6 days for differentiation
This section covers the differentiation of bone marrow-derived macrophages (BMDMs).
-
1.
Set up a clean workbench with an isoflurane chamber, dissection tray (a piece of polystyrene foam covered with aluminum foil), and two beakers containing water and 70% ethanol (Figure 1A). Clean all the tools with 70% ethanol (EtOH). Place a 50 mL centrifuge tube on a rack and filled with 30 mL of ice-cold PBS for bone collection.
-
2.
Sacrifice mice in prepared isoflurane containing chamber (5% isoflurane concentration) with lid covered for about 5 min. An appropriate alternative method is euthanasia by CO2 asphyxiation. In both cases, cervical dislocation may be used as a secondary means to assure the death of the animal. Assess the death of the animal by and lack of lack of heartbeat and by pedal reflex (firm toe pinch). Sacrificing and assessing the death of the animal should follow ethical guidelines as published by individual institution. Spray the deceased animal with 70% EtOH and place in dissection tray. Secure limbs to tray with 25G x 5/8 inch needles (Figure 1B).
-
3.
The skin, fur, and muscle are removed first to facilitate the bone collection. Isolate the femur and tibia using a pair of tweezers and scissor. Use tweezers to grasp bone, and if necessary, cut away excess tissue from around the ends of the bone. Avoid cutting or breaking the femurs. Cut down each leg along the separated skin with the foot still attached to the leg (Figure 1C). Remove the foot and place the leg in the PBS-filled centrifuge tube (Figure 1D).
-
4.
Inside the tissue culture hood, fill a 10 mL syringe with 10 mL of ice-cold PBS and attach a 26G x 5/8 inch needle. One syringe per leg is recommended for maximum bone marrow recovery (Figure 1E). Place the legs on a sterile 10 cm tissue culture dish. It will be necessary to cut the distal end of the tibia in order to flush the marrow.
-
5.
Place the isolated femur and tibia on a separate 10 cm tissue culture dish (Figure 1F).
-
6.
Place a 40 μM cell strainer on a 50 mL centrifuge tube. Pierce bone at the ends with syringe and flush bone with PBS into the cell strainer (Figure 1G). Bones should be flushed until they become a translucent white color. Each bone should require between 3 – 5 mL of PBS.
-
7.
Rinse the cell strainer with 5 mL of ice-cold PBS for maximum bone marrow recovery.
-
8.
Spin down the bone marrow at 365 × g for 7 min at room temperature (RT, 20°C) and aspirate off the PBS (Figure 1H).
-
9.
Resuspend the bone marrow in 3 mL of red blood cell (RBC) lysis buffer (see Key resources table) and place at RT for 3 min. Neutralize the RBC lysis buffer by adding 7 mL of ice-cold PBS.
-
10.
Spin down the bone marrow at 365 × g for 5 min at RT.
-
11.
Aspirate off the PBS and resuspend the bone marrow in 20 mL of BMDM medium for cell counting. Use a hematocytometer or an automated cell counter to determine the number of live macrophages. Cells can be stained with trypan blue for cell counting with a hematocytometer. Live cell count can be assessed with an automated cell counter using the acridine orange (AO) and propidium iodide (PI) dyes.
-
12.
To prevent macrophages from overgrowing by day 6 of differentiation, seed 16 – 20 million monocyte precursors per 15 cm tissue culture-treated culture dishes for differentiation (Figure 2A). Add 25 mL of macrophage media per dish.
-
13.
On day 4 of macrophage differentiation (Figures 2B and 2C), change the medium by aspirating 20 mL of spent macrophage media and replacing with 20 mL of fresh macrophage media.
Figure 1.

Extraction of bone marrow for macrophage differentiation
Figure 2.

Stages of macrophage differentiation and plating density
Macrophage plating for lipidomics experiment
This section covers plating of BMDMs for lipidomics experiments.
-
14.
On day 6, remove 23 mL of macrophage media and replenish it with 15 mL of fresh media (Figures 2D and 2E).
-
15.
Tilt the plate on an angle without spilling the medium and gently scrape the tissue culture plate with cell lifters.
-
16.
Transfer the macrophages into a 50 mL centrifuge tube. Use a serological pipette to pipette the cell suspension to ensure cell dissociation. Use a hematocytometer or an automated cell counter to determine the number of live macrophages. Macrophages should be relatively similar in sizes.
-
17.
Using a P1000 micropipette, pipette 9 × 105 BMDMs into each well of a 6 well plate. Top-up the media to a total of 2 mL per well. Disperse the cell equally by gently rocking the plate back and forth.
-
18.
Place the plates on a flat surface in a 5% CO2 incubator at 37°C.
Activation of BMDM
This section covers plating of BMDMs for lipidomics experiments.
-
19.
48 h post-plating, visually inspect individual wells to ensure all wells have reached 85% to 90% confluency (Figure 2F). Figures 2G and 2H illustrate suboptimal cell density due to improper plating.
-
20.
Warm up the BMDM experimental media 15 min prior to macrophage treatment.
-
21.
Label 50 mL centrifuge tubes. Pipette 20 mL of macrophage experimental media to each tube.
-
22.
Add the stimuli or vehicle controls (e.g., TLR ligands) into the labeled tubes. Vortex the tubes to ensure proper mixing.
-
23.
After aspirating the media, use a P1000 micropipette to transfer 2 mL of the experimental media containing the appropriate stimulus to each well. Gently rock the plate to ensure proper mixing.
-
24.
Place the plates on a flat surface in a 5% CO2 incubator at 37°C.
Imaging for cell count and sample collection
This section details live cell imaging for cell count and the collection of lipidomics samples.
-
25.
Prior to live cell staining for cell counts, visually inspect each well under the microscope. Figures 3A and 3B shows the ideal density for imaging and collection. Figures 3C–3F show suboptimal confluency for lipidomics sample collection due to initial plating issues.
-
26.
To image the plate for live cell count, stain each well with 1.25 mM Calcein-AM dissolved in DMEM (final centration 1.45 μM). Calcein staining allows the identification of live cells within a well. The plates are then imaged on a Molecular Devices ImageXpress XL instrument. 20 high magnification fluorescence images were captured for sections of each well (covering 24% of total well surface area of a 6 well) using a 10× objective (Nikon Plan Fluor, 0.3 NA). Total live cell number was assessed with MetaXpress Software with Powercore using the Multi-wavelength cell scoring module. (see Materials and equipment for alternative imaging devices compatible with Calcein-AM staining).
-
27.
Prior to sample collection, pre-chill all the glass tubes, PBS and collection tube rack (Figure 4A). Samples can be collected directly into Kimble tubes (direct storage) or glass tubes (stored in the aluminum tube rack). Two wells of cells were combined as a single replicate (Figure 2B). After removal of culturing media, 1 mL of ice-cold PBS was added into each well and cells were scraped with cell lifters and spun down in glass tubes or 10 mL Kimble tubes (Figures 4B and 4C). Collected samples are spun at 365 × g for 10 min at 4°C (Figure 4D).
-
28.
Aspirate the PBS and the tubes can be snap frozen for −80°C storage (up to 6 months as tested) or proceed directly for lipid extraction (Figure 4E).
Figure 3.

Effects of plating density on cell count
Figure 4.

Lipidomics sample collection
Lipid extraction
This section of the protocol covers Bligh and Dyer lipid extraction for subsequent lipidomics analysis using the Sciex Lipidyzer Mass Spectrometry Platform (Bligh and Dyer, 1959). The protocol is a modified version of the Serum Extraction Protocol published in the Lipidyzer Platform user guide (Sciex, 2016). For internal standard preparation, see Chemical preparations of the Lipidyzer Platform user guide (Sciex, 2016) and (Ubhi, 2018).
-
29.
Warm frozen cell pellet for 30 min to ambient temperature by placing them on a clean bench top (Figure 5A).
-
30.
To each Kimble tube contain the sample add, 0.9 mL of water, 2 mL of methanol, and 0.9 mL of dichloromethane in that order.
-
31.
Add 0.1 mL of the Lipidyzer Internal Standard Mixture to the extracts. Addition of the internal standard mixture helps correct for variation in the extraction efficiency (Figure 5B). See Materials and equipment for alternative internal standards.
-
32.
Invert the 10 mL Kimble tubes 4 to 8 times.
-
33.
The extracts are then set on the benchtop at room temperature for 30 min (Figure 5C).
-
34.
An additional 1 mL of water and 0.9 mL of dichloromethane are added to the extracts.
-
35.
The extracts are inverted 4 to 8 times and then centrifuged at 1,200 × g for 10 min at room temperature to separate the extract into a bilayer (Figure 5D).
-
36.
The bottom organic layer is transferred using a glass Pasteur pipette to a new 10 mL Kimble tube for each extract.
-
37.
To increase the amount of lipids that can be extracted, add another 1.8 mL of dichloromethane to the original extract Kimble tubes.
-
38.
The original extracts are inverted 4 to 8 times and centrifuged at 1,200 × g for 10 min at room temperature to separate the extract into a bilayer.
-
39.
The bottom layers are taken again and added to the previous aliquots in step 35.
-
40.
Kimble tubes containing organic bottom layers are capped and gently vortexed for 3 to 5 s. This will help separate any residual unwanted material (from the upper layers) from desired bottom layer by trapping material onto upper glass tube walls.
-
41.
The combined bottom layers for each sample are dried using an EZ-2 Elite evaporator (low boiling point, heat off, room temperature, fixed speed) and reconstituted in 0.25 mL of the lipidomics running solution (dichloromethane [50]:methanol [50] with 10 mM ammonium acetate). Alternatively, samples can be dried using a nitrogen evaporator.
-
42.
Briefly vortex the Kimble tubes. Gently pipette the mixture up and down using a glass Pasteur pipette to wash off samples stuck onto the tube.
-
43.
Use glass Pasteur pipettes to transfer samples to 1 mL Robovials with 100 μL fixed glass insert.
-
44.
Centrifuge the Robovials in 931 × g for 10 min at 10°C before loading into autosampler for Lipidyzer mass spectrometry analysis.
Figure 5.

Bligh and Dyer lipid extraction
Expected outcomes
This method can be used to examine how different immune stimuli (e.g., TLR agonists or cytokines) reprogram macrophage lipid metabolism. Downstream bioinformatic analysis — using for instance, heatmaps and PCA analysis (Hsieh et al., 2020) — will allow the user to identify lipid species of interests for further investigation.
The Lipidyzer platform was originally designed to measure composition of complex lipids using 100 μL of plasma or serum (Sciex, 2016; Ubhi, 2018). The results displayed by the Lipidyzer has an embedded 10× multiplier to the lipid content measured (e.g., conversion of nmoles of lipids per 100 μL sample to nmoles of lipids/mL). To obtain usable data for macrophage and for other cell types, the lipid concentration from the output is first divided by a factor of 10 (e.g., XX nmoles becomes XX/10 nmol). This value is subsequently divided by the live cell number and scaled up to 107 cells (e.g., XX nmol / cell count X 107). This calculation must be carried out for all “Concentration” data sets reported by the Lipidyzer.
Figure 6 provides a simplified example of Lipidyzer results generated from a macrophage shotgun lipidomics experiment. There are 6 data sheet tabs in total, with the blue arrow illustrating the relationship between the tabs:
Figure 6.

Macrophage lipidomics results overview generated by the Lipidyzer
Lipid Species Concentration details the nanomolar values of each lipid species measured in this experiment and are normalized to the cell number (i.e., reported as XX nmoles per 107 macrophages). All subsequent tables can be mathematically derived from the Lipid Species Concentration.
Fatty Acid Concentration details the sum of a specific acyl tail (i.e., nmol of 16:0 FA per 107 macrophages) measured within a lipid class. These values are normalized to the cell number and have the same units as Species Normalization.
Lipid Class Concentration details the sum of all lipid species within a lipid class (i.e., nmoles of phosphatidylcholine [PC] per 107 macrophages). These values also have the same units as Species Normalization.
In Species Composition, each lipid species (e.g., CER [14:0]) within a lipid class (e.g., class ceramides) is calculated as a percentage of the total lipid in the class. This calculation is not dependent on normalization to sample size and can be useful when normalization values are uncertain (e.g., due to inaccurate cell count). This also allows comparison between species within the same class.
In Fatty Acid Composition, the sum of lipids within a class that contain a specific fatty acid acyl tail (e.g., DAG [16:0]) and calculated as a percentage of the total lipid in the class (e.g., DAG).
Limitations
This protocol details the profiling of the macrophage lipidome using the direct infusion shotgun mass spectrometry on the Lipidyzer platform. While the sample preparation method could be utilized with other mass spectrometry platforms, this change might require modifications to the amount of sample needed and the running buffers used.
Mass spectrometry (MS)-based “shotgun lipidomics” can provide quantitative and qualitative analysis of the complex lipids in a biological system. The strengths and limitations of this technology have been extensively reviewed (Hsu, 2018). From our experience, the greatest strength of a shotgun lipidomics workflow is highly quantitative measurements facilitated by the simultaneous measurement of analytes and standards during direct infusion. The differential elution of analytes and standards during a conventional LC-MS runs result in poor quantification as ionization efficiency varies due to the solvent gradient and inconsistent ion suppression. The Lipidyzer platform leverages this strength with an extensive standard set for the best possible quantitative measurements. The limitation of this shotgun lipidomics workflow is that low abundance lipid species will not be detected due to high ion suppression in direct infusion. To compensate for this limitation, we use a higher amount of starting material (e.g., a higher cell number); 3 to 4 million of BMDMs per replicated is required to achieve the sensitivity illustrated in (Hsieh et al., 2020). A LC-MS based approach might be more suitable to detect a low abundance lipid species with less starting material.
Lipid extraction methods can also influence detection efficiencies of lipid species. The Bligh and Dyer extraction has been shown to efficiently and consistently extract all major lipid classes, including phospholipids and glycerol lipid classes. However, measurement of low abundant lipid species, such as oxysterols, is incompatible with this a general lipid extraction protocol. This analysis would require a more specific extraction such as neutral lipid extraction (Tsui et al., 2019), or solid phase extraction (Meriwether et al., 2016), in conjunction with a targeted LC-MS analysis.
Troubleshooting
Problem
Low bone marrow yield.
Potential solution
Increase the number of animal usage. In our experience, an animal (C57BL6, male, between 8 and 12 weeks of age) should yield between 40 to 50 million progenitor cells. Minimum of two animals per experiment is recommended.
Problem
Low macrophage yield at day 6 of differentiation.
Potential solution
In our experience, an animal (C57BL6, male, between 8 and 12 weeks of age) should yield between 50 to 80 million BMDMs by day 6 of differentiation. A low yield can either due to environmental contamination as mentioned in the introduction or a low concentration of M-CSF in the growth media. Increasing the percentage volume of macrophage conditioned media (from 5% to 20% in some studies) or switching to recombinant M-CSF would improve the yield.
Problem
Poor cell count or large deviation of cell counts between replicates.
Potential solution
This issue is primarily associated with poor plating of cells. Prior to executing step 16, pipette the cell suspension 5 to 10 times to ensure cell dissociate. Use a pipette instead of a pipette-aid when plating will reduce deviation between replicates.
Problem
On step 31, over mixing/vortexing of sample, water, methanol, and dichloromethane will lead to poor phase partitioning, reducing extraction efficiencies (see Figure 5D right tube).
Potential solution
Do not over mix the solution. Reduce the amount of bilayer extracted (step 35) to increase sample purity.
Problem
On step 35, formation of thick aggregates (a cake layer) due to too much starting material, resulted in poor phase separations and extract purity (see Figure 5D middle tube).
Potential solution
This issue is primarily associated with extraction of lipids from tissues or a large quantity of serum, rather than from cells. Reducing the amount of organic layer extracted in step 35 will improve sample purity. Increase the amount of dichloromethane in step 36 will allow a larger volume of organic layer to be extracted.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Steven J. Bensinger (SBensinger@mednet.ucla.edu).
Materials availability
No new mouse lines are generated in this study.
Data and code availability
See (Hsieh et al., 2020) for data and code availability.
Acknowledgments
S.J.B. was supported by NIH HL146358. We thank the UCLA Lipidomics Lab for providing services technical support and project consultations.
Author contributions
W.Y.H. conceptualized, designed/implemented experiments, analyzed data, and constructed the protocol. K.J.W. and B.L.S. provided lipidomics and computational analysis. S.J.B. provided resources and supervision, and contributed to conceptualization, design of experiments, analysis of data, and construction of the protocol.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Wei-Yuan Hsieh, Email: waynehsieh@ucla.edu.
Steven J. Bensinger, Email: sbensinger@mednet.ucla.edu.
References
- Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Hsieh W.Y., Zhou Q.D., York A.G., Williams K.J., Scumpia P.O., Kronenberger E.B., Hoi X.P., Su B., Chi X., Bui V.L. Toll-like receptors induce signal-specific reprogramming of the macrophage lipidome. Cell Metab. 2020;32:128–143.e5. doi: 10.1016/j.cmet.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F.-F. ‘Mass spectrometry-based shotgun lipidomics – a critical review from the technical point of view’. Anal. Bioanal. Chem. 2018;410:6387–6409. doi: 10.1007/s00216-018-1252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriwether D., Sulaiman D., Wagner A., Grijalva V., Kaji I., Williams K.J., Yu L., Fogelman S., Volpe C., Bensinger S.J. Transintestinal transport of the anti-inflammatory drug 4F and the modulation of transintestinal cholesterol efflux. J. Lipid Res. 2016;57:1175–1193. doi: 10.1194/jlr.M067025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciex . Sciex; 2016. LipidyzerTM Platform. [Google Scholar]
- Tsui H.S., Pham N.V.B., Amer B.R., Bradley M.C., Gosschalk J.E., Gallagher-Jones M., Ibarra H., Clubb R.T., Blaby-Haas C.E., Clarke C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019;60:1293–1310. doi: 10.1194/jlr.M093534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi B.K. Direct infusion-tandem mass spectrometry (DI-MS/MS) analysis of complex lipids in human plasma and serum using the lipidyzerTM platform. Methods Mol. Biol. 2018;1730:227–236. doi: 10.1007/978-1-4939-7592-1_15. [DOI] [PubMed] [Google Scholar]
- Yao C.-H., Liu G.Y., Yang K., Gross R.W., Patti G.J. Inaccurate quantitation of palmitate in metabolomics and isotope tracer studies due to plastics. Metabolomics. 2016;12:143. doi: 10.1007/s11306-016-1081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
See (Hsieh et al., 2020) for data and code availability.